

Abstract
The epitope of a monoclonal antibody raised against human thrombin has been determined by hydrogen/deuterium exchange coupled to MALDI mass spectrometry. The antibody epitope was identified as the surface of thrombin that retained deuterium in the presence of the monoclonal antibody compared to control experiments in its absence. Covalent attachment of the antibody to protein G beads and efficient elution of the antigen after deuterium exchange afforded the analysis of all possible epitopes in a single MALDI mass spectrum. The epitope, which was discontinuous, consisting of two peptides close to anion-binding exosite I, was readily identified. The epitope overlapped with, but was not identical to, the thrombomodulin binding site, consistent with inhibition studies. The antibody bound specifically to human thrombin and not to murine or bovine thrombin, although these proteins share 86% identity with the human protein. Interestingly, the epitope turned out to be the more structured of two surface regions in which higher sequence variation between the three species is seen.
Keywords: Epitope mapping, MALDI, thrombin, hydrogen/deuterium exchange, BIACORE
Epitope mapping is the process of identification of the molecular determinants for antibody–antigen recognition. Identification of the epitope is a key step in the characterization of monoclonal antibodies, especially those used in therapeutic strategies (Nelson et al. 2000). It has also been essential in the design of vaccines against toxins (Kazemi and Finkelstein 1991; Logan et al. 1991) or against enzymes (Gonzalez et al. 1994; for review, see Saint-Remy 1997) and in the mapping of ligand binding sites in receptors using monoclonal antibodies (Lorenzo et al. 1991).
Several methods have been developed for mapping protein epitopes of monoclonal antibodies. A popular strategy involves probing the antibody-binding ability of synthetic peptides derived from the amino acid sequence of the antigen. Binding is assessed using BIACORE or ELISA (Reineke et al. 1999). More recent methods involve the combinatorial synthesis of peptide libraries onto a planar support (i.e., a protein chip), followed by exposure of the surface to the antibody. Identification of the epitope peptides is done on the chip by any of the multiple methods for solid-phase screening (Reineke et al. 2001). Another combinatorial method involves the phage display of a library of peptides derived from the sequence of the protein antigen. Selection of the epitope peptides takes place after several rounds of biopanning (Burritt et al. 2001). One major limitation of these peptide-based techniques is that epitope identification is feasible only for antibodies that have a linear, continuous epitope. In other words, peptide-based methods are not useful in identifying discontinuous epitopes, a category to which most protein epitopes belong (Klein and Horejsi 1997).
The application of MALDI mass spectrometry to the analysis of proteins has triggered the development of new strategies for the identification of discontinuous epitopes. In limited proteolysis, the antigen is cleaved by different proteases, in the presence and in the absence of the antibody, and the fragments are identified by mass spectrometry. The epitope is the region of the antigen that becomes protected from proteolysis upon binding of the antibody (Suckau et al. 1990). In selective chemical modification, the antigen reacts with chemical tags specific for Arg, Tyr, and Lys residues. The sites of modification in the free antigen and in the antibody-bound antigen are determined by mass-spectrometric peptide mapping (Fiedler et al. 1998). In epitope excision, antibody beads are incubated with the antigen protein and subjected to extensive proteolysis. The beads are then washed several times to get rid of the nonepitope fragments, whereas the epitope fragments remain bound to the antibody. After acid elution from the antibody beads, the epitope peptides are identified by MALDI mass spectrometry (Van de Water et al. 1997).
The major limitation of these MALDI-based methods is resolution. Both epitope excision and limited proteolysis involve proteolytic cleavage with trypsin. This limits the mapping to epitope regions that contain cleavage sites and results in the identification of long peptide sequences (typically 30–60 residues) that poorly define the epitopes. Similarly, the effectiveness of chemical modification methods depends on whether label-reactive residues are located in the epitope as much as it depends on the spacing between these reactive residues.
In this work we have mapped the epitope of a monoclonal antibody (mAb) from mouse, generated against human thrombin, by measuring deuterium retention of backbone amides in thrombin upon complexation with the mAb in a variation of the method developed by Mandell et al. (1998a,b). In the experiments described here, the mAb was first immobilized on protein G beads and covalently linked. Then, both the thrombin and the mAb beads were allowed to become deuterium-labeled in the free state and subsequently incubated together to form the deuterated immunocomplex. Exchanging the complex back into water allowed the nonepitope region to become protonated, while the epitope region retained deuterium due to protection by the mAb. The thrombin and mAb were then separated by an acidic organic solution that was compatible with pepsin digestion. Finally, the deuterium retention of the peptic fragments was measured using MALDI mass spectrometry. The epitope was found to be two discontinuous surface strands on thrombin. The sequence diversity of the region is high compared to the overall high similarity of 86% identical to mouse, explaining the high selectivity of the antibody for foreign (human) versus self (mouse) antigen.
Results
The mAb inhibits the activity of thrombin toward protein C
Previous studies had already shown that mAb binding to human thrombin resulted in the inhibition of clotting activity but not in the inhibition of amidolytic activity (Dawes et al. 1984). Inhibition of clotting could be caused in two distinct ways: The mAb could be binding in the vicinity of the active site and disallowing the entrance of fibrinogen by steric hindrance, or the mAb could be binding to the fibrinogen recognition exosite, which also serves as the primary binding site for thrombomodulin (TM). To distinguish between these two possibilities, we measured the ability of the thrombin:TM complex to cleave and activate protein C. The protein C activation assay was done in the presence of increasing amounts of the mAb (Fig. 1 ▶). A decrease in protein C activation was observed after incubating the thrombin with mAb for 10 min, indicating that the mAb binds to a region of thrombin involved in protein C activation. The antibody did not interfere with protein C binding, as shown by the fact that the TM-independent activity of thrombin toward protein C was not affected by the binding of the mAb. Finally, the mAb was shown to completely block thrombin binding to TMEGF456 on a BIACORE surface (Fig. 2 ▶).
