

Abstract
The structure of the sucrose-specific porin (ScrY) from Salmonella typhimurium has been elucidated by X-ray crystallography to consist of 18 antiparallel β-strands, associated as a trimer complex similar to ion-transport channels. However, the 71-amino-acid-residue N-terminal periplasmic domain was not determined from the crystal structure due to the absence of sufficient electron density. The N-terminal polypeptide contains a coiled-coil structural motif and has been assumed to play a role in the sugar binding of ScrY porin. In this study the proteolytic stability and a specific proteolytic truncation site at the N-terminal domain were identified by the complete primary structure characterization of ScrY porin, using MALDI mass spectrometry and post-source-decay fragmentation. The secondary structure and supramolecular association of the coiled-coil N-terminal domain were determined by chemical synthesis of the complete N-terminal polypeptide and several partial sequences and their spectroscopic, biophysical, and mass spectrometric characterization. Circular dichroism spectra revealed predominant α-helical conformation for the putative coiled-coil domain comprising residues 4–46. Specific association to both dimer and trimer complexes was identified by electrospray ionization mass spectra and was ascertained by dynamic light scattering and electrophoresis data. The role of the N-terminal domain in sugar binding was examined by comparative TR-NOE-NMR spectroscopy of the complete ScrY porin and a recombinant mutant, ScrY(Δ1–62), lacking the N-terminal polypeptide. The TR-NOE-NMR data showed a strong influence of ScrY porin on the sugar-binding affinity and suggested a possible function of the periplasmic N terminus for supramolecular stabilization and low-affinity sugar binding.
Keywords: ScrY porin, N-terminal periplasmic domain, coiled-coil, circular dichroism, mass spectrometry, TR-NOE-NMR spectroscopy, supramolecular association
A sucrose-specific uptake channel, originally discovered by Schmid et al. (1982), has been found on plasmid pUR400 in Salmonella typhimurium and Escherichia coli that confers to the cells, under growth conditions, the ability to grow on sucrose as a sole carbon source. The structure of the sucrose-specific porin (ScrY) from S. typhimurium has been elucidated by X-ray crystallography to consist of 18 antiparallel β-strands (Fig. 1 ▶; Forst et al. 1998). However, the 71-residue N-terminal periplasmic polypeptide sequence of ScrY porin could not be traced and modeled in the crystal structure (Schmid et al. 1982; Forst et al. 1998) due to the absence of electron density. Mature ScrY porin consists of 483 residues. A comparison of the amino acid sequence of ScrY porin with that of maltoporin (LamB) revealed a remarkable homology for the 411 residues of the C-terminal domain of ScrY porin (see Fig. 4 ▶, below; Hardesty et al. 1991; Schmid et al. 1991). LamB transports maltose by means of a specific periplasmic-binding protein to the phosphoenol transferase system (PTS). A corresponding periplasmic-binding protein is unknown at present for ScrY, but its N-terminal 71 residues outside the barrel, which have no equivalent in LamB, may have a possible function for sugar transport in the periplasmic space. The N-terminal sequence indicates a high probability for a coiled-coil structure within residues 4–46 (Forst et al. 1993). Furthermore, the crystal structure showed that ScrY porin forms a trimer complex of identical subunits (Fig. 1 ▶). These results suggested a possible role of the periplasmic domain in sugar transport and supramolecular association of ScrY porin.
Fig. 1.
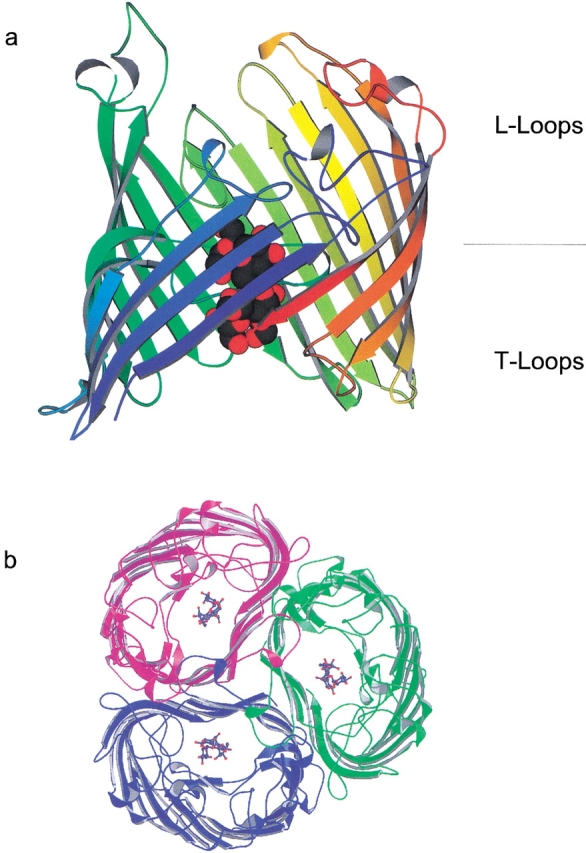
X-ray crystal structure of ScrY porin, top view (a) and membrane-plane view (b). The enhanced structures show maltotetraose inside of the porin channel.
Fig. 4.

Topology scheme with coiled-coil N-terminal periplasmic sequence of ScrY porin (a) and sequences of synthetic N-terminal polypeptides (b).
