

Abstract
Germination protease (GPR) plays an important role in the germination of spores of Bacillus and Clostridium species. A few very similar GPRs form a singleton group without significant sequence similarities to any other proteins. Their active site locations and catalytic mechanisms are unclear, despite the recent 3-D structure determination of Bacillus megaterium GPR. Using structural comparison and sequence analysis, we show that GPR is homologous to bacterial hydrogenase maturation protease (HybD). HybD's activity relies on the recognition and binding of metal ions in Ni–Fe hydrogenase, its substrate. Two highly conserved motifs are shared among GPRs, hydrogenase maturation proteases, and another group of hypothetical proteins. Conservation of two acidic residues in all these homologs indicates that metal binding is important for their function. Our analysis helps localize the active site of GPRs and provides insight into the catalytic mechanisms of a superfamily of putative metal-regulated proteases.
Keywords: Metalloprotease, homology, structural comparison, sequence motif, catalytic mechanism
Genome sequencing projects result in vast amounts of sequence data. The enlarged sequence space greatly facilitates structural–functional annotations by similarity searches. Profile methods (Gribskov et al. 1987), such as PSI-BLAST (Altschul et al. 1997) and HMMER (Eddy 1996), make effective use of evolutionary information and can often result in very sensitive homology detection. However, many remote homology relationships still cannot be detected easily (Murzin 1998). For example, evolutionary information seems to be lost for the so-called singleton sequence groups that contain only one sequence or a few very close homologs. 10%–40% of the sequences in a genome are estimated to be singletons (Cort et al. 2000). Many of them may actually turn out to be distant homologs of well-characterized protein families. Accumulation of structural information can help functional inference (Koppensteiner et al. 2000); structural genomics projects attempt to shed light on the functions of uncharacterized proteins in this manner (Kim 1998; Sali 1998; Terwilliger et al. 1998; Zarembinski et al. 1998; Eisenstein et al. 2000). Structural comparison, in many cases, can reveal distant homology that is beyond detection by automatic similarity searches (Murzin 1998,1999). Distinct structural features, usually important for function, are manifested in sequence motifs that might be too short or too divergent to be detected by sequence analysis alone. Effective integration of evolutionary and structural information can result in improved homology detection and provide functional insight (Aravind and Koonin 1999). We report such an example by tracing the evolutionary history and locating the active site of germination protease.
Certain gram-positive bacteria develop spores under severe physical or chemical conditions. In spores of Bacillus and Clostridium species, the DNA is protected by a group of small acid-soluble proteins (SASP; Setlow 1988,1995). Spore germination requires degradation of these proteins through the action of a specific germination protease (GPR; Setlow 1988). GPRs from a few Bacillus or Clostridium species form a typical singleton group of very similar homologs, showing no detectable sequence similarity to other protease families. The crystal structure has been determined for GPR from Bacillus megaterium (Ponnuraj et al. 2000). However, little information has been derived regarding the enzyme's active site location and catalytic mechanism (Ponnuraj et al. 2000).
By structural comparison and sequence analysis, we show that GPR is homologous to the bacterial Ni–Fe hydrogenase processing protease HybD, an enzyme with a known structure (Fritsche et al. 1999). GPR is likely to be regulated by metal ions and its active site location is predicted to be similar to that of HybD.
Results and Discussion
Structural comparison results from FSSP (Fold classification based on Structure–Structure alignment of Proteins) classification and the Dali server (Holm and Sander 1995; Dietmann et al. 2001) reveal readily that B. megaterium GPR (Protein Data Bank (PDB) entry 1c8b, chain A) shows significant structural similarities to several proteins. Among the top hits are hydrogenase maturation protease (PDB entry 1cfz, chain A; Z-score 8.2; Fritsche et al. 1999), peptidyl-tRNA hydrolase (PDB entry 2pth; Z-score 7.9; Schmitt et al. 1998), and 5`-deoxy-5`-methylthioadenosine phosphorylase (PDB entry 1cb0, chain A; Z-score 6.2; Appleby et al. 1999). All have the phosphorylase/hydrolase-like fold, according to the Structural Classification of Proteins (SCOP; Murzin et al. 1995). The GPR structure does include the core structural elements with the same topology as the phosphorylase/hydrolase-like fold: it has 3 layers αβα; the central beta sheet consists of 5 strands in the order of baced, and strand d is antiparallel to the rest of the four strands (Fig. 1a,b ▶). These results are in contrast to the claim that GPR has a novel fold (Ponnuraj et al. 2000). GPR has a long N-terminal extension to the core structure, adding three antiparallel strands and several peripheral helices (Fig. 1a ▶). Hydrogenase maturation protease HybD (Fritsche et al. 1999; Fig. 1b ▶) from Escherichia coli has the highest Z-score (8.2), shares 20% sequence identity, and has a RMSD of 2.6 Å over 123 residues with GPR according to Dali. The superposition of the two structures over the core structural elements is shown in Figure 1c ▶.