Fig. 1.

The activity of the thrombin:TMEGF456 complex toward protein C was measured after incubating the thrombin with increasing concentrations of the mAb for 10 min (black bars). The mAb had no effect on the TMEGF456-independent activation of protein C (gray bars).
Fig. 2.

Binding of thrombin to a TMEGF456 surface was monitored in real time using a BIACORE assay. Sensorgrams were collected both in the presence (black) and in the absence (gray) of mAb. Binding of mAb to thrombin completely abolished the latter's ability to bind to TMEGF456.
Mapping of the antibody binding site on thrombin
First, a traditional epitope mapping experiment was performed in which a 20-fold molar excess of thrombin was digested to completion with pepsin. The resulting digest mixture was used in a competition experiment in which 135 ng of thrombin was immunoprecipitated with mAb covalently linked to protein G beads. The same amount of thrombin was immunoprecipitated in the presence or absence of the digest mixture, suggesting that the epitope may consist of more than one segment on thrombin.
Thinking that the epitope might be discontinuous, we thought to use amide hydgrogen/deuterium (H/D) exchange following the same strategy that was previously applied to identifying the TMEGF45 binding site on thrombin and the inhibitor binding site on protein kinase A (Mandell et al. 1998b). The strategy involves deuteration of the free proteins prior to complexation, back exchange into water, and quantitation of the amount of deuterium retained by the different regions of thrombin upon binding to the mAb. Because the mAb, a large protein, produced many peptides that interfered with analysis of the thrombin by mass spectrometry, the mAb was first covalently immobilized onto a solid support. This allowed us to eliminate the mAb from the reaction mixture by quickly incubating the thrombin-bound mAb beads with 50% 1-propanol, 50% 0.1% TFA at pH 2.5 prior to digestion with pepsin. Upon centrifugation, the thrombin was separated from the mAb beads and the resulting mass spectra contained only peptides arising from thrombin digestion.
Much of thrombin becomes deuterated after 10 min incubation in buffered D2O, but after complexation with the antibody, most of the thrombin peptides retained little or no deuterium after back exchange for equivalent times into water (Table 1). Among these antibody-inert regions of thrombin were some that previously showed significant deuterium retention upon binding to the thrombomodulin fragment TMEGF45 (Mandell et al. 2001). This was the case for residues 167–180 (residues 133–144 in chymotrypsin numbering), shown in Figure 3A ▶, which corresponds to the loop containing W141. Also in this same category were residues 117–132 (residues 85–101 in chymotrypsin numbering [Fig. 3B ▶]), which correspond to a surface connection between the fibrinogen recognition site and the active site of thrombin. Finally, the C-terminal tail of thrombin, which also retained deuterium upon TMEGF45 binding, showed no retention upon binding of the mAb (Fig. 4 ▶).
Table 1.
Quantitative differences for those regions of thrombin that previously showed differences upon thrombomodulin binding
| Thrombin regiona | Residue no. | Centroidb free thrombin | Centroid thrombin/+ MAb | ΔMass |
| TM-binding site | 97–117 | 2589.1 | 2591.3 | 2.2 |
| TM-binding site | 96–112 | 2129.8 | 2130.5 | 0.7 |
| TM-binding site | 54–61 | 1006.1 | 1006.2 | 0.1 |
| TM-binding peripheral site | 139–149 | 1265.2 | 1266.7 | 1.5 |
| W141-segment | 167–180 | 1508.7 | 1509.1 | 0.4 |
| W148-segment | 181–196 | 1717.4 | 1717.6 | 0.2 |
| C-terminal helix | 281–293 | 1704.0 | 1704.1 | 0.1 |
| Allosteric site/active site loop | 117–132 | 2147.1 | 2147.2 | 0.1 |
a Thrombin regions as defined in Mandell et al. (2001): peptides corresponding to the thrombomodulin (TM) binding site, peripheral site, and allosteric sites including the W141-W148 loop and the C-terminal helix. All other regions that were covered quantitatively as indicated in Figure 4 ▶ showed no difference in the presence or absence of mAb.
b Weighted average of the isotopic mass envelope for a given peptide. These experiments were performed in duplicate, and the error between determinations is ∼0.2 deuterons.
Fig. 3.
