

Abstract
Analytical ultracentrifugation (AU) is reemerging as a versatile tool for the study of proteins. Monitoring the sedimentation of macromolecules in the centrifugal field allows their hydrodynamic and thermodynamic characterization in solution, without interaction with any matrix or surface. The combination of new instrumentation and powerful computational software for data analysis has led to major advances in the characterization of proteins and protein complexes. The pace of new advancements makes it difficult for protein scientists to gain sufficient expertise to apply modern AU to their research problems. To address this problem, this review builds from the basic concepts to advanced approaches for the characterization of protein systems, and key computational and internet resources are provided. We will first explore the characterization of proteins by sedimentation velocity (SV). Determination of sedimentation coefficients allows for the modeling of the hydrodynamic shape of proteins and protein complexes. The computational treatment of SV data to resolve sedimenting components has been achieved. Hence, SV can be very useful in the identification of the oligomeric state and the stoichiometry of heterogeneous interactions. The second major part of the review covers sedimentation equilibrium (SE) of proteins, including membrane proteins and glycoproteins. This is the method of choice for molar mass determinations and the study of self-association and heterogeneous interactions, such as protein–protein, protein–nucleic acid, and protein–small molecule binding.
Keywords: Sedimentation velocity, sedimentation equilibrium, protein interactions, reversible association, hydrodynamic shape, membrane proteins
Although analytical ultracentrifugation (AU) played a notable role in the history of the characterization of proteins and protein complexes (Schachman 1992), this methodology has suffered a decline in use for many years, in considerable part because of a lack of new instrumentation capable of digital data acquisition. Fortunately, the capabilities of AU have been transformed by the combination of new instrumentation and major developments in computational software for data analysis. Very important new approaches for the determination of sedimentation coefficients and the deconvolution of sedimenting species have been introduced with respect to the analysis of boundary sedimentation velocity (SV) data. With regard to sedimentation equilibrium (SE) analysis, investigators now have the ability to determine association constants for many homogeneous and heterogeneous interacting systems from such measurements. Although there is an abundant literature using these new developments for characterizing protein systems, numerous investigators, and particularly new investigators, are unfamiliar with AU methodologies. The objective of this review is to assist protein scientists to gain a greater understanding of AU and the power of SV and SE for the characterization of proteins and protein complexes. This seems timely as the interest in methodology for characterizing the proteome and the interactome are increasingly important. Although AU is typically not conducted with high throughput, it is firmly based on equilibrium and nonequilibrium thermodynamics and does represent the gold standard for characterizing the hydrodynamic properties of proteins and protein complexes, as well as molar-mass and binding-constant determinations.
The limitations of a short tutorial review require a high degree of selectivity of the topics that can be covered. The basic principles are treated only briefly, but we refer to appropriate textbooks and other literature that will allow the reader to gain sufficient knowledge for the successful application of AU. We give considerable attention to software resources for AU data analysis, because this is of great importance when attempting to apply AU. As investigators of the Molecular Interactions Resource of NIH, we have emphasized both SV and SE methodologies as implemented in our laboratory. With respect to the former, we emphasize our approach to modeling boundary SV and size-distribution analysis of sedimenting proteins and compare our new developments with past contributions of other investigators that have focused on this issue. In addition to boundary SV, we also review the principles and methodology for band SV analysis. The second part of this review focuses on SE analysis of proteins and protein complexes.
Instrumentation for analytical ultracentrifugation
Beckman-Coulter Instruments has introduced two analytical ultracentrifuges, the XLA and the XLI. The former has UV and visible absorption optics for the detection of biopolymers, and the latter has integrated absorbance and interference optics. The enhanced features of interference optics are as follows: (1) All biological macromolecules can be detected through refractive index changes, and consequently, nonabsorbing biopolymers, such as polysaccharides, can be investigated. (2) Ligand- or drug-induced changes in protein conformation or association (e.g., ATP- or GTP-binding proteins) can be analyzed without concern that the UV absorbance of the ligand/drug will obscure the protein absorbance. (3) Macromolecular solute concentrations can be increased well beyond the range of the absorbance system, thereby allowing a much greater concentration range for SE. (4) Because interference patterns are recorded from the entire cell at once, large data sets can be rapidly accumulated for SV experiments, significantly improving the computational analysis for the detection of sedimenting species. The disadvantage of interference optics is that more care has to be taken in matching the volume and chemical composition of the sample and reference columns and ensuring that the optics are correctly adjusted. The choice of the appropriate optical detection, if one has an XLI, is dependent on the experimental system under investigation (Schuck and Braswell 2000).
Sedimentation velocity characterization of proteins and protein complexes
Principles of sedimentation velocity and basic hydrodynamics
The application of a centrifugal force causes the depletion of macromolecules at the meniscus and the formation of a concentration boundary that moves toward the bottom of the centrifuge cell as a function of time, Figure 1 ▶. The definition of the sedimentation coefficient of a macromolecule, s, and the molecular parameters that determine the s-value are given by the well-known Svedberg equation:
 |
(1) |
where u is the observed radial velocity of the macromolecule, ω is the angular velocity of the rotor, r is the radial position, ω2r is the centrifugal field, M is the molar mass, v is the partial specific volume, ρ is the density of the solvent, NA is Avogadro's number, f is the frictional coefficient, D is the diffusion coefficient, and R is the gas constant. The relationship D = RT/NAf was used to obtain the right-hand version of the Svedberg equation. The s-values are commonly reported in Svedberg (S) units, which correspond to 10−13 sec.
Fig. 1.

SV data of a bovine serum albumin sample. Shown are the concentration versus radius distributions at different times after start of the sedimentation at 50,000 rpm. Concentrations are in units of fringe displacement in the interference optical system, which corresponds to ∼0.3 mg/mL per fringe.
The Stokes equation can be used to determine the f-value for smooth, compact spherical proteins:
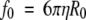 |
(2) |
where f0 is the frictional coefficient of the spherical particle, η is the viscosity of the solution, and R0 is the radius of the sphere. One can combine the Svedberg equation and Stokes equation (Teller et al. 1979; van Holde et al. 1998), in which the R0 of the sphere is expressed as
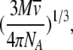 |
to obtain
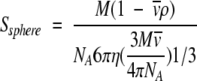 |
(3) |
Substituting the values for all the constants (η for water at 20°C) yields equation 4, the s-value for a sphere in terms of M, v , and ρ only (where M is in units of daltons, s in S units, v in milliliters per gram, and ρ in grams per milliliter).
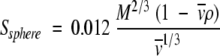 |
(4) |
Using equation 4, one can predict the sedimentation velocity coefficients for smooth compact spherical proteins in water at 20°C. This ssphere-value is the maximum s-value that can be obtained for a protein of a given mass, because a compact sphere has the minimum surface area in contact with solvent and consequently the protein would have a minimum frictional coefficient, f0. A correction of the experimental s-value to a standard state of water at 20°C is necessary for comparative purposes of data obtained from different laboratories as well as under different experimental conditions. The standard correction equation is given as:
 |
(5) |
where T and B designate the values at the temperature and under the buffer conditions of the experiment, and the index 20,w indicates standard conditions. The ratio of the maximum s-value to the observed s-value, ssphere/s20,w, is equal to the ratio of the experimental frictional coefficient to the minimum frictional coefficient (f/f0), which measures the maximum shape asymmetry from a sphere. The frictional ratio can also be determined from s-values under experimental conditions as the ratio of the ssphere from equation 3 (substituting appropriate constants) divided by the experimental s-value.
Characterization of proteins using boundary sedimentation velocity
For the sake of simplicity, let us first consider a single component system. The motion of the boundary as a function of time determines the s-value. Depending on the optical system chosen, this experiment typically requires 0.05 to 0.5 mg of material. The software for the determination of s-values is described below. The evaluation of s20,w from the experimental s-value via equation 5 can be rapidly accomplished using the public domain software program SEDNTERP (http://www.rasmb.bbri.org/) developed by Hayes, Laue, and Philo. SEDNTERP also provides rapid determination of the η and ρ for a large variety of solutions. In addition, SEDNTERP calculates  from the amino acid composition of the protein. The latter can be imported from a data bank in either the one-letter or three-letter code for amino acids. If one has access to an Anton-Paar DMA 5000 density meter or comparable precise density measuring instrumentation, then
from the amino acid composition of the protein. The latter can be imported from a data bank in either the one-letter or three-letter code for amino acids. If one has access to an Anton-Paar DMA 5000 density meter or comparable precise density measuring instrumentation, then  values can be determined experimentally (Kratky et al. 1973; although this requires ∼5 mg, which may not be available). Once s20,w has been determined, one can ask the following question: Is the s20,w consistent with the sequence molar mass of a monomer? Equation 4 can be used to estimate the maximum s20,w-value for a monomer. If the experimental s20,w is significantly higher, one can conclude that the quaternary state is not monomeric, whereas a lower value would indicate an extended shape of the monomer caused by a larger frictional coefficient. For example, let us assume that you have a protein with a molar mass of 50 kD and a
values can be determined experimentally (Kratky et al. 1973; although this requires ∼5 mg, which may not be available). Once s20,w has been determined, one can ask the following question: Is the s20,w consistent with the sequence molar mass of a monomer? Equation 4 can be used to estimate the maximum s20,w-value for a monomer. If the experimental s20,w is significantly higher, one can conclude that the quaternary state is not monomeric, whereas a lower value would indicate an extended shape of the monomer caused by a larger frictional coefficient. For example, let us assume that you have a protein with a molar mass of 50 kD and a 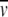 of 0.730 mL/g and you perform a boundary SV experiment in phosphate-buffered saline and obtain an experimental s-value of 5.67 S. This corrects to an s20,w of 5.87 S. Using equation 4, you would predict a maximal s-value of 4.93 S based on the monomeric molar mass. Clearly, the experimental s20,w is much higher than the predicted value for a spherical monomer, pointing to self-association. A dimer would have a theoretical s20,w value of 7.77 S. As indicated above, ssphere/s20,w measures the maximum shape asymmetry of the protein, f/f0, and for the above example f/f0 would be 1.33, which supports the formation of a dimer with a moderately extended shape.