Fig. 1.
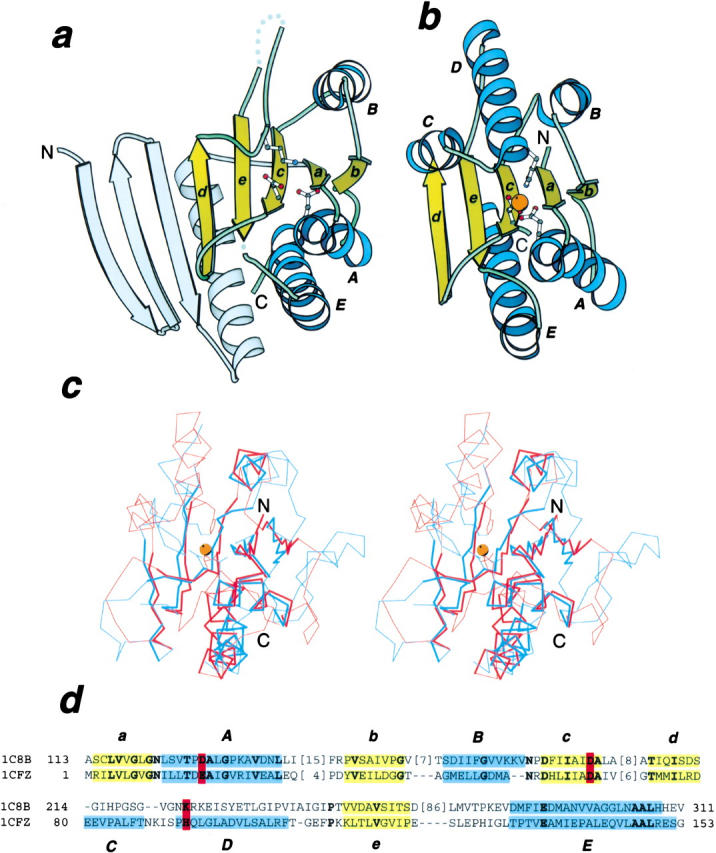
Ribbon diagram and structure superposition of GPR and HybD. (a) Ribbon diagram of B. megaterium GPR, PDB entry 1c8b, chain A, residues 143 to 460. (b) Ribbon diagram of E. coli hydrogenase maturation protease HybD, pdb entry 1cfz, chain A, residues 2 to 154. Secondary structure elements forming the phosphorylase/hydrolase fold are labeled. Side-chains of the metal binding residues in HybD and their structural counterparts in GPR are shown in ball-and-stick representation. The metal ion in HybD is shown as an orange ball. (c) Stereo pair of superimposed Cα traces of GPR (blue) and HybD (red). The side-chain of the (putative) metal ligands and the metal are also shown. Unconserved regions not used in RMSD minimization are shown as thin lines. (d) Structure-based sequence alignment of GPR and HybD. Coloring and labeling are as in (a) and (b). The (putative) metal ligands are in bold letters and are shaded in red. Positions with identical residues are in bold letters. All structure diagrams are drawn with Bobscript (Esnouf 1997).
