

Abstract
The experimental material accumulated in the literature on the conformational behavior of intrinsically unstructured (natively unfolded) proteins was analyzed. Results of this analysis showed that these proteins do not possess uniform structural properties, as expected for members of a single thermodynamic entity. Rather, these proteins may be divided into two structurally different groups: intrinsic coils, and premolten globules. Proteins from the first group have hydrodynamic dimensions typical of random coils in poor solvent and do not possess any (or almost any) ordered secondary structure. Proteins from the second group are essentially more compact, exhibiting some amount of residual secondary structure, although they are still less dense than native or molten globule proteins. An important feature of the intrinsically unstructured proteins is that they undergo disorder–order transition during or prior to their biological function. In this respect, the Protein Quartet model, with function arising from four specific conformations (ordered forms, molten globules, premolten globules, and random coils) and transitions between any two of the states, is discussed.
Keywords: Intrinsically unfolded protein, intrinsically disordered protein, unfolded protein, molten globule, premolten globule, partially folded intermediate, random coil, conformational transition
This review introduces an intriguing protein family of natively unfolded proteins, whose existence questions one of the cornerstones in protein biology, chemistry and physics, that is, the structure–function paradigm. This concept claims that a specific function of a protein is determined by its unique and rigid three-dimensional (3D) structure. This idea, formulated more than 100 years ago as a lock-and-key model for explaining the amazing specificity of the enzymatic hydrolysis of glucosides (Fischer 1894), proved to be extremely fruitful. Figuratively speaking, the protein structure–function paradigm may be considered as the big bang, creating the universe of modern protein science. Figure 1 ▶ attempts to illustrate the most obvious scientific consequences of this concept.
Fig. 1.

Protein structure–function paradigm could be considered as the big bang created universe of the modern protein science.
However, a reappraisal of the protein structure–function paradigm is now warranted based on systematic studies of intrinsically unfolded/disordered proteins (Wright and Dyson 1999; Dunker et al. 2001; Uversky 2002). There are two major reasons for such a reappraisal: the results of the amino acid sequence analyses and the accumulation of experimental evidence for the existence of a rather large amount of protein domains and even entire proteins, lacking ordered structure under physiological conditions. Intriguingly, both of these sets of evidences were gathered using scientific concepts and approaches originating from the protein structure–function paradigm, namely, proteomics and protein self-organization (see Fig. 1 ▶).
Proteomics versus protein structure–function paradigm
The application of neuronal network predictors for protein disorder using primary sequence information to the Swiss Protein Database has predicted that more than 15,000 proteins may contain disordered regions of at least 40 consecutive amino acid residues, with more than 1050 of them having high scores indicating disorder (Dunker et al. 1998; Romero et al. 1998b). This observation helped to conclude that "a large portion of gene sequences appear to code not for folded, globular proteins, but for long stretches of amino acids that are likely to be either unfolded in solution or adopt non-globular structures of unknown conformation. . . . The high proportion of gene sequences in the genomes of all organisms argues for important, as yet unknown functions, since there could be no other reason for their persistence throughout evolution" (Wright and Dyson 1999). Intriguingly, recent predictions on 29 genomes have established that proteins from eucaryotes have more intrinsic disorder than those from bacteria and archaea, with more than 30% of eucaryotic proteins having disordered regions greater than 50 consecutive residues (Dunker et al. 2001).
Protein self-organization and protein structure–function paradigm
There is a rapidly growing set of proteins, which have been shown to be disordered or have profound disordered regions under physiological conditions. Several experimental approaches sensitive to the intrinsic disorder of a given protein or its part have been used to provide this evidence based on studies of protein self-organization problems.
It is known that the unique 3D structure of a globular protein is stabilized by noncovalent interactions (conformational forces) of different natures, such as hydrogen bonds, hydrophobic forces, van der Vaals interactions, etc. It was established long ago that high concentrations of strong denaturants (such as urea or guanidinium chloride [GdmCl]) lead to the complete disruption of all these interactions and, as a consequence, to the transformation of an initially folded protein molecule into a highly disordered random coil (Anson and Mirsky 1932; Mirsky and Pauling 1936; Neurath et al. 1944; Tanford 1968). In other words, such conditions cause the complete unfolding of proteins. However, sometimes changes in the environment can reduce (or even completely shut down) part of the conformational interactions, while the rest remain unchanged (or be even intensified). In these cases proteins will usually lose their biological activity, that is, they will be denatured. Denaturation is not necessarily accompanied by the complete unfolding of a protein, but rather results in the appearance of new conformations with properties intermediate between those of the native and the completely unfolded states.
It is known that globular proteins may exist in at least four different conformations: native (ordered), molten globule, premolten globule, and unfolded (Uversky and Ptitsyn 1994, 1996a; Ptitsyn 1995; Uversky 1997, 1998). The structural properties of the molten globule are well known, and have been systematized in a number of reviews (e.g., Ptitsyn 1995). It has been established that the protein molecule in this intermediate state has no (or has only a trace of) rigid cooperatively melted tertiary structure, that is, it is denatured. Small-angle X-ray scattering showed that the protein molecule in this intermediate state has a globular structure typical of native globular proteins (Eleiser et al. 1993; Kataoka et al. 1993, 1997; Semisotnov et al. 1996; Uversky et al. 1998). 2D NMR coupled with hydrogen-deuterium exchange showed that the protein molecule in the molten globule state is characterized not only by the native-like secondary structure content, but also by the native-like folding pattern (Baum et al. 1989; Bushnel et al. 1990; Jeng et al. 1990; Chyan et al. 1993; Wu et al. 1993; Eliezer et al. 1998; Bose et al. 1999; Bracken 2001). A considerable increase in the accessibility of a protein molecule to proteases was noted as a specific property of the molten globule state (Merrill et al. 1990; Fontana et al. 1993). It was also shown that transformation into this intermediate state is accompanied by a considerable increase in the affinity of a protein molecule to the hydrophobic fluorescence probes (such as 8-anilinonaphthalene-1-sulfonate, ANS) and this behavior should be considered as a characteristic property of the molten globule state (Semisotnov et al. 1991; Uversky et al. 1996). Finally, it was established that the averaged value for the increase in the hydrodynamic radius in the molten globule state compared with the native state is no more than 15%, which corresponds to volume increase of ∼50%.
The structural peculiarities of a polypeptide chain in the premolten globule state are summarized below. The protein molecule in this state is denatured, that is, it has no rigid tertiary structure. It is characterized by a considerable secondary structure, although much less pronounced than that of the native or the molten globule protein (protein in the premolten globule state has ∼50% native secondary structure, whereas in the molten globule state the corresponding value is close to 100%). The protein molecule in the premolten globule state is considerably less compact than in the molten globule or native states, but it is still more compact than the random coil (its hydrodynamic volume in the molten globule, the premolten globule, and the unfolded states, in comparison to that of the native state, increases 1.5, ∼3, and ∼12 times, respectively). The protein molecule in the premolten globule state can effectively interact with the hydrophobic fluorescent probe ANS, although essentially weaker than in the molten globule state. This means that at least part of the hydrophobic clusters of polypeptide chain accessible to the solvent is already formed in the premolten globule state (Uversky and Ptitsyn 1994, 1996a; Ptitsyn 1995; Uversky 1997, 1998). It has also been established that in the premolten globule state the protein molecule has no globular structure (Uversky 1997, 1998; Uversky et al. 1998). The last observation indicates that the premolten globule probably represents a "squeezed" and partially ordered form of a coil. Finally, it has been shown that the premolten globule is separated from the molten globule state by an all-or-none transition, which represents an intramolecular analog of the first-order phase transition (Uversky and Ptitsyn 1994, 1996a; Ptitsyn 1995; Uversky 1997, 1998). This means that the molten globule and premolten globule represent divers thermodynamic (phase) states.
As native, molten globule, premolten globule, and unfolded conformations possess defined structural differences along with increasing amounts of disorder, they may be easily discriminated from one another by several physico-chemical methods (Uversky 1999). These techniques are briefly considered below.
X-ray crystallography defines missing electron density in many protein structures, which may correspond to disordered region(s). The increased flexibility of atoms in such a region leads to the noncoherent X-ray scattering, making them unobserved (Bloomer et al. 1978; Bode et al. 1978; Huber 1979, 1987; Schulz 1979; Alber et al. 1982; Spolar and Record 1994; Lewis et al. 1996; Muchmore et al. 1996; Dunker et al. 1997, 2001; Worbs et al. 2000).
Heteronuclear multidimensional NMR is an extremely powerful technique for protein 3D structure determination in solution and for the characterization of protein dynamics. Recent advances in this technology have allowed the complete assignment of resonances for several unfolded and partially folded proteins, as well as the disordered fragments of folded proteins (Alexandrescu et al. 1994; Logan et al. 1994; Zhang et al. 1994; Cho et al. 1996; Kriwacki et al. 1996; Lisse et al. 1996; Donne et al. 1997; Gillespie and Shortle 1997a, 1997b; Penkett et al. 1997; Daughdrill et al. 1998; Eliezer et al. 1998; Fletcher et al. 1998; Liu et al. 1998; Mogridge et al. 1998; Zhang and Matthews 1998; Bose et al. 1999; Fiebig et al. 1999; Hazzard et al. 1999; Hershey et al. 1999; Love 1999; Bracken 2001; see also Wright and Dyson 1999, and references cited therein).
There are two types of optically active chromophores in proteins: side groups of aromatic amino acid residues, and peptide bonds (Adler et al. 1973; Fasman 1996). CD spectra in the near ultraviolet region (250–350 nm), also called the aromatic region, reflect the symmetry of the environment of aromatic amino acid residues and, consequently, are characteristic of protein tertiary structure. Protein denaturation may be easily detected by the simplification of near-UV CD spectrum.
Diminishing of ordered secondary structure may be detected by several spectroscopic techniques including far-UV CD (Adler et al. 1973; Provencher and Glöckner 1981; Johnson 1988; Woody 1995; Fasman 1996; Kelly and Price 1997; Uversky et al. 2000a), ORD, FTIR (see Uversky et al. 2000a, and reference cited therein) and Raman optical activity (Smyth et al. 2001).
Hydrodynamic parameters obtained from techniques such as gel-filtration, viscometry, SAXS, SANS, sedimentation, and dynamic and static light scattering may help in determining whether a protein is compact, or it has became unfolded. The unfolding of a protein molecule results in an essential increase in its hydrodynamic volume. For instance, there is a well-documented 15–20% increase in the hydrodynamic radius of globular proteins upon their transformation into the molten globule state (Uversky 1993, 1994; Ptitsyn 1995), while the hydrodynamic volume of the premolten globule is even larger (Uversky and Ptitsyn 1994, 1996a; Ptitsyn 1995; Uversky 1997, 1998). Moreover, it has been shown that native and unfolded conformations of globular proteins possess very different molecular mass dependencies of their hydrodynamic radii, RS (Tanford 1961, 1968; Uversky 1993). As a result, intrinsically disordered proteins will have an increased hydrodynamic volume relative to native proteins, leading to an increase in their apparent molecular mass (summarized in Uversky et al. 2000a).
Another very important structural parameter is the degree of globularity, which reflects the presence or absence of a tightly packed core in a protein molecule. This information may be extracted from the analysis of SAXS data in the form of a Kratky plot, whose shape is sensitive to the conformational state of the scattering protein molecules (Glatter and Kratky 1982; Feigin and Svergun 1987; Semisotnov et al. 1996; Uversky et al. 1998). It has been shown that a scattering curve in the Kratky coordinates has a characteristic maximum for globular proteins in either their native or molten globule states (i.e., states with globular structure). However, if a protein is completely unfolded or in a premolten globule conformation (i.e., with no globular structure), such a maximum will be absent (Glatter and Kratky 1982; Feigin and Svergun 1987; Semisotnov et al. 1996; Uversky 1997, 1998; Uversky et al. 1998; Tcherkasskaya and Uversky 2001).
Additional knowledge on the intramolecular mobility and compactness of a protein may be extracted from the analysis of different fluorescence characteristics. This includes FRET, shape and position of the intrinsic fluorescence spectrum, fluorescence anisotropy and lifetime, accessibility of the chromophore groups to external quenchers, and steady-state and time-resolved parameters of the fluorescent dyes. Overall, these techniques add important information to the conformational description of a polypeptide.
Increased proteolytic degradation in vitro of intrinsically disordered proteins indirectly confirmed by their increased flexibility (Markus 1965; Mikhalyi 1978; Fontana et al. 1993; Hubbard et al. 1994, 1998; Kriwacki et al. 1996; Lisse et al. 1996; Horiuchi et al. 1997; Hubbard 1998; Hershey et al. 1999; Ratnaswamy et al. 1999; Bouivier and Stafford 2000; Iakoucheva et al. 2001; see Dunker et al. 2001, for recent review).
Immunochemical methods may also be applied toward the elucidation of protein disorder. Important to this discussion, the immunoglobulins obtained against a given protein may be specific for different levels of macromolecule: the primary structure (Amit et al. 1985; Wilson et al. 1985), the secondary structure (Fujio et al. 1985), or the tertiary structure (Amit et al. 1985; Fujio et al. 1985; Wilson et al. 1985). In the latter case, the antigenic determinants may reside on either the neighboring residues in the chain (loops) (Amit et al. 1985; Wilson et al. 1985) or on spatially distant residues (Fujio et al. 1985). Furthermore, it has been shown that antibodies in the immune serum may possess a high affinity to the internal elements of an antigen (Fujio et al. 1985). Thus, antibodies may be successfully used to study the structural changes, which a protein-immunogen undergoes upon changes of the experimental conditions. For example, antibodies obtained against the Ca2+-saturated F1-fragment of prothrombin did not interact with the calcium-free apo-form of this protein (Furie and Furie 1979). An analogous effect was also observed in the case of osteocalcine (Delmas et al. 1984).