of 0.730 mL/g and you perform a boundary SV experiment in phosphate-buffered saline and obtain an experimental s-value of 5.67 S. This corrects to an s20,w of 5.87 S. Using equation 4, you would predict a maximal s-value of 4.93 S based on the monomeric molar mass. Clearly, the experimental s20,w is much higher than the predicted value for a spherical monomer, pointing to self-association. A dimer would have a theoretical s20,w value of 7.77 S. As indicated above, ssphere/s20,w measures the maximum shape asymmetry of the protein, f/f0, and for the above example f/f0 would be 1.33, which supports the formation of a dimer with a moderately extended shape.
We are now in position to do some basic hydrodynamic modeling of the putative dimer. The total shape asymmetry f/f0 can be separated into two factors, a geometrical shape asymmetry and a hydration expansion:
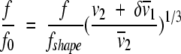 |
(6) |
The symbol δ denotes the hydration of the protein in grams of water per gram of protein, for which a consensus value is commonly taken to be 0.3 g/g (Perkins 2001). The partial specific volume subscripts 1 and 2 denote solvent and protein, respectively. The right-hand term in parentheses is the volume asymmetry due to hydration, or a hydration frictional ratio f/fhyd. In our example, because f/f0 is 1.33, we obtain, with δ of 0.3 g/g, frictional ratios f/fhyd of 1.12 and f/fshape of 1.19. The simplest hydrodynamic shape analysis consists in the approximation of the protein shape by a prolate or oblate ellipsoid. SEDNTERP can determine f/fshape and the axial ratio of the respective ellipsoid model. Our hypothetical protein dimer would be hydrodynamically equivalent to a prolate ellipsoid with an axial ratio of 4, with axial dimensions of 17.3 nm (2a) and 4.3 nm (2b).
It is evident from the above example that we have gained considerable information from a boundary SV experiment. It is also evident that the SEDNTERP software greatly facilitates the ability to determine all the experimental parameters relevant in SV and performs basic hydrodynamic calculations. SEDNTERP has a companion paper covering many of the points discussed above in greater depth (Laue et al. 1992). The help menu of SEDNTERP tersely covers hydrodynamic concepts with references. For more advanced hydrodynamic concepts, the reader is referred to the reviews of Teller et al. (1979) and Garcia de la Torre (1992).
The above discussion of basic hydrodynamic principles opens for consideration the questions of how the experimental s-value is extracted from the measured data, possible deconvolution of multiple sedimenting components, and other advanced topics. These are discussed below.
Determination of sedimentation coefficients and interpretation of sedimentation velocity experiments
The data measured in AU are concentration profiles in the radial direction as a function of time (Fig. 1 ▶). Hence, conceptually the simplest determination of a macromolecular sedimentation coefficient is based on the formation of a sedimentation boundary in a high centrifugal field, where the s-value might be determined, for example, by the displacement of the boundary midpoint (Fig. 1 ▶; Svedberg and Pedersen 1940). However, this method is mainly of historical relevance, as modern computational techniques have enabled much more powerful approaches, such as modeling the data directly with the underlying transport equation (the Lamm equation):
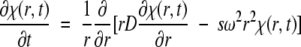 |
(7) |
Equation 7 describes the evolution of the concentration distribution of macromolecular species χ as a function of time and radial position under the influence of sedimentation and diffusion in the sector-shaped ultracentrifugal sample cell (Lamm 1929).
Modeling the sedimentation data with the Lamm equation 7 takes full advantage of the rich data basis of the full sedimentation process (Fig. 1 ▶), which can typically consist of 105 data points with a signal-to-noise ratio between 100 and 1000. The precision of the sedimentation coefficients increases with rotor speed, and typically is between 0.1% and 1%. Nonlinear least-squares regression can be performed with several software packages operating on the Windows platform, including LAMM (authored by J. Behlke and O. Ristau; see ftp://ftp.rasmb.bbri.org/rasmb/spin/), SVEDBERG (by J. Philo; http://www.jphilo.mailway.com/svedberg.htm), and Sedfit (by P. Schuck; http://www.analyticalultracentrifugation.com), or for the Unix environment UltraScan (by B. Demeler; see http://www.ultrascan.uthscsa.edu/ ). Lamm and Svedberg use approximate analytical solutions to equation 7 (Behlke and Ristau 1997; Philo 1997), whereas UltraScan and Sedfit use different numerical finite element solutions (Demeler and Saber 1998; Schuck 1998). An advantage of the latter approach is its generality, which allows sedimentation coefficients to be determined even where no clearly visible boundary is formed, theoretically only requiring any molecular redistribution as a result of the applied centrifugal force. In practice, Sedfit can be used to model a wide range of sedimentation processes, from the redistribution of salts (typically <0.1 S) or the sedimentation of small molecules, peptides, proteins, and protein complexes, to large particles such as virus capsids or dispersion particles (>1000 S). Further, it allows determination of both sedimentation and flotation coefficients (e.g., for detergents or lipoproteins with 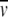 > 1/ρsol), and modeling of experimental configurations generated with synthetic boundaries where the initial distribution is not uniform (such as analytical zone centrifugation; Lebowitz et al. 1998; see below). Sedfit also has a comprehensive set of tools adjusting the analysis for the special noise structure of interference optical data (Schuck and Demeler 1999). A detailed online help system and several introductory tutorials are available. Recently, Sedphat, a version of Sedfit for global modeling of multiple experiments (including sedimentation equilibrium and dynamic light scattering), has been introduced and applied for the simultaneous fit of sedimentation velocity experiments at different rotor speeds, with improved resolution of different species.
> 1/ρsol), and modeling of experimental configurations generated with synthetic boundaries where the initial distribution is not uniform (such as analytical zone centrifugation; Lebowitz et al. 1998; see below). Sedfit also has a comprehensive set of tools adjusting the analysis for the special noise structure of interference optical data (Schuck and Demeler 1999). A detailed online help system and several introductory tutorials are available. Recently, Sedphat, a version of Sedfit for global modeling of multiple experiments (including sedimentation equilibrium and dynamic light scattering), has been introduced and applied for the simultaneous fit of sedimentation velocity experiments at different rotor speeds, with improved resolution of different species.
As usual when modeling experimental data, the analysis results in measures of goodness of fit (e.g., the rms error of the fit and the distribution of the residuals), which allow assessment if the model satisfactorily describes the sedimentation process. If a solution to the Lamm equation 7 (or a superposition of a small number of Lamm equation solutions) is not a good model, it is possible that either a larger number of different sedimenting species is present, and/or that chemical reactions are present (reversible interactions) on the time scale of the sedimentation experiment. Both cases can be distinguished from the details of the time course of sedimentation, and from the concentration-dependent sedimentation behavior shown by interacting proteins.
Analysis of size distributions
The analysis of multicomponent protein mixtures or protein samples with possible contamination by peptides or aggregates can be of considerable importance for a complete characterization of a protein system under investigation. Such systems may have resolvable sedimentation species, and one would observe two or more boundaries as a function of time. However, diffusional spreading of components often leads to only a single observable boundary composed of nonresolved multiple sedimenting components. Figure 1 ▶ shows a single boundary for a BSA sample that was composed of a partial proteolytic breakdown product, monomers, and oligomeric species based on size exclusion chromatography. We use this sedimentation boundary data to illustrate the analysis of size distributions by different computational treatments.