Multiple alignments of the GPRs (Fig. 2 ▶, top group) and hydrogenase-processing proteases (Fig. 2 ▶, bottom group) were made and merged in accordance with the structural superposition. Despite the low overall sequence similarity between GPR and HybD, sequence and structural analysis indicates strongly that they are homologous. The most prominent features are two highly conserved motifs (Fig. 2 ▶). Each motif contains an important acidic residue that acts as a metal (cadmium) ligand in HybD (Glu 16 and Asp 62; Fritsche et al. 1999). It has been proposed that metals are important for the recognition and binding of the substrates to HybD (Fritsche et al. 1999; Theodoratou et al. 2000a). The first motif has a consensus sequence of shGNhhhsX[DE]shs (s: mainly small residues, in red bold letters in Figure 2 ▶; h: mainly hydrophobic residues, shaded in yellow). In the structures, this motif corresponds to the loop connection between strand a and helix A. The loop conformations are rather similar in the two structures. The sequence Nhhh forms a type I turn (Crawford et al. 1973) with the side-chain of the conserved Asn making a hydrogen bond to the backbone nitrogen two residues after it. Helix A in both GPR and HybD structures starts with two turns of 310 helix. Strand a and helix A form an angle of ∼30° with each other. The second motif is located at the end of strand c, with an invariant Asp and a small residue following it (Fig. 2 ▶). Several positions in the two motifs are occupied mainly by small residues. The Gly immediately before the conserved Asn in the first motif is invariant, showing similar main-chain conformations with phi and psi angles of 138° and 163° in GPR and 138° and 188° in HybD. The main reason for the conservation of the other small residues is the steric hindrance caused by the unique tight packing of the N-terminus of helix A to strands a and c. Another possible reason is to make room for metal or substrate binding, as proposed by Frische et al. (1999).
Fig. 2.
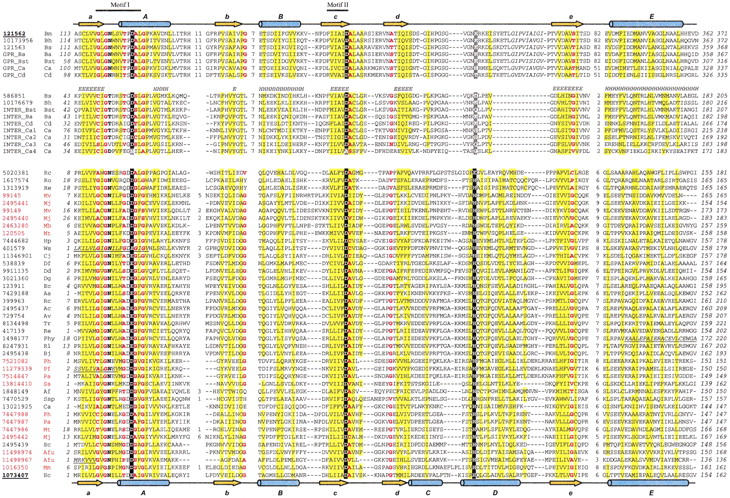
Multiple sequence alignment of the GPR/hydrogenase maturation protease superfamily. Groups of sequences are separated by empty lines. GPRs and hydrogenase maturation proteases are the top and the bottom groups, respectively, with the intermediate sequences in between. Each sequence is identified by the NCBI gene identification number (gi), except for those obtained from the ERGO database or NCBI unfinished genomes DNA databases, which use the group name abbreviations followed by the species abbreviations. Sequence identifiers are followed by abbreviations of the species names. The gi numbers of the sequences with known structures are underlined and in bold. The archaeal identifiers are marked with red. The first and the last residue numbers of the sequences are indicated, and the total length of the proteins is shown at the end (italicized numbers). Residues in unconserved loop regions are not displayed, with the number of omitted residues shown instead. The metal-binding residues are shown in white bold letters on black background. The putative third metal ligand in the GPRs and intermediate sequences is shaded in gray. Uncharged residues at mainly hydrophobic positions are shaded yellow. Conserved small residues (G, A, S, C, T, P, V, D, and N) are in red bold letters. Sequences with apparently incorrect starting or ending positions are corrected, with the added N-terminal fragments in underlined italicized letters. One loop that is not modeled in the structure of GPR is shown in italicized letters. Diagrams of the secondary structural elements are shown at the top (GPR: 1c8b, chain A) and bottom (HybD: 1cfz, chain A). β-strands and α-helices are shown as yellow arrows and blue cylinders, respectively. The labeling associated with the secondary structure elements corresponds to Figure 1 ▶. Predicted secondary structure by Jpred2 is shown for the intermediate sequences (E indicates a strand; H indicates a helix). The species name abbreviations are: Aa, Aquifex aeolicus; Ac, Azotobacter chroococcum; Af, Acetomicrobium flavidum; Afu, Archaeoglobus