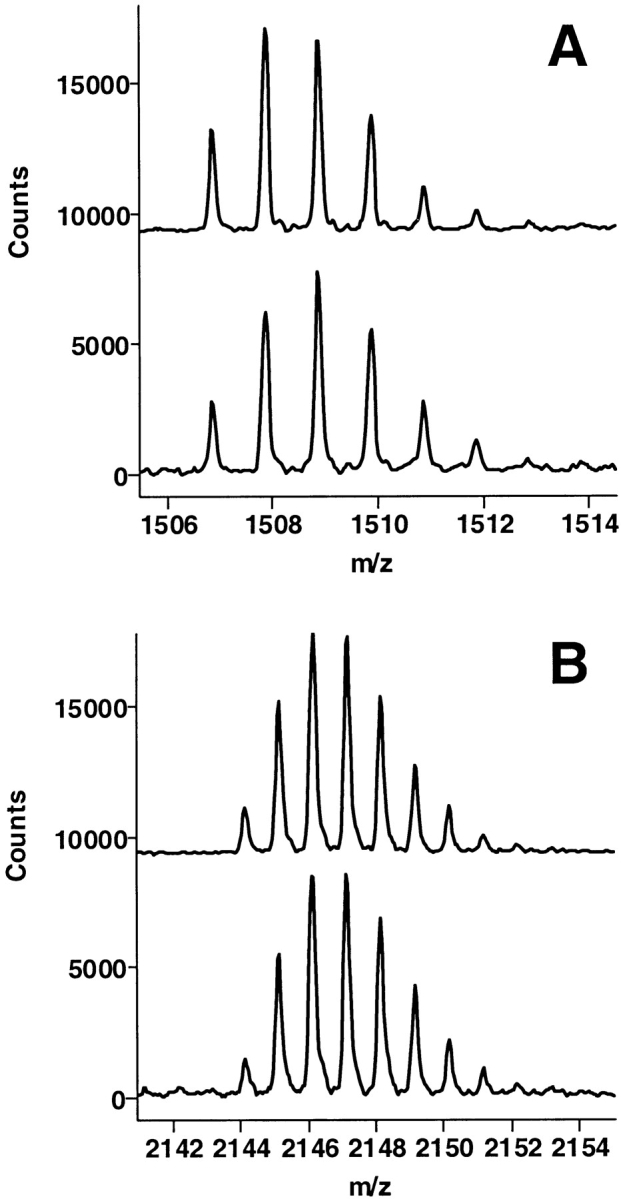
Several regions of thrombin that showed retention of deuterium upon binding of thrombomodulin (Mandell et al. 2001) did not show any significant mass difference upon binding of mAb (bottom spectrum) compared to the no-mAb control (top spectrum). (A) Thrombin residues 167–180 (residues 133–144 in chymotrypsin numbering). (B) Thrombin residues 117–132 (residues 85–101 in chymotrypsin numbering).
Fig. 4.

Sequence of thrombin showing the different peptides that were obtained from pepsin digestion and for which quantitative hydrogen/deuterium (H/D) exchange data could be obtained. Key residues are identified above the sequentially numbered sequence in the chymotrypsin numbering scheme. The black bars denote peptides that did not shift in the presence of either TMEGF45 or mAb. The green bars correspond to regions that shifted in mass in the presence of TMEGF45 but not in the presence of the mAb. The red bars show the regions that shifted in mass in the presence of both TMEGF45 and the mAb.
The fibrinogen recognition site itself, residues 97–117 (residues 67–85 in the chymotrypsin numbering), was significantly protected by the mAb, although the portion of this peptide that is actually involved in binding to mAb is different from the portion contacting TMEGF45 (Fig. 5 ▶). Comparing the shift in mass for this peptide, 97–117 (Δmass = 2.2), with the mass shift of the shorter overlapping peptide, 96–112 (Δmass = 0.7), we observed that most of the protection was taking place in residues 113–117. This observation comes in direct contrast with the protection of this region by TMEGF45, which showed the same degree of protection of both peptides, indicating that the binding site of TMEGF45 is in the 96–112 segment.
Fig. 5.

The region of thrombin corresponding to amino acids 97–117 (residues 67–85 in the chymotrypsin numbering) retained a significant amount of amide deuteration in the presence of the mAb (bottom spectrum) relative to the control (top spectrum).
Residues 139–149 (residues 108–116 in chymotrypsin numbering) were significantly protected by the mAb (Fig. 6 ▶). This region was only marginally protected upon binding of TMEGF45. Based on these mass spectrometric measurements of deuterium retention, we propose that peptides corresponding to the adjacent residues 139–149 and 113–117 (Fig. 7 ▶) comprise the discontinuous epitope for binding of the mAb.
Fig. 6.

The other region of thrombin that was significantly protected in the presence of the mAb was the one corresponding to amino acids 139–149 (residues 108–116 in chymotrypsin numbering). This region also showed some protection upon contact with TMEGF45 (Mandell et al. 2001), but the amount of protection was less.
Fig. 7.
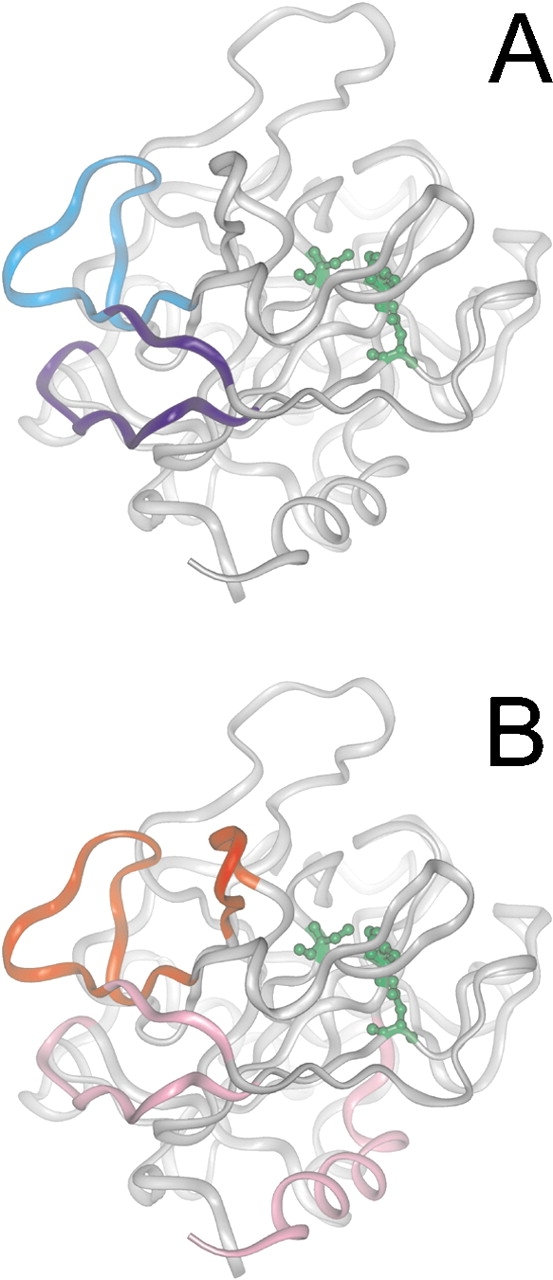
Comparison of the binding maps generated by hydrogen/deuterium (H/D) exchange experiments for the interaction of thrombin with mAb (A) and TMEGF45 (B; Mandell et al. 2001). Surface segments of thrombin that were highly protected (blue) are distinguished from those that were only slightly protected (cyan) upon binding of the mAb. Similarly, segments of thrombin that were highly protected (red) are distinguished from those that were only slightly protected (magenta) upon binding of TMEGF45. The binding sites of MAb and TMEGF45 are overlapping but not identical. The thrombin structures are shown in the same orientation and the active site residues are indicated in green.