For the past decade, two approaches have been used extensively for unraveling sedimenting components: the integral sedimentation coefficient distribution G(s) (van Holde and Weischet 1978); and the dc/dt method for calculating a differential apparent sedimentation coefficient distribution g(s*) (Stafford 1992). The G(s) method is based on a geometric division of the sedimentation boundary and attempts to resolve sedimenting species by extrapolating each boundary division to infinite time to eliminate the effect of diffusion on sedimentation. The extrapolated s-values for each boundary fraction produce an integral sedimentation coefficient distribution G(s) (for review see Hansen et al. 1994). An adaptation of this methodology to the noise structure of interference optical data has been described (Schuck et al. 2002). It is implemented in the software UltraScan and Sedfit. The G(s) method has great diagnostic value for the presence of heterogeneity and of attractive or repulsive interactions (Hansen et al. 1994; Demeler et al. 1997). Characterization of the lipid free form of the apolipoprotein A-1 used the van Holde-Weischet G(s) method to show the conformational plasticity of the protein and how N-terminal deletions reduced conformational transitions significantly (Rodgers et al. 1998a,b).
Stafford's dc/dt approach subtracts closely spaced boundary scans to approximate a set of time-derivative dc/dt versus radius profiles. Based on the equation for sedimentation of an ideal nondiffusing species, the radial coordinate is transformed into an s-value, designated s*, and the time derivative dc/dt is transformed into dc/ds, which is a differential sedimentation coefficient distribution, designated g(s*). g(s*) distributions can be computed with the software Dcdt+ developed by J. Philo (http://www.jphilo.mailway.com/dcdt+.htm). This method has been very successful, in part because of the invariance of dc/dt with respect to the systematic noise structure of interference optical ultracentrifuge data. However, no correction for diffusion is made, which limits the resolution (see below). Furthermore, the approximation of dc/dt with finite differences of sequential scans causes constraints in the rotor speed and the number of scans that can be analyzed (Philo 2000a). More recently, a least-squares variant of the apparent sedimentation coefficient distribution from direct boundary modeling (with equation 7 and taking D = 0) has been introduced, termed ls-g*(s) and implemented in Sedfit, which eliminates some of these constraints and can be applied equally to absorbance and interference data (Schuck and Rossmanith 2000). The g*(s) method has been reviewed by Laue (2001). An example from our work is the use of the g*(s) analysis to monitor the binding of the SmtB repressor to different DNA target sequences and the hydrodynamic characterization of the binding data. These results were coupled with SE for determining the protein–DNA stoichiometry, which provided a working model for the formation of the repression complex (Kar et al. 2001).
More recently, a differential sedimentation coefficient distribution that deconvolutes diffusion effects, based on direct boundary modeling with a distribution of Lamm equation solutions, has been implemented. In brief, a sedimentation coefficient distribution c(s) can be defined as
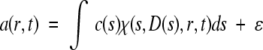 |
(8) |
with a(r,t) denoting the observed sedimentation data, c(s) the concentration of species with sedimentation coefficients between s and s + ds, and χ(s,D(s),r,t) the Lamm equation solution described above. Recently, mathematical methods for solving equation 8 have been described, using maximum entropy regularization and implemented in SEDFIT (Schuck 2000). Different variants for estimating the relationship of s and D for mixtures of globular proteins are available, with the most general one based on a weight-average shape factor f/f0 that can also be extracted from the experimental data. Conversion of the c(s) distribution into a molar mass distribution c(M) is possible. Because of the assumption of a weight-average shape factor f/f0, c(M) may not lead to correct molar mass values for all species. In contrast, the approximation of diffusion by a weight-average shape factor f/f0 has virtually no effect on the peak value of the sedimentation coefficient distribution c(s). For cases where the molar mass of the main species is known, this can be used as prior knowledge in the calculation of c(s). More details are described in Schuck (2000) and Schuck et al. (2002).
The result of a c(s) sedimentation coefficient distribution for a SV experiment of bovine serum albumin is shown in Figure 2 ▶. It is apparent that the sedimentation coefficient distribution that has been deconvoluted is conceptually similar to a chromatogram from gel filtration. However, it should be noted that the separation is achieved without resorting to interactions with a matrix, that it is based on differences in mass and friction, with the former more strongly size-dependent (∼M2/3) than the Stokes radius (∼M1/3), and that sedimentation coefficient distributions have a much wider dynamic range. Although the diffusional broadening in the raw data is less in gel filtration, the database in sedimentation is very large and its solid foundation on first principles allows for diffusional deconvolution. The s-value of each peak can be interpreted as described above. It is apparent from the c(s) curve in Figure 2 ▶ that the BSA monomer, dimer, and trimer are baseline separated (and resolved from a small degradation product at 2.7 S). Figure 2 ▶ also compares results from using the G(s) and g*(s) approaches. Although both indicate the presence of different BSA oligomers, neither technique is successful in resolving them. It has been argued that the results from g(s*) are conceptually similar to those of c(s) (Laue 2001), but a very clear qualitative difference in the resolution is apparent when comparing a g(s*) analysis on our BSA data (dotted line) with the c(s) curve (solid). A detailed analysis and comparison of the different size-distribution methods in theory and practice can be found in Schuck et al. (2002).
Fig. 2.

Sedimentation coefficient distributions calculated from a SV experiment of bovine serum albumin. Shown are the distribution of Lamm equation solution c(s) (solid line), the apparent sedimentation coefficient distribution g(s*) (dotted line), and the integral sedimentation coefficient distribution G(s), scaled to the loading concentration. The differential distributions c(s) and g(s*) are in units of fringes per svedberg, and the integral distribution G(s) is in units of loading concentration. The inset shows a chromatogram of BSA in size-exclusion HPLC (Toso Haas, TSK-gel Super SW3000, 4.6 mm × 30 cm).
In our laboratory, the determination of the number of species present by c(s) usually precedes the modeling with a discrete number of Lamm equation solutions as described above. Many examples for the use of c(s) in the study of protein oligomeric state, self-association, and conformational changes have been published (Perugini et al. 2000; Schuck et al. 2000; Cole and Garsky 2001; Hatters et al. 2001a; Lacroix et al. 2001; Arthos et al. 2002; Chang et al. 2002).
Analysis of interacting systems using sedimentation velocity
For the study of the thermodynamic aspects of self-association or hetero-association, SE is usually the method of choice (see below). However, SV can be applied when the proteins do not show sufficient stability for the extended time required in SE, and in many cases SV gives a wealth of complementary information beyond the thermodynamics of molecular interactions. The study of self-associating systems by SV can give information about the association scheme, sedimentation coefficients, and hydrodynamic shape of the reversibly formed oligomers in solution, which can be used to build simple geometric models for assembly of the oligomers (among recent examples are Rivas et al. 2000; Schuck et al. 2000; Correia et al. 2001; Kar et al. 2001). Furthermore, the comparison of the equilibrium constant obtained in SV and SE shows the pressure dependence of the association, and can indicate volume changes associated with the interaction.
A traditional strategy for the sedimentation analysis of rapid self-associating and hetero-associating systems is the determination of the weight-average sedimentation coefficient sw(c) as a function of concentration (sw can be derived, for example, via integration of any of the differential sedimentation coefficient distributions c(s), g(s*), or ls-g*(s); see above). If the experiments are conducted at sufficiently high rotor speeds to generate a solution plateau, sw is only a function of the chemical composition at the plateau concentration, and the concentration dependence of sw(c) can be analyzed by fitting the binding isotherm with mass action law models (Rivas et al. 1999; Correia 2000).
For slow self-associations, all the size-distribution methods described above can still be used, with c(s) usually resolving sedimentation coefficients of monomers, dimers, and higher oligomers (Perugini et al. 2000), while separating possible degradation products and aggregates. However, if there are any molecular interactions on the time scale of sedimentation, the sedimentation behavior will be dependent on concentration and rotor speed, and the sedimentation/reaction/diffusion process becomes significantly more complex. In this case, the application of the size-distribution methods involving diffusional deconvolution, c(s) and G(s), can only give qualitative and diagnostic information. In particular, the modeling of the sedimentation boundary as a superposition of Lamm equation solutions for noninteracting species, c(s), can lead to misleading or artifactual details on the size distribution. However, the weight-average sedimentation coefficient, as obtained by integration of the differential sedimentation coefficient distributions, is a correct representation of the system.
Another approach for rapid interactions, which has only recently become generally available, is based on the direct modeling of the sedimentation profiles (similar to Fig. 1 ▶) with equation 7, modified to allow concentration-dependent sedimentation and diffusion coefficients (Cox 1969). For self-associating proteins with unknown oligomeric states, this allows taking advantage of the distinct sedimentation patterns generated by different self-association schemes. Sedfit has several models for proteins in rapid reversible self-association equilibrium (Schuck 1998). A strategy for the treatment of hetero-associations has been reported (Stafford 2000). One difficulty can be that the number of free parameters required for the complete characterization of protein self-association can exceed the information content of a single SV experiment. Therefore, prior knowledge can be inserted in the form of the monomer molar mass from amino acid sequence, monomer or oligomer sedimentation coefficient from SV under very dilute or highly concentrated conditions, or association constants known from SE (if there is no pressure dependence of the association). However, global modeling techniques (e.g., in Sedphat) allow one to reduce the need for additional prior knowledge. Another form of interaction that can be globally modeled in this way is caused by thermodynamic and hydrodynamic nonideal solutions, which has been used to measure the second virial coefficient in the study of crystallization conditions (Solovyova et al. 2001). Because the direct modeling approach has more stringent requirements for sample purity than an analysis of sw(c) but can require fewer experiments, the choice of method may be dictated by practical considerations.