In this study, we have pursued two complementary analytical concepts to characterize the structure and possible function of the N-terminal periplasmic domain. The complete N-terminal sequence and four model peptides comprising overlapping N-terminal domains within and beyond the coiled-coil motif were prepared by chemical synthesis to examine the supramolecular association and possible sugar-binding structure. Previous studies have shown that a synthetic 25-residue peptide was able to form a stable two-stranded coiled-coil (Hodges et al. 1981). Hence, peptides of varying chain lengths should be suitable to determine the minimum-size requirement for a supramolecular association. The structures of the N-terminal polypeptides were characterized by spectroscopic methods and by MALDI mass spectrometry (MALDI-MS) and, particularly, electrospray ionisation-MS (ESI-MS), which is capable of identifying noncovalent associations (Przybylski and Glocker 1996; Loo 1997; Przybylski et al. 1998). Both dimeric and trimeric complexes were identified by ESI-MS and ascertained by dynamic light-scattering analysis, suggesting a possible supramolecular periplasmic association of ScrY porin. In a second approach, the complete primary structures of ScrY porin and a recombinant deletion mutant, ScrY(Δ1–62), lacking the N-terminal domain were characterized and a specific proteolytic cleavage yielding the complete N-terminal domain was found. Nuclear magnetic resonance (NMR) spectroscopy using transferred nuclear Overhauser enhancement (TR-NOE) analysis was employed as an efficient tool to study sugar-binding affinities of ScrY porin and the ScrY deletion mutant. NOE spectroscopy has been previously employed for characterizing sugar-binding affinities of lectins (Ni and Scheraga 1994; Poppe et al. 1997; Poveda and Jiminenez-Barbero 1998) and is demonstrated here for the first time for a transport protein. The TR-NOE data for the two porins revealed a low affinity for the periplasmic domain and are consistent with a recent study (Dumas et al. 2000) showing a sugar-slide function for the N terminus of ScrY.
Results and Discussion
Structural characterization and proteolytic stability of ScrY porin
The detailed primary structure characterization of ScrY porin was performed as a first step in this study because the X-ray crystal structure did not provide information of the N-terminal periplasmic domain and preliminary data suggested the possible proteolytic cleavage of the N-terminal polypeptide outside the β-barrel structure. The exact molecular weight determination of ScrY porin, as well as other porins, by MALDI-MS has been a considerable challenge because of high salt and detergent concentrations required for preparation of suitable solution of the protein. The development of specific sample preparation techniques for membrane proteins using a microultrasonic device has been previously reported (Schnaible et al. 1997; Bühler et al. 1998) and has yielded considerably improved molecular ion signals in MALDI spectra. Under these conditions, MALDI mass spectra of ScrY porin provided precise molecular weight determinations, as shown in Figure 2 ▶, for samples in two different detergents, β-octylglucoside and dodecylmaltoside. MALDI-MS in 0.8% β-octylglucoside (above the critical micelle concentration [cmc]) provided a single homogeneous molecular ion (average molecular weight, Mr = 53165 ± 14 daltons) consistent with the complete amino acid sequence 1–483; the intact protein was observed even after several months of storage. In contrast, the sample in 0.04% dodecylmaltoside showed the progressive formation of a truncated form (ScrY-tr; Mr = 46241 ± 21 daltons), corresponding to cleavage of the N-terminal domain, after just 2 wk (Fig. 2a ▶). Analysis by native gel electrophoresis provided identical results with a band due to the truncated form in dodecylmaltoside (data not shown). MALDI-MS of the deletion mutant, ScrY(Δ1–62) (Mr = 46240 daltons), yielded a nearly identical molecular ion to ScrY-tr, 46234 ± 14 daltons (data not shown). Edman sequence analysis of the partially truncated form of ScrY provided a mixture of the intact N-terminal sequence and that of the deletion mutant beginning at Leu 63 (Q/L, T/E, and D/K for the first three sequencing cycles), thus confirming the specific cleavage of the Arg 62–Leu 63 peptide bond.
Fig. 2.

MALDI-MS analyses of (a) intact ScrY porin and (b) partially truncated ScrY-tr. Spectrum a was obtained in 0.04% dodecylmaltoside, spectrum b in 0.15 % n-octylglucoside as described in Materials and Methods. The β-barrel scheme MN indicates full-length intact ScrY (M.W. = 53164 daltons), Mtr indicated the truncated ScrY (M.W. = 46240 daltons).
The complete primary structures of both ScrY porin and ScrY(Δ1–62) were ascertained by MALDI-MS peptide mapping analyses (Bühler et al. 1998) using trypsin and endoprotease AspN digestion (tryptic fragments; see Table 1). The peptide masses identified cover a major part (∼85%) of the sequences for both porins. In addition, MALDI-post-source decay (PSD), yielding sequence-specific fragment ions (Fligge et al. 1999), was used to verify partial sequences of tryptic peptides and identified some carbamoylated fragments that originated from the required use of urea during the proteolytic digestion, as shown in Figure 3 ▶, for the tryptic peptide 217–224. No posttranslational modifications of porins were found by MALDI-MS molecular weight and PSD analysis. The precise truncation site of ScrY porin upon storage in dodecylmaltoside (cf. Fig. 2 ▶) between residues R62 and L63 was confirmed by the N-terminal tryptic fragments 63–66, 63–70, 71–80, and 71–93 observed for ScrY-tr (data not shown), whereas none of the N-terminal fragments of intact ScrY were found.
Table 1.