Synthetic epitope peptide corresponding to region 139–149 disrupts thrombin:mAb binding
The peptides corresponding to regions 97–117 and 139–149 were synthesized to see if the peptides alone were sufficient for antibody binding. The association of mAb to the thrombin on a BIACORE surface was monitored both in the presence and in the absence of a 2000- and 10,000-fold molar excess of either or both peptides. Figure 8 ▶ shows that the presence of the peptide corresponding to amino acids 97–117 did not affect recognition of thrombin by the mAb even at a 10,000-fold molar excess. This shows that this peptide alone is not sufficient for recognition. It is possible that the recognition of this peptide by the antibody requires the peptide to be structured in a native-like conformation.
Fig. 8.
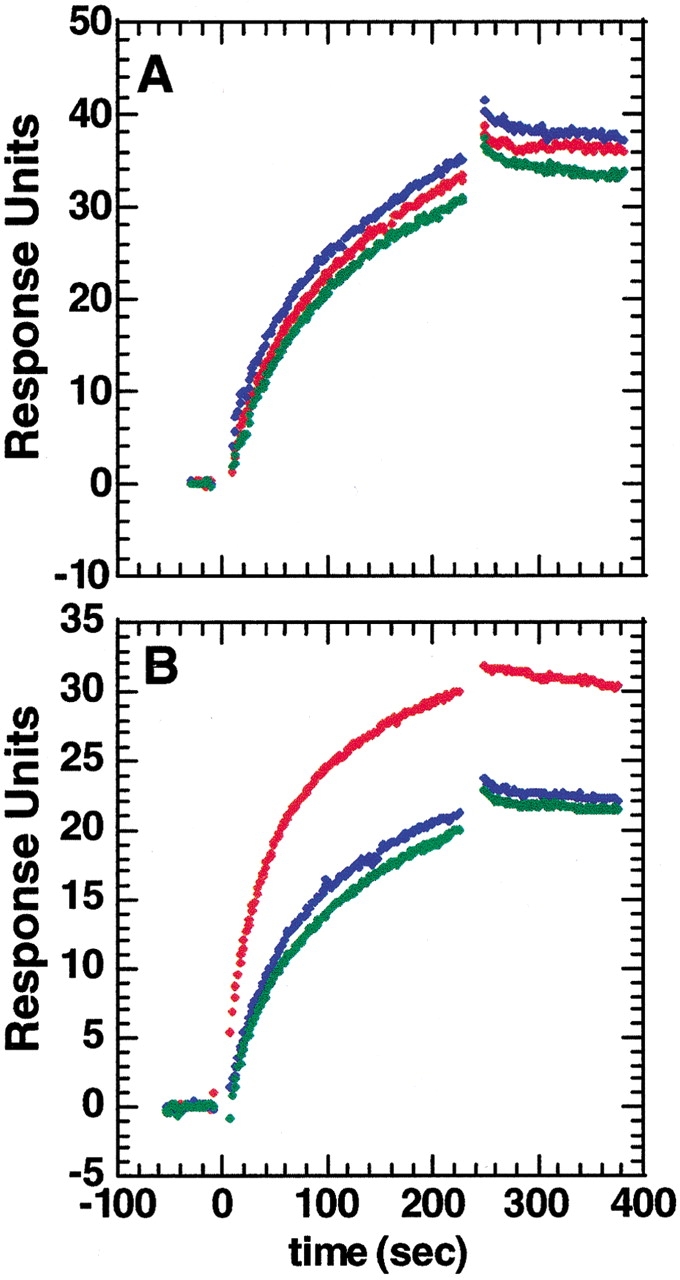
Binding of the mAb (10 nM) to a surface containing biotinylated thrombin was monitored in real time both by itself (red) or in the presence of 20 mM (blue) and of 100 mM (green) of the peptide corresponding to residues 97–117 of thrombin (A) and residues 139–149 of thrombin (B). Although the peptide corresponding to residues 97–117 did not affect binding of mAb, the peptide corresponding to residues 139–149 disrupted binding partially at high concentration.
The peptide corresponding to amino acids 139–149 competed for binding to the antibody at a 2000-fold molar excess, indicating that this stretch of amino acids is the primary epitope for the thrombin:mAb interaction. The fact that no peptides were found in a classical epitope competition experiment (see above) is consistent with the observation that even this peptide binds to mAb with a low affinity when not in its native conformation.
Discussion
In recent years, the use of mass spectrometry to interrogate protein structure and function has become widespread (Chambers et al. 2000). In this work we have used amide H/D exchange to identify the epitope of thrombin that binds a monoclonal antibody. The covalent immobilization of the mAb onto protein G beads simplified the experiment threefold. First, the mAb was eliminated from the solution; this simplified the analysis of the mass spectra, which can get overcrowded with peaks rather easily. Second, the thrombin:mAb beads were washed in water once during the back-exchange reaction, ensuring that all the thrombin that had not bound to mAb was washed away, so that we only measure deuterium retention in thrombin molecules that were in complex with the mAb. This modification probably resulted in an increase in the sensitivity of the method. Third, the use of mass spectrometry combined with short deuteration times allows preferential analysis of the surface of the antigen. In the past, amide H/D-exchange experiments using NMR, which mainly detects slowly exchanging amides, were complicated by long-range conformational changes that were observed upon antibody binding (Williams et al. 1997).