fulgidus; Av, Azotobacter vinelandii; Ba, Bacillus anthracis; Bh, Bacillus halodurans; Bj, Bradyrhizobium japonicum; Bm, Bacillus megaterium; Bs, Bacillus subtilis; Bst, Bacillus stearothermophilus; Ca, Clostridium acetobutilycum; Cd, Clostridium difficile; Cj, Campylobacter jejuni; Dd Desutobacterium dehalogenans; Df, Desulfovibrio fructosovorans; Dg, Desulfovibrio gigas; Ec, Escherichia coli; Hp, Helicobacter pylori; Mb, Methanosarcina barkeri; Mj, Methanococcus jannaschii; Mm, Methanosarcina mazei; Mt, Methobacterium thermoautotrophicum; Mv, Methanococcus voltae; Pa, Pyrococcus abyssi; Pf, Pyrococcus furiosus; Ph, Pyrococcus horikoshii; Phy, Pseudomonas hydrogenovora; Rc, Rhodobacter capsulatus; Re, Ralstonia eutropha; Rl, Rhizobium leguminosarum; Ro, Rhodococcus opacus; Ss, Sulfolobus solfataricus; Ssp, Synechocystis sp.; Tr, Thiocapsa roseopersicina; Ws, Wolinella succinogenes. The alignment was generated using T-Coffee (Notredame et al. 2000), followed by manual adjustment.
In HybD, His 93 at the N-terminus of helix D (between strands d and e; Fig. 1 ▶) serves as the third ligand to the metal. It is highly conserved within the group of hydrogenase maturation proteases (Fig. 2 ▶). Main-chain conformations differ drastically in the connection between strands d and e in HybD and GPR (Fig. 1a,b ▶). In HybD, there are two helices (C and D), whereas no corresponding regular secondary structure elements are present in GPR. The side-chain amine of Lys 324 in GPR superimposes well with the liganded nitrogen of His 93 (Fig. 1c ▶), indicating that it might function similarly. However, this lysine is conserved only in the Bacillus species (Fig. 2 ▶). Thus, the third ligand might be less important for GPRs. Alternatively, main-chain oxygen atoms in the loop region could serve as the ligands, much like those in some calcium-binding proteins (Scott et al. 1990; Burger et al. 1994).
Despite the structural similarity and the conservation of two functional motifs in GPR and HybD, PSI-BLAST searches were not able to identify their similarities at significant levels. Interestingly, during similarity searches, we identified another distant group homologous to GPRs (Fig. 2 ▶, middle group). For example, PSI-BLAST searches starting from C. acetabutylicum GPR identified a hypothetical protein (gi586851) at e-value 0.81, with both aforementioned motifs conserved. In the non-redundant database (June 2001), there are only two hypothetical proteins in this group (gi586851 and gi10176679). We call them "intermediate" sequences. To alleviate the restrictions of small sequence numbers, we also tried to retrieve sequences from the ERGO database (http://igweb.integratedgenomics.com/ERGO/) and the complete and unfinished genome DNA databases at NCBI (http://www.ncbi.nlm.nih.gov/Microb_blast/unfinishedgenome.html). A total of seven GPRs and nine intermediate sequences from different Bacillus and Clostridium species were found (Fig. 2 ▶). Additional sequences improved the profile and helped homology detection. For example, we used the profile generated from the alignment of the nine intermediate sequences for PSI-BLAST searches and detected GPR sequences at more significant e-value levels (<0.1). The succession of the predicted secondary structure elements in the intermediate group is consistent with the two known structures (Figure 2 ▶). All the intermediate sequences are also from the Bacillus or Clostridium species, apparently forming another singleton group. It is highly likely that they are proteases that may also participate in some process unique for Bacillus and Clostridium species, such as sporulation. Indeed, there is evidence of a second SASP-specific protease in B. megaterium besides GPR (Postemsky et al. 1978; Setlow 1988). Whereas each Bacillus species has only one intermediate sequence, Clostridium acetabutylicum has four, one of which (INTER_Ca4; Fig. 2 ▶) is predicted to have no activity because both the key acidic residues are mutated. Like the GPRs, intermediate sequences do not have a conserved residue corresponding to the third metal ligand of HybD.