Peptide fragments identified by MALDI-MS for ScrY-porin and ScrY(Δ1-62) by trypsin digestion
| Molecular Mass | ||||
| Peptide fragment | Sequence/PSDa | Calculated | Measured (ScrY) | Measured (ScrY(Δ1-62)) |
| T1 | QTDISTIEAR (1-10) | 1133.58 | 1133.38 | — |
| T2–6 | LNALEKRLQEAENRAQTAENR (11–37) | 2982.3 | 2980.8 | — |
| T4 | LQEAENR (18–24) | 859.4 | 859.3 | — |
| T5 | AQTAENR (25–31) | 789.3 | 789.2 | — |
| T5–6 | AQTAENRAGAAEK (25–37) | 1316.7 | 1317.0 | — |
| T6–7 | AGAAEKK (32–38) | 673.8 | 674 | — |
| T7–9 | KVQQLTAQQQKNQNSTQEVAQR (38–59) | 2555.8 | 2555.0 | — |
| T9 | NQNSTQEVAQRa (49–59) | 1274.6 | 1274.4 | — |
| T9–10 | NQNSTQEVAQRTAR (49–62) | 1602.8 | 1603.6 | — |
| T10–11 | TARLEK (60–65) | 717.4 | 717.8 | — |
| T12–13 | KADDK (66–70) | 576.3 | 576.0 | — |
| T14 | SGFEFHGYARa (71–80) | 1170.5 | 1170.9 | — |
| T14–15 | SGFEFHGYARSGVIMNDSGASTK (71–93) | 2418.1 | 2418.0 | — |
| T16 | SGAYITAGETGGAIGa (94–110) | 1577.7 | 1577.4 | 1577.8 |
| T17 | LGNQADTYVEMNLEHKa (111–126) | 1861.8 | 1861.6 | — |
| T19–20 | FKVMVADGQTSYNDWTASTSDLNVR (137–161) | 2805.3 | 2806.1 | — |
| T20 | VMVADGQTSYNDWTASTSDLNVR (139–161) | 2530.1 | 2529.8 | 2530.1 |
| T21 | QAFVELGNLPTFAGPFK (162–178) | 1835.9 | 1834.6 | 1836.0 |
| T26 | WNDGLRa (217–222) | 760.3 | 760.2 | 760.4 |
| T27 | SNFSLYGRa (223–230) | 943.4 | 943.2 | 943.1 |
| T28–29 | NFGDIDDSSNSVQNYILTMNHFAGPLQMMVSGLRAK (231–266) | 3972.5 | 3974.0 | — |
| T30–31 | DNDERK (267–272) | 776.3 | 775.9 | — |
| T34–35 | DGSSKTALLYGHGLGAEVK (304–322) | 1902.0 | 1903.1 | — |
| T36 | GIGSDGALRPGADTWR (323–338) | 1628.8 | 1628.7 | 1629.1 |
| T38–39 | SKDR (362–365) | 505.3 | 505.3 | 505.2 |
| T40 | YADGDSYQWATFNLR (366–380) | 1806.8 | 1806.4 | — |
| T40–41 | YADGDSYQWATFNLRIQAINQNFALAYEGSYQYMDLKPEGYNDR (366–410) | 5312.5 | 5313.0 | — |
| T42–43 | QAVNGSKFYKLTFAPTFK (411–427) | 1919.0 | 1918.2 | 1918.0 |
| T43–44 | LTFAPTFKVGSIGDFFSRPEIR (420–441) | 2485.3 | 2485.0 | 2483.9 |
| T44 | VGSIGDFFSRPEIR (428–441) | 1579.8 | 1579.7 | 1578.1 |
| T45–46 | FYTSWMDWSKK (442–452) | 1478.6 | 1478.3 | 1479.1 |
a PSD-sequencing was performed on the underlined fragments.
Fig. 3.

MALDI-PSD analysis of tryptic peptide fragment N-carbanoyl-ScrY (217–222). Signal of sequence-specific fragments y1–y6 and corresponding fragments by loss of NH3 are denoted; b-ions (except b1) and other fragments are not denoted.
Structural characterization of synthetic N-terminal peptides of ScrY porin
Several model peptides comprising different partial sequences and the complete N-terminal domain 1–71 of ScrY porin were synthesized using Fmoc and side-chain-protecting procedures as described in Materials and Methods (see Fig. 4 ▶). Peptide 2, ScrY(1–48)ox was prepared with the N-terminal sequence 1–45 of ScrY porin and additional insertion of a Gly-Gly-Cys sequence, which enabled oxidative formation of a disulfide linkage to yield a stable coiled-coil model peptide. All peptides contained the characteristic seven-residue repeating pattern (Fig. 4a ▶), indicating a high probability for coiled-coil formation within residues 4–46 of ScrY (Schülein et al. 1995).
All peptides were purified by semipreparative HPLC and characterized by mass spectrometric and spectroscopic methods (data not shown). The HPLC analyses showed single major peaks indicating homogeneous peptides, which were ascertained by MALDI-MS and ESI-MS determinations. The primary structures with correct sequences were further confirmed by MALDI-MS peptide-mapping analyses after trypsin digestion and by high-resolution ESI-FTICR-MS, providing exact mass determinations with isotopic resolution for the (M + nH)n+ ions (data not shown). The ESI spectra of the ScrY peptides provided evidence for the formation of dimeric and trimeric complexes as described below.
CD spectroscopy was carried out in different solvents and by concentration- and temperature-dependent studies to characterize secondary structures and a possible conformation-dependent formation of coiled-coil complexes. CD spectra revealed substantial extents of α-helical conformations; estimations of relative α-helix contents according to Yang et al. (1986) provided values ranging from ∼20% to quantitative helix formation (Table 2). According to Lau et al. (1984) and Rozzelle et al. (1995) the ratio of the mean residue ellipticity of the bands near 222 nm and 208 nm ([θ222]/([θ208]) was taken to indicate coiled-coil formation, with values close to 1 indicating an α-helical coiled-coil and values near 0.8 indicating an isolated α-helix. The corresponding CD data suggested significant coiled-coil formation within a wide temperature and pH range (data not shown; Michels 1999). For example, the thermal denaturation of peptide 1, ScrY(1–30) showed a stabilized coiled-coil formation at temperatures between 10°C and 40°C. In contrast, peptides with increasing chain length extending beyond the N-terminal coiled-coil region showed reduced relative α-helicity, and at the lowest α-helicities no factors indicative of coiled-coil formation were found. Consistent with these results, addition of trifluoroethanol (TFE) to the peptide solutions yielded significantly enhanced α-helicity (data not shown) with a maximum of conformational induction observed at ∼50% TFE.
Table 2.