Finally, intrinsic disorder may be detected by the analysis of protein conformational stability. For example, the presence or absence of a cooperative transition on the calorimetric melting curve for a given protein is a simple and convenient criterion indicating the presence or absence of a rigid tertiary structure (Privalov 1979; Ptitsyn 1995; Uversky 1999). Furthermore, it has been shown that the steepness of urea- or guanidinium chloride-induced unfolding curves depends strongly on whether a given protein has a rigid tertiary structure (i.e., it is native) or is already denatured and exists as a molten globule (Ptitsyn and Uversky 1994; Uversky and Ptitsyn 1996b). To extend this type of analysis, the values of Δνeff (which is the difference in the number of denaturant molecules "bound" to one protein molecule in its two states) should be determined. Then this quantity should be compared to the ΔνeffN→U and ΔνeffMG→U values corresponding to the native to coil and molten globule to coil transitions in globular protein of a given molecular mass, respectively (Uversky and Ptitsyn 1996b).
Application of several techniques mentioned above to a given protein provides the most unambiguous evidence for the presence of partially folded intermediates.
It has been shown that a considerable number of proteins possess some amount of disorder rather than rigid structure. A special term, "natively denatured," was introduced in 1994 (Schweers et al. 1994) to emphasize the existence of a drastic structural difference between "normal" globular protein, with rigid tertiary structure, and an "abnormal" extremely flexible tau protein. Two years later, a new term "natively unfolded" originated as a result of conformational analysis of α-synuclein, which under physiological conditions appeared to lack any secondary structure (Weinreb et al. 1996). Two alternative terms, "intrinsically unstructured" (Wright and Dyson 1999) and "intrinsically disordered" (Dunker et al. 2001), have also been suggested to describe these proteins. Because "abnormal" proteins show an extremely wide diversity in their structural properties, the meaning of the above terms should be clarified. Thus, the terms denatured and disordered may be considered as synonyms, and indicate any set of nonrigid conformations of polypeptide chains including different compact partially folded conformations: molten globules and premolten globules, and random coil. The terms unstructured and unfolded may be considered synonymous, and should only be applied to the subset of disordered proteins characterized by the absence of any (or almost any) ordered structure. For the remaining of this review, only natively unfolded proteins will be considered, excluding "native molten globules".
Are natively unfolded proteins common?
The number of proteins and protein domains that have been shown in vitro to have little or no ordered structure under physiological conditions is rapidly expanding. For example, over the past 10 years there has been a significant increase in publications describing structural properties of intrinsically unstructured (natively unfolded) proteins, starting from two papers in 1991 and ending with more than 30 in 2000. During the same time other interesting aspects of natively unfolded proteins have also been investigated. For example, 2382, 1960, and 370 papers were published concerning different aspects of amyloid-beta peptide, tau protein, and α-synuclein, respectively.
The current list of different natively unfolded proteins includes more than 100 entries, with information on 91 of them presented in our recent work (Uversky et al. 2000a) and Table 1. Only full-length proteins or domains with chain length greater than 50 amino acid residues have been considered. This list would probably be doubled if shorter polypeptides 30 to 50 residues long were included. Finally, the set of 100 proteins described in the literature as "natively unfolded" have at least 250 homologs, which are also expected to be natively unfolded. Additionally, a large number of proteins and protein domains have been predicted to be disordered based on the results of the analysis of amino acid sequences using the neuronal network predictors (Dunker et al. 1998, 2001; Romero et al. 1998b). All this shows that polypeptides without ordered structure under physiological are common, rather than exceptions.
Table 1.
Major structural characteristics of the "natively unfolded" proteins
| Protein | Length, a. a. r. | [θ]200, deg cm2 dmol−1 | [θ]222, deg cm2 dmol−1 | Vh, Å3 | Reference |
| Thymosin α1 | 28 | −15,700 | −2000 | Grottesi et al. 1998 | |
| c-Jun oncoprotein, basic subdomain | 29 | −19,200 | −1100 | Krebs et al. 1995 | |
| P19Arf tumor suppressor protein, N-terminal fragment | 37 | −19,000 | −1500 | DiGiammarino et al. 2001 | |
| Bacteriophage Pf1 gene 5 protein, C-terminal domain | 38 | −20,500 | −800 | Fox et al. 2001 | |
| WW domain of Yes-associated protein, W17F mutant | 38 | −16,400 | −2000 | Koepf et al. 1999 | |
| 50S ribosomal protein L34 | 46 | −25,300 | −2100 | Venyaminov et al. 1981 | |
| α-Tubulin, 404–451 fragment | 48 | −17,300 | −1500 | Jimenez et al. 1999 | |
| β-Tubulin, 394–445 fragment | 52 | −16,600 | −2000 | Jimenez et al. 1999 | |
| Calpastatin, domain I | 53 | −23,600 | −2500 | Konno et al. 1997 | |
| 50S ribosomal protein L33 | 54 | −21,300 | −2000 | Venyaminov et al. 1981 | |
| HIV-1 integrase, N-terminal domain | 55 | −17,500 | −2000 | Zheng et al. 1996 | |
| RNA polymerase II, C-terminal domain | 56 | −15,000 | −1500 | Bienkiewicz et al. 2000 | |
| Galline | 61 | −25,000 | −1000 | Nakano et al. 1989 | |
| B-cell specific transcription coactivator Bob1, N-terminal domain | 65 | −17,500 | −1700 | Chang et al. 1999 | |
| Nonhistone chromosomal protein HMG-H6 | 69 | −22,000 | −1000 | Cary et al. 1981 | |
| 50S ribosomal protein L31 | 70 | −20,000 | −1500 | Venyaminov et al. 1981 | |
| 30S ribosomal protein S21 | 70 | −20,300 | −3000 | Venyaminov et al. 1981 | |
| Subtilisin BPN` pro-domain | 75 | −17,500 | −2000 | Tangrea et al. 2001 | |
| Phosphodiesterase γ-subunit | 83 | −14,940 | −1690 | 73,622 | Uversky et al. 2002 |
| 50S ribosomal protein L27 | 84 | −17,700 | −2000 | Venyaminov et al. 1981 | |
| Vmw65, carboxyl-terminal transactivation domain | 87 | −21,155 | −1840 | 91,952 | Donaldson and Capone 1992 |
| Nonhistone chromosomal protein HMG-17 | 89 | −21,500 | −1300 | 93,097 | Abercrombie et al. 1978 |
| 30S ribosomal protein S19 | 91 | −17,000 | −1200 | Venyaminov et al. 1981 | |
| Em protein | 93 | −18,340 | −2440 | 93,937 | McCubbin and Kay 1985 |
| Nonhistone chromosomal protein HMG-14 | 100 | −17,800 | −1950 | Cary et al. 1980 | |
| Human sperm protamine P2 | 102 | −20,000 | −1200 | Gatewood et al. 1990 | |
| Human sperm protamine P3 | 103 | −21,000 | −1000 | Gatewood et al. 1990 | |
| Apo-cytochrome c | 104 | 103,097 | Stellwagen et al. 1972 | ||
| Prothymosin α | 110 | −22,000 | −500 | 60,105 | Gast et al. 1995; Uversky et al. 1999 |
| 50S ribosomal protein L22 | 110 | −22,500 | −3000 | Venyaminov et al. 1981 | |
| Fibronectin-binding domain B-type, S. dysgalactiae | 112 | −19,200 | −1500 | 121,200 | House-Pompco et al. 1996 |
| 4E-binding protein 1 | 118 | −16,500 | −2250 | Fletcher et al. 1998 | |
| NF-κB transcription factor, p65 subunit | 120 | −18,000 | −800 | Schmitz et al. 1994 | |
| Fibronectin-binding domain A-type, S. dysgalactiae | 124 | −22,000 | −1200 | 134,434 | House-Pompeo et al. 1996 |
| Fibronectin-binding domain, D-type, S. aureus | 134 | −21,000 | −1200 | 134,701 | House-Pompeo et al. 1996 |
| β-Synuclein | 134 | −15,495 | −2124 | 137,258 | |
| γ-Synuclein | 134 | −15,560 | −2031 | 117,681 | |
| α-Synuclein | 140 | −15,309 | −2073 | 141,155 | Weinreb et al. 1996; Uversky et al. 2001b |
| Helix destabilizing protein (Pf1 gene 5 protein) | 144 | −17,500 | −700 | Bogdarina et al. 1998 | |
| Stathmin | 148 | 150,533 | Belmont and Mitchinson, 1996 | ||
| Calpastatin, domain I | 148 | −16,800 | −2500 | Konno et al. 1997 | |
| Dessication-related protein | 155 | −19,000 | −1000 | Lisse et al. 1996 | |
| Thyroid transcription factor-1, N-terminal domain | 156 | −16,700 | −1350 | Tell et al. 1998 | |
| DFF45 N-terminal domain | 116 | −23,000 | −500 | Zhou et al. 2001 | |
| C-terminal domain of bZIP transcription factor Fos | 164 | −16,000 | −1000 | 179,594 | Campbell et al. 2000 |
| Myelin-binding protein | 196 | −15,200 | −1000 | Polverini et al. 1999 | |
| Nonhistone chromosomal protein HMG-T | 204 | −18,000 | −2000 | Cary et al. 1981 | |
| Extracellular domain of the low affinity NGF receptor | 222 | −21,500 | −500 | Timm et al. 1992 | |
| CREB, truncated form (ACT265) | 265 | −15,300 | −2000 | Richards et al. 1996 | |
| Salivary proline-rich glycoprotein | 293 | −25,000 | −3500 | Loomis et al. 1985 | |
| Histidine-rich protein II | 327 | −16,000 | −2000 | Lynn et al. 1999 | |
| GAGA factor, glutamine-rich domain | 383 | −18,500 | −2300 | Agianian et al. 1999 | |
| Chromogranin A | 439 | −16,000 | −2500 | 838,603 | Yoo and Albanesi 1990 |
| Caldesmon | 771 | 3,156,551 | Lynch et al. 1987 | ||
| Microtubule-associated protein MAP2 | 1827 | −19,500 | −2980 | 7,606,206 | Hernández et al. 1986 |
| Parathyroid hormone-related protein, N-terminal fragment | 34 | −13,500 | −4000 | Willis 1994 | |
| Naturally occurring peptide L1-37 | 37 | −11,400 | −3000 | Johansson et al. 1998 | |
| DNA polymerase I, substrate-binding peptide | 50 | −11,000 | −4000 | Mullen et al. 1993 | |
| Osteocalcin | 50 | −10,000 | −4500 | 26,094 | Isbell et al. 1993; Dowd et al. 2001 |
| 50S ribosomal protein L32 | 56 | −12,400 | −4050 | Venyaminov et al. 1981 | |
| GCN4, DNA-binding domain | 57 | −9800 | −4750 | Weiss et al. 1990 | |
| RNase HI, C-terminal domain (88–155) | 68 | −12,500 | −5100 | Kanaya and Kanaya 1995 | |
| Rhodostomin | 68 | −11,629 | −2350 | Guo et al. 2001 | |
| Lysozyme 1-40/98-127 | 69 | −12,500 | −5500 | 28,731 | Demarest et al. 2001 |
| Heat stable protein kinase inhibitor | 72 | −8500 | −2,662 | 46,452 | Thomas et al. 1991 |
| 30S ribosomal protein S18 | 74 | −12,000 | −4500 | Venyaminov et al. 1981 | |
| SMK killer toxin, β-subunit | 77 | −11,000 | −2500 | Suzuki et al. 1997 | |
| Tropomodulin, N11 fragment | 91 | −8500 | −3500 | Kostyukova et al. 2000 | |
| Cyclin-dependent kinase inhibitor p21Waf1/Cip1/Sdi1, N-terminal domain | 94 | −10,000 | −2500 | Kriwacki et al. 1996 | |
| β-Dystroglycan, extracellular region | 96 | −11,500 | −3,780 | Di Stasio et al. 1999 | |
| HPV16 E7 protein | 98 | −10,800 | −5200 | Pahel et al. 1993 | |
| 50S ribosomal protein L23 | 99 | −12,700 | −7100 | Venyaminov et al. 1981 | |
| 50S ribosomal protein L24 | 103 | −9900 | −2700 | Venyaminov et al. 1981 | |
| Reduced RNase T1 | 104 | −10,700 | −2900 | Baskakov and Bolen 1998 | |
| DNA-binding domain of 1,25-Dihydroxyvitamin D3 receptor | 110 | −10,000 | −5,000 | Creig et al. 1997 | |
| RNase P | 116 | −11,000 | −3,000 | Henkels et al. 2001 | |
| 50S ribosomal protein L14 | 120 | −9300 | −3000 | Venyaminov et al. 1981 | |
| 30S ribosomal protein S12 | 123 | −12,000 | −3500 | Venyaminov et al. 1981 | |
| Steroidogenic acute regulatory protein, StAR, N-terminal domain | 131 | −11,000 | −3000 | Song et al. 2001 | |
| SNase, Δ131Δ fragment | 131 | −12,000 | −3600 | 65,450 | Alexandrescu and Shortle 1994; Shortle and Meeker 1989 |
| Caldesmon, 636–771 fragment | 136 | −12,200 | −3440 | 89,656 | Uversky, unpubl commun |
| cGMP-dependent protein kinase inhibitor | 155 | 124,788 | Aswad and Greengard 1981 | ||
| MAX-protein | 163 | −10,500 | −6,100 | Horiuchi et al. 1997 | |
| Cyclin-dependent kinase inhibitor p21Waf1/Cip1/Sdi1 | 164 | −11,000 | −3060 | Kriwacki et al. 1996 | |
| Heat-shock transcription factor, N-terminal activation domain (S. cerevisiae) | 167 | −9500 | −3500 | Cho et al. 1996 | |
| Protein phosphatase inhibitor-1 | 171 | 141,092 | Nimmo and Cohen 1978 | ||
| Glucocorticoid receptor, 77–262 fragment | 186 | −8000 | −3200 | Baskakov et al. 1999 | |
| Heat-shock transcription factor, N-terminal activation domain (K. lactis) | 194 | −10,000 | −4000 | Cho et al. 1996 | |
| αs-Casein | 199 | −11,000 | −4500 | Bhattacharyya and Das 1999 | |
| Dopamine- and cAMP-regulated neuronal phosphoprotein, DARPP-32 | 202 | 164,636 | Nemmings et al. 1984 | ||
| 50S ribosomal protein L3 | 209 | −10,100 | −3300 | Venyaminov et al. 1981 | |
| Histone H1 | 224 | −9450 | −3150 | Tarkka et al. 1997 | |
| Manganese stabilizing protein, L245E mutant | 247 | −10,000 | −3500 | 146,464 | Lydakis-Simantiris et al. 1999 |
| Sec9, SNAP-25-like domain | 251 | −11,000 | −5500 | Rice et al. 1997 | |
| 50S ribosomal protein L2 | 272 | −10,400 | −2500 | Venyaminov et al. 1981 | |
| SPARC, BM-40, osteonectin | 285 | −12,000 | −5150 | Engel et al. 1987 | |
| GAGA factor, central domain | 307 | −9000 | −3300 | Agianian et al. 1999 | |
| Calreticulin, human−41C fragment | 359 | 413,061 | Bouvier and Stafford 2000 | ||
| Calsequestrin | 367 | −8500 | −4000 | 381,704 | Cozens and Reithmeier 1984; He et al. 1993 |
| Calreticulin, human | 400 | 413,061 | Bouvier and Stafford 2000 | ||
| Calreticulin, bovine | 407 | 361,706 | Bouvier and Stafford 2000 | ||
| Soluble transducer HtrXI | 451 | −10,300 | −4500 | Larsen et al. 1999 | |
| Taka-amylase A, reduced | 477 | 335,367 | Tanford 1968 | ||
| SdrD protein, B1–B5 fragment | 562 | −10,560 | −4,120 | 685,568 | Josefsson et al. 1998 |
| Secretogranin (chromatogranin B) | 657 | −10,500 | −4250 | 533,080 | Yoo 1995 |
| DNA topoisomerase I | 765 | 830,031 | Stewart et al. 1996 | ||
| Fibronectin | 4772 | 6,370,626 | Pelta et al. 2000 |
How is unfoldedness encoded in a protein amino acid sequence?