High sequence divergence makes it difficult to build a reliable evolutionary tree for all these homologous proteins. Instead, we used a distance diagram (V.N. Grishin and N.V. Grishin, unpubl.; see Materials and Methods for details) to illustrate the similarities among them. The distance diagram shown in Figure 3 ▶ is a two-dimensional projection of the multi-dimensional Euclidian space, with the points representing individual sequences. The distance between two points reflects the dissimilarity of the two sequences. Three well-separated groups of points are obvious in the diagram, corresponding to the three distantly related sequence groups. The group of hydrogenase maturation proteases has many representatives. The GPRs form a typical singleton group with the seven sequences being very close to each other, whereas the group of intermediate sequences is more dispersed (the predicted inactive protein from C. acetabutylicum shows the largest deviation from other sequences).
Fig. 3.

Distance diagram of GPRs (triangles), hydrogenase maturation proteases (rectangles), and the intermediate sequences (circles). Coordinates 1 and 2 are the dimensions of maximal scatter of points in a multidimensional Euclidean space. The points represent all the sequences in Figure 2 ▶.
Functional studies indicate that metal ions (nickel) play an important role in the catalytic activities of hydrogenase maturation proteases (Menon and Robson 1994; Rossmann et al. 1994, Maier and Bock 1996). Nickel needs to be present in the substrate (hydrogenase precursor) to allow proteolysis to occur (Rossmann et al. 1994). There is evidence that nickel in the hydrogenase precursor serves as a recognition and binding motif for the protease (Theodoratou et al. 2000a). The hydrogenases are cleaved after a histidine or an arginine in the C-terminal consensus sequence DPCXXCXXH(R)h (h is usually a hydrophobic residue) (Theodoratou et al. 2000b). The two cysteines are ligands for the metals (nickel and iron) in the mature form of hydrogenases (Volbeda et al. 1995). The protease might recognize and bind to the substrate through interaction with the metals, as seen in the structure (Fritsche et al. 1999; Theodoratou et al. 2000a). The precise catalytic mechanism is still unclear. However, metals could also be involved in the catalytic processes. Typically, metalloproteases use a general base (in most cases a conserved acidic residue) to activate a metal-bound water molecule for its nucleophilic attack on the scissile peptide bond (Christianson et al. 1989; Bode et al. 1992, Makarova and Grishin 1999). The metal can help polarize the water to lower its pKa (Christianson et al. 1987). We did not identify any other conserved acidic residues in these proteases except for the two metal ligands. However, such a general base could be provided by the substrates or come from solution, as exemplified by the bicarbonate ion in the structure of dizinc leucine aminopeptidase (Strater et al. 1999). Alternatively, one of the conserved cysteines at the cleavage site could attack the scissile bond directly. The use of metal-activated groups in the substrates for catalysis is not unusual, as seen in farnesyl transferases (Long et al. 2000) and geranylgeranyl transferases (Zhang et al. 2000).
Although there is no direct evidence that metals function in the catalysis of GPRs, the homology to hydrogenase maturation proteases and the abundance of divalent metal ions in the spores (Ponnuraj et al. 2000; Setlow 1994) indicate strongly that they do. The GPR substrates are the SASPs that are classified into two types, α/β and γ (Setlow 1988). Analysis of their cleavage sites revealed a short motif with two conserved acidic residues, [ED]hXXE. Cleavage was found to occur after the first acidic residue. The acidic residues could act as metal ligands in substrate recognition or as the general base in catalysis. There seems to be no similarity around the cleavage sites of the Ni–Fe hydrogenases and the substrates of GPRs, except that in both cases the position immediately after the cleavage site is usually occupied by a hydrophobic residue. This residue was shown to be important for the processing of the E. coli hydrogenase (HycE; Theodoratou et al. 2000b). Difference of cleavage site signatures implies that the exact mechanisms for GPRs and hydrogenase maturation proteases could be different. The exact roles of metals and the key residues involved in the catalysis of GPR still need to be addressed by experimental studies, but this work points to the location of the active site in GPR and residues that should be important for its function.