Boundary shape analysis for the determination of molar mass
As pointed out above, the Svedberg equation 1 relates the sedimentation and diffusion coefficients with the buoyant molar mass, and it is therefore possible to determine the molar mass from the boundary shape of SV profiles. (Or conversely, knowledge of the buoyant molar mass of the protein from amino acid composition or from SE can be used as prior knowledge to improve the resolution of the data analysis.) In contrast to the sedimentation coefficients, the precision of the molar mass determination increases with lower rotor speed, converging to the approach to SE (see below). Although the molar mass from the boundary shape is more susceptible to artifacts arising from the sedimentation process, it has the important practical advantage of its relative rapidity.
When determining the molar mass from the boundary shape, one difficulty is that heterogeneous populations of macromolecules with slightly different s-values can cause boundary spreading. For the latter case the uncritical application of equation 1 using an average s-value and the apparent diffusion coefficient will lead to an apparent molar mass significantly smaller than the true (average) molar mass. This problem can be diagnosed by a sedimentation coefficient exceeding the maximum s-value for the apparent molar mass using equation 3. Conversely, repulsive macromolecular interactions decrease s but can also reduce boundary spreading, leading to an erroneous apparent molar mass. When working with globular proteins, these repulsive interactions can usually be avoided at concentrations below 1 mg/mL and sufficient supporting electrolyte (for most proteins 50–100 mM). If the s-value decreases with increasing concentration, in the absence of charge effects, then hydrodynamic interactions, which are governed by the frictional asymmetry of the particle, are affecting the sedimentation process (Rowe 1992). Both of these factors lead to sedimentation profiles distinctly different from those shown in Figure 1 ▶, and can be detected by critical inspection of the residuals of a direct boundary model. A detailed description of sedimentation under nonideal conditions can be found in Solovyova et al. (2001), where sedimentation boundary profiles were modeled with modified transport equations for the purpose of exploring protein crystallization conditions.
In practice, the most reliable determination of the molar mass is obtained by direct boundary modeling with a small number of discrete (preferably a single) Lamm equation solutions (eq. 7). This analysis can be accomplished, for example, using Lamm, Svedberg, and Sedfit, or the modeling of the time-derivative dc/dt with the approximate Lamm equation solutions with Dcdt+ (as recently introduced by Philo 2000a). An older technique of modeling of the apparent sedimentation coefficient distributions g*(s) with Gaussians also gives a molar mass estimate, but this is considerably less precise and requires more constrained experimental conditions (Schuck and Rossmanith 2000; Philo 2000a). As indicated above, size-distribution methods for molar mass distributions have been recently described and are implemented in Sedfit (Schuck 2000), but these are inherently more difficult to determine than the sedimentation coefficient distributions. An example for its application to small nucleic acid complexes has been reported by Hatters et al. (2001b).
Analytical zone centrifugation or band centrifugation
An alternative SV methodology was developed by Vinograd et al. (1963) 39 years ago (originally designated band centrifugation) and has recently been reexamined (Lebowitz et al. 1998). Instead of starting the experiment with a uniform loading of the sample solution, in the analytical zone centrifugation (AZC) method, upon initiation of the centrifugal field, the macromolecules are transferred from a small well containing 20–30 μL on top of a column of solvent of greater density (e.g., by a difference in NaCl concentration or the presence of D2O) than the macromolecular solution. A self-generating density gradient occurs by diffusion of small molecules from the main liquid column into the transferred macromolecular lamella. Although this diffusional density gradient is very small (and distinctly different from the more commonly used density gradients that are preformed or formed by sedimentation), it is sufficient to prevent convection and stabilizes the sedimenting zone or band of macromolecules. Very recent results from this laboratory have shown that AZC data can be modeled with the transport equation 7 implemented in Sedfit. More details and applications can be found in Lebowitz (1994), Lebowitz et al. (1994, 1998), and Kar et al. (2001).
Analytical zone centrifugation (AZC) can represent an attractive approach to conventional boundary SV of proteins and protein complexes with the major advantages of using ∼1/20th as much material as conventional boundary analysis, and the potential for physical separation of sedimenting species. Hence, sedimentation coefficients are easily obtained with only microgram quantities of protein. Because the protein zones rapidly sediment into the bulk column solution, AZC also allows for either ion or pH exchanges. Consequently, investigators can study the effects of environmental changes on proteins or protein complexes with small amounts of material. For the case of DNA, the AZC characterization of pH-induced conformational transitions of polyoma DNA played a key role toward the discovery of supercoiled DNA (Vinograd et al. 1965). Also, the economy in sample volumes of AZC can be advantageous when studying the effects of temperature. For example, the thermal stability of the human immunodeficiency virus-1 reverse transcriptase heterodimer was monitored using AZC (Lebowitz et al. 1994).
Sedimentation equilibrium measurements
At centrifugal fields lower than those generally used for SV, sedimentation is balanced by diffusional transport, and SE is achieved when the net transport vanishes throughout the solution. It can be easily established by running the centrifuge until the concentration distribution appears to be invariant with time. In equilibrium, the concentration distribution generally approaches an exponential (for derivations, see van Holde et al. 1998), and for a mixture of noninteracting ideally sedimenting solutes, the measured signal as a function of radial position, a(r), takes the following form:
 |
(9) |
where the summation is over all species n; cn,0 denotes the molar concentration of species n at a reference position r0; Mn, vn, and ɛn denote the molar mass, partial specific volume, and the molar extinction coefficient, respectively; d is the optical path length (usually 1.2 cm); and δ is a baseline offset, which compensates for differences in nonsedimenting absorbing solutes between sample and reference compartments and small nonidealities in the cell assemblies and data acquisition. Similar to SV, repulsive interactions between proteins will lead to nonideal sedimentation equilibrium, which can usually be avoided at concentrations below 1 mg/mL and with supporting electrolyte of 100 mM. When using the interference optics, the extinction coefficient in equation 9 should be replaced by a specific signal increment, and the baseline offset is usually radial-dependent, requiring separate experimental determination (Ansevin et al. 1970). For a detailed description of practical aspects of planning, conducting, and analyzing an SE experiment, such as choice of optical system, buffer conditions, rotor speeds, experimental time, sample purity, sample volume, etc., see Schuck and Braswell (2000).
Equation 9 states that the exponential distribution at SE is the sum of the exponentials of the macromolecular species present in solution. The concentration of each component varies exponentially with r2/2 as a function of Mn(1 −  nρ)ω2/2RT. The term M(1 −
nρ)ω2/2RT. The term M(1 − 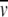 nρ) is the buoyant or reduced molar mass; that is, following Archimedes' principle, the mass of a macromolecule acted on in solution by the centrifugal field is reduced by the mass of the displaced solvent. For a single protein, only one exponential distribution will be present, which readily allows for the determination of the buoyant molar mass of this molecule, and with knowledge of the
nρ) is the buoyant or reduced molar mass; that is, following Archimedes' principle, the mass of a macromolecule acted on in solution by the centrifugal field is reduced by the mass of the displaced solvent. For a single protein, only one exponential distribution will be present, which readily allows for the determination of the buoyant molar mass of this molecule, and with knowledge of the  of the protein, the molar mass is readily evaluated. Classically, equation 9 was converted to a linear form, and the weight-average molar mass was determined from the slope. Presently, computational software can readily fit an exponential model to determine the molar mass. In fact, the power of global nonlinear regression fitting of multiple data sets has enormously extended the applications of SE to complex systems. A number of these applications are discussed below.
of the protein, the molar mass is readily evaluated. Classically, equation 9 was converted to a linear form, and the weight-average molar mass was determined from the slope. Presently, computational software can readily fit an exponential model to determine the molar mass. In fact, the power of global nonlinear regression fitting of multiple data sets has enormously extended the applications of SE to complex systems. A number of these applications are discussed below.
Self-associating systems
As indicated above, interacting systems are of particular interest to protein scientists. With some modifications to introduce equilibrium constants and mass action law, equation 9 also describes the SE of reversibly formed protein complexes. For self-associating systems, we can relate the molar concentration at the reference point of all oligomeric species via cn,0 = Kn(c1,0)n (with the subscript 1 denoting the monomer). In addition, for Mn we substitute nM1 in each exponential term. If no change in the volume of the protomers accompanies the self-association ( is constant), we obtain
is constant), we obtain
 |
(10) |
It should be noted that association constants Kn are defined from the monomer to the n-mer. Intermediate association constants are readily calculated once the Kn values have been determined. Using equation 10, we are in position to perform global nonlinear regression fitting of multiple SE data sets at different loading concentrations and rotor speeds to determine the monomer molar mass, Kn-values, and stoichiometries for selected association models that best fit the data. An excellent Beckman-Coulter Instruments monograph on the analysis of self-associating systems has been prepared by McRorie and Voelker (1993). It offers the reader very practical steps for self-association modeling, including error analysis. As pointed out above, SV measurements provide a good basis for selecting the association model.