The existence of at least three different disordered equilibrium conformations, molten globule (MG), premolten globule (PMG), and unfolded (random coil-like, U), has been established for typical globular proteins (Uversky and Ptitsyn 1994, 1996a; Ptitsyn 1995; Uversky 1997, 1998). Apparently, the ability of a protein to adopt different stable conformations is an intrinsic property of a polypeptide chain. Although the correct folding of a protein into its rigid biologically active conformation is thought to be determined by its amino acid sequence (Anfinsen et al. 1961), the absence of rigid structure in natively unfolded proteins may be reflected in specific features of their amino acid sequences.
In an attempt to understand the relationship between sequence and disorder, Dunker and coauthors have developed several neuronal network predictors (Romero et al. 1997, 1998a, 1998b, 2001a; Dunker et al. 1998, 2001; Garner et al. 1998; Li et al. 1999, 2000). They assumed that if a protein structure has evolved to have a functional disordered state then a propensity for disorder might be predictable from its amino acid sequence and composition. The results of such analysis were impressive. It was established that disordered regions shared at least some common sequence features between many proteins, and that more than 15,000 proteins in the Swiss Protein database were identified as having long regions of sequence that shared these features (Romero et al. 1998b). Interestingly, the Top 20 proteins (i.e., proteins with the highest scores) were shown to have low sequence complexity, as defined by Wootton (1993, 1994; Wootton and Federhen 1996). In other words, sequences of natively unfolded proteins may be essentially degenerate. Figure 2A ▶ illustrates this idea, comparing the s-antigen from Plasmodium (which was shown to be at the head of the Top 20) with that of human serum albumin (a rigid globular protein of similar molecular mass) in terms of their amino acid composition scaled according to McCaldon and Argos (1988). Interestingly, it was later established that the distributions of the complexity values for ordered and disordered sequences overlapped (Romero et al. 2001b), suggesting that low sequence complexity did not represent the only characteristic feature of intrinsically disordered proteins. However, some general sequence peculiarities of natively unfolded proteins have been recognized long ago. These include the presence of numerous uncompensated charged groups, resulting in a large net charge at neutral pH (Hemmings et al. 1984; Gast et al. 1995; Weinreb et al. 1996) and a low content of hydrophobic amino acid residues (Hemmings et al. 1984; Gast et al. 1995).
Fig. 2.

How the unfoldedness is encoded in protein amino acid sequence. (A) Comparison of sequences of rigid globular protein, human serum albumin (top) and intrinsically disordered protein s-antigen from Plasmodium, which was shown to be at the head of the Top 20 proteins with highest disorder scores estimated by neuronal network predictors (Romero et al. 1998b) (bottom) in terms of their composition scaled according to McCaldon and Argos (1988). (B) The unique property of natively unfolded protein sequences is a combination of low overall hydrophobicity and large net charge. Comparison of the mean hydrophobicity and the mean net charge for a set of 275 folded (black circles) and 102 natively unfolded proteins (open circles). The solid line represents the border between intrinsically unstructured and native proteins calculated using equation 1. Data for this plot are taken from Uversky et al. (2000a).
Recently, we have established that the combination of low mean hydrophobicity and relatively high net charge represents an important prerequisite for the absence of compact structure in proteins under physiological conditions, leading to natively unfolded proteins (Uversky et al. 2000a). Figure 2B ▶ shows that natively unfolded proteins are specifically localized within a unique region of the charge-hydrophobicity phase space. The solid line in this figure represents the border between intrinsically unstructured and native proteins, satisfying the following relationship:
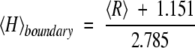 |
(1) |
This equation gives the estimation of the "boundary" mean hydrophobicity value, 〈H〉boundary, below which a polypeptide chain with a given mean net charge 〈R〉 will most probably be unfolded. Thus, sequences of natively unfolded proteins may be characterized by a low sequence complexity and/or high net charge coupled with low mean hydrophobicity.
How unfolded are intrinsically unstructured proteins? A classification attempt
It is well known that in the presence of large concentrations of strong denaturants, such as 8 M urea or 6 M GdmCl, normal proteins lose the majority of their specific structure, that is, become essentially unfolded (Anson and Mirsky 1932; Mirsky and Pauling 1936; Neurath et al. 1944; Tanford 1968). One can expect that under these conditions unfolded proteins will obey the theoretical and empirical rules that apply to linear random coils (Tanford 1968). In accordance with Tanford, a polymer molecule is randomly coiled when internal rotation can take place at about every single bond of the molecule with the same freedom with which it would take place in a molecule of low molecular weight containing the same kind of bonds (Tanford 1968). The properties of linear random coils are well understood, as synthetic polymers frequently adopt this conformation (Flory 1953; Tanford 1961). Because the dimensions of random coils depend only on the backbone rotational angles, the dependence of the hydrodynamic dimensions on molecular mass (length of polypeptide chain) represents the most effective diagnostic tool for recognition of linear random coils (Tanford 1961, 1968). Results of early studies on hydrodynamic dimensions of proteins in the presence of 6 M GdmCl were consistent with the conclusion that unfolded proteins could be described as random coils (Tanford 1961, 1968). However, it was later established by heteronuclear NMR that even in high concentrations of strong denaturants, when the native state of globular proteins breaks down, the polypeptide chains contained some amount of residual structure, that is, the polypeptide chain did not reach a random coil conformation (Dill and Shortle 1991; Logan et al. 1994; Zhang et al. 1994; Shortle 1996; Pappu et al. 2000; Baldwin and Zimm 2000). These findings raised several compelling biophysical questions related to the structural characteristics of natively unfolded proteins. How unfolded are these proteins? Are they random coils, or do they possess residual structure? If they have residual structure, how then should they be classified? Fortunately, the information accumulated to date on natively unfolded proteins allows us to make an initial structural classification of these intriguing members of the polypeptide kingdom.
As it follows from their definition, intrinsically unstructured proteins show complete (or almost complete) loss of any ordered structure under physiological conditions in vitro; that is, they should behave as random coils. Structurally, this may be manifested by (1) larger hydrodynamic dimensions compared to typical native globular proteins with corresponding molecular mass, (2) low content of ordered secondary structure, and (3) high intramolecular flexibility. Such anomalous behavior is usually detected by numerous hydrodynamic techniques (gel-filtration, viscometry, SAXS, SANS, sedimentation, and dynamic and static light scattering), far-UV CD, ORD, FTIR, and NMR spectroscopy (one-dimensional and heteronuclear multidimensional). These techniques may also be used for the identification of residual structure (if any) in an unfolded protein molecule. Once again, simultaneous application of several approaches should permit one to make more reliable conclusion.
Flexibility and residual structure by NMR spectroscopy
NMR spectroscopy of natively unfolded proteins has established that they contain varied amounts of residual structure. Examples of proteins which are essentially unfolded under in vitro physiological conditions include: DFF45 N-terminal domain (Zhou et al. 2001), DNA-binding domain of vitamin D receptor (Craig et al. 1997), C-terminal domain of anti-sigma factor FlgM (Daughdrill et al. 1998), p53 regulatory domain of p19Arf tumor suppressor (DiGiammarino et al. 2001), substrate-binding peptide from DNA polymerase I (Mullen et al. 1993), poplar apo-plastocyanin (Bai et al. 2001), N-terminal domain of StAR (Song et al. 2001), bone sialoprotein and osteopontin (Fisher et al. 2001), C-terminal domains of α- and β-tubulins (Jimenez et al. 1999), N-terminal activation domain of heat-shock transcription factors (Cho et al. 1996); 4E-binding proteins I and II (Fletcher et al. 1998), cyclin-dependent kinase inhibitor p21Waf1/Cip1/Sdi1 (Kriwacki et al. 1996), SNase, Δ131Δ fragment (Alexandrescu et al. 1994; Gillespie and Shortle 1997a, 1997b), dessication-related protein (Lisse et al. 1996), functional domain of eIF4G1 (Hershey et al. 1999), cytoplasmic domain of synaptobrevin (Hazzard et al. 1999), N-terminal domain of prion protein (Donne et al. 1997), C-terminal HMG domain of LEF-1 (Love 1999), N-terminal region of TAFII-23011–77 (Liu et al. 1998), antitermination protein N (Mogridge et al. 1998), cytoplasmic domain of Snc1 (Fiebig et al. 1999), prothymosin α (Uversky et al. 1999), nonhistone chromosomal proteins HMG-14 (Cary et al. 1980), HMG-17 (Abercrombie et al. 1978), HMG-T and HMG-H6 (Cary et al. 1981), fibronectin-binding domains (Penkett et al. 1998), DNA-binding domain of GCN4 (Weiss et al. 1990), EMB-1 protein (Eom et al. 1996), NEF protein (Geyer et al. 1999), osteocalcin (Isbell et al. 1993), two-domain fragment of neutral zinc finger factor 1 (Berkovits and Berg 1999), and several other proteins. On the other hand, heteronuclear multidimensional NMR analysis provided evidence of some ordered structure in several natively unfolded proteins, although the amount and quality of residual structure varied tremendously. In some cases the authors were unable to detect any secondary or tertiary contacts (e.g., Abercrombie et al. 1978; Cary et al. 1980, 1981; Cho et al. 1996; Eom et al. 1996; Lisse et al. 1996; Penkett et al. 1997; Fletcher et al. 1998; Zhang and Matthews 1998; Bose et al. 1999; Uversky et al. 1999; Campbell et al. 2000; Fisher et al. 2001; DiGiammarino et al. 2001). In other cases it was concluded that the proteins contained mostly dynamic structure favoring helical or β-structural conformation (e.g., Mullen et al. 1993; Schmitz et al. 1994; Kriwacki et al. 1996; Gillespie and Shortle 1997a, 1997b; Daughdrill et al. 1998; Bai et al. 2001, and many others). Thus, NMR analysis clearly showed that natively unfolded proteins do not possess uniform structural properties, as expected for members of a single thermodynamic entity.
Hydrodynamic dimensions
As previously discussed, the most unambiguous characteristic of the conformational state of a globular protein is its hydrodynamic dimension. In fact, it has been shown that equilibrium conformations of a globular protein (native, molten globule, premolten globule, and unfolded states) may easily be discriminated by the degree of compactness of the polypeptide chain (Uversky 1993, 1994, 1997, 1998; Uversky and Ptitsyn 1994, 1996a; Ptitsyn 1995). The equilibrium conformations were characterized by very different dependencies of their hydrodynamic dimensions on molecular mass (length of polypeptide chain) (Tanford 1961, 1968; Uversky 1993; Tcherkasskaya and Uversky 2001).
To clarify the physical nature of natively unfolded proteins, Figure 3 ▶ represents the dependencies of their hydrodynamic dimensions on the length of their polypeptide chains (see Table 1). The same trends determined for globular proteins in their native, molten globule, premolten globule, and urea or GdmCl unfolded states are shown for comparison. The values of the hydrodynamic volumes, Vh, were calculated from the corresponding Stokes radii, RS, as Vh = 4/3 π RS3. Data for the different conformations of globular proteins were taken from Tcherkasskaya and Uversky (2001). For these species, the dependencies of Vh on the length of the polypeptide chains, N, were described by a set of straight lines (using a logarithmic scale):
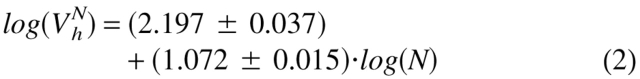 |
(2) |
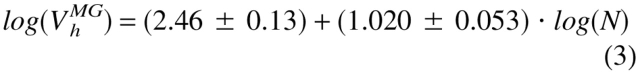 |
(3) |
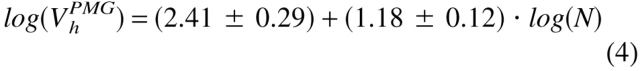 |
(4) |
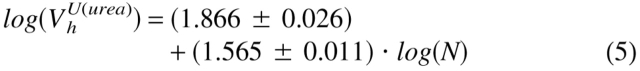 |
(5) |
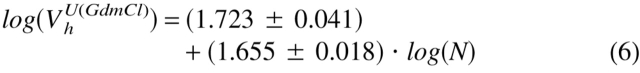 |
(6) |
Here, N, MG, PMG, U(urea), and U(GdmCl) correspond to the native, molten globule, premolten globule, urea, and GdmCl unfolded globular proteins, respectively.