Association of ScrY-peptides from dynamic light scattering dataa
| Peptide no. | pH | α-helix (%)b | RH (nm)c | Vsph (Å3)d | m.w.exp (kDa)e | m.w.corr. (kDa)f | m.w.seq. (kDa)g | Association |
| 1 | 6 | 100 | 1.71 | 20944 | 10.5 | 3.59 | 3.41 | 3 |
| 10 | 67 | 1.74 | 22066 | 11.5 | 3.80 | 3.41 | 3 | |
| 2 | 6 | 14 | 2.39 | 57185 | 24.0 | 5.56 | 10.28 | 2 |
| 10 | — | 2.31 | 51632 | 22.1 | 5.48 | 10.28 | 2 | |
| 3 | 6 | 40 | 2.17 | 42802 | 19.5 | 4.14 | 5.14 | 4 (2) |
| 10 | — | 2.37 | 55761 | 23.5 | 5.53 | 5.14 | 4 (2) | |
| 4 | 6 | 37 | 2.09 | 38240 | 17.6 | 5.53 | 6.05 | 3 |
a Solution concentrations of 1.4–4 × 10−3 M were determined.
b Estimation of % α-helicity from CD spectra.
c Average of 10 single determinations.
d Volume estimated for a spherical polypeptide.
e Molecular weight estimated from RH for a spherical polypeptide.
f Molecular weight estimated from RH with ellipticity correction according to α-helical coiled-coil shape.
g Sequence molecular weight.
The possible coiled-coil association of ScrY polypeptides was investigated by dynamic light scattering in aqueous solution and by ESI-MS under near-native solution conditions, and the results were compared with native gel electrophoresis data. ESI-MS has been successfully applied in recent years to the identification of noncovalent biopolymer interactions, including coiled-coil complexes such as leucine zipper polypeptides (Przybylski and Glocker 1996; Loo 1997; Przybylski et al. 1998). The ESI-MS data provided evidence for the presence of dimeric and trimeric complexes at different desolvation conditions (declustering potential) used to cause dissociation of noncovalent interactions (Przybylski and Glocker 1996). Evidence for the presence of dimers was obtained for all peptides 1–4 by the corresponding fivefold protonated, 5+ charged ions (2M + 5H)5+ and additional 7+ charged, sevenfold protonated ions corresponding to trimers were observed for ScrY(1–30) (data not shown). In the case of the disulfide-bridged peptide 2, ScrY(1–48)ox, ions due to the dimer were detected only with low abundance, consistent with the relatively low α-helix content found for this peptide. The oligomeric association of the N-terminal ScrY peptides was further ascertained by light scattering data (Table 2). The degrees of oligomer associations were derived from molecular weight estimations from values for the hydrodynamic radius (RH) of a spherical polypeptide, compared to values obtained by ellipticity corrections assuming an α-helical shape. These results indicated the formation of both dimers and trimers. Hence, the light-scattering data corroborated the coiled-coil association of ScrY polypeptides as observed by ESI-MS and are consistent with the recent observation of a three-stranded complex for a recombinant N-terminal ScrY polypeptide by analytical ultracentrifugation (Dumas et al. 2000).
NMR spectroscopic characterization of sugar binding of ScrY porin
TR-NOE-NMR studies allow the assessment of ligand-receptor interactions and provide details about the protein-bound conformation of oligosaccharides (Ni and Scheraga 1994; Poppe et al. 1997; Poveda and Jiminenez-Barbero 1998). TR-NOEs are observed only for equilibria with fast exchangement between free and receptor-bound ligands. This prerequisite restricts applications of the method to dissociation constants KD in the millimolar range. In this study maltotetraose was used instead of sucrose for the binding experiments. The binding affinity of maltotetraose was previously estimated with a KD of ∼1 mM in black-lipid-bilayer experiments (Schülein et al. 1995; Schnaible et al. 1997) in the presence of a membrane potential, whereas no membrane potential is required for the TR-NOE experiments.
In complexes involving large proteins such as ScrY porin, cross-relaxation rates of the bound ligand have an opposite sign relative to the rate of the free ligand, yielding negative NOEs. Therefore, ligand binding can be easily identified from the NOESY spectrum because NOEs for low-molecular-weight ligands are positive, as shown in Figure 5 ▶, for a ScrY-porin-maltotetraose-binding experiment. The porin–sugar complex (PL) was clearly detected by its positive NOESY cross-relaxation rates, whereas a negative cross peak indicates a shift of the equilibrium of protein and sugar (excess) to the sugar-free state. The dissociation constant for the complex between porin and maltotetraose was determined from the cross-signal rates according to Cooke et al. (1994). Comparative results for the binding of maltotetraose to intact ScrY porin and to the deletion mutant ScrY(Δ1–62) were determined from the linear regression plot of the ligand-binding curve according to equation 3 (see Materials and Methods), which yields −KD from the ordinate (Fig. 6 ▶; Table 3). The TR-NOE data provided a dissociation constant of 1.1 mM for intact ScrY porin but a five times higher KD of ∼5 mM for the ScrY(Δ1–62) deletion mutant. These results suggest a significant role of the N-terminal domain for sugar binding.
Fig. 5.

TR-NOE effect of ScrY porin and maltotetraose in 1H-NMR and NOESY spectra. The 1H-NMR spectra show the mixture of ScrY porin and maltotetraose in the sugar-free (negative cross signal) and sugar-bound state (positive cross signal). The NOESY spectrum shows the sugar-free state. The sign inversion of the NOE intensity identifies the protein binding of the ligand.
Fig. 6.

Ligand binding curves of ScrY porin and ScrY(Δ1–62) (a) and scheme of protein–ligand complex (b).
Table 3.