Materials and methods
Similarity searches
The PSI-BLAST program (Altschul et al. 1997) was used to search for homologs of GPRs and hydrogenase maturation proteases against the NCBI non-redundant database (June 2001; 691,806 sequences; 217,936,898 letters). The e-value threshold for inclusion of sequences into a profile was 0.01. Default parameters were used otherwise. Found homologs were grouped by single-linkage clustering as implemented in the SEALS package (Walker and Koonin 1997); the representative sequences from each group were used as queries for further searches. To retrieve more sequences for the GPR group and the intermediate group, gapped BLAST (Altschul et al. 1997) was used for searching the ERGO database (http://igweb.integratedgenomics.com/ERGO/) and tblastn was used for searching the complete and unfinished genome DNA databases at NCBI (http://www.ncbi.nlm.nih.gov/Microb_blast/unfinishedgenome.html).
Multiple alignments, secondary structure predictions, and generation of sequence profile
Multiple sequence alignments were constructed using the T-Coffee program (Notredame et al. 2000) for each sequence group. These alignments were merged manually based on the structural superposition of GPR (PDB entry 1c8b, chain A) and HybD (PDB entry 1cfz, chain A), in conjunction with sequence conservation patterns and secondary structure predictions from the Jpred2 server (Cuff and Barton 2000). The sequence alignment of the intermediate group was used as an input to construct a profile (-B option in the blastpgp program; Altschul et al. 1997) for a new round of PSI-BLAST searches starting from each intermediate sequence.
Euclidian space mapping and distance diagram
The conserved segments of each sequence shown in Figure 2 ▶ were used to calculate pairwise identity fractions qij between each sequence pair i and j. The identity fractions were converted to evolutionary distances with the formula
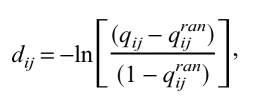 |
in which qranij is an expected identity percentage of two random sequences with the same amino acid composition as the sequences i and j. Each sequence was represented as a point in a multi-dimensional Euclidian space in such a way that Euclidian distances dij between the points optimally approximated the estimated distances dij between the sequences
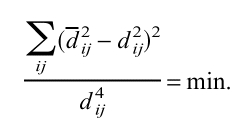 |
These points were grouped using the following procedure. Each point representing a sequence generated a Gaussian density in the Euclidian space in which the mean of each density was the point's coordinates and the variance σ2 of each density was identical for all points. Starting from each point, the local maximum of the sum of such Gaussians was found. The points giving rise to the same local maximum were grouped together.
Acknowledgments
We thank James Wrabl and Lisa Kinch for reading the manuscript critically and making helpful comments. We thank Vyacheslav Grishin for providing the distance diagram program before publication. The work was supported in part by the Welch foundation grant I-1505 to N.V.G.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.27302.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleby, T.C., Erion, M.D., and Ealick, S.E. 1999. The structure of human 5`-deoxy-5`-methylthioadenosine phosphorylase at 1.7 Å resolution provides insights into substrate binding and catalysis. Structure Fold Des. 7 629–641. [DOI] [PubMed] [Google Scholar]
- Aravind, L. and Koonin, E.V. 1999. Gleaning non-trivial structural, functional, and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 287 1023–1040. [DOI] [PubMed] [Google Scholar]
- Bode, W., Gomis-Ruth, F.X., Huber, R., Zwilling, R., and Stocker, W. 1992. Structure of astacin and implications for activation of astacins and zinc-ligation of collagenases. Nature 358 164–167. [DOI] [PubMed] [Google Scholar]