Fig. 3.

Dependencies of the hydrodynamic volume, VS, on protein polypeptide length, N, for native (open circles, N), molten globule (gray reversed triangles, MG), premolten globule (open squares, PMG), 8 M urea-unfolded (open triangles, Uurea) and 6 M GdmCl-unfolded (black triangles, UGdmCl) conformational states of globular proteins and natively unfolded proteins with coil-like (gray triangles, NUcoil) and PMG-like properties (gray squares, NUPMG). Data used to plot the dependencies for native, molten globule, premolten globule, and GdmCl-unfolded states of globular proteins are taken from Tcherkasskaya and Uversky (2001); data for natively unfolded proteins are summarized in Table 1.
The existence of significant differences in the molecular mass dependencies of the Vh measured for urea and GdmCl unfolded proteins should be emphasized. It is well established that the hydrodynamic dimensions of random coils essentially depend on the quality of solvent (Flory 1953; Tanford 1961, 1968). A poor solvent induces the attraction of macromolecular segments, resulting in the squeezing of a chain. On the other hand, in a good solvent repulsive forces occur between segments, leading to the formation of a loose fluctuating coil (Grossberg and Khokhlov 1989). It is assumed that solutions of urea and GdmCl are rather good solvents for polypeptide chains, with GdmCl being closer to the ideal one (Tanford 1961, 1968). This difference in solvent quality could account for the observed divergence in log(Vh) versus log(N) dependencies for the globular proteins unfolded by urea and GdmCl.
Figure 3 ▶ shows that natively unfolded proteins are much less compact compared to native and molten globule proteins of similar molecular mass. Surprisingly, under physiological conditions in vitro (i.e., in aqueous solution, a poor solvent for a polypeptide chain), natively unstructured proteins are split in two different subclasses (see also Table 1). Proteins from the first subclass, which consists of 17 representatives, behave as random coils in poor solvent, whereas the 18 proteins of the second subclass are essentially more compact, being close to premolten globules as it follows from their hydrodynamic parameters (cf. equations 3–5):
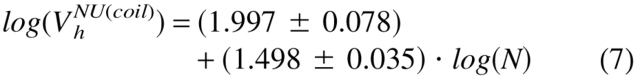 |
(7) |
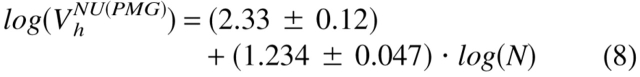 |
(8) |
where NU(coil) and NU(PMG) correspond to the natively unfolded proteins with coil-like and premolten globule-like hydrodynamic characteristics, respectively. This very intriguing observation, confirming conclusion on the structural diversity of intrinsically disordered proteins drawn from their NMR analysis, may be further verified by the analysis of far-UV CD spectra.
Residual secondary structure from far-UV CD spectra
Unfolded polypeptide chains are characterized by very specific shapes of their far-UV CD spectrum, with an intensive minimum in the vicinity of 200 nm and an ellipticity close to zero in the vicinity of 222 nm (Adler et al. 1973; Provencher and Glöckner 1981; Johnson 1988; Woody 1995; Fasman 1996; Kelly and Price 1997; Uversky 1999). This is a very useful graphical criterion for the selection of natively unfolded proteins (see Table 1). To date, a coil-like shape of far-UV CD spectrum has been reported for ∼100 proteins (see Table 1), which is almost threefold larger than the number of proteins shown to be unfolded in accordance with their hydrodynamic dimensions (35).
Figure 4 ▶ represents a "double wavelength" plot, [θ]222 versus [θ]200 that may be used to assort natively unfolded proteins into two nonoverlapping groups. Fifty-one proteins were characterized by far-UV CD spectra characteristic of almost completely unfolded polypeptide chains: with [θ]200 = υ(18,900 ± 2800) deg•cm2•dmol−1 and [θ]222 = −(1700 ± 700) deg • cm2 • dmol−1. On the other hand, 44 other protein spectra were consistent with the existence of some residual secondary structure, possessing shape typical of the premolten globule state of globular proteins (with [θ]200 =−(10,700 ± 1300) deg•cm2•dmol−1 and [θ]222 = −(3900±1100) deg • cm2 • dmol−1).
Fig. 4.

Analysis of far-UV CD spectra in terms of double wavelength plot, [θ]222 versus [θ]200, allows the natively unfolded proteins division on coil-like (gray circles) and premolten globule-like subclasses (black circles). Intrinsic premolten globules and intrinsic coils for which the hydrodynamic parameters were measured (see Table 1) are marked by white-dotted and black-dotted symbols, respectively.
Definitely, the difference in the shape of far-UV CD spectra alone does not allow the unambiguous discrimination between the two conformations. However, among more than 100 reported cases, 23 proteins were simultaneously characterized by CD and hydrodynamic methods, making classification more certain (see Table 1). Intrinsic premolten globules and intrinsic coils studied by both techniques are indicated in Figure 4 ▶ as white-dotted and black-dotted symbols, respectively. These data are consistent with the important conclusions that more compact polypeptides (with PMG-like hydrodynamic characteristics) possess larger amounts of ordered secondary structure than less compact coil-like natively unfolded proteins. Thus, the simultaneous application of CD and hydrodynamic techniques leaves no doubts that natively unfolded proteins should be subdivided into two structurally distinct groups: intrinsic coils and intrinsic premolten globules.
Amino acid composition of native coils and native premolten globules
Figure 5 ▶ compares the amino acid compositions of intrinsic coils and intrinsic premolten globules. Protein sets analyzed in Figure 4 ▶ were used to create these graphs. The inset to Figure 5 ▶ shows that proteins from both subclasses occupy the same region of the charge-hydrophobicity phase space. Native coils were more dispersed, whereas intrinsic premolten globules were localized closer to the border between intrinsically unstructured and native proteins (cf. Fig. 2 ▶). To confirm this idea, the corresponding distances between the given sequence and the border between intrinsically unstructured and native proteins were calculated. Results of this comparison are shown in Figure 5 ▶ as Δ〈H〉 = (〈H〉boundary − 〈H〉) plots. The mean "boundary" hydrophobicity, 〈H〉boundary, for a given polypeptide chain with a mean net charge 〈R〉 has been calculated using equation 1. One can see that intrinsic coils are essentially more distant from the border than intrinsic premolten globules. Statistical analysis shows that the averaged Δ〈H〉 values are −(0.089 ± 0.086) and −(0.037 ± 0.033) for the native coils and native premolten globule, respectively. However, because the sequence characteristics of the two subclasses overlap, it is difficult to differentiate these proteins by taking into account their mean hydrophobicity and mean net charge only. Probably, some other sequence features, such as propensity to form secondary structure should also be considered.
Fig. 5.

Comparison of the amino acid compositions of intrinsic coils (gray circles) and intrinsic premolten globules (black circles) in terms of the distance of a given sequence from the border between rigid and intrinsically unstructured proteins. The distances were calculated as Δ〈H〉 = (〈H〉boundary − 〈H〉). The mean "boundary" hydrophobicity, 〈H〉boundary, for a given polypeptide chain with a mean net charge 〈R〉 has been calculated using equation 1. Inset shows that proteins from both subclasses occupy the same region of the charge-hydrophobicity phase space, with intrinsic coils being more dispersed.
Function-related folding of the intrinsically unstructured proteins
The functional importance of being disordered has been intensively analyzed (Schulz 1979; Pontius 1993; Dunker et al. 1997, 2001; Plaxco and Gross 1997; Wright and Dyson 1999). It has been established that increased intrinsic plasticity represents an important prerequisite for effective molecular recognition (Plaxco and Gross 1997; Wright and Dyson 1999; Dunker et al. 2001). The variety diapason of biological functions for intrinsically disordered proteins is very wide, including cell cycle control, transcriptional and translational regulation, modulation of activity and/or assembly of other proteins, and even regulation of nerve cell function (reviewed in Wright and Dyson 1999; Dunker et al. 2001). Importantly, it must be emphasized that the majority of intrinsically disordered proteins undergo a disorder-to-order transition upon functioning (Schulz 1979; Pontius 1993; Spolar and Record 1994; Rosenfeld et al. 1995; Plaxco and Gross 1997; Dunker et al. 1997, 2001; Wright and Dyson 1999). It has been suggested that the persistence of natively unfolded proteins throughout evolution may reside in advantages of flexible structure during disorder–order transitions in comparison with rigid proteins (Dunker et al. 1997, 1998, 2001; Romero et al. 1998b; Wright and Dyson 1999). Among the potential advantages of intrinsic lack of structure and function-related disorder–order transitions are (1) the possibility of high specificity coupled with low affinity (Schulz 1979; Kriwacki et al. 1996; Dunker et al. 1998, 2001); (2) the ability of binding to several different targets (Wright and Dyson 1999; Dunker et al. 2001), known as one to many signaling (Romero et al. 1998b); (3) the capability to overcome steric restrictions, enabling essentially larger interaction surfaces in the complex than could be obtained for the rigid partners (Meador et al. 1992; Choo and Schwabe 1998; Dunker et al. 2001); (4) the precise control and simple regulation of the binding thermodynamic (Schulz 1979; Spolar and Record 1994; Rosenfeld et al. 1995; Wright and Dyson 1999; Dunker et al. 2001); (5) the increased rates of specific macromolecular association (Pontius 1993; Dunker et al. 2001); and (6) the reduced lifetime of intrinsically disordered proteins in the cell, possibly representing a mechanism of rapid turnover of the important regulatory molecules (Wright and Dyson 1999). There is, however, an alternative explanation for the involvement of intrinsic disorder in protein function. By computer modeling it has been shown that selective pressure for functionality is rather unrelated to that for stability and foldability. In this view, a protein that is successfully folded into one structure would likely be as functional as a protein that successfully folded into an alternative structure. Including functionality in the model does not greatly alter the distribution of the observed structures (Williams et al. 2001). This could mean that metastable proteins are favored during evolution because there is a tremendously larger amount of sequences coding for these proteins compared to the very rigid ones. In other words, the involvement of intrinsic disorder in protein function may be related to history and evolution rather than to functional needs (Dunker et al. 2001).
In their excellent review, Dunker et al. (2001) formulated the idea that the protein structure–function paradigm (which emphasizes that ordered 3D structures represent the indispensable prerequisite to the effective protein functioning) should be altered as The Protein Trinity paradigm (see Fig. 6A ▶). According to The Protein Trinity model, native intracellular proteins (or their functional regions) can exist in any of the three thermodynamic states, ordered, molten globule, and random coil. Function can arise from any of the three conformations and transitions between them. "In this view, not just the ordered state, but any of the three states can be the native state of a protein" (Dunker et al. 2001). Experimental results on the conformational behavior of intrinsically unstructured (natively unfolded) proteins indicated, however, that these proteins did not possess uniform structural properties, as expected for members of one thermodynamic group, random coils. They were split into two structurally different subclasses, which, by analogy with conformational states of globular proteins, may be designated as intrinsic coils and intrinsic premolten globules. Moreover, it was already noted that molten globule and premolten globule might represent different phase states of the protein, as they are separated by the first-order phase transition (Uversky and Ptitsyn 1994, 1996a; Ptitsyn 1995; Uversky 1997, 1998). These observations bring a new player, the native premolten globule, on the protein functioning field. In other words, The Protein Trinity should be extended to The Protein Quartet model, with function arising from four specific conformations (ordered forms, molten globules, premolten globules, and random coils) and transitions between any two of the states (see Fig. 6B ▶). Experimental evidences for the validity of this extension are presented below.
Fig. 6.

Extension of The Protein Trinity (A, Dunker et al. 2001) to the Protein Quartet model of protein functioning (B). In accordance with this model, function arises from four specific conformations of the polypeptide chain (ordered forms, molten globules, premolten globules, and random coils) and transitions between any of the states.
Function-related coil–premolten globule transitions
It has been established that the binding of Zn2+ induces partial folding of intrinsically unfolded proteins, such as thymosin α1 (Grottesi et al. 1998), prothymosin α (Uversky et al. 2000b), human sperm protamines P2 and P3 (Gatewood et al. 1996), and phosphodiesterase γ-subunit (Uversky et al. 2002). Human α-synuclein was also shown to be partially folded in the presence of several divalent and trivalent metal ions (Uversky et al. 2001a). Analysis of structural changes associated with the cation binding showed that the transformation of intrinsic coils into premolten globule-like conformations took place in these cases.
Function-related coil–molten globule transitions
The myelin basic protein, MBP, is a major protein of myelin, the multilamellar membranous sheath surrounding nerve axons. MBP was isolated in water-soluble or detergent-soluble form together with endogenous myelin lipids. The water-soluble form is a member of the intrinsic coil family. Binding of lipids transformed this protein into the molten globule-like conformation (Polverini et al. 1999). Similar structural rearrangements were induced in the coil-like 77–262 fragment of the glucocorticoid receptor (Baskakov et al. 1999) and in the N-terminal domain of HIV-1 integrase (Zheng et al. 1996) by TMAO and by Zn2+ binding, respectively. Self-association of an intrinsically unfolded γ-subunit of phosphodiesterase induced folding of this protein into a molten globule-like conformation (Uversky et al. 2002).
Function-related coil–rigid structure transitions
The N-terminal domain of the caspase-activated DNA fragmentation factor DFF45 was unfolded in solution. Its folding into the rigid 3D structure was induced upon interaction with the N-terminal domain of DFF40 (Zhou et al. 2001). Structural analysis revealed that the isolated 50S ribosomal proteins, L22 and L27, and 30S ribosomal protein S19 were essentially unfolded in solution (Venyaminov et al. 1981). However, they transform into a rigid well-folded conformation in the functional ribosome (Yusupov et al. 2001).