Our method for epitope mapping can be applied to any protein–protein interaction, in which one protein can be covalently immobilized and the other "pulled down". Typically for a pull-down experiment to work, the kd of the interaction has to be <0.01 min−1, and this is generally true for interactions with Kd <10 nM (Mandell et al. 2001). The method works especially well for epitope mapping because the binding site on the antibody is far from the protein-G-bound constant region. Subsequent amide H/D exchange surface mapping gives higher resolution of the epitope than existing methods. Because pepsin cleaves at many sites, overlapping peptides are generated, allowing identification of exact binding sites and of discontinuous sites.
Most, if not all, epitopes of antibodies produced by immunizing with native proteins are of the discontinuous type (Klein and Horejsi 1997). Protein structure is so convoluted that there are virtually no contiguous regions on the molecular surface large enough to form an epitope. Initial experiments for finding the epitope showed that no peptides generated by pepsin digestion of thrombin competed with the antibody for thrombin binding, suggesting that the identification of an epitope comprised of a single peptide would not be possible. The epitope did indeed turn out to be discontinuous, consisting primarily of two adjacent regions of thrombin: residues 113–117 and 139–149 (Fig. 7A ▶).
Other regions that were previously found to be protected by TMEGF45 showed little or no retention of deuterium, including residues 167–180, residues 117–132, and the C-terminal tail of thrombin (Figs. 4, 7B ▶ ▶). One explanation for this is that the mAb recognizes a smaller region than the cofactor TMEGF45. This explanation is consistent with the results from theoretical studies of comparison of different protein–protein interfaces, which show that, in general, antibody–antigen interfaces consist of fewer atoms than the average protein–protein interface (Lo Conte et al. 1999). Also antibody–antigen interactions have a lower-than-average fraction of interface atoms completely buried and a higher-than-average fraction of interface atoms still in contact with the solvent.
It has also been observed that, in general, antibodies bind like rigid molecules and require that their antigen be in the proper conformation and have optimal curvature for binding (Rees et al. 1994). Our findings are consistent with this notion because the antibody appears to bind to a small region and only cause changes in solvent accessibility in the vicinity of the binding site, whereas TMEGF45 binding appeared to have a significant influence over the dynamic behavior of remote regions of thrombin (Ye et al. 1991; Mandell et al. 2001). It is interesting to note that despite the fact that the antibody–antigen interaction most likely involves primarily the interaction of amino acid side chains across the interface, we were able to detect the epitope based on decreases in amide exchange of the backbone. The most probable reason for this observation is that the epitopes were in loops that were solvent-exposed on the surface of thrombin and became less exposed in the antibody-bound complex. It is likely that most but not all antibody–antigen interactions would involve some decrease in solvent exposure of the binding site, even if it is mainly side chains that are directly involved in the interaction.
The sequences of bovine, mouse, and human thrombin show remarkable similarity. Over 85% of the sequence is identical for all three species of mammals, and we know the similarity between human and bovine results in various forms of cross-reactivity. Bovine thrombin cleaves human fibrinogen and human protein C and binds to human TMEGF456 with the same affinity and kinetics as human thrombin (Baerga-Ortiz et al. 2000). The mAb was absolutely specific for human thrombin and did not cross-react with bovine thrombin. No binding was observed between mAb and bovine thrombin in BIACORE assays. The mAb was, of course, selected in mice and would not bind mouse thrombin because thrombin is an essential self-protein. Of the 17 positions in thrombin sequence (out of 293) that differed between human, mouse, and cow (Fig. 9 ▶), only eight are on the surface of thrombin. Of these, three residues were located close to the region that we identified as the epitope (108, 136, and 143). Interestingly, five variable sites clustered around loop 181–196 (residues 145–160 in the chymotrypsin numbering) of thrombin (Fig. 9 ▶), and this loop therefore would have been identified as the best epitope candidate by sequence alignments. Two peptides covered this region and neither showed retention of deuterium upon mAb binding (Fig. 4 ▶). This loop is known to be highly unstructured, so perhaps it is not a good candidate for antibody recognition. It is very interesting that the antibody selected the most highly variable structured surface on thrombin. The combination of sequence variation and structural conservation at this surface probably provided the highest selectivity of binding for human over self.
Fig. 9.