Sedimentation equilibrium with glycoproteins
Many eukaryotic and viral proteins are heavily glycosylated with a heterogeneous distribution of carbohydrates. This situation often leads to anomalous behavior on size exclusion chromatography and SDS-PAGE. In contrast, AU offers a more rigorous methodology for the determination of the molar mass and the oligomeric state in solution. A complication is the partial specific volume of a glycoprotein, which leads to a buoyancy that is different from that of nonglycosylated proteins, and is strictly dependent on the carbohydrate composition. Ideally, one could measure the v of the glycoprotein using a density meter; however, material limitations often preclude this approach. For determination of the extent of glycosylation, ultracentrifugal methods have been described (Shire 1992; Lewis and Junghans 2000). Alternatively, it is frequently possible to determine the extent of glycosylation for a monomer by mass spectrometry (MS), if combined with the molar mass of the protein from the amino acid composition (Fairman et al. 1999). This allows the use of ultracentrifugal data to determine the oligomeric state in solution. In this case, the partial specific volume can be expressed in terms of weight fractions for the protein and carbohydrate moiety, wp and wc, and the protein and carbohydrate partial specific volumes 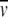 p and
p and 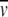 c, respectively (Shire 1992). If carbohydrate composition data are available, the
c, respectively (Shire 1992). If carbohydrate composition data are available, the  c can be determined for different N-linked oligosaccharides (Shire 1992; Fairman et al. 1999; Lewis and Junghans 2000). However, if the carbohydrate composition is unknown, one can use estimates for the average
c can be determined for different N-linked oligosaccharides (Shire 1992; Fairman et al. 1999; Lewis and Junghans 2000). However, if the carbohydrate composition is unknown, one can use estimates for the average 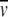 c of carbohydrates, which translates to uncertainties in the value of molar mass of the glycoprotein of usually only a few percent (Lewis and Junghans 2000).
c of carbohydrates, which translates to uncertainties in the value of molar mass of the glycoprotein of usually only a few percent (Lewis and Junghans 2000).
This approach was used by Center et al. (2000) for the analysis of the HIV-1 recombinant gp120. Obviously, for a monomeric glycoprotein there should be good agreement between the weight-average molar mass determined by MS and SE, and this was the case for gp120 cited above. For self-associating proteins, the SE results can reveal the oligomeric state and association constants. Fairman et al. (1999) applied the above principles to the characterization of T-cell and B-cell receptors using SE and electrospray MS analysis to obtain 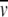 c.
c.
Characterization of membrane proteins using sedimentation equilibrium
Because of their hydrophobic nature, integral membrane proteins require nonionic detergents for solubilization in a functional state. The solubilization process involves the formation of protein–detergent micelle complexes. Consequently, a molar mass determination by SE would yield the sum of the protein mass and the mass of bound detergent. Although this is, in principle, not a problem for the study of hetero-associations of membrane proteins, it provides a significant complication for measuring the molar mass and the self-association properties. Reynolds and Tanford (1976) developed an ingenious methodology for the determination of the protein molar mass in protein–detergent complexes without direct knowledge of detergent binding. The buoyant molar mass of a protein–detergent complex can be decomposed into a term for the protein and a term for the detergent, respectively:
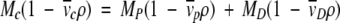 |
(11) |
The subscripts p, D, and c denote protein, detergent, and protein–detergent complex, respectively. If the density of the solution is adjusted with an appropriate solute to match the density of the detergent (ρ = 1/ D), then the detergent becomes gravitationally transparent and the term MD(1 −
D), then the detergent becomes gravitationally transparent and the term MD(1 − 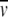 Dρ) will vanish. Hence, the SE concentration distribution only reflects the molar mass of the protein.
Dρ) will vanish. Hence, the SE concentration distribution only reflects the molar mass of the protein.
A review of the interaction of membrane proteins and lipids with solubilizing detergents has recently been published (le Maire et al. 2000). It lists the properties of detergents commonly used for the solubilization of membrane proteins, such as the critical micelle concentration and 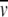 D. Past SE work has relied almost exclusively on using H2O:D2O mixtures for density matching, with considerable success (Reynolds and Tanford 1976; Tanford and Reynolds 1976; Reynolds and McCaslin 1985; Schubert and Schuck 1991). Recently sucrose, glycerol, and Nycodenz solutions have been used successfully for density matching (Mayer et al. 1999; Lustig et al. 2000). Preferential hydration of the micelles (and also the protein) can occur in these solutions, which can significantly decrease the matching density in sucrose, glycerol, and Nycodenz solutions compared with H2O:D2O mixtures. Therefore, these solute additives can provide greater experimental versatility for detergent selection and the solution preparation. In addition, sucrose and glycerol are known to stabilize proteins, which is advantageous for membrane proteins that may be labile. For a description of methods for determining the density of the detergent, see Mayer et al. (1999) and Lustig et al. (2000). In these studies, the density additives discussed above did not introduce nonideal sedimentation behavior or significant changes in the partial specific volume of the membrane protein (Mayer et al. 1999; Lustig et al. 2000). The effects of different sugars on (1 −
D. Past SE work has relied almost exclusively on using H2O:D2O mixtures for density matching, with considerable success (Reynolds and Tanford 1976; Tanford and Reynolds 1976; Reynolds and McCaslin 1985; Schubert and Schuck 1991). Recently sucrose, glycerol, and Nycodenz solutions have been used successfully for density matching (Mayer et al. 1999; Lustig et al. 2000). Preferential hydration of the micelles (and also the protein) can occur in these solutions, which can significantly decrease the matching density in sucrose, glycerol, and Nycodenz solutions compared with H2O:D2O mixtures. Therefore, these solute additives can provide greater experimental versatility for detergent selection and the solution preparation. In addition, sucrose and glycerol are known to stabilize proteins, which is advantageous for membrane proteins that may be labile. For a description of methods for determining the density of the detergent, see Mayer et al. (1999) and Lustig et al. (2000). In these studies, the density additives discussed above did not introduce nonideal sedimentation behavior or significant changes in the partial specific volume of the membrane protein (Mayer et al. 1999; Lustig et al. 2000). The effects of different sugars on (1 −  ρ) of muscle adolase have recently been explored to probe protein–sugar interactions (Ebel et al. 2000). This study allows an estimate of the error in the molar mass due to the change in
ρ) of muscle adolase have recently been explored to probe protein–sugar interactions (Ebel et al. 2000). This study allows an estimate of the error in the molar mass due to the change in 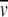 of the protein by the addition of sucrose. At a sucrose density of 1.04 (9.7%), the error in molar mass would be ∼4%. Although this error estimate may vary with different proteins and in the presence of detergents, it supports the evaluation of the stoichiometry of a membrane protein or complex using sugar additives. If density matching of a detergent requires large concentrations of an additive, significant errors may result because of large changes in the partial specific volume, nonideal behavior, and density gradient formation of the additive (Mayer et al. 1999). An alternative to direct density matching for high additive concentrations is to perform a series of sedimentation equilibrium experiments at different solvent densities and to extrapolate the results to the matching density of the detergent (Tanford and Reynolds 1976; Schubert and Schuck 1991; Lustig et al. 2000).
of the protein by the addition of sucrose. At a sucrose density of 1.04 (9.7%), the error in molar mass would be ∼4%. Although this error estimate may vary with different proteins and in the presence of detergents, it supports the evaluation of the stoichiometry of a membrane protein or complex using sugar additives. If density matching of a detergent requires large concentrations of an additive, significant errors may result because of large changes in the partial specific volume, nonideal behavior, and density gradient formation of the additive (Mayer et al. 1999). An alternative to direct density matching for high additive concentrations is to perform a series of sedimentation equilibrium experiments at different solvent densities and to extrapolate the results to the matching density of the detergent (Tanford and Reynolds 1976; Schubert and Schuck 1991; Lustig et al. 2000).
Very recently we have measured the molar mass of SIV-1 envelope complex purified directly from the virus. After cross-linking of the surface-exposed membrane proteins gp120 and gp41, hydrogenated Triton X-100 was used for solubilization of the complex from virion membranes, and the molar mass was measured using density matching with sucrose. The stoichiometry of the env complex was found to be trimeric, and this could be visualized in the electron microscope (Center et al. 2001). The mass measurements from SE were in very good agreement with mass measurements from scanning transmission electron microscopy (Center et al. 2001). The density matching methodology allows for the characterization of membrane protein self-association (Schubert and Schuck 1991; Fleming et al. 1997; Fleming and Engelman 2001). It should be pointed out that association may be dependent on the type of solubilizing detergent (Musatov et al. 2000). Density matching has also been extended to the analysis of the oligomeric state of the erythrocyte band 3 protein reconstituted in small unilamellar lipid vesicles (Lindenthal and Schubert 1991). For a review of the quaternary structure and function of transport proteins with numerous citations to analytical ultracentrifugation characterization, see Veenhoff et al. (2002).