Function-related premolten globule–molten globule transitions
The human antibacterial peptide LL-37 existed in the premolten globule like conformation at micromolar concentrations in aqueous solution. A cooperative transition from a disordered to a helical molten globule-like structure was observed in the presence of several anions or with increasing protein concentration. The extent of α-helicity correlated well with the antibacterial activity of LL-37 against both Gram-positive and Gram-negative bacteria (Johansson et al. 1998). Comparably the degree of folding was induced in osteocalcin as a result of binding of Ca2+, Lu3+ (Isbell et al. 1993) or Pb3+ (Dowd et al. 2001). Premolten globule to molten globule transitions accompanied Ca2+ binding to skeletal muscle sarcoplasmic reticulum calsequestrin (Cozens and Reithmeier 1984; He et al. 1993) and SPARC, an extracellular glycoprotein expressed in mineralized and nonmineralized tissues (Engel et al. 1987).
The DNA-binding domain of the 1,25-dihydroxyvitamin D3 receptor was shown to undergo a premolten globuleto-molten globule transition as a result of specific Zn2+ binding. This cation-induced folding was an important prerequisite to the formation of functional complex with osteopontin and several vitamin D response elements (Craig et al. 1997). The first step in steroidogenesis is the movement of cholesterol from the outer to inner mitochondrial membrane, which is facilitated by the steroidogenic acute regulatory protein StAR. The interaction of premolten globule-like StAR with dodecylphosphocholine and phospholipid liposomes was accompanied by transition of the protein into the molten globule conformation (Song et al. 2001). The E7 gene of the human papillomaviruses encodes a 98-amino acid chain of a multifunctional nuclear phosphoprotein, E7 protein, which cooperates with an activated ras oncogene to transform primary rodent cells. CD spectroscopy indicated that Zn2+ and Cd2+ binding by the HPV16 E7 protein induced structural transformations consistent with premolten globule–molten globule transition (Pahel et al. 1993).
Transitions of intrinsic premolten globules to rigid conformation
It was shown that self-dimerization and DNA binding induce rigid 3D structure in the intrinsic premolten globule-like Max protein (Ferre-D'Amare et al. 1993; Horiuchi et al. 1997). Specific Ca2+ binding initiated folding of the premolten globule-like B-repeat segment of SdrD (Josefsson et al. 1998). Similarly, the 50S ribosomal proteins L2, L3, L14, L23, L24, and L32, as well as the 30S ribosomal proteins S12 and S18 were native premolten globules in their free forms (Venyaminov et al. 1981), but adopted rigid well-folded conformations during the formation of a functional ribosome (Yusupov et al. 2001).
The Escherichia coli RNase HI variant, with the K86A mutation, was purified in two forms: nicked and intact. The nicked protein, resulting from the cleavage of a Lys87–Arg88 peptide bond, was enzymatically active. The N-terminal fragment possessed characteristics of molten globule, whereas the C-terminal fragment was essentially disordered. The premolten globule-like C-fragment underwent a transition to the rigid 3D structure as a result of RNase HI reconstitution (Kanaya and Kanaya 1995). Comparably, the intrinsically unstructured β-subunit of SMK killer toxin folded into a rigid conformation as a result of interaction with the α-subunit (Suzuki et al. 1997). Finally, the formation of a yeast SNARE complex was accompanied by a complete folding of the two of its components, Snc1 and Sec9 (Rice et al. 1997).
RNase P is the endoribonuclease responsible for the 5`-maturation of precursor tRNA transcript. Intriguingly, RNase P from Bacillus subtilis, being predominantly unfolded in 10 mM sodium cacodilate at neutral pH, folded into a native α/β structure upon addition of various small molecular anions (Henkels et al. 2001). This protein (Henkels et al. 2001), as well as the reduced RNase T1 (Baskakov and Bolen 1998), also underwent a cooperative folding transition upon addition of the osmolyte TMAO.
The cyclin-dependent kinase (Cdk) inhibitor p21Waf1/Cip1/Sdi1 and its N-terminal fragment lack stable secondary and tertiary structure in the free solution state. In sharp contrast to the disordered free solution state, these proteins adopted an ordered stable conformation when bound to Cdk2 (Kriwacki et al. 1996).
Conclusions
Conformational analysis shows that natively unfolded proteins do not represent a uniform family, but rather two structurally different groups. Proteins from the first group have hydrodynamic dimensions typical of random coils in poor solvent (i.e., they behave as slightly squeezed coils) and do not possess any (or almost any) ordered secondary structure. Proteins from the second group are essentially more compact (but still significantly less compact than native or molten globule proteins). They exhibit some amount of ordered secondary structure being characterized by far-UV CD spectra as typical essentially disordered polypeptide chain, with a pronounced minimum in the vicinity of 200 nm. By analogy with the conformational classification of "normal" globular proteins, intrinsically unstructured proteins could be divided in intrinsic coils and intrinsic premolten globules.
Because the amino acid sequences of native coils are similar to native premolten globules (only slightly less hydrophobic and slightly more charged), some other sequence features (e.g., propensity to form secondary structure) have to be taken into account for the unambiguous sequence-based separation of the intrinsic coils from the intrinsic premolten globules.
An intriguing property of intrinsically unstructured proteins is their capability to undergo disorder-to-order transition upon functioning. The degree of these structural rearrangements varies over a very wide range, from coil–premolten globule transitions to formation of rigid ordered structures. Thus, protein functioning may be described by the Protein Quartet model, with biological activity arising from four unique conformations of the polypeptide chain (ordered forms, molten globules, premolten globules, and random coils) and transitions between any of them.
Acknowledgments
I am grateful to Prof. A.K. Dunker for the valuable discussions. I thank Dr. P. Souillac for the careful reading and editing of the manuscript. I appreciate Prof. J. Goers for his invaluable help with the manuscript improvement.
Abbreviations
NMR, nuclear magnetic resonance
CD, circular dichroism
UV, ultraviolet
ORD, optical rotatory dispersion
FTIR, Fourier-transform infrared
SAXS, small-angle X-ray scattering
SANS, small-angle neutron scattering
FRET, fluorescence resonance energy transfer
N, native
MG, molten globule
PMG, premolten globule
U, unfolded
NU, natively unfolded
NUcoil, natively unfolded proteins with coil-like properties
NUPMG, natively unfolded proteins with PMG-like properties
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4210102.
References
- Abercrombie, B.D., Kneale, G.G., Crane Robinson, C., Bradbury, E.M., Goodwin, G.H., Walker, J.M., and Johns, E.W. 1978. Studies on the conformational properties of the high-mobility-group chromosomal protein HMG 17 and its interaction with DNA. Eur. J. Biochem. 84 173–177. [DOI] [PubMed] [Google Scholar]
- Adler, A.J., Greenfield, N.J., and Fasman, G.D. 1973. Circular dichroism and optical rotatory dispersion of proteins and polypeptides. Methods Enzymol. 27 675–735. [DOI] [PubMed] [Google Scholar]
- Agianian, B., Leonard, K., Bonte, E., Van der Zandt, H., Becker, P.B., and Tucker, P.A. 1999. The glutamine-rich domain of the Drosophila GAGA factor is necessary for amyloid fiber formation in vitro, but not for chromatin remodelling. J. Mol. Biol. 285 527–544. [DOI] [PubMed] [Google Scholar]
- Alber, T., Gilbert, W.A., Ponzi, D.R., and Petsko, G.A. 1982. The role of mobility in the substrate binding and catalytic machinery of enzymes. Ciba Found. Symp. 93 4–24. [DOI] [PubMed] [Google Scholar]
- Alexandrescu, A.T., Abeygunawardana, C., and Shortle, D. 1994. Structure and dynamics of a denatured 131-residue fragment of staphylococcal nuclease: A heteronuclear NMR study. Biochemistry 33 1063–1072. [DOI] [PubMed] [Google Scholar]
- Amit, A.G., Mariuzza, R.A., Phillips, S.E.V., and Dolyak, R.J. 1985. Three-dimensional structure of an antigen–antibody complex at 6 Å resolution. Nature 313 156–158. [DOI] [PubMed] [Google Scholar]
- Anfinsen, C.B., Haber, E., Sela, M., and White, F.N. 1961. Kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. 47 1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson, M.L. and Mirsky, A.E. 1932. The effect of denaturation on the viscosity of protein systems. J. Gen. Physiol. 15 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aswad, D.W. and Greengard, P. 1981. A specific substrate from rabbit cerebellum for guanosine 3`:5`-monophosphate-dependent protein kinase. I. Purification and characterization. J. Biol. Chem. 256 3487–3493. [PubMed] [Google Scholar]
- Bai, Y., Chung, J., Dyson, H.J., and Wright, P.E. 2001. Structural and dynamic characterization of an unfolded state of poplar apo-plastocyanin formed under nondenaturing conditions. Protein Sci. 10 1056–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, R.L. and Zimm, B.H. 2000. Are denatured proteins ever random coils? Proc. Natl. Acad. Sci. 97 12391–12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskakov, I. and Bolen, D.W. 1998. Forcing thermodynamically unfolded proteins to fold. J. Biol. Chem. 273 4831–4834. [DOI] [PubMed] [Google Scholar]
- Baskakov, I.V., Kumar, R., Srinivasan, G., Ji, Y.S., Bolen, D.W., and Thompson, E.B. 1999. Trimethylamine N-oxide-induced cooperative folding of an intrinsically unfolded transcription-activating fragment of human glucocorticoid receptor. J. Biol. Chem. 274 10693–10696. [DOI] [PubMed] [Google Scholar]
- Baum, J., Dobson, C.M., Evans, P.A., and Hanly, C. 1989. Characterization of a partly folded protein by NMR methods: Studies on the molten globule state of guinea pig alpha-lactalbumin. Biochemistry 28 7–13. [DOI] [PubMed] [Google Scholar]
- Belmont, L.D. and Mitchison, T.J. 1996. Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 84 623–631. [DOI] [PubMed] [Google Scholar]
- Berkovits, H.J. and Berg, J.M. 1999. Metal and DNA binding properties of a two-domain fragment of neural zinc finger factor 1, a CCHC-type zinc binding protein. Biochemistry 38 16826–16830. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya, J. and Das, K.P. 1999. Molecular chaperone-like properties of an unfolded protein, alpha(s)-casein. J. Biol. Chem. 274 15505–15509. [DOI] [PubMed] [Google Scholar]
- Bienkiewicz, E.A., Moon Woody, A., and Woody, R.W. 2000. Conformation of the RNA polymerase II C-terminal domain: Circular dichroism of long and short fragments. J. Mol. Biol. 297 119–133. [DOI] [PubMed] [Google Scholar]
- Bloomer, A.C., Champness, J.N., Bricogne, G., Staden, R., and Klug, A. 1978. A protein disk of tobacco mosaic virus at 2.8 Å resolution showing the interactions within and between subunits. Nature 276 362–368. [DOI] [PubMed] [Google Scholar]