Calculated and experimentally determined data for 1H-NMR-NOESY-spectra of ScrY-porin and ScrY(Δ1–62)
| ScrY-porin | ScrY(Δ1–62) | ||||||||||||||||
| Exp. No.a | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| MMb | 1.6 | 2.4 | 3.6 | 4.8 | 6.8 | 8.8 | 12 | 16 | 1.32 | 1.98 | 2.64 | 3.63 | 4.62 | 5.61 | 7.26 | 10.56 | 15.5 |
| Ic/Idc | 83.7 | 63.2 | 41.7 | 33.5 | 25.3 | 19.3 | 14.7 | 10.9 | 11.1 | 6.5 | 5.2 | 4.2 | 3.3 | 2.6 | 1.3 | 0.5 | −0.6 |
| PL/Ld | 84.0 | 63.8 | 42.6 | 34.5 | 26.5 | 20.6 | 16.0 | 12.3 | 12.5 | 8.0 | 6.7 | 5.7 | 4.8 | 4.1 | 3.0 | 2.0 | 1.0 |
| xe | 0.12 | 0.16 | 0.235 | 0.29 | 0.38 | 0.485 | 0.625 | 0.81 | 0.8 | 1.25 | 1.5 | 1.75 | 2.1 | 2.4 | 3.3 | 5.0 | 10.0 |
a Number of experiment.
b Molarity of maltotetraose.
c Cross/diagonal signal of maltotetraose (%).
d Protein–ligand complex (%).
e [P]t[L]/[PL].
Similar results for sugar binding, and supportive of a possible transport function of the N-terminal domain, were derived from lipid-bilayer experiments (data not shown) and from determinations of single-channel conductivities, which have been extensively employed for the characterization of functional states of porins (Hancock et al. 1990; Cooke et al. 1994). Single-porin conductivities were measured from aliquots of porins in 1% Genapol solutions, dissolved in1 M KCl at 25°C (see Materials and Methods), and yielded values of 1.18 nS for the intact ScrY porin but a significantly higher 1.35 nS for the ScrY(Δ1–62) deletion mutant, respectively. Hence, these data and comparative studies with chemically modified ScrY derivatives (data not shown; Michels et al. 1999) further corroborate a functional role of the N-terminal polypeptide domain for the sugar transport.
Conclusions
In this study we present evidence for the 70-amino-acid N-terminal domain of ScrY porin to form α-helical coiled-coils with stable dimeric, as well as triple-stranded complexes. The oligomerization of peptides to dimers and trimers was identified by two different methods, ESI-MS and dynamic light scattering. These results obtained with synthetic polypeptides comprising the predicted coiled-coil domain ScrY(4–46) (Lupas 1996; Forst et al. 1998) are in agreement with recent data for a recombinant periplasmic ScrY domain analyzed by analytical ultracentrifugation (Dumas et al. 2000). However, the chemically synthesized peptides in this study having the ability to form coiled-coils may facilitate the design of suitable models for further structure–function studies.
TR-NOE-NMR studies comparing ScrY porin and the ScrY(Δ1–62) deletion mutant suggest the N-terminal domain as a potential binding structure for the transport of oligosaccharides into the periplasmic space. Binding affinities determined by NMR were consistent with results from black-lipid-bilayer experiments (Schülein et al. 1991; Michels 1999), thus confirming the efficiency of TR-NOE as a molecular analytical tool. A particular advantage of the TR-NOE method is that it can be performed in micellar solutions of the porin, that is, no electrical potential is required to analyze sugar-binding affinities. Although the detailed mechanism of periplasmic sugar transport is still unclear (as KD values give no information of transport direction through the pore and could apply to both high and low koff and kon values), the significant difference of KD for ScrY and ScrY(Δ1–62) porins clearly suggests a functional role of the N terminus for sugar binding. A structural model of the N-terminal domain may include formation of a trimer complex with the barrel domain of ScrY porin inserted into the outer membrane, which might provide an equilibrium between the single chains for low affinity binding of sugar and with flexibility for vertical movement. Corresponding future studies of sugar interactions and/or other possible periplasmic interaction partners can utilize the N-terminal peptides in this study.
Materials and methods
Expression and isolation of ScrY and deletion mutants
ScrY porin and the deletion mutant ScrY(Δ1–62) were expressed and purified essentially as previously described (Schülein et al. 1995) by using the E. coli strains KS-26 for ScrY and PUSL-112 for ScrY(Δ1–62). The expression procedure for the deletion mutant ScrY(Δ1–62) differed from that for ScrY in that carbenillicin (100 mg/mL) was employed in the growth medium instead of spectinomycin (Forst et al. 1998). β-octylglucoside (β-OG; 1.2%) was used as a detergent instead of the previously employed LDAO because β-OG has been found to be well feasible with MALDI-MS analysis (Schnaible et al. 1997).
Proteolytic digestion
Digestion of ScrY porin and ScrY(Δ1–62) with trypsin in solution was performed with a 20 μL aliquot of isolated porin (100–120 μg) containing 0.8% β-octylglucoside or 0.04% dodecylmaltoside, which was mixed with 20 μL of an 8 M urea solution. The solution was heated to 95°C for 5 min and after cooling to 25°C 40 μL digestion buffer (50 mM NH4HCO3 at pH 8) and 2.5 μL solution of trypsin (1 μg/μL) were added. The digestion was quenched after 4 h at 37°C by freezing. The resulting digestion mixture was separated by reversed-phase HPLC and the isolated fractions analyzed by MALDI-MS. For digestion of ScrY and ScrY(Δ1–62) porins by AspN protease, identical denaturation conditions and digestion times were employed as for the tryptic digestion. After denaturation of the porin solution, 40 μL digestion buffer and 10 μL AspN solution (0.04 μg) were added, and the resulting digestion mixture was separated by reversed-phase HPLC.