Heterogeneous interactions
In this discussion, heterogeneous interactions are defined as interactions in which two (or more) reactants reversibly form a complex with a specific stoichiometry. Such interactions would follow association schemes like A + B ⇔ AB; A + 2B ⇔ AB2; AB + B ⇔ AB2; and so on. Equilibrium constants over the range of 104 to 108 are readily measured, and, under certain circumstances, both lower or higher equilibrium constants can be obtained. Additionally, the stoichiometry of the interaction can usually be determined. For the present purpose, let us consider the SE characterization of the simple reaction A + B ⇔ AB. This requires the study of the components A and B alone, as well as mixtures of A and B at different molar ratios (including equimolar). As above, it is convenient to introduce the buoyant molar mass as M* = M(1 − 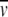 ρ), and SE experiments of solutions containing the separate components will allow us to determine the values of MA* and MB*. These values may include contributions from glycosylation or detergent micelles, which do not need to be known or further specified for the study of hetero-associations. For the AB complex, we make the reasonable assumption that the partial specific volume of the AB complex can be calculated from the respective weight fractions of the partial specific volumes of components A and B, hence, MAB* = MA* + MB*. For the A + B ⇔ AB reaction, we have three species in solution, free A and B and the AB complex, which in chemical equilibrium obey the mass action law cAB = cAcBKAB. At equilibrium in the centrifugal field, the radial distribution is (analogous to eq. 10):
ρ), and SE experiments of solutions containing the separate components will allow us to determine the values of MA* and MB*. These values may include contributions from glycosylation or detergent micelles, which do not need to be known or further specified for the study of hetero-associations. For the AB complex, we make the reasonable assumption that the partial specific volume of the AB complex can be calculated from the respective weight fractions of the partial specific volumes of components A and B, hence, MAB* = MA* + MB*. For the A + B ⇔ AB reaction, we have three species in solution, free A and B and the AB complex, which in chemical equilibrium obey the mass action law cAB = cAcBKAB. At equilibrium in the centrifugal field, the radial distribution is (analogous to eq. 10):
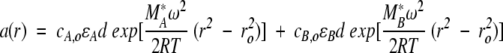 |
(12) |
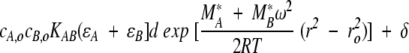 |
The major difficulty in fitting the parameters of this heterogeneous interaction model is the ability to evaluate the contribution of the complex formed and the uncomplexed reactants to the total signal. This usually requires global modeling of data acquired at different loading concentrations and rotor speeds. However, one of the great advantages of the absorption optical system is the ability to scan at a variety of wavelengths, which greatly simplifies the analysis for spectrally distinct proteins and makes it possible to study heterogeneous interactions that would otherwise be impossible.
The characterization of protein–DNA interactions represents an important area where multiwavelength absorbance analysis of SE data has been successfully applied. A target oligonucleotide and DNA-binding protein have significantly different absorption spectra, and one can select a range of wavelengths such that the resulting scans vary from the signal being dominated by the oligonucleotide to the signal being dominated by the protein. This can be combined with global analysis using independently determined extinction coefficients (Lewis et al. 1994; Bailey et al. 1996; Cole et al. 1997; Lee et al. 2001). It should be pointed out that the above multiple wavelength analysis assumes that there is no change in the extinction coefficients of the reactants upon binding, that is, no hypo- or hyperchromic shift at the scanned wavelengths, and ɛAB = ɛA + ɛB. This can be experimentally examined by measuring the sum of the absorbance of each component using a dual-compartment cuvette in comparison to the absorbance of the mixture of reactants under conditions where substantial complex formation occurs (Bailey et al. 1996). For the situation in which there is a substantial extinction coefficient change upon complex formation, a methodology has been developed that combines absorption spectra scanned at multiple radii and radial profiles scanned at multiple wavelengths (Schuck 1994). This provides a two-dimensional data surface that can be analyzed to achieve a simultaneous detection of the components extinction spectra and the other variables in the SE model.
Another example of multiple wavelength SE is the study of the association of small peptides with much larger proteins. The problem here is that the mass of the peptide–protein complex differs so little from that of the protein alone that it is difficult to discriminate between them from the radial distribution alone. However, if the peptide can be synthesized with 5-hydroxy tryptophan incorporated into the molecule (Laue et al. 1993), it is spectrally distinguishable from the protein. The extinction coefficients at the different wavelengths can be calculated from the ratios of the observed scans of the reactants, and the equilibrium constant can be obtained by globally fitting appropriate models to the scans (Yoo and Lewis 2000).
SE analysis of both self-associating and hetero-associating proteins can be greatly facilitated if the total amount of soluble protein remains constant during the time course of the experiment (see, e.g., Becerra et al. 1991). This approach has recently been reviewed by Philo (2000b). Binding constants of small absorbing molecules to proteins have very recently been determined using the multiwavelength strategy coupled with conservation of mass methodology (Arkin and Lear 2001).
Software for sedimentation equilibrium analyses
Nonlinear least-squares parameter estimation is the major numerical method for SE data analysis. An examination of the biophysical/biochemical literature will reveal the use of diverse software. Because computational skills vary considerably, we believe that investigators should evaluate the software outlined below based on their particular research applications and individual computational data-analysis expertise. With respect to commercial mathematical modeling programs, we cannot endorse one program over another.
For molar mass analysis and self-associating systems, the program Nonlin, developed by Yphantis and Johnson at the National Analytical Ultracentrifuge Facility (NAUF) of the University of Connecticut Biotechnology Center, has been applied extensively. Beckman-Coulter Instruments supplies a data acquisition and analysis software package that includes a version of the Nonlin program based on the Origin software by MicroCal Inc with each XLA and XLI purchased. It also includes a subtraction of data utility to allow for the determination of when SE has been reached. (A more advanced software for testing attainment of equilibrium is WinMatch, developed at NAUF.) There are additional useful utilities including a data simulator. Investigators can obtain from NAUF the most current program, WinNonlin, and other programs for editing and examining the SE data from single-component and self-associating systems. There is an organization entitled Reversible Associations in Structural and Molecular Biology (RASMB; http://www.bbri.org/RASMB/rasmb.html) that has an archive of AU software. Very recently the SE data analysis software UltraSpin has been developed by the Center for Protein Engineering, Medical Research Council, University of Cambridge (available from the Web site http://www.mrc-cpe.cam.ac.uk/ ultraspin_intro free to academic/nonprofit laboratories and for a small charge to commercial investigators). UltraSpin can fit 20 different models. Multiwavelength fitting is provided for several protein–DNA interaction models and for several heterogeneous protein interactions. For the Unix or Linux environment, SE analysis can be performed with the software UltraScan from Borries Demeler at http://www.ultrascan.uthscsa.edu./.
Curve-fitting tools of commercial software such as SigmaPlot, IgorPro or KaleidaGraph have been used. The MLAB software (Civilized Software) is a command line advanced mathematical and statistical modeling system that has been used by this laboratory for many years. There are many other commercial programs for nonlinear regression modeling that can be found on the Web.
Conclusions
As mentioned above, the scope of this short review does not allow descriptions of many topics and applications of AU. Other reviews cited should be consulted as well as SV and SE methodological treatments that have been published in Methods in Enzymology. For example, see Laue and Stafford (1999), Rivas et al. (1999), and Schuck and Braswell (2000). We have provided a tutorial review building from the basic concepts to advanced AU approaches for the characterization of protein systems. We have focused on what can be learned about proteins and their interactions using AU, and have given information to allow protein scientists to take the practical steps toward reaching these goals. Investigators should be encouraged to use both SV and SE to characterize their systems to gain the maximum information possible. It should be noted that Beckman-Coulter Instruments offers customers a three-day course on the operation of the XLA/XLI. This course is intended for beginner operators with minimal background in AU. For more advanced training, the NAUF of the University of Connecticut Biotechnology Center offers a workshop in SV and SE data analysis. Finally, there is a discussion group in the RASMB network of investigators who are interested in the reversible interactions of macromolecules, to which one can readily subscribe at http://www.rasmb.bbri.org/.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0207702.