- Bode, W., Schwager, P., and Huber, R. 1978. The transition of bovine trypsinogen to trypsin-like state upon strong ligand binding. The refined crystal structures of the bovine trypsinogen–pancreatic trypsin inhibitor complex and of its ternary complex with Ile-Val at 1.9 Å resolution. J. Mol. Biol. 118 99–112. [DOI] [PubMed] [Google Scholar]
- Bogdarina, I., Fox, D.G., and Kneale, G.G. 1998. Equilibrium and kinetic binding analysis of the N-terminal domain of the Pf1 gene 5 protein and its interaction with single-stranded DNA. J. Mol. Biol. 275 443–452. [DOI] [PubMed] [Google Scholar]
- Bose, H.S., Whittal, R.M., Baldwin, M.A., and Miller, W.L. 1999. The active form of the steroidogenic acute regulatory protein, StAR, appears to be a molten globule. Proc. Natl. Acad. Sci. 96 7250–7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier, M. and Stafford, W.P. 2000. Probing the three-dimensional structure of human calreticulin. Biochemistry 39 14950–14959. [DOI] [PubMed] [Google Scholar]
- Bracken, C. 2001. NMR spin relaxation methods for characterization of disorder and folding in proteins. J. Mol. Graph. Model. 19 3–12. [DOI] [PubMed] [Google Scholar]
- Bushnell, G.W., Louie, G.V., and Brayer, G.D. 1990. High-resolution three-dimensional structure of horse heart cytochrome c. J. Mol. Biol. 214 585–595. [DOI] [PubMed] [Google Scholar]
- Campbell, K.M., Terrell, A.R., Laybourn, P.J., and Lumb, K. J. 2000. Intrinsic structural disorder of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry 39 2708–2713. [DOI] [PubMed] [Google Scholar]
- Cary, P.D., Crane Robinson, C., Bradbury, E.M., and Dixon, G.H. 1981. Structural studies of the non-histone chromosomal proteins HMG-T and H6 from trout testis. Eur. J. Biochem. 119 545–551. [DOI] [PubMed] [Google Scholar]
- Cary, P.D., King, D.S., Crane Robinson, C., Bradbury, E.M., Rabbani, A., Goodwin, G.H., and Johns, E.W. 1980. Structural studies on two high-mobility-group proteins from calf thymus, HMG-14 and HMG-20 (ubiquitin), and their interaction with DNA. Eur. J. Biochem. 112 577–580. [DOI] [PubMed] [Google Scholar]
- Chang, J.F., Phillips, K., Lundback, T., Gstaiger, M., Ladbury, J.E., and Luisi, B. 1999. Oct-1 POU and octamer DNA co-operate to recognise the Bob-1 transcription co-activator via induced folding. J. Mol. Biol. 288 941–952. [DOI] [PubMed] [Google Scholar]
- Cho, H.S., Liu, C.W., Damberger, F.F., Pelton, J.G., Nelson, H.C.M., and Wemmer, D.E. 1996. Yeast heat shock transcription factor N-terminal activation domains are unstructured as probed by heteronuclear NMR spectroscopy. Protein Sci. 5 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo, Y. and Schwabe, J.W. 1998. All wrapped up. Nat. Struct. Biol. 5 253–255. [DOI] [PubMed] [Google Scholar]
- Chyan, C.-L., Wormald, C., Dobson, C.M., Evans, P.A., and Baum, J. 1993. Structure and stability of the molten globule state of guinea-pig alpha-lactalbumin: A hydrogen exchange study. Biochemistry 32 5681–5691. [DOI] [PubMed] [Google Scholar]
- Cozens, B. and Reithmeier, R.A. 1984. Size and shape of rabbit muscle calsequestrin. J. Biol. Chem. 259 6248–6252. [PubMed] [Google Scholar]
- Craig, T.A., Veenstra, T.D., Naylor, S., Tomlinson, A.J., Johnson, K.L., Macura, S., Juranic, N., and Kumar, R. 1997. Zinc binding properties of the DNA binding domain of the 1,25-dihydroxyvitamin D3 receptor. Biochemistry 36 10482–10491. [DOI] [PubMed] [Google Scholar]
- Daughdrill, G.W., Hanely, L.J., and Dahlquist, F.W. 1998. The C-terminal half of the anti-sigma factor FlgM contains a dynamic equilibrium solution structure favoring helical conformations. Biochemistry 37 1076–1082. [DOI] [PubMed] [Google Scholar]
- Delmas, P.D., Stenner, D.D., Romberg, R.W., Riggs, B.L., and Mann, K.G. 1984. Immunochemical studies of conformational alterations in bone gamma-carboxyglutamic acid containing protein. Biochemistry 23 4720–4725. [DOI] [PubMed] [Google Scholar]
- Demarest, S.J., Zhou, S.Q., Robblee, J., Fairman, R., Chu, B., and Raleigh, D.P. 2001. A comparative study of peptide models of the alpha-domain of alpha-lactalbumin, lysozyme, and alpha-lactalbumin/lysozyme chimeras allows the elucidation of critical factors that contribute to the ability to form stable partially folded states. Biochemistry 40 2138–2147. [DOI] [PubMed] [Google Scholar]
- DiGiammarino, E.L., Filippov, I., Weber, J.D., Bothner, B., and Kriwacki, R.W. 2001. Solution structure of the p53 regulatory domain of the p19Arf tumor suppressor protein. Biochemistry 40 2379–2386. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. and Shortle, D. 1991. Denatured states of proteins. Annu. Rev. Biochem. 60 795–825. [DOI] [PubMed] [Google Scholar]
- Di Stasio, E., Sciandra, F., Maras, B., Di Tommaso, F., Petrucci, T.C., Giardina, B., and Brancaccio, A. 1999. Structural and functional analysis of the N-terminal extracellular region of beta-dystroglycan. Biochem. Biophys. Res. Commun. 266 274–278. [DOI] [PubMed] [Google Scholar]
- Donaldson, L. and Capone, J.P. 1992. Purification and characterization of the carboxyl-terminal transactivation domain of Vmw65 from herpes simplex virus type 1. J. Biol. Chem. 267 1411–1414. [PubMed] [Google Scholar]
- Donne, D.G., Viles, J.H., Groth, D., Mehlhorn, I., James, T. L., Cohen, F.E., Prusiner, S.B., Wright, P.E., and Dyson, H.J. 1997. Structure of the recombinant full-length hamster prion protein PrP(29–231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. 94 13452–13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd, T.L., Rosen, J.F., Mints, L., and Gundberg, C.M. 2001. The effect of Pb(2+) on the structure and hydroxyapatite binding properties of osteocalcin. Biochim. Biophys. Acta 1535 153–163. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Garner, E., Guilliot, S., Romero, P., Albercht, K., Hart, J., Obradovic, Z., Kissinger, C., and Villafranca, J.E. 1998. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 3 473–484. [PubMed] [Google Scholar]
- Dunker, A.K., Lawson, J.D., Brown, C.J., Williams, R.M., Romero, P., Oh, J.S., Oldfield, C.J., Campen, A.M., Ratliff, C.M., Hipps, K.W., Ausio, J., Nissen, M.S., Reeves, R., Kang, C.-H., Kissinger, C.R., Bailey, R.W., Griswold, M.D., Chiu, W., Garber, E.C., and Obradovic, Z. 2001. Intrinsically disordered proteins. J. Mol. Graph. Model. 19 26–59. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Obradovic, Z., Romero, P., Kissinger, C., and Villafranca, J.E. 1997. On the importance of being disordered. PDB Newslett. 81 3–5. [Google Scholar]
- Eliezer, D., Chiba, K., Tsuruta, H., Doniach, S., Hodgson, K.O., and Kihara, H. 1993. Evidence of an associative intermediate on the myoglobin refolding pathway. Biophys. J. 65 912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliezer, D., Yao, J., Dyson, H.J., and Wright, P.E. 1998. Structural and dynamic characterization of partially folded states of myoglobin and implications for protein folding. Nat. Struct. Biol. 5 148–155. [DOI] [PubMed] [Google Scholar]
- Engel, J., Taylor, W., Paulsson, M., Sage, H., and Hogan, B. 1987. Calcium binding domains and calcium-induced conformational transition of SPARC/BM-40/osteonectin, an extracellular glycoprotein expressed in mineralized and nonmineralized tissues. Biochemistry 26 6958–6965. [DOI] [PubMed] [Google Scholar]
- Eom, J.W., Baker, W.R., Kintanar, A., and Wurtele, E.S. 1996. The embryo-specific EMB-1 protein of Daucus carota is flexible and unstructured in solution. Plant Sci. 115 17–24. [Google Scholar]
- Fasman, G.D. 1996. Circular dichroism and conformational analysis of biomolecules. Plenum Press, New York.
- Feigin, L.A. and Svergun, D.I. 1987. Structural snalysis by small-angle X-ray and neutron scattering. Plenum Press, New York.
- Ferre-D'Amare, A.R., Prendergast, G.C., Ziff, E.B., and Burley, S.K. 1993. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 363 38–45. [DOI] [PubMed] [Google Scholar]
- Fiebig, K.M., Rice, L.M., Pollock, E., and Brunger, A.T. 1999. Folding intermediates of SNARE complex assembly. Nat. Struct. Biol. 6 117–123. [DOI] [PubMed] [Google Scholar]
- Fischer, E. 1894. Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges. 27 2985–2993. [Google Scholar]
- Fisher, L.W., Torchia, D.A., Fohr, B., Young, M.F., and Fedarko, N.S. 2001. Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochem. Biophys. Res. Commun. 280 460–465. [DOI] [PubMed] [Google Scholar]
- Fletcher, C.M., McGuire, A.M., Gingras, A.C., Li, H., Matsuo, H., Sonenberg, N., and Wagner, G. 1998. 4E binding proteins inhibit the translation factor eIF4E without folded structure. Biochemistry 37 9–15. [DOI] [PubMed] [Google Scholar]
- Flory, P.J. 1953. Principles of polymer chemistry. Cornell University Press, Ithaca, NY.
- Fontana, A., Polverino de Laureto, P., and De Philipps, V. 1993. Molecular aspects of proteolysis of globular proteins. In Protein stability and stabilization (eds., W. van den Tweel, A. Harder, and M. Buitelear), pp. 101–110. Elsevier Science, Amsterdam.
- Fox, D.G., Cary, P.D., and Kneale, G.G. 1999. Conformational studies of the C-terminal domain of bacteriophage Pf1 gene 5 protein. Biochim. Biophys. Acta 1435 138–146. [DOI] [PubMed] [Google Scholar]
- Fujio, H., Takagaki, Y., Ha, Y. M., Doi, E.M., Soebandrio, A., and Sakato, N. 1985. Native and non-native conformation-specific antibodies directed to the loop region of hen egg-white lysozyme. J. Biochem. (Tokyo) 98 949–962. [DOI] [PubMed] [Google Scholar]
- Furie, B. and Furie, B.C. 1979. Conformation-specific antibodies as probes of the gamma-carboxyglutamic acid-rich region of bovine prothrombin. Studies of metal-induced structural changes. J. Biol. Chem. 254 9766–9771. [PubMed] [Google Scholar]
- Garner, E., Cannon, P., Romero, P., Obradovic, Z., and Dunker, A.K. 1998. Predicting disordered regions from amino acid sequence: Common themes despite different structural characterization. Genome Informatics 9 201–213. [PubMed] [Google Scholar]
- Gast, K., Damaschun, H., Eckert, K., Schulze-Foster, K., Maurer, H.R., Müller-Frohne, M., Zirwer, D., Czarnecki, J., and Damaschun, G. 1995. Prothymosin α: A biologically active protein with random coil conformation. Biochemistry 34 13211–13218. [DOI] [PubMed] [Google Scholar]
- Gatewood, J.M., Schroth, G.P., Schmid, C.W., and Bradbury, E.M. 1990. Zinc-induced secondary structure transitions in human sperm protamines. J. Biol. Chem. 265 20667–20672. [PubMed] [Google Scholar]
- Geyer, M., Munte, C. E., Schorr, J., Kellner, R., and Kalbitzer, H.R. 1999. Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J. Mol. Biol. 289 123–138. [DOI] [PubMed] [Google Scholar]
- Gillespie, J.R. and Shortle, D. 1997a. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J. Mol. Biol. 268 158–169. [DOI] [PubMed] [Google Scholar]
- Gillespie, J.R. and Shortle, D. 1997b. Characterization of long-range structure in the denatured state of staphylococcal nuclease. II. Distance restraints from paramagnetic relaxation and calculation of an ensemble of structures. J. Mol. Biol. 268 170–184. [DOI] [PubMed] [Google Scholar]
- Glatter, O. and Kratky, O. 1982. Small angle X-ray scattering. Academic Press, London.
- Grossberg, A.Yu. and Khohlov, A.R. 1989. Statistical physics of macromolecules. Nauka, Moscow.