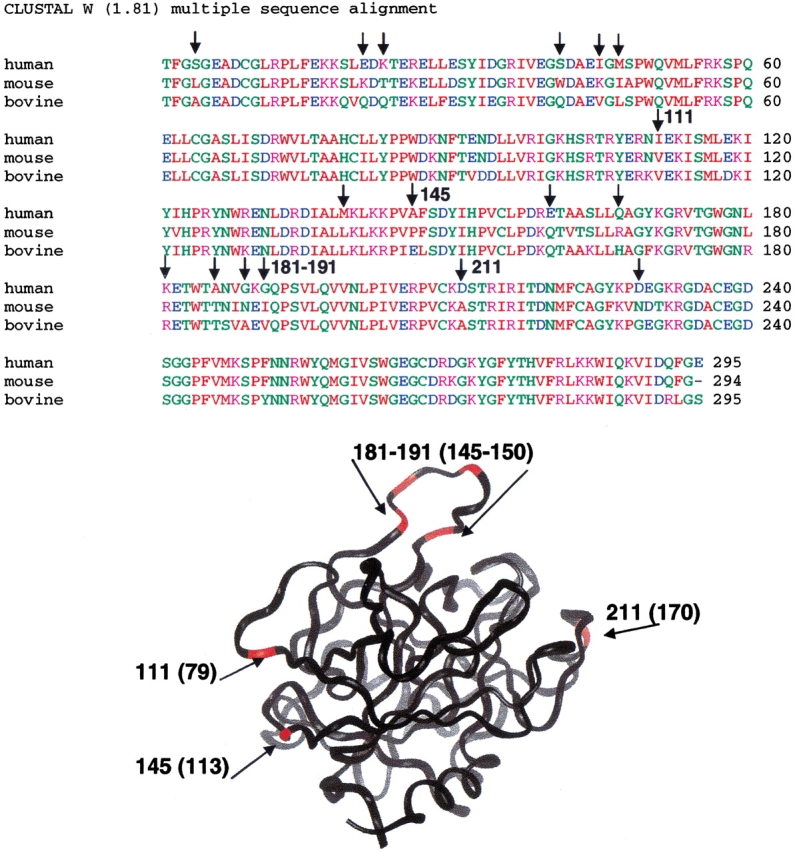
Alignment of the sequences for human, bovine, and mouse thrombin shows possible sites for antibody recognition. Of 17 sequence differences (vertical arrows above the sequence) between human and mouse thrombin, 8 were on the surface of thrombin and were viable epitope candidates (red in the structure). The residue numbers for these are shown on both the sequence and the structure and they map to different regions of thrombin. Two of them, 111 and 145, were in the identified epitope (arrows), whereas 4 were located near the 181–196 loop, which did not retain deuterium upon binding of mAb.
Materials and methods
Proteins
Human thrombin was purified from plasma barium citrate eluate (Sigma) according to previously published procedures (Ni et al. 1990). Essentially, the eluate powder was redisolved and the 70% ammonium sulfate fraction containing prothrombin was obtained. After gel filtration, the prothrombin was activated by incubating with Echis carinatus venom and the protein purified on a Mono S FPLC column equilibrated with 25 mM KPhos (pH 6.5), 100 mM NaCl. The thrombin was eluted with a linear gradient increasing to 500 mM NaCl. Active thrombin was identified by fibrinogen clotting assay.
For the mass spectrometry experiments, thrombin was exchanged into buffer (50 mM NaH2PO4 [pH 6.6], 50 mM NaCl) using a centricon 10 (Millipore). The final solution (1 mg/mL) was lyophilized in 750-pmol portions and stored at −70°C. The mouse monoclonal antibody (Dawes et al. 1984) was purchased from Haematologic Technologies.
Protein C activation assay
The protein C activation assay has also been previously described (Lougheed et al. 1995). Essentially, thrombin was mixed with TMEGF456 in a 96-well plate and human protein C was added. After 20 min, the reaction was quenched with 40 μL of a solution of heparin/antithrombin III and the amount of activated protein C was determined using chromogenic substrate S2366 (Chromogenix).
To assay whether the mAb competed with TMEGF456 in the protein C activation assay, 0.04 mg/mL thrombin was incubated with 1.66 mg/mL, 0.33 mg/mL, 0.17 mg/mL, 0.08 mg/mL, 0.04 mg/mL, and 0 mg/mL mAb for 30 min. The samples were then diluted 1:10 right before adding them to the assay mixture. To assay whether the mAb inhibited the TM-independent activation of protein C, the thrombin:mAb mixtures were diluted 2-fold instead of 10-fold, and the assay was performed in the manner already described except that no TMEGF456 was added to the well.
BIACORE measurements
BIACORE measurements were carried out on a BIACORE 3000 instrument as described previously (Baerga-Ortiz et al. 2000). The surface was prepared by flowing TMEGF456 (TM residues 346–465) that was biotin labeled at the N terminus over a surface containing immobilized streptavidin. TMEGF456 (430 response units) was coupled to the surface and thrombin binding was assayed in 10 mM HEPES (pH 7.4), 150 mM NaCl, 2.5 mM CaCl2, 0.005% P-20 by flowing a single concentration of thrombin 12.5 nM over the surface after incubation at room temperature for 30 min. In previous experiments we showed that the smallest active fragment of TM, TMEGF45 (TM residues 346–426), has a 10-fold faster kd, which makes BIACORE measurements on this fragment difficult. The BIACORE experiments presented here were therefore performed using the larger fragment, TMEGF456 (TM residues 346–465). To assay for binding competition by the mAb, a mixture containing 12.5 nM thrombin and 40 nM mAb was prepared in the flowing buffer and allowed to complex for 30 min prior to injection.
Preparation of antibody beads
Protein G beads were purchased from Sigma and incubated (200 μL) with 2 mg of antibody (bead capacity: 20 mg of IgG/1mL beads) in 50 mM sodium borate (pH 9) and 3 M NaCl overnight while mixing. After washing with borate buffer, 20 mM dimethylpimelimidate (DMP) was added to covalently cross-link the mAb to the protein G on the solid support. After 2 h the cross-linking was quenched with 1 M ethanolamine (pH 8.5) for 20 min. The beads were washed extensively with borate buffer and then with a solution of 10 mM glycine (pH 1.7) to eliminate the mAb that did not get covalently bound to the beads. Finally, the beads were buffer exchanged into 50 mM NaH2PO4 (pH 6.6), 50 mM NaCl, and stored in 30 μL aliquots. Thrombin-binding to the mAb beads and to control (protein G) beads was assessed by pull-down experiments in which various concentrations of thrombin were incubated with 30 μL portions of beads. These experiments revealed that thrombin did not bind nonspecifically to the protein G alone, even under conditions in which thrombin was in a 20-fold excess (i.e., <10% of the thrombin bound to the beads; data not shown). These control experiments were also used to ascertain the binding capacity of the beads.