References
- Ansevin, A.T., Roark, D.E., and Yphantis, D.A. 1970. Improved ultracentrifuge cells for high-speed sedimentation equilibrium studies with interference optics. Anal. Biochem. 34 237–261. [DOI] [PubMed] [Google Scholar]
- Arkin, M. and Lear, J.D. 2001. A new data analysis method to determine binding constants of small molecules using equilibrium analytical ultracentrifugation with absorption optics. Anal. Biochem. 299 98–107. [DOI] [PubMed] [Google Scholar]
- Arthos, J., Cicala, C., Steenbeke, T.D., VanRyk, D., Dela Cruz, C., Khazanie, P., Selig, S.M., Hanback, D.B., Nam, D., Schuck, P., et al. 2002. Efficient inhibition of HIV-1 viral replication by a novel modification of sCD4. J. Biol. Chem. 277 11456–11464. [DOI] [PubMed] [Google Scholar]
- Bailey, M.F., Davidson, B.E., Minton, A.P., Sawyer, W.H., and Howlett, G.J. 1996. The effect of self-association on the interaction of the Escherichia coli regulatory protein TyrR with DNA. J. Mol. Biol. 263 671–684. [DOI] [PubMed] [Google Scholar]
- Becerra, S.P., Kumar, A., Lewis, M.S., Widen, S.G., Abbots, J., Karawya, E.M., Hughes, S.H., Shiloach, J., and Wilson, S.H. 1991. Protein–protein interactions of HIV-1 reverse transcriptase: Implication of central and C-terminal regions in subunit binding. (Lewis, MS. Appendix: Ultracentrifuge analysis of a mixed association.). Biochemistry 30 11707–11719. [DOI] [PubMed] [Google Scholar]
- Behlke, J. and Ristau, O. 1997. Molecular mass determination by sedimentation velocity experiments and direct fitting of the concentration profiles. Biophys. J. 72 428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center, R.J., Earl, P.L., Lebowitz, J., Schuck, P., and Moss, B. 2000. The human immunodeficiency virus type 1 gp120 V2 domain mediates gp41-independent intersubunit contacts. J. Virol. 74 4448–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center, R.J., Schuck, P., Leapman, R.D., Arthur, L.O., Earl, P.L., Moss, B., and Lebowitz, J. 2001. Oligomeric structure of virion-associated and soluble forms of the simian immunodeficiency virus envelope protein in the pre-fusion activated conformation. Proc. Natl. Acad. Sci. 98 14877–14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, H.-C., Chou, W.-Y., and Chang, G.-G. 2002. Effect of metal binding on the structural stability of pigeon liver malic enzyme. J. Biol. Chem. 277 4663–4671. [DOI] [PubMed] [Google Scholar]
- Cole, J.L. and Garsky, V.M. 2001. Thermodynamics of peptide inhibitor binding to HIV-1 gp41. Biochemistry 40 5633–5641. [DOI] [PubMed] [Google Scholar]
- Cole, J.L., Carroll, S.S., Blue, E.S., Viscount, T., and Kuo, L. 1997. Activation of RNase by 2`,5`-oligoadenylates. Biophysical characterization. J. Biol. Chem. 272 19187–19192. [DOI] [PubMed] [Google Scholar]
- Correia, J.J. 2000. Analysis of weight average sedimentation velocity data. Meth. Enzymol. 321 81–100. [DOI] [PubMed] [Google Scholar]
- Correia, J.J., Chacko, B.M., Lamm, S.S., and Lin, K. 2001. Sedimentation studies reveal a direct role of phosphorylation in Smad3:Smad4 homo- and hetero-trimerization. Biochemistry 40 1473–1482. [DOI] [PubMed] [Google Scholar]
- Cox, D.J. 1969. Computer simulation of sedimentation in the ultracentrifuge. IV. Velocity sedimentation of self-associating solutes. Arch. Biochem. Biophys. 129 106–123. [DOI] [PubMed] [Google Scholar]
- Demeler, B. and Saber, H. 1998. Determination of molecular parameters by fitting sedimentation data to finite element solutions of the Lamm equation. Biophys. J. 74 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeler, B., Saber, H., and Hansen, J.C. 1997. Identification and interpretation of complexity in sedimentation velocity boundaries. Biophys. J. 72 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebel, C., Eisenberg, H., and Ghirlando, R. 2000. Probing protein–sugar interactions. Biophys. J. 78 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman, R., Fenderson, W., Hail, M.E., Wu, Y., and Shaw, S.-Y. 1999. Molecular weights of CTLA-4 and CD80 by sedimentation equilibrium ultracentrifugation. Anal. Biochem. 270 286–295. [DOI] [PubMed] [Google Scholar]
- Fleming, K.G. and Engelman, D.M. 2001. Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc. Natl. Acad. Sci. 98 14340–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, K.G., Ackerman, A.L., and Engelman, D.M. 1997. The effect of point mutations on the free energy of transmembrane α-helix dimerization. J. Mol. Biol. 272 266–275. [DOI] [PubMed] [Google Scholar]
- Garcia de la Torre, J.G. 1992. Sedimentation coefficients of complex biological particles. In Analytical ultracentrifugation in biochemistry and polymer science. (eds. S.E. Harding et al.), pp. 333–358. The Royal Society of Chemistry, Cambridge, UK.
- Hansen, J.C., Lebowitz, J., and Demeler, B. 1994. Analytical ultracentrifugation of complex macromolecular systems. Biochemistry 33 13155–13163. [DOI] [PubMed] [Google Scholar]
- Hatters, D.M., Lindner, R.A., Carver, J.A., and Howlett, G.J. 2001a. The molecular chaperone, α-crystallin, inhibits amyloid formation by apolipoprotein C-II. J. Biol. Chem. 276 33755–33761. [DOI] [PubMed] [Google Scholar]
- Hatters, D.M., Wilson, L., Atcliffe, B.W., Mulhern, T.D., Guzzo-Pernell, N., and Howlett, G.J. 2001b. Sedimentation analysis of novel DNA structures formed by homo-oligonucleotides. Biophys. J. 81 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar, S.R., Lebowitz, J., Blume, S., Taylor, K.B., and Hall, L.M. 2001. SmtB–DNA and protein–protein interactions in the formation of the cyanobacterial metallothionein repression complex: Zn2+ does not dissociate the protein–DNA complex in vitro. Biochemistry 40 13378–13389. [DOI] [PubMed] [Google Scholar]
- Kratky, O., Leopold, H., and Stabinger, H. 1973. The determination of the partial-specific volume of proteins by the mechanical oscillator technique. Methods Enzymol., 27 98–110. [DOI] [PubMed] [Google Scholar]
- Lacroix, M., Ebel, C., Kardos, J., Dobo, J., Gal, P., Zavodszky, P., Arlaud, G.J., and Thielens, N.M. 2001. Assembly and enzymatic properties of the catalytic domain of human complement protease C1r. J. Biol. Chem. 276 36233–36240. [DOI] [PubMed] [Google Scholar]
- Lamm, O. 1929. Die Differentialgleichung der Ultrazentrifugierung. Ark. Mat. Astr. Fys. 21B(2): 1–4. [Google Scholar]
- Laue, T. 2001. Biophysical studies by ultracentrifugation. Curr. Opin. Struct. Biol. 11 579–583. [DOI] [PubMed] [Google Scholar]
- Laue, T.M. and Stafford, W.F. 1999. Modern applications of analytical ultracentrifugation. Anal. Rev. Biophys. Biomol. Struct. 28 75–100. [DOI] [PubMed] [Google Scholar]
- Laue, T.M., Shah, B.D., Ridgeway, T.M., and Pelletier, S.L. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. In Analytical ultracentrifugation in biochemistry and polymer science (eds. S.E. Harding et al.), pp. 90–125. The Royal Society of Chemistry, Cambridge, UK.
- Laue, T.M., Senear, D.F., Eaton, S., and Ross, J.B. 1993. 5-Hydroxytryptophan as a new intrinsic probe for investigating protein–DNA interactions by analytical ultracentrifugation. Study of the effect of DNA on self-assembly of the bacteriophage λ cI repressor. Biochemistry 32 2469–2472. [DOI] [PubMed] [Google Scholar]
- Lebowitz, J. 1994. Stability of human immunodeficiency virus-1 reverse transcriptase heterodimer. In Application information A-1807A solution interaction analysis, pp. 1–8. Beckman Instruments, Palo Alto, CA. http://www.beckman-coulter.com/Literature/BioResearch/a_1807a.pdf.
- Lebowitz, J., Kar, S.R., Braswell, E., McPherson, S., and Richard, D.L. 1994. Human immunodeficiency virus-1 reverse transcriptase heterodimer stability. Protein Sci. 3 1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebowitz, J., Teale, M., and Schuck, P. 1998. Analytical band centrifugation of proteins and protein complexes. Biochem. Soc. Transact. 26 745–749. [DOI] [PubMed] [Google Scholar]
- Lee, S.P., Fuior, E., Lewis, M.S., and Han, M.K. 2001. Analytical ultracentrifugation studies of translin: analysis of protein–DNA interactions using a single-stranded fluorogenic oligonucleotide. Biochemistry 40 14081– 14088. [DOI] [PubMed] [Google Scholar]
- le Maire, M., Champeil, P., and Moller, J.V. 2000. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta 1508 86–111. [DOI] [PubMed] [Google Scholar]
- Lewis, M.S. and Junghans, R.P. 2000. Ultracentrifugal analysis of the molecular mass of glycoproteins of unknown or ill-defined carbohydrate composition. Methods Enzymol. 321 136–149. [DOI] [PubMed] [Google Scholar]
- Lewis, M.S., Shrager, R.I., and Kim, S.-J. 1994. Analysis of protein-nucleic acid and protein–protein interactions using multi-wavelength scans from the XL-A analytical ultracentrifuge. In Modern analytical ultracentrifugation. (eds. T.M. Schuster, and T.M. Laue), pp. 94–115. Birkhäuser, Boston.