- Grottesi, A., Sette, M., Palamara, T., Rotilio, G., Garaci, E., and Paci, M. 1998. The conformation of peptide thymosin alpha 1 in solution and in a membrane-like environment by circular dichroism and NMR spectroscopy. A possible model for its interaction with the lymphocyte membrane. Peptides 19 1731–1738. [DOI] [PubMed] [Google Scholar]
- Guo, R.T., Chou, L.J., Chen, Y.C., Chen, C.Y., Pari, K., Jen, C.J., Lo, S.J., Huang, S.L., Lee, C.Y., Chang, T.W., and Chaung, W.J. 2001. Expression in Pichia pastoris and characterization by circular dichroism and NMR of rhodostomin. Proteins Struct. Funct. Genet. 43 499–508. [DOI] [PubMed] [Google Scholar]
- Hazzard, J., Sudhol, T.C., and Rizo, J. 1999. NMR analysis of the structure of synaptobrevin and of its interaction with syntaxin. J. Biomol. NMR 14 203–207. [DOI] [PubMed] [Google Scholar]
- He, Z., Dunker, A.K., Wesson, C.R., and Trumble, W.R. 1993. Ca(2+)-induced folding and aggregation of skeletal muscle sarcoplasmic reticulum calsequestrin. The involvement of the trifluoperazine-binding site. J. Biol. Chem. 268 24635–24641. [PubMed] [Google Scholar]
- Hemmings, H.G., Jr., Nairin, A.C., Aswad, D.W., and Greengard, P. 1984. DARPP-32, a dopamine-and adenosine 3`:5`-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. II. Purification and characterization of the phosphoprotein from bovine caudate nucleus. J. Biol. Chem. 4 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkels, C.H., Kurz, J.C., Fierke, C.A., and Oas, T.G. 2001. Linked folding and anion binding of the Bacillus subtilis ribonuclease P protein. Biochemistry 40 2777–2789. [DOI] [PubMed] [Google Scholar]
- Hernández, M.A., Avila, J., and Andreu, J.M. 1986. Physicochemical characterization of the heat-stable microtubule-associated protein MAP2. Eur. J. Biochem. 154 41–48. [DOI] [PubMed] [Google Scholar]
- Hershey, P.E., McWhirter, S.M., Gross, J.D., Wagner, G., Alber, T., and Sachs, A.B. 1999. The Cap-binding protein eIF4E promotes folding of a functional domain of yeast translation initiation factor eIF4G1. J. Biol. Chem. 274 21297–21304. [DOI] [PubMed] [Google Scholar]
- Horiuchi, M., Kurihara, Y., Katahira, M., Maeda, T., Saito, T., and Uesugi, S. 1997. Dimerization and DNA binding facilitate alpha-helix formation of Max in solution. J. Biochem. (Tokyo) 122 711–716. [DOI] [PubMed] [Google Scholar]
- House-Pompeo, K., Xu, Y., Joh, D., Speziale, P., and Hook, M. 1996. Conformational changes in the fibronectin binding MSCRAMMs are induced by ligand binding. J. Biol. Chem. 271 1379–1384. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.J. 1998. The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta 1382 191–206. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.J., Beynon, R.J., and Thornton, J.M. 1998. Assessment of conformational parameters as predictors of limited proteolytic sites in native protein structures. Protein Eng. 11 349–359. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.J., Eisenmenger, F., and Thornton, J.M. 1994. Modeling studies of the change in conformation required for cleavage of limited proteolytic sites. Protein Sci. 3 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, R. 1979. Conformational flexibility and its functional significance in some protein molecules. Trends Biochem. Sci. 4 271–276. [Google Scholar]
- Huber, R. 1987. Flexibility and rigidity, requirements for the function of proteins and protein pigment complexes. Biochem. Soc. Trans. 15 1009–1020. [DOI] [PubMed] [Google Scholar]
- Hughson, F.M., Wright, P.E., and Baldwin, R.L. 1990. Structural characterization of a partly folded apomyoglobin intermediate. Science 249 1544–1548. [DOI] [PubMed] [Google Scholar]
- Iakoucheva, L.M., Kimzey, A.L., Masselon, C.D., Bruce, J.E., Garner, E.C., Brown, C.J., Dunker, A.K., Smith, R.D., and Ackerman, E.J. 2001. Identification of intrinsic order and disorder in the DNA repair protein XPA. Protein Sci. 10 560–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbell, D.T., Du, S., Schroering, A.G., Colombo, G., and Shelling, J.G. 1993. Metal ion binding to dog osteocalcin studied by 1H NMR spectroscopy. Biochemistry 32 11352–11362. [DOI] [PubMed] [Google Scholar]
- Jeng, M.F., Englander, S.W., Elöve, G.A., Wang, A.I., and Roder, H. 1990. Structural description of acid-denatured cytochrome c by hydrogen exchange and 2D NMR. Biochemistry 29 10433–10437. [DOI] [PubMed] [Google Scholar]
- Jimenez, M.A., Evangelio, J.A., Aranda, C., Lopez-Brauet, A., Andreu, D., Rico, M., Lagos, R., Andreu, J.M., and Monasterio, O. 1999. Helicity of alpha(404–451) and beta(394–445) tubulin C-terminal recombinant peptides. Protein Sci. 8 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, J., Gudmundsson, G.H., Rottenberg, M.E., Berndt, K.D., and Agerberth, B. 1998. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 273 3718–3724. [DOI] [PubMed] [Google Scholar]
- Johnson, W.C., Jr. 1988. Secondary structure of proteins through circular dichroism spectroscopy. Annu. Rev. Biophys. Chem. 17 145–166. [DOI] [PubMed] [Google Scholar]
- Josefsson, E., O'Connell, D., Foster, T.J., Durussel, I., and Cox, J.A. 1998. The binding of calcium to the B-repeat segment of SdrD, a cell surface protein of Staphylococcus aureus. J. Biol. Chem. 273 31145–31152. [DOI] [PubMed] [Google Scholar]
- Kanaya, E. and Kanaya, S. 1995. Reconstitution of Escherichia coli RNase HI from the N-fragment with high helicity and the C-fragment with a disordered structure. J. Biol. Chem. 270 19853–19860. [DOI] [PubMed] [Google Scholar]
- Kataoka, M., Hagihara, Y., Mihara, K., and Goto, Y. 1993. Molten globule of cytochrome c studied by small angle X-ray scattering. J. Mol. Biol. 229 591–596. [DOI] [PubMed] [Google Scholar]
- Kataoka, M., Kuwajima, K., Tokunaga, F., and Goto, Y. 1997. Structural characterization of the molten globule of alpha-lactalbumin by solution X-ray scattering. Protein Sci. 6 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, S.M. and Price, N.C. 1997. The application of circular dichroism to studies of protein folding and unfolding. Biochim. Biophys. Acta 1338 161–185. [DOI] [PubMed] [Google Scholar]
- Koepf, E.K., Petrassi, H.M., Ratnaswamy, G., Huff, M.E., Sudol, M., and Kelly, J.W. 1999. Characterization of the structure and function of W → F WW domain variants: Identification of a natively unfolded protein that folds upon ligand binding. Biochemistry 38 14338–14351. [DOI] [PubMed] [Google Scholar]
- Konno, T., Tanaka, N., Kataoka, M., Takano, E., and Maki, M. 1997. A circular dichroism study of preferential hydration and alcohol effects on a denatured protein, pig calpastatin domain I. Biochim. Biophys. Acta 1342 73–82. [DOI] [PubMed] [Google Scholar]
- Kostyukova, A., Maeda, K., Yamauchi, E., Krieger, I., and Maeda, Y. 2000. Domain structure of tropomodulin: Distinct properties of the N-terminal and C-terminal halves. Eur. J. Biochem. 267 6470–6475. [DOI] [PubMed] [Google Scholar]
- Krebs, D., Dahmani, B., el Antri, S., Monnot, M., Convert, O., Mauffret, O., Troalen, F., and Fermandjian, S. 1995. The basic subdomain of the c-Jun oncoprotein. A joint CD, Fourier-transform infrared and NMR study. Eur. J. Biochem. 231 370–380. [DOI] [PubMed] [Google Scholar]
- Kriwacki, R.W., Hengst, L., Tennant, L., Reed, S.I., and Wright, P.E. 1996. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. 93 11504–11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, R.W., Yang, J., Hou, S., Helms, M.K., Jameson, D.M., and Alam, M. 1999. Spectroscopic characterization of two soluble transducers from the Archaeon Halobacterium salinarum. J. Protein Chem. 18 269–275. [DOI] [PubMed] [Google Scholar]
- Lewis, M., Chang, G., Horton, N.C., Kercher, M.A., Pace, H.C., Schumacher, M.A., Brennan, R.G., and Lu, P. 1996. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science 271 1247–1254. [DOI] [PubMed] [Google Scholar]
- Li, X., Obradovic, Z., Brown, C.L., Garner, E., and Dunker, A.K. 2000. Comparing predictors of disordered protein. Genome Informatics 11 172–184. [PubMed] [Google Scholar]
- Li, X., Romero, P., Rani, M., Dunker, A.K., and Obradovic, Z. 1999. Predicting protein disorders for N-, C-, and internal regions. Genome Informatics 10 30–40. [PubMed] [Google Scholar]
- Lisse, T., Bartels, D., Kalbitzer, H.R., and Jaenicke, R. 1996. The recombinant dehydrin-like desiccation stress protein from the resurrection plant Craterostigma plantagineum displays no defined three-dimensional structure in its native state. Biol. Chem. 377 555–561. [DOI] [PubMed] [Google Scholar]
- Liu, D., Ishima, R., Tong, K.I., Bagby, S., Kokubo, T., Muhandiram, D. R., Kay, L.E., Nakatani, Y., and Ikura, M. 1998. Solution structure of a TBP-TAF(II)230 complex: Protein mimicry of the minor groove surface of the TATA box unwound by TBP. Cell 94 573–583. [DOI] [PubMed] [Google Scholar]
- Logan, T.M., Theriault, Y., and Fesik, S.W. 1994. Structural characterization of the FK506 binding protein unfolded in urea and guanidine hydrochloride. J. Mol. Biol. 236 637–648. [DOI] [PubMed] [Google Scholar]
- Loomis, R.E., Bergey, E.J., Levine, M.J., and Tabak, L.A. 1985. Circular dichroism and fluorescence spectroscopic analyses of a proline-rich glycoprotein from human parotid saliva. Int. J. Pept. Protein Res. 26 621–629. [DOI] [PubMed] [Google Scholar]
- Love, J.J. 1999. Biophysical characterization of HMG-1 box domain of the lymphoid enhancer binding factor-1. PhD. Thesis, University of California, San Diego.
- Lydakis-Simantiris, N., Betts, S.D., and Yocum, C.F. 1999. Leucine 245 is a critical residue for folding and function of the manganese stabilizing protein of photosystem II. Biochemistry 38 15528–15535. [DOI] [PubMed] [Google Scholar]
- Lynch, W.P., Riseman, V.M., and Bretscher, A. 1987. Smooth muscle caldesmon is an extended flexible monomeric protein in solution that can readily undergo reversible intra- and intermolecular sulfhydryl cross-linking. A mechanism for caldesmon's F-actin bundling activity. J. Biol. Chem. 262 7429–7437. [PubMed] [Google Scholar]
- Lynn, A., Chandra, S., Malhotra, P., and Chauhan, V.S. 1999. Heme binding and polymerization by Plasmodium falciparum histidine rich protein II: Influence of pH on activity and conformation. FEBS Lett. 459 267–271 [DOI] [PubMed] [Google Scholar]
- Markus, G. 1965. Protein substrate conformation and proteolysis. Proc. Natl. Acad. Sci. 54 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaldon, P. and Argos, P. 1988. Oligopeptide biases in protein sequences and their use in predicting protein coding regions in nucleotide sequences. Proteins Struct. Funct. Genet. 4 99–122. [DOI] [PubMed] [Google Scholar]
- McCubbin, W.D. and Kay, C.M. 1985. Hydrodynamic and optical properties of the wheat EM protein. Can. J. Biochem. 63 803–810. [Google Scholar]
- Meador, W.E., Means, A.R., and Quiocho, F.A. 1992. Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin–peptide complex. Science 257 1251–1255. [DOI] [PubMed] [Google Scholar]
- Merrill, A.R., Cohen, F.S., and Cramer, W.A. 1990. On the nature of the structural change of the colicin E1 channel peptide necessary for its translocation-competent state. Biochemistry 29 5829–5836. [DOI] [PubMed] [Google Scholar]
- Mikhalyi, E. 1978. Application of proteolytic enzymes to protein structure studies. CRC Press, Boca Raton, FL.
- Mirsky, A.E. and Pauling, L. 1936. On the structure of native, denatured and coagulated proteins. Proc. Natl. Acad. Sci. 22 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogridge, J., Legault, P., Li, J., Van Oene, M. D., Kay, L.E., and Greenblatt, J. 1998. Independent ligand-induced folding of the RNA-binding domain and two functionally distinct antitermination regions in the phage lambda N protein. Mol. Cell 1 265–275. [DOI] [PubMed] [Google Scholar]
- Muchmore, S.W., Sattler, M., Liang, H., Meadows, R.P., Harlan, J.E., Yoon, H.S., Nettesheim, D., Chang, B.S., Thompson, C.B., Wong, S.L., Ng, S.L., and Fesik, S.W. 1996. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381 335–341. [DOI] [PubMed] [Google Scholar]
- Mullen, G.P., Vaughn, J.B., Jr, and Mildvan, A.S. 1993. Sequential proton NMR resonance assignments, circular dichroism, and structural properties of a 50-residue substrate-binding peptide from DNA polymerase I. Arch. Biochem. Biophys. 301 174–183 [DOI] [PubMed] [Google Scholar]
- Nakano, M., Kasai, K., Yoshida, K., Tanimoto, T., Tamaki, Y., and Tobita, T. 1989. Conformation of the fowl protamine, galline, and its binding properties to DNA. J. Biochem. (Tokyo) 105 133–137. [DOI] [PubMed] [Google Scholar]
- Neurath, H., Greenstein, J.P., Putnam, F.W., and Erickson, J.O. 1944. The chemistry of protein denaturation. Chem. Rev. 34 157–265. [Google Scholar]
- Nimmo, G.A. and Cohen, P. 1978. The regulation of glycogen metabolism. Purification and characterisation of protein phosphatase inhibitor-1 from rabbit skeletal muscle. Eur. J. Biochem. 87 341–351. [DOI] [PubMed] [Google Scholar]
- Pahel, G., Aulabaugh, A., Short, S.A., Barnes, J.A., Painter, G.R., Ray, P., and Phelps, W.C. 1993. Structural and functional characterization of the HPV16 E7 protein expressed in bacteria. J. Biol. Chem. 268 26018–26025. [PubMed] [Google Scholar]
- Pappu, R.V., Srinivasan, R., and Rose, G.D. 2000. The Flory isolated-pair hypothesis is not valid for polypeptide chains: Implications for protein folding. Proc. Natl. Acad. Sci. 97 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelta, J., Berry, H., Fadda, G.C., Pauthe, E., and Lairez, D. 2000. Statistical conformation of human plasma fibronectin. Biochemistry 39 5146–5154. [DOI] [PubMed] [Google Scholar]
- Penkett, C.J., Redfield, C., Dodd, I., Hubbard, J., McBay, D.L., Mossakowska D.E., Smith, R.A.G., Dobson, C.M., and Smith, L.J. 1997. NMR analysis of main chain conformational preferences in an unfolded fibronectin-binding protein. J. Mol. Biol. 274 152–159. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W. and Gross, M. 1997. The importance of being unfolded. Nature 386 657–659. [DOI] [PubMed] [Google Scholar]
- Polverini, E., Fasano, A., Zito, F., Riccio, P., and Cavatorta, P. 1999. Conformation of bovine myelin basic protein purified with bound lipids. Eur. Biophys. J. 28 351–355. [DOI] [PubMed] [Google Scholar]
- Pontius, B.W. 1993. Close encounters: Why unstructured, polymeric domains can increase rates of specific macromolecular association. Trends Biochem. Sci. 18 181–186. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. 1979. Stability of proteins: Small globular proteins. Adv. Protein Chem. 33 167–241. [DOI] [PubMed] [Google Scholar]
- Provencher, S.W. and Glöckner, J. 1981. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20 33–37. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47 83–229. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. and Uversky, V.N. 1994. The molten globule is a third thermodinamical state of protein molecules. FEBS Lett. 341 15–18. [DOI] [PubMed] [Google Scholar]
- Ratnaswamy, G., Koepf, E., Bekele, H., Yin, H., and Kelly, J.W. 1999. The amyloidogenicity of gelsolin is controlled by proteolysis and pH. Chem. Biol. 6 293–304. [DOI] [PubMed] [Google Scholar]
- Rice, L.M., Brennwald, P., and Brünger, A.T. 1997. Formation of a yeast SNARE complex is accompanied by significant structural changes. FEBS Lett. 415 49–55. [DOI] [PubMed] [Google Scholar]
- Richards, J.P., Bächinger, H.P., Goodman, R.H., and Brennan, R.G. 1996. Analysis of the structural properties of cAMP-responsive element-binding protein (CREB) and phosphorylated CREB. J. Biol. Chem. 271 13716–13723. [DOI] [PubMed] [Google Scholar]
- Romero, P., Obradovic, Z., and Dunker, A.K. 1998a. Sequence data analysis for long distorted regions prediction in the calcineurin family. Genome Informatics 8 110–124. [PubMed] [Google Scholar]
- Romero, P., Obradovic, Z., and Dunker, A.K. 2001a. Intelligent data analysis for protein disorder prediction. Artif. Intell. Rev. 14 447–484. [Google Scholar]
- Romero, P., Obradovic, Z., Kissinger, C., Villafranca, J.E., and Dunker, A.K. 1997. Identifying disordered regions in proteins from amino acid sequence. Proc IEEE Int. Conf. Neuronal Networks 1997 1 90–95. [Google Scholar]
- Romero, P., Obradovic, Z., Kissinger, C., Villafranca, J.E, Garner, E., Guilliot, S., and Dunker, A.K. 1998b. Thousands of proteins likely to have long disordered regions. Pac. Symp. Biocomput. 3 437–448. [PubMed] [Google Scholar]
- Romero, P., Obradovic, Z., Li, X., Garner, E.C., Brown, C.J., and Dunker, A.K. 2001b. Sequence complexity of disordered proteins. Protein Struct. Funct. Genet. 42 38–48. [DOI] [PubMed] [Google Scholar]
- Rosenfeld, P., Vajda, S., and DeLisi, C. 1995. Flexible docking and design. Annu. Rev. Biophys. Biomol. Struct. 24 677–700. [DOI] [PubMed] [Google Scholar]
- Schmitz, M.L., dos Santos Silva, M.A., Altmann, H., Czisch, M., Holak, T.A., and Baeuerle, P.A. 1994. Structural and functional analysis of the NF-kappa B p65 C terminus. An acidic and modular transactivation domain with the potential to adopt an alpha-helical conformation. J. Biol. Chem. 269 25613–25620 [PubMed] [Google Scholar]
- Schulz, G.E. 1979. Nucleotide binding proteins. In Molecular mechanism of biological recognition (ed., M. Balaban), pp. 79–94. Elsevier/North-Holland Biomedical Press, New York.