- Burger, A., Voges, D., Demange, P., Perez, C.R., Huber, R., and Berendes, R. 1994. Structural and electrophysiological analysis of annexin V mutants. Mutagenesis of human annexin V, an in vitro voltage-gated calcium channel, provides information about the structural features of the ion pathway, the voltage sensor, and the ion selectivity filter. J. Mol. Biol. 237 479–499. [DOI] [PubMed] [Google Scholar]
- Christianson, D.W., David, P.R., and Lipscomb, W.N. 1987. Mechanism of carboxypeptidase A: Hydration of a ketonic substrate analogue. Proc. Natl. Acad. Sci. 84 1512–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson, D.W., Mangani, S., Shoham, G., and Lipscomb, W.N. 1989. Binding of D-phenylalanine and D-tyrosine to carboxypeptidase A. J. Biol. Chem. 264 12849–12853. [PubMed] [Google Scholar]
- Cort, J.R., Yee, A., Edwards, A.M., Arrowsmith, C.H., and Kennedy, M.A. 2000. Structure-based functional classification of hypothetical protein MTH538 from Methanobacterium thermoautotrophicum. J. Mol. Biol. 302 189–203. [DOI] [PubMed] [Google Scholar]
- Crawford, J.L., Lipscomb, W.N., and Schellman, C.G. 1973. The reverse turn as a polypeptide conformation in globular proteins. Proc. Natl. Acad. Sci. 70 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuff, J.A. and Barton, G.J. 2000. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40 502–511. [DOI] [PubMed] [Google Scholar]
- Dietmann, S., Park, J., Notredame, C., Heger, A., Lappe, M., and Holm, L. 2001. A fully automatic evolutionary classification of protein folds: Dali Domain Dictionary version 3. Nucleic Acids Res. 29 55–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy, S. R. 1996. Hidden Markov models. Curr. Opin. Struct. Biol. 6 361–365. [DOI] [PubMed] [Google Scholar]
- Eisenstein, E., Gilliland, G.L., Herzberg, O., Moult, J., Orban, J., Poljak, R.J., Banerjei, L., Richardson, D., and Howard, A.J. 2000. Biological function made crystal clear - annotation of hypothetical proteins via structural genomics. Curr. Opin. Biotechnol. 11 25–30. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 15, 132–134, 112–113. [DOI] [PubMed] [Google Scholar]
- Fritsche, E., Paschos, A., Beisel, H.G., Bock, A., and Huber, R. 1999. Crystal structure of the hydrogenase maturating endopeptidase HYBD from Escherichia coli. J. Mol. Biol. 288 989–998. [DOI] [PubMed] [Google Scholar]
- Gribskov, M., McLachlan, A.D., and Eisenberg, D. 1987. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. 84 4355–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1995. Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 20 478–480. [DOI] [PubMed] [Google Scholar]
- Kim, S. H. 1998. Shining a light on structural genomics. Nat. Struct. Biol. (Suppl.) 5 643–645. [DOI] [PubMed] [Google Scholar]
- Koppensteiner, W.A., Lackner, P., Wiederstein, M., and Sippl, M.J. 2000. Characterization of novel proteins based on known protein structures. J. Mol. Biol. 296 1139–1152. [DOI] [PubMed] [Google Scholar]
- Long, S.B., Casey, P.J., and Beese, L.S. 2000. The basis for K-Ras4B binding specificity to protein farnesyltransferase revealed by 2 Å resolution ternary complex structures. Structure Fold. Des. 8 209–222. [DOI] [PubMed] [Google Scholar]
- Maier, T. and Bock, A. 1996. Generation of active [NiFe] hydrogenase in vitro from a nickel-free precursor form. Biochemistry 35 10089–10093. [DOI] [PubMed] [Google Scholar]
- Makarova, K.S. & Grishin, N.V. 1999. The Zn-peptidase superfamily: Functional convergence after evolutionary divergence. J. Mol. Biol. 292 11–17. [DOI] [PubMed] [Google Scholar]
- Menon, A.L. and Robson, R.L. 1994. In vivo and in vitro nickel-dependent processing of the [NiFe] hydrogenase in Azotobacter vinelandii. J. Bacteriol. 176 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin, A.G. 1998. How far divergent evolution goes in proteins. Curr. Opin. Struct. Biol. 8 380–387. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G. 1999. Structure classification-based assessment of CASP3 predictions for the fold recognition targets. Proteins (Suppl.) 3 88–103. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302 205–217. [DOI] [PubMed] [Google Scholar]