Deuterium retention experiments
Thrombin was resuspended in 3 μL D2O (Cambridge Isotopes) for 10 min to allow deuteration of surface amides. The mAb beads (30 μL) were resuspended in 270 μL of deuterated phosphate buffer (50 mM KH2PO4 at pH 6.6, 50 mM NaCl) for 10 min and then centrifuged and decanted to 30 μL. The mAb and thrombin were mixed together for 10 min in a volume of 33 μL.
Back exchange was carried out by diluting the mixture of thrombin plus beads in 270 μL of H2O. The beads were centrifuged, the supernatant was discarded, and the beads (in ∼30 μL) were resuspended in 270 μL H2O, centrifuged again, and then decanted to 30 μL. The time from the first dilution into H2O until completion of the final centrifugation was 2 min. During this 2 min, back exchange of all the amides that were not in the interface of the thrombin:mAb complex was occurring. To measure the remaining deuterium content in the amides of thrombin, the reaction was quenched by mixing with 30 μL of a chilled (4°C) solution of equal parts of 0.1% TFA (pH 2.25) and 1-propanol. This solution not only quenched the exchange but also eluted the thrombin from the beads, which were removed by centrifugation and discarded. The supernatant (30 μL) was mixed with 100 μL of a slurry of immobilized pepsin (Pierce Chemicals) at 4°C in 0.1% TFA (pH 2.25) for 10 min. After 10 min of digestion, the immobilized pepsin was removed by centrifugation and the sample was rapidly frozen in liquid nitrogen and stored at −70°C until analysis within 1–2 d.
A control experiment (no mAb) was carried out by resuspending the thrombin in 3 μL of D2O and subjecting it to back exchange with 27 μL of H2O. The rest of the experiment was performed exactly the same way as already described for the mAb experiment. This control experienced a 90% dilution of the D2O with H2O, whereas the thrombin:mAb sample on the beads experienced a 99% dilution. Despite this difference, retention of deuterium in the thrombin:mAb sample was clearly observed over that of the free thrombin. In MALDI-MS analysis of H/D exchange, a small correction is required for the residual D2O present at the time of mass spectrometry and this correction (for 0.05% remaining for mAb experiments or 5% remaining for control experiments after dilution into matrix) was applied to the data shown in Table 1 (Mandell et al. 2001).
Mass spectrometry
MALDI-TOF mass spectrometry was done on a Voyager DE STR (PE Biosystems) and the matrix was 5 mg/mL alpha-Cyano-4-hydroxycinnamic acid (Sigma) in 1:1:1 (0.1%TFA:ethanol:acetonitrile) that was adjusted to pH 2.2 after mixing all the ingredients. Pepsin-digested thrombin samples were thawed and mixed 1:1 with cold matrix solution. The mixture (1 μL) was quickly spotted onto a cold MALDI plate and analyzed. The identities of each peak in the spectrum of pepsin-digested thrombin have been previously determined and reported (Mandell et al. 2001). The spectra were displayed and analyzed using the GRAMS-MS software and calibration of the spectra was done using the theoretical mass of two prominent previously identified thrombin peptides (1048.5328 and 2144.1406). The centroid of each isotopic mass envelope was measured with the CAPP software written by Jeff Mandell (Mandell et al. 1998a).
Sequence alignment
Sequences were aligned using the CLUSTALW module for multiple-sequence alignments (Thompson et al. 1994).
Peptide synthesis
Peptides corresponding to regions 97–117 (VRIGKHSRTRYER NIEKISML) and 139–149 (MKLKKPVAFSD) were synthesized using FMOC chemistry on a MilliGen 9050 solid-state peptide synthesizer and purified as previously described (Hunter and Komives 1995). The purity of each peptide was verified by mass spectrometry.
Inhibition of thrombin:mAb binding by epitope peptides
A BIACORE surface containing thrombin was prepared as follows. Thrombin was biotin-labeled at the active site by reaction with the covalent active site inhibitor PPACK that was biotin-labeled (Haematologic Technologies). The amount of biotinylated thrombin coupled to the streptavidin surface was 250 response units (SA-chip, BIACORE). Coupling was carried out at a flow rate of 5 μL/min in 10 mM Hepes (pH 7.4), 300 mM NaCl, 2.5 mM CaCl2, 0.005% P-20. The remaining biotin binding sites were blocked by injecting 100 μL of a solution of biotin (0.02 mg/mL in the same buffer). A solution of 100 nM mAb in HBS (10 mM Hepes [pH 7.4], 150 mM NaCl, 2.5 CaCl2, 0.005% P-20) was incubated for 4 h in the presence of a 2000-fold and a 10,000-fold molar excess of peptide corresponding to amino acids 97–117 or 139–149 of thrombin or a control peptide not derived from the thrombin sequence. Each sample was diluted 1:10 in HBS prior to injection at 5 μL/min.
Acknowledgments
Funding for this project was provided by NIH and NSF. A.B.-O. acknowledges support from an individual NRSA predoctoral fellowship, C.A.H. from the Molecular Biophysics Training Grant, and J.G.M. from the Growth Regulation and Oncogenesis Training Grant and from the La Jolla Interfaces in Science Program (Burroughs Wellcome).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4670102.