- Schweers, O., Schönbrunn Hanebeck, E., Marx, A., and Mandelkow, E. 1994. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 269 24290–24297. [PubMed] [Google Scholar]
- Semisotnov, G.V., Kihara, H., Kotova, N.V., Kimura, K., Amemiya, Y., Wakabayashi, K., Serdyuk, I.N., Timchenko, A.A., Chiba, K., Nikaido, K., Ikura, T., and Kuwajima. K. 1996. Protein globularization during folding. A study by synchrotron small-angle X-ray scattering. J. Mol. Biol. 262 559–574. [DOI] [PubMed] [Google Scholar]
- Semisotnov, G.V., Rodionova, N.V., Razgulyaev, O.I., Uversky, V.N., Gripas, A.F., and Gilmanshin, R.I. 1991. Study of the "molten globule" intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31 119–128. [DOI] [PubMed] [Google Scholar]
- Shortle, D. 1996. The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J. 10 27–34. [DOI] [PubMed] [Google Scholar]
- Shortle, D. and Meeker, A.K. 1989. Residual structure in large fragments of staphylococcal nuclease: Effects of amino acid substitutions. Biochemistry 28 936–944. [DOI] [PubMed] [Google Scholar]
- Smyth, E., Syme, C.D., Blanch, E.W., Hecht, L., Vašák, M., Barron, L.D. 2001. Solution structure of native proteins with irregular folds from Raman optical activity. Biopolymers 58 138–151. [DOI] [PubMed] [Google Scholar]
- Song, M., Shao, H., Mujeeb, A., James, T.L., and Miller, W.L. 2001. Molten-globule structure and membrane binding of the N-terminal protease-resistant domain (63–193) of the steroidogenic acute regulatory protein (StAR). Biochem. J. 356 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spolar, R.S. and Record II, M.T. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science 263 777–784. [DOI] [PubMed] [Google Scholar]
- Stellwagen, E., Rysary, R., and Babul, G. 1972. The conformation of horse heart apocytochrome c. J. Biol. Chem. 247 8074–8077. [PubMed] [Google Scholar]
- Stewart, L., Ireton, G.C., Parker, L.H., Madden, K.R., and Champoux, J.J. 1996. Biochemical and biophysical analyses of recombinant forms of human topoisomerase I. J. Biol. Chem. 271 7593–7601. [DOI] [PubMed] [Google Scholar]
- Suzuki, C., Kashiwagi, T., Tsuchiya, F., Kunishima, N., Morikawa, K., Nikkuni, S., and Arata, Y. 1997. Circular dichroism analysis of the interaction between the alpha and beta subunits in a killer toxin produced by a halotolerant yeast, Pichia farinosa. Protein Eng. 10 99–101. [DOI] [PubMed] [Google Scholar]
- Tanford, C. 1961. Physical chemistry of macromolecules. Wiley, New York.
- Tanford, C. 1968. Protein denaturation. Adv. Protein Chem. 23 121–282. [DOI] [PubMed] [Google Scholar]
- Tangrea, M.A., Alexander, P., Bryan, P.N., Eisenstein, E., Toedt, J., and Orban, J. 2001. Stability and global fold of the mouse prohormone convertase 1 pro-domain. Biochemistry 40 5488–5495. [DOI] [PubMed] [Google Scholar]
- Tarkka, T., Oikarinen, J., and Grundström, T. 1997. Nucleotide and calcium-induced conformational changes in histone H1. FEBS Lett. 406 56–60. [DOI] [PubMed] [Google Scholar]
- Tcherkasskaya, O. and Uversky, V.N. 2001. Denatured collapsed states in protein folding: Example of apomyoglobin. Proteins Struct. Funct. Genet. 44 244–254. [DOI] [PubMed] [Google Scholar]
- Tell, G., Perrone, L., Fabbro, D., Pellizzari, L., Pucillo, C., De Felice, M., Acquaviva, R., Formisano, S., and Damante, G. 1998. Structural and functional properties of the N transcriptional activation domain of thyroid transcription factor-1: Similarities with the acidic activation domains. Biochem. J. 329 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, J., Van Patten, S.M., Howard, P., Day, K.H., Mitchell, R.D., Sosnick, T., Trewhella, J., Walsh, D.A., and Maurer, R.A. 1991. Expression in Escherichia coli and characterization of the heat-stable inhibitor of the cAMP-dependent protein kinase. J. Biol. Chem. 266 10906–10911. [PubMed] [Google Scholar]
- Timm, D.E., Vissavajjhala, P., Ross, A.H., and Neet, K.E. 1992. Spectroscopic and chemical studies of the interaction between nerve growth factor (NGF) and the extracellular domain of the low affinity NGF receptor. Protein Sci. 1 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V.N. 1993. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry 32 13288–13298. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. 1994. Gel-permeation chromatography as a unique instrument for quantitative and qualitative analysis of protein denaturation and unfolding. Int. J. Bio-Chromatogr. 1 103–114. [Google Scholar]
- Uversky, V.N. 1997. Diversity of compact forms of denatured globular proteins Protein Pept. Lett. 4 355–367. [Google Scholar]
- Uversky, V.N. 1998. How many molten globule states there exist? Biofizika (Moscow) 43 416–421. [PubMed] [Google Scholar]
- Uversky, V.N. 1999. A multiparametric approach to studies of self-organization of globular proteins. Biochemistry (Moscow) 64 250–266. [PubMed] [Google Scholar]
- Uversky, V.N. 2002. What does it mean to be natively unfolded? Eur. J. Biochem. 269 2–12. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. and Ptitsyn O.B. 1994. "Partly folded" state, a new equilibrium state of protein molecules: Four-state guanidinium chloride-induced unfolding of β-lactamase at low temperature. Biochemistry 33 2782–2791. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. and Ptitsyn, O.B. 1996a. Further evidence on the equilibrium "pre-molten globule state": Four-state GdmCl-induced unfolding of carbonic anhydrase B at low temperature. J. Mol. Biol. 255 215–228. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. and Ptitsyn, O.B. 1996b. All-or-none solvent-induced transitions between native, molten globule and unfolded states in globular proteins. Fold. Design 1 117–122. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Gillespie, J.R., and Fink, A.L. 2000a. Why are "natively unfolded" proteins unstructured under the physiological conditions? Proteins Struct. Funct. Genet. 41 415–427. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Gillespie, J.R., Millett, I.S., Khodyakova, A.V., Vasiliev, A.M., Chernovskaya, T.V., Vasilenko, R.N., Kozlovskaya, G.D., Dolgikh, D.A., Doniach, S., Fink, A.L., and Abramov, V.M. 1999. "Natively unfolded" human prothymosin α adopts partially-folded conformation at acidic pH. Biochemistry 38 15009–15016. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Gillespie, J.R., Millett, I.S., Khodyakova, A.V., Vasilenko, R.N., Vasiliev, A.M., Rodionov, I.L., Kozlovskaya, G.D., Dolgikh, D.A., Doniach, S., Fink, A.L., Permyakov, E.A., and Abramov, V.M. 2000b. Zn2+-mediated structure formation and compaction of the "natively unfolded" human prothymosin α. Biochem. Biophys. Res. Comunun. 267 663–668. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Karnoup, A.S., Segel, D.J., Seshadri, S., Doniach, S., and Fink, A.L. 1998. Anion-induced folding of Staphylococcal nuclease: Characterization of multiple partially folded intermediates. J. Mol. Biol. 278 879–894. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Li, J., and Fink, A.L. 2001a. Metal-triggered structural transformations, aggregation and fibril formation of human α-synuclein. A possible molecular link between Parkinson's disease and heavy metal exposure. J. Biol. Chem. 276 44284–44296. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Evidence for a partially-folded intermediate in α-synuclein fibrillation. J. Biol. Chem. 276 10737–10744. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Permyakov, S.E., Zagranichny, V.E., Rodionov, I.L., Fink, A.L., Cherskaya, A.M., Wasserman, L.A., and Permyakov, E.A. 2002. Effect of zinc and temperature on the conformation of the γ subunit of retinal phosphodiesterase: A natively unfolded protein. J. Proteome Res. In press. [DOI] [PubMed]
- Uversky, V.N., Winter, S., and Löber, G. 1996. Use of fluorescence decay times of 8-ANS–protein complexes to study the conformational transitions in proteins which unfold through the molten globule state. Biophys. Chem. 60 79–88. [DOI] [PubMed] [Google Scholar]
- Venyaminov, S.Yu., Gudkov, A.T., Gogia, Z.V., and Tumanova, L.G. 1981. Absorption and Circular Dichroism Spectra of Individual Proteins from Escherichia Coli Ribosomes. Pushchino, Russia.
- Weinreb, P.H., Zhen, W., Poon, A.W., Conway, K.A., and Lansbury, P.T., Jr. 1996. NACP, a protein implicated in Alzheimer's diseases and learning, is natively unfolded. Biochemistry 35 13709–13715. [DOI] [PubMed] [Google Scholar]
- Weiss, M.A., Ellenberger, T., Wobbe, C.R., Lee, J.P., Harrison, S.C., and Struhl, K. 1990. Folding transition in the DNA-binding domain of GCN4 on specific binding to DNA. Nature 347 575–578. [DOI] [PubMed] [Google Scholar]
- Williams, P.D., Pollock, D.D., and Goldstein, R.A. 2001. Evolution of functionality in lattice proteins. J. Mol. Graph. Model. 19 150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, K.J. 1994. Interaction with model membrane systems induces secondary structure in amino-terminal fragments of parathyroid hormone related protein. Int. J. Pept. Protein Res. 43 23–28. [DOI] [PubMed] [Google Scholar]
- Wilson, I.A., Haft, D.H., Getzoff, E.D., Tainer, J.A., Lerner, R.A., and Brenner, S. 1985. Identical short peptide sequences in unrelated proteins can have different conformations: A testing ground for theories of immune recognition. Proc. Natl. Acad. Sci. 82 5255–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woody, R.W. 1995. Circular dichroism. Methods Enzymol. 246 34–71. [DOI] [PubMed] [Google Scholar]
- Wootton, J.C. 1993. Statistics of local complexity in amino acid sequence and sequence databases. Comput. Chem. 17 149–163. [Google Scholar]
- Wootton, J.C. 1994. Non-globular domains in protein sequences: Automated segmentation using complexity measures. Comput. Chem. 18 269–285. [DOI] [PubMed] [Google Scholar]
- Wootton, J.C. and Federhen, S. 1996. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 266 554–571. [DOI] [PubMed] [Google Scholar]
- Worbs, M., Huber, R., and Wahl, M.C. 2000. Crystal structure of ribosomal protein L4 shows RNA-binding sites for ribosome incorporation and feedback control of the S10 operon. EMBO J. 19 807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, P.E. and Dyson, H.J. 1999. Intrinsically unstructured proteins: Re-assessing the protein structure–function paradigm. J. Mol. Biol. 293 321–331. [DOI] [PubMed] [Google Scholar]
- Wu, L.C., Laub, P.B., Elöve, G.A., Carey, J., and Roder, H. 1993. A noncovalent peptide complex as a model for an early folding intermediate of cytochrome c. Biochemistry 32 10271–10276. [DOI] [PubMed] [Google Scholar]
- Yoo, S.H. 1995. pH- and Ca(2+)-induced conformational change and aggregation of chromogranin B. Comparison with chromogranin A and implication in secretory vesicle biogenesis. J. Biol. Chem. 270 12578–12583. [DOI] [PubMed] [Google Scholar]
- Yoo, S.H. and Albanesi, J.P. 1990. Ca2(+)-induced conformational change and aggregation of chromogranin A. J. Biol. Chem. 265 14414–14421. [PubMed] [Google Scholar]
- Yusupov, M.M., Yusupova, G.Z., Baucom, A., Lieberman, K., Earnest, T.N., Cate, J.H., and Noller, H.F. 2001. Crystal structure of the ribosome at 5.5 A resolution. Science 292 883–896. [DOI] [PubMed] [Google Scholar]
- Zhang, J. and Matthews, C.R. 1998. Ligand binding is the principal determinant of stability for the p21H-ras protein. Biochemistry 37 14881–14890. [DOI] [PubMed] [Google Scholar]
- Zhang, O., Kay, L.E., Olivier, J.P., and Forman-Kay, J.D. 1994. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR 4 845–858. [DOI] [PubMed] [Google Scholar]
- Zheng, R., Jenkins, T.M., and Craigie, R. 1996. Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc. Natl. Acad. Sci. 93 13659–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P., Lugovskoy, A.A., McCarty, J.S., Li, P., and Wagner, G. 2001. Solution structure of DFF40 and DFF45 N-terminal domain complex and mutual chaperone activity of DFF40 and DFF45. Proc. Natl. Acad. Sci. 98 6051–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]