

Abstract
Using hydrogen-deuterium exchange (HX) and electrospray ionization mass spectrometry, we have investigated the stability and structural changes of recombinant human interferon-γ (IFN-γ) during aggregation induced by guanidine hydrochloride (GdnHCl) and potassium thiocyanate. First, HX labeling was initiated after the amorphous aggregates were formed to probe the tertiary structure of the aggregated state. Second, labeling was performed at low protein concentrations to assess stability under aggregation prone conditions. In 1 M GdnHCl, the stability of IFN-γ was greatly reduced and much less protection from HX in solution was observed. Exchange under these conditions was slower in helix C than in the rest of the protein. Aggregates formed in 1 M GdnHCl showed a HX pattern consistent with a partially unfolded state with an intact helix C. Although aggregates formed in 0.3 M KSCN exhibited a HX pattern similar to those formed in GdnHCl, the solution phase HX pattern in 0.3 M KSCN was surprisingly comparable to that of the native state. Varying the aggregation time before performing HX revealed that KSCN first precipitated native protein and then facilitated partial unfolding of the precipitated protein. These results show that helix C, which forms the hydrophobic core of the IFN-γ dimer, is highly protected from HX under native conditions, is more stable in GdnHCl than the rest of the protein and remains intact in both GdnHCl- and KSCN-induced aggregates. This suggests that native-state HX patterns may presage regions of the protein susceptible to unfolding during aggregation.
Keywords: Recombinant human interferon-γ, protein aggregation, conformational changes, chaotropic salts, hydrogen exchange, electrospray mass spectrometry
Misfolding and association of proteins into soluble or insoluble aggregates is a phenomenon widely observed in biotechnology. From bench scale to commercial scale, it is an undesired side reaction in processes such as the renaturation of protein from inclusion bodies (De Bernardez-