References
- Baerga-Ortiz, A., Rezaie, A.R., and Komives, E.A. 2000. Electrostatic dependence of the thrombin–thrombomodulin interaction. J. Mol. Biol. 296 651–658. [DOI] [PubMed] [Google Scholar]
- Burritt, J., DeLeo, F., McDonald, C., Prigge, J., Dinauer, M., Nakamura, M., Nauseef, W., and Jesaitis, A. 2001. Phage display epitope mapping of human neutrophil flavocytochrome b558. J. Biol. Chem. 276 2053–2061. [DOI] [PubMed] [Google Scholar]
- Chambers, G., Lawrie, L., Cash, P., and Murray, G. 2000. Proteomics: A new approach to the study of disease. J. Pathol. 192 280–288. [DOI] [PubMed] [Google Scholar]
- Dawes, J., James, K., Micklem, L., Pepper, D., and Prowse, C. 1984. Monoclonal antibodies directed against human α-thrombin and the thrombin–antithrombin III complex. Throm. Res. 36 397–409. [DOI] [PubMed] [Google Scholar]
- Fiedler, W., Borchers, C., Macht, M., Deininger, S., and Przybylski, M. 1998. Molecular characterization of a conformational epitope of hen egg white lysozyme by differential chemical modification of immune complexes and mass spectrometric peptide mapping. Bioconjug. Chem. 9 236–241. [DOI] [PubMed] [Google Scholar]
- Gonzalez, G., Orn, A., Cazzulo, J., and Grönvik, K. 1994. Production and characterization of monoclonal antibodies against the major cysteine proteinase of Trypanosoma cruzi. Scand. J. Immunol. 40 389–394. [DOI] [PubMed] [Google Scholar]
- Hunter, M.J. and Komives, E.A. 1995. Thrombin binding affinities of different disulfide bonding isomers of the fifth EGF-like domain of thrombomodulin. Protein Sci. 4 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi, M. and Finkelstein, R. 1991. Mapping epitopic regions of cholera toxin B-subunit protein. Mol. Immunol. 28 865–876. [DOI] [PubMed] [Google Scholar]
- Klein, J. and Horejsi, V. 1997. Antigens, superantigens and other lymphocyte-activating substances. In Immunology (ed. Oxford), 2nd ed. Chapter 13, p. 402. Blackwell Science Ltd., Malden, MA.
- Lo Conte, L., Chothia, C., and Janin, J. 1999. The atomic structure of protein–protein recognition sites. J. Mol. Biol. 285 2177–2198. [DOI] [PubMed] [Google Scholar]
- Logan, A.J., Williamson, E.D., Titball, R.W., Percival, D.A., Shuttleworth, A.D., Conlan, J.W., and Kelly, D.C. 1991. Epitope mapping of the α-toxin of Clostridium perfringens. Infect. Immun. 59 4338–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo, F., Jaulin, C., Vita, N., Froussard, P., Ferrara, P., Jankovic, D., and Theze, J. 1991. Structure–function study of the p55 subunit of murine IL-2 receptor by epitope mapping. J. Immunol. 147 2970–2977. [PubMed] [Google Scholar]
- Lougheed, J.C., Bowman, C.L., Meininger, D.P., and Komives, E.A. 1995. Thrombin inhibition by cyclic peptides from thrombomodulin. Protein Sci. 4 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell, J., Falick, A., and Komives, E. 1998a. Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal. Chem. 70 3987–3995. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. Identification of protein–protein interfaces by decreased amide proton solvent accessibility. Proc. Nat. Acad. Sci. 95 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell, J., Baerga-Ortiz, A., Akashi, S., Takio, K., and Komives, E. 2001. Solvent accessibility of the thrombin–thrombomodulin interface. J. Mol. Biol. 306 575–589. [DOI] [PubMed] [Google Scholar]
- Nelson, P.N., Reynolds, G.M., Waldron, E.E., Ward, E., Giannopoulos, K., and Murray, P.G. 2000. Monoclonal antibodies. Mol. Pathol. 53 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, F., Konishi, Y., and Scheraga, H.A. 1990. Thrombin-bound conformation of the C-terminal fragments of hirudin determined by transferred nuclear Overhauser effects. Biochemistry 29 4479–4489. [DOI] [PubMed] [Google Scholar]
- Rees, A., Staunton, D., Webster, D., Searle, S., Henry, A., and Pedersen, J. 1994. Antibody design: Beyond the natural limits. Trends Biotechnol. 12 199–206. [DOI] [PubMed] [Google Scholar]
- Reineke, U., Kramer, A., and Schneider-Mergener, J. 1999. Antigen sequence- and library-based mapping of linear and discontinuous protein–protein-interaction sites by spot synthesis. Curr. Top. Microbiol. Immunol. 243 23–36. [DOI] [PubMed] [Google Scholar]
- Reineke, U., Volkmer-Engert, R., and Schneider-Mergener, J. 2001. Applications of peptide arrays prepared by the SPOT-technology. Curr. Opin. Biotechnol. 12 59–64. [DOI] [PubMed] [Google Scholar]
- Saint-Remy, J. 1997. Epitope mapping: A new method for biological evaluation and immunotoxicology. Toxicology 119 77–81. [DOI] [PubMed] [Google Scholar]
- Suckau, D., Köhl, J., Karwath, G., Schneider, K., Casaretto, M., Bitter-Suermann, D., and Przybylski, M. 1990. Molecular epitope identification by limited proteolysis of an immobilized antigen–antibody complex and mass spectrometric peptide mapping. Proc. Nat. Acad. Sci. 87 9848–9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J., Higgins, D., and Gibson, T. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Water, J., Deininger, S., Macht, M., Przybylski, M., and Gershwin, M. 1997. Detection of molecular determinants and epitope mapping using MALDI-TOF mass spectrometry. Clin. Immunol. Immunopathol. 85 229–235. [DOI] [PubMed] [Google Scholar]
- Williams, D.C., Rule, G.S., Poljak, R.J., and Benjamin, D.C. 1997. Reduction in the amide hydrogen exchange rates of an anti-lysozyme Fv fragment due to formation of the Fv-lysozyme complex. J. Mol. Biol. 270 751–762. [DOI] [PubMed] [Google Scholar]
- Ye, J., Esmon, N.L., Esmon, C.T., and Johnson, A.E. 1991. The active site of thrombin is altered upon binding to thrombomodulin. J. Biol. Chem. 266 23016–23021. [PubMed] [Google Scholar]