- Lindenthal, S. and Schubert, D. 1991. Monomeric erythrocyte band 3 protein transports anions. Proc. Natl. Acad. Sci. 88 6540–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig, A., Engel, A., Tsiotis, G., Landau, E.M., and Baschong, W. 2000. Molecular weight determination of membrane proteins by sedimentation equilibrium at the sucrose or Nycodenz-adjusted density of the hydrated detergent micelle. Biochim. Biophys. Acta 1464 199–206. [DOI] [PubMed] [Google Scholar]
- Mayer, G., Ludwig, B., Muller, H.W., van den Broek, J.A., Friesen, R.H.E., and Schubert, D. 1999. Studying membrane proteins in detergent solution by analytical ultracentrifugation: Different methods for density matching. Prog. Colloid Polymer Sci. 113 176–181. [Google Scholar]
- McRorie, D.K. and Voelker, P.J. 1993. Self-associating systems in the analytical ultracentrifuge. Beckman Instruments, Fullerton, CA.
- Musatov, A., Ortega-Lopez, J., and Robinson, N.C. 2000. Detergent-solubilized bovine cytochrome c oxidase: Dimerization depends on the amphilic environment. Biochemistry 39 12996–13004. [DOI] [PubMed] [Google Scholar]
- Perkins, S.J. 2001. X-Ray and neutron scattering analyses of hydration shells: A molecular interpretation based on sequence predictions and modelling fits. Biophys. Chem. 93 129–139. [DOI] [PubMed] [Google Scholar]
- Perugini, M.A., Schuck, P., and Howlett, G.J. 2000. Self-association of human apolipoprotein E3 and E4 in the presence and absence of phospholipid. J. Biol. Chem. 275 36758–36765. [DOI] [PubMed] [Google Scholar]
- Philo, J.S. 1997. An improved function for fitting sedimentation velocity data for low molecular weight solutes. Biophys. J. 72 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2000a. A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal. Biochem. 279 151–163. [DOI] [PubMed] [Google Scholar]
- ———. 2000b. Sedimentation equilibrium analysis of mixed associations using numerical constraints to impose mass or signal conservation. Methods Enzymol. 321 100–120. [DOI] [PubMed] [Google Scholar]
- Reynolds, J.A. and McCaslin, D.R. 1985. Determination of protein molecular weight in complexes with detergent without knowledge of binding. Methods Enzymol. 117 41–53. [DOI] [PubMed] [Google Scholar]
- Reynolds, J.A. and Tanford, C. 1976. Determination of molecular weight of protein moiety in protein–detergent complexes without prior knowledge of detergent binding. Proc. Natl. Acad. Sci. 73 4467–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas, G., Lopez, A., Minogrance, J., Ferrandiz, M.J., Zorrilla, S., Minton, A.P., Vincente, M., and Andreau, J.M. 2000. Magnesium-induced linear self-association of the FtsZ bacterial cell division protein monomer. The primary steps for FtsZ assembly. J. Biol. Chem. 275 11740–11749. [DOI] [PubMed] [Google Scholar]
- Rivas, G., Stafford, W., and Minton, A.P. 1999. Characterization of heterologous protein–protein interactions via analytical ultracentrifugation. Methods: A companion to methods in enzymology 19 194–212. [DOI] [PubMed] [Google Scholar]
- Robinson, N.C., Gomez, B., Musatov, A., and Ortega-Lopez, J. 1998. Analysis of detergent-solubilized membrane proteins in the analytical ultracentrifuge. ChemTracts—Biochem. Mol. Biol. 11 960–968. [Google Scholar]
- Rodgers, D.P., Roberts, L.M., Lebowitz, J., Datta, G., Anantharamaiah, G.M., Engler, J.A., and Brouillette, C.G. 1998a. The lipid free structure of apolipoprotein A-1: Effects of amino-terminal deletions. Biochemistry 37 11714–11725. [DOI] [PubMed] [Google Scholar]
- Rodgers, D.P., Roberts, L.M., Lebowitz, J., Engler, J.A., and Brouillette, C.G. 1998b. Structural analysis of apolipoprotein A-1: Effects of amino- and carboxy-terminal deletions on the lipid free structure. Biochemistry 37 945–955. [DOI] [PubMed] [Google Scholar]
- Rowe, A.J. 1992. The concentration dependence of sedimentation. In Analytical ultracentrifugation in biochemistry and polymer science (eds. S.E. Harding et al.), pp. 394–406. The Royal Society of Chemistry, Cambridge, UK.
- Schachman, H.K. 1992. Is there a future for the ultracentrifuge? In Analytical ultracentrifugation in biochemistry and polymer science (eds. S.E. Harding et al.), pp. 3–15. The Royal Society of Chemistry, Cambridge, UK.
- Schubert, D. and Schuck, P. 1991. Analytical ultracentrifugation as a tool for studying membrane proteins. Progr. Colloid Polym. Sci. 86 12–22. [Google Scholar]
- Schuck, P. 1994. Simultaneous radial and wavelength analysis with the Optima XL-A analytical ultracentrifuge. Progr. Colloid Polym. Sci. 94 1–13. [Google Scholar]
- ———. 1998. Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys. J. 75 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2000. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck, P. and Braswell, E.H. 2000. Measurement of protein interactions by equilibrium ultracentrifugation. In Current protocols in immunology (eds. J.E. Coligan et al.), pp. 18.18.11–18.18.22. Wiley, New York.
- Schuck, P. and Demeler, B. 1999. Direct sedimentation analysis of interference optical data in analytical ultracentrifugation. Biophys. J. 76 2288–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck, P. and Rossmanith, P. 2000. Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers 54 328–341. [DOI] [PubMed] [Google Scholar]
- Schuck, P., Taraporewala, Z., McPhie, P., and Patton, J.T. 2000. Rotavirus nonstructural protein NSP2 self-assembles into octamers that undergo ligand-induced conformational changes. J. Biol. Chem. 276 9679–9687. [DOI] [PubMed] [Google Scholar]
- Schuck, P., Perugini, M.A., Gonzales, N.R., Howlett, G.J., and Schubert, D. 2002. Size-distribution analysis of proteins by analytical ultracentrifugation: Strategies and application to model systems. Biophys. J. 82 1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shire, S. 1992. Determination of molecular weight of glycoproteins by analytical ultracentrifugation. Beckman Instruments, Palo Alto, CA.
- Solovyova, A., Schuck, P., Constenaro, L., and Ebel, C. 2001. Non-ideality by sedimentation velocity of halophilic malate dehydrogenase in complex solvents. Biophys. J. 81 1868–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford, W.F. 1992. Boundary analysis in sedimentation transport experiments: A procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal. Biochem. 203 295–301. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Analysis of reversibly interacting macromolecular systems by time derivative sedimentation velocity. Methods Enzymol. 323 302–325. [DOI] [PubMed] [Google Scholar]
- Svedberg, T. and Pedersen, K.O. 1940. The ultracentrifuge. Oxford University Press, London.
- Tanford, C. and Reynolds, J.A. 1976. Characterization of membrane proteins in detergent solutions. Biochim. Biophys. Acta 457 133–170. [DOI] [PubMed] [Google Scholar]
- Teller, D.C., Swanson, E., and DeHaen, C. 1979. The translation frictional coefficient of proteins. Methods Enzymol. 61 103–124. [DOI] [PubMed] [Google Scholar]
- van Holde, K.E. and Weischet, W.O. 1978. Boundary analysis of sedimentation velocity experiments with monodisperse and paucidisperse solutes. Biopolymers 17 1387–1403. [Google Scholar]
- van Holde, K.E., Johnson, W.C., and Ho, P.S. 1998. Principles of physical biochemistry. Prentice Hall, Upper Saddle River, NJ.
- Veenhoff, L.M., Heuberger, E.H.M.L., and Poolman, B. 2002. Quaternary structure and function of transport proteins. Trends Biochem. Sci 27 242–249. [DOI] [PubMed] [Google Scholar]
- Vinograd, J., Bruner, R., Kent, R., and Weigle, J. 1963. Band centrifugation of macromolecules and viruses in self-generating density gradients. Proc. Natl. Acad. Sci. 49 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinograd, J., Lebowitz, J., Radloff, R., Watson, R., and Laipis, P. 1965. The twisted circular form of polyoma viral DNA. Proc. Natl. Acad. Sci. 53 1104–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo, S.H. and Lewis, M.S. 2000. Interaction of chromogranin B and the near N-terminal region of chromogranin B with an intraluminal loop peptide of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem 275 30293–30300. [DOI] [PubMed] [Google Scholar]