Standard tryptic digestion of the synthetic ScrY peptides was performed at pH 8 with TPCK-treated trypsin at 37°C for 4 h and the reaction mixture quenched by freezing. Synthetic peptides were dissolved in 50 mM NH4HCO3 buffer and a 2% trypsin solution was added. Proteolytic mixtures were directly analyzed by MALDI-MS.
In-gel digestion of ScrY and ScrY(Δ1–62) porins was performed in excised gel plugs as previously described (Schnaible et al. 1997; Bühler et al. 1998). Staining and destaining times of the gel were kept to a minimum because both steps led to partial fixing of the protein in the gel (Mortz et al. 1994; Shevchenko et al. 1996). The excised gel plugs were washed with 50 mM NH4HCO3:acetonitrile (60:40) and dried by vacuum centrifugation. TPCK-treated trypsin (12 ng/μL; sequencing grade) in digestion buffer (50 mM NH4HCO3) was added to the dry gel pieces and incubated for 60 min on ice for rehydration. After removing the supernatant, 20–40 μL digestion buffer was added and the digestion was continued for 8 h at 37°C. The peptide mixture was extracted, dried in vacuum, centrifuged, and dissolved in 10–20 μL of 5% formic acid for mass spectrometric analysis.
High-performance liquid chromatography
Chromatographic analyses were performed on a Waters-Millipore gradient HPLC system consisting of two high-performance pumps (M600A and M45) and a multiwavelength detector (490E) operating at 220, 254, and 280 nm. Separations were carried out on a 10 × 0.4 cm Vydac C18-column at 25°C at an eluent flow rate of 1 mL/min. Peptide solutions of 1 mg/mL in 0.1% TFA (eluent A) were employed with an injection volume of 20 μL. Mobile phase A was 0.1% TFA in water; mobile phase B was 0.07% TFA in acetonitrile. Retention times were determined using a linear gradient of 10%–95% B in 65 min.
Synthesis and characterization of N-terminal peptides
The N-terminal ScrY peptides 1–4 (Fig. 4 ▶) were synthesised on a semiautomated peptide synthesizer (EPS 221, Abimed) using standard SPPS Fmoc chemistry methods. All chemicals were of analytical grade or highest available purity. Fmoc amino acids, NovaSyn TGR-resin, PyBOB, and other reagents used in the peptide synthesis were obtained from Novabiochem. Fluka supplied DMF, TFA, and piperidine. HPLC-grade acetonitrile and acetic anhydride were obtained from Merck. Water was purified with a Milli-Q system (Millipore). To synthesize C-terminal carboxamide peptides the NovaSyn resin was employed with 40-min coupling time and 5-min deprotection time in a 20% (v/v) piperidine solution in DMF. Final cleavage from the resin was performed with 95% TFA/5% water for 2 h. Purification of the crude peptides was carried out with preparative HPLC on a Grom-Sil ODS-4Me column (250 × 20 mM; Grom) using an eluent gradient of 10%–95% acetonitrile in 0.1% aqueous TFA over 65 min. Approximately 10–15 mg of purified peptides were obtained.
Mass spectrometry
MALDI time-of-flight mass spectometry was performed with a Bruker Biflex-DE mass spectrometer equipped with a Scout MALDI source and video system (Bruker Daltonics), a nitrogen UV laser (λmax = 337 nm), and a dual-channel plate detector. Sample preparation was performed with 1 μL of a freshly prepared saturated solution of HCCA in isopropanol/water/HCOOH (v/v 3:2:1), which was directly mixed with 0.5 μL of the peptide solution (dried droplet sample preparation; Przybylski et al. 1998). Spectra were recorded at an acceleration voltage of 25 kV and were averaged over five single laser shots. Calibration was carried out using the singly and doubly protonated ions of bovine insulin as internal standard.
Low-resolution ESI-MS was performed on a Vestec 201A single quadrupole mass spectrometer (Vestec Corp.) equipped with a Teknivent Vector 2 data system. The employed nanoelectrospray device and insertion probe described previously (Fligge et al. 1999) was custom-made in our laboratory. Gold-coated nanospray needles were prepared from borosilicate glass capillaries GC120F-10 (Clark Electromedical Instruments). The gold coating was carried out with a Polaron SC7610 Sputter-Coater (VG Microtech) using a gold/palladium alloy in an Ar atmosphere of ∼0.1 mbar and an electric current of 10–15 mA (Fligge et al. 1999). A desolvation potential of 10 V at a needle voltage of 1.6–2.4 kV and temperature of 40°–55°C in the spraying chamber was used. Sample preparation was performed in 5 mM ammonium acetate solution and methanol (v/v 9:1) at pH 2, 6.5, 7, and 9. All sample solutions were filtered through a 0.45 μm syringe filter (Millipore) prior to use. FTICR-ESI mass spectra were recorded with an Apex II 7T FT-ICR instrument (Bruker Daltonik) equipped with an Apollo II electrospray/nanoelectrospray multiport ion source. Typical ESI conditions were ∼2 kV needle voltage and 100 nA spray current. Ions were accumulated in a hexapole for 2 sec and then transferred into the cylindrical ICR cell.
Single-channel conductivity determination
Single-channel conductivity measurements of porins were performed with a black-lipid-bilayer instrument custom-built in our laboratory, as described previously (Michels 1999), using 1,2-diphytanoyl-sn-glycero-3-phosphocholine membranes. Porin solutions (3–5 μL) in 0.1 M and 1 M KCl were added with a micropipette and measurements performed at 10–20 mV.
CD spectroscopy
CD spectra were recorded on a Jasco J-715 spectral polarimeter at 25°C in the range 178–260 nm and calibrated with an aqueous solution of (+)-10-camphersulfonic acid. Peptide solutions of 10−4 M in NH4Ac (5 mM) or phosphate buffer (10 mM) were employed. All CD measurements were performed within a pH range of 6–11, determined after dissolving the peptides in the aqueous solvent. Measurements were taken at 50-nm/min scan speed and a 1-nm bandwidth and were averaged over five scans.