- Ponnuraj, K., Rowland, S., Nessi, C., Setlow, P., and Jedrzejas, M. J. 2000. Crystal structure of a novel germination protease from spores of Bacillus megaterium: Structural arrangement and zymogen activation. J. Mol. Biol. 300 1–10. [DOI] [PubMed] [Google Scholar]
- Postemsky, C.J., Dignam, S.S., and Setlow, P. 1978. Isolation and characterization of Bacillus megaterium mutants containing decreased levels of spore protease. J. Bacteriol. 135 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann, R., Sauter, M., Lottspeich, F., and Bock, A. 1994. Maturation of the large subunit (HYCE) of Escherichia coli hydrogenase 3 requires nickel incorporation followed by C-terminal processing at Arg537. Eur. J. Biochem. 220 377–384. [DOI] [PubMed] [Google Scholar]
- Sali, A. 1998. 100,000 protein structures for the biologist. Nat. Struct. Biol. 5 1029–1032. [DOI] [PubMed] [Google Scholar]
- Schmitt, E., Panvert, M., Blanquet, S., and Mechulam, Y. 1998. Crystal structure of methionyl-tRNAfMet transformylase complexed with the initiator formyl-methionyl-tRNAfMet. EMBO J. 17 6819–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, D.L., Otwinowski, Z., Gelb, M.H., and Sigler, P.B. 1990. Crystal structure of bee-venom phospholipase A2 in a complex with a transition-state analogue. Science 250 1563–1566. [DOI] [PubMed] [Google Scholar]
- Setlow, P. 1988. Small, acid-soluble spore proteins of Bacillus species: Structure, synthesis, genetics, function, and degradation. Annu. Rev. Microbiol. 42 319–338. [DOI] [PubMed] [Google Scholar]
- Setlow, P. 1994. Mechanisms which contribute to the long-term survival of spores of Bacillus species. Soc. Appl. Bacteriol. Symp. Ser. 23 49S–60S. [DOI] [PubMed] [Google Scholar]
- Setlow, P. 1995. Mechanisms for the prevention of damage to DNA in spores of Bacillus species. Annu. Rev. Microbiol. 49 29–54. [DOI] [PubMed] [Google Scholar]
- Strater, N., Sun, L., Kantrowitz, E.R., and Lipscomb, W.N. 1999. A bicarbonate ion as a general base in the mechanism of peptide hydrolysis by dizinc leucine aminopeptidase. Proc. Natl. Acad. Sci. 96 11151–11155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C., Waldo, G., Peat, T.S., Newman, J.M., Chu, K., and Berendzen, J. 1998. Class-directed structure determination: Foundation for a protein structure initiative. Protein Sci. 7 1851–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoratou, E., Paschos, A., Magalon, A., Fritsche, E., Huber, R., and Bock, A. 2000a. Nickel serves as a substrate recognition motif for the endopeptidase involved in hydrogenase maturation. Eur. J. Biochem. 267 1995–1999. [DOI] [PubMed] [Google Scholar]
- Theodoratou, E., Paschos, A., Mintz, W., and Bock, A. 2000b. Analysis of the cleavage site specificity of the endopeptidase involved in the maturation of the large subunit of hydrogenase 3 from Escherichia coli. Arch. Microbiol. 173 110–116. [DOI] [PubMed] [Google Scholar]
- Volbeda, A., Charon, M. H., Piras, C., Hatchikian, E. C., Frey, M., and Fontecilla-Camps, J.C. 1995. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 373 580–587. [DOI] [PubMed] [Google Scholar]
- Walker, D.R. and Koonin, E.V. 1997. SEALS: A system for easy analysis of lots of sequences. Intelligtent Systems for Molecular Biology 5 333–339. [PubMed] [Google Scholar]
- Zarembinski, T.I., Hung, L.W., Mueller-Dieckmann, H.J., Kim, K.K., Yokota, H., Kim, R., and Kim, S.H. 1998. Structure-based assignment of the biochemical function of a hypothetical protein: A test case of structural genomics. Proc. Natl. Acad. Sci. 95 15189–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Seabra, M.C., and Deisenhofer, J. 2000. Crystal structure of Rab geranylgeranyltransferase at 2.0 Å resolution. Structure Fold. Des. 8 241–251. [DOI] [PubMed] [Google Scholar]