For determining fractional ellipticities fH for each peptide, the mean residue ellipticity [Θ] at 222 nm was used from the equation from Chen et al. (Chen et al. 1972; Schmid et al. 1991). Thermal denaturation was monitored at 222 nm. Each solution was cooled to 10°C, allowed to equilibrate for 5 min, and scanned twice for an average time of 15 sec. This procedure was repeated at 10°C intervals until complete denaturation was indicated by a constant signal.
Dynamic light scattering
Diffusion coefficients of ScrY peptides were measured by dynamic light using a DynaPro instrument (DynaPro). Peptide concentrations of 2.2–4.4 10−4 M in 5 mM sodium phosphate buffer were prepared and measurements carried out at 25°C and pH 6, 7, and 10, respectively. Solutions were filtered through a 20-nm syringe filter (Anotec Separation) and samples illuminated at 780 nm using a solid-state laser.
The hydrodynamic radius (RH) was determined from the Stokes-Einstein equation
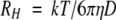 |
(1) |
where k is Bolzmann's constant and T the absolute temperature. The hydrodynamic radius RH is empirically related to the volume Vk and the molecular weight MWexp of a spherical molecule according to
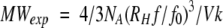 |
(2) |
where NA is Avogadro's number, Vk a partial volume set to 0.73 for a spherical protein, and f/f0 the ratio of frictional coefficients set to 1.2 for spherical proteins. Estimations of the oligomerization degree of the synthetic peptides were based on the molecular weights MWexp calculated for a spherical protein.
NMR spectroscopy
NMR spectra were acquired with a 600-MHz Bruker DRX 600 instrument (Bruker). Evaluation of spectra was carried out with the program UX NMR 1.3 on a SGI Indy workstation using tetramethylsilane or the resonance signal of deuterated solvent as internal standard. Two TR-NOE experiments were performed using maltotetraose for ScrYand ScrY(Δ1–62) porins, respectively. Different aliquots of a 0.4-mM maltotetraose standard solution were pipetted to a 0.1-mM ScrY- and ScrY (Δ1–62)-porin solution (see Table 3) containing 1.2% β-octylglucoside. After a mixing time of 500 msec, 1H-NMR and NOESY spectra of the three anomeric maltotetraose protons were measured. KD values for maltotetraose and porins were determined according to Cooke et al. (1994) using the equations
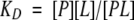 |
where [P] = porin; [L] = maltotetraose ligand; [PL] = complex.
With [P] = [P]t − [PL] (t = total concentration), and KD = [P]t[L] − [L][PL]/[PL] and KD = [P]t[L]/[PL] − [L], this yields
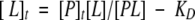 |
(3) |
In equation 3, used for the determination of KD, [L] is approximately equal to [L]t, which is only valid for [PL] ≪ [L] or [P]t ≪ [L]. NOE values of 1 for 100%-bound maltotetraose and of −1.6 for free maltotetraose were used in the equation
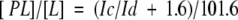 |
(Ich/Id, % cross/diagonal signal of maltotetraose). In equation (3) the x-axis equals [P]t[PL] = 0.1/[PL/L].
Acknowledgments
We gratefully acknowledge the expert assistance by Dr. Jörg Freygang in the purification of ScrY porin, and by Klaus Hägele with the FTICR-ESI mass spectrometric studies. This work has been supported by grants from the Deutsche Forschungsgemeinschaft (Biopolymer mass spectrometry), the Fonds der Chemischen Industrie, and the Ministery of Science Baden-Württemberg (Proteomics Competence Centre).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.2760102.
References
- Bühler, S., Michels, J., Wendt, S., Rück, A., Brdiczka, D., Welte, W., and Przybylski, M. 1998. Mass spectrometric mapping of ion channel proteins (porins) and identification of their supramolecular membrane assembly. Proteins Suppl. 2 63–73. [DOI] [PubMed] [Google Scholar]
- Chen, J.H., Yang, J.T., and Martinez, H.M. 1972. Determination of the secondary structures of proteins by circular dichroism and optical rotary dispersion. Biochem. 11 4120–4131. [DOI] [PubMed] [Google Scholar]
- Cooke, R.M., Hale, R.S., Lister, S.G., Shah, G., and Weir, M.P. 1994. The confirmation of the sialyl Lewis X ligand changes upon binding to E-selectin. Biochemistry 33 10591–10596. [DOI] [PubMed] [Google Scholar]
- Dumas, F., Frank, S., Koebnik, R., Maillet, E., Lustig, A., and Van Gelder, P. 2000. Extended sugar slide function for the periplasmic coiled coil domain of ScrY. J. Mol. Biol. 300 687–695. [DOI] [PubMed] [Google Scholar]
- Fligge, T.A., Kast, J., Bruns, K., and Przybylski, M. 1999. Direct monitoring of protein–chemical reactions utilizing nanoelectrospray mass spectrometry. J. Am. Soc. Mass Spectrom. 10 112–118. [DOI] [PubMed] [Google Scholar]
- Forst, D., Schülein, K., Wacker, T., Diederichs, K., Kreutz, W., Benz, R., and Welte, W. 1993. Crystallisation and preliminary X-ray diffraction analysis of ScrY, a specific bacterial outer membrane porin. J. Mol. Biol. 229 228–262. [DOI] [PubMed] [Google Scholar]
- Forst, D., Welte, W., Wacker, T., and Diederichs, K. 1998. Structure of the sucrose-specific porin ScrY from Salmonella typhimurium and its complex with sucrose. Nat. Struct. Biol. 5 37–46. [DOI] [PubMed] [Google Scholar]
- Hancock, R.E., Siehnel, R., and Martin, N. 1990. Outer membrane proteins of Pseudomonas. Mol. Microbiol. 4 1069–1075. [DOI] [PubMed] [Google Scholar]
- Hardesty, C., Ferran, C., and DiRienzo, J.M. 1991. Plasmid-mediated sucrose metabolism in Escherichia coli: Characterization of ScrY, the structural gene for a phosphoenolpyruvate-dependent sucrose phosphotransferase system outer membrane porin. J. Bacteriol. 173 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges, R.S., Saund, A.K., Chong, P.C.S., St. Pierre, S.A., and Reid, R.E. 1981. Synthetic model for two-stranded α-helical coiled coils. Design, synthesis, and characterization of an 86-residue analog of tropomyosin. J. Biol. Chem. 256 1214–1224. [PubMed] [Google Scholar]
- Lau, S.Y.M., Taneja, A.K., and Hodges, R.S. 1984. Synthesis of a model protein of defined secondary and quarternary structure. Effect of chain length on the stabilization and formation of two-stranded α-helical coiled-coils. J. Biol. Chem. 259 13253–13261. [PubMed] [Google Scholar]
- Loo, J.A. 1997. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 16 1–23. [DOI] [PubMed] [Google Scholar]
- Lupas, A. 1996. Coiled coils: New structures and new functions. TIBS 21 375–382. [PubMed] [Google Scholar]
- Michels, J. 1999. Struktur-und Funktionscharakterisierung von Ionenkanal-Proteinen mit proteinchemischen, biophysikalischen und massenspektrometrischen Methoden. In Faculty of Chemistry, pp. 1–243. University of Konstanz, Konstanz, Germany.
- Michels, J., Przybylski, M., Geyer, A., and Welte, W. 1999. The sucrose-porin (ScrY) from S. thyphimurium: Synthesis, structure characterisation and supramolecular association of the N-terminal polypeptide domain. Protein Science 8 128. Structural and spectroscopic characterization of native ScrY-porin and its sugar-complexes. [Google Scholar]
- Mortz, E., Vorm, O., Mann, M., and Roepstorff, P. 1994. Identification of proteins in polyacrylamide gels by mass spectrometric peptide mapping combined with database search. Biol. Mass Spectrom. 23 249–261. [DOI] [PubMed] [Google Scholar]
- Ni, F. and Scheraga, H.A. 1994. Use of the transferred nuclear Overhauser effect to determine the conformations of ligands bound to proteins. Acc. Chem. Res. 27 257–264. [Google Scholar]
- Poppe, L., Brown, S.G., Philo, J.S., Nikrad, P.V., and Shah, B.H. 1997. Conformation of sLex tetrasaccharide, free in solution and bound to E-, P-, and L-Selectin. J. Am. Chem. Soc. 119 1727–1736. [Google Scholar]
- Poveda, A. and Jiminenez-Barbero, J. 1998. NMR studies of carbohydrate–protein interactions in solution. Chem. Soc. Rev. 27 257–264. [Google Scholar]
- Przybylski, M. and Glocker, M.O. 1996. Electrospray mass spectrometry of biomacromolecules complexes with non-covalent interactions—New analytical perspectives for supramolecular chemistry and molecular recognition processes. Angew. Chem. Int. Ed. Engl. 35 806–826. [Google Scholar]
- Przybylski, M., Schnaible, V., Kast, J., Bühler, S., Michels, J., Wattenberg, A., Fligge, T.A., Forst, D., Diederichs, K., Zeth, C., et al. 1998. Approaches to the characterization of tertiary and supramolecular protein structures by combination of protein chemistry and mass spectrometry. In New Methods for the Study of Biomolecular Complexes (eds. W.E. et al.), pp. 17–43. Kluwer Academic Publishers, Amsterdam.
- Rozzelle, J.E., Jr., Wang, J.-G., Wagner, D.S., and Erickson, B.W. 1995. Self-association of a synthetic peptide fromthe N-terminus of the hepatitis d virus protein into an immunoreactive α-helical multimer. Proc. Natl. Acad. Sci. 92 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, K., Schupfner, M., and Schmitt, R. 1982. Plasmid-mediated uptake and metabolism of sucrose by Escherichia coli K-12. J. Bacteriol. 151 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, K., Ebner, R., Jahreis, K., Lengeler, J.W., and Titgemeyer, F. 1991. A sugar-specific porin, ScrY, is involved in sucrose uptake in enteric bacteria. Mol. Microbiol. 5 941–950. [DOI] [PubMed] [Google Scholar]
- Schnaible, V., Michels, J., Zeth, K., Freigang, J., Welte, W., Bühler, S., Glocker, M.O., and Przybylski, M. 1997. Approaches to the characterization of membrane channel proteins (porins) by UV MALDI-MS. Int. J. Mass Spectrom. Ion Process. 169/170 165–177. [Google Scholar]
- Schülein, K., Schmid, K., and Benz, R. 1991. The sugar-specific outer membrane channel ScrY contains functional characteristics of general diffusion pores and substrate-specific porins. Mol. Microbiol. 5 2233–2241. [DOI] [PubMed] [Google Scholar]
- Schülein, K., Andersen, C., and Benz, R. 1995. The deletion of 70 amino acids near the N-terminal end of the sucrose-specific porin ScrY causes its functional similarity to LamB in vivo and in vitro. Mol. Microbiol. 17 757–767. [DOI] [PubMed] [Google Scholar]
- Shevchenko, A., Jensen, O.N., Podtelejnikov, A.V., Sagliocco, F., Wilm, M., Vorm, O., Mortensen, P., Shevchenko, A., Boucherie, H., and Mann, M. 1996. Linking genome and proteome by mass spectrometry; large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. USA 93 1440–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J.T., Wu, C.S., and Martinez, H.M. 1986. Calculation of protein conformation from circular dichroism. Methods Enzymol. 130 208–269. [DOI] [PubMed] [Google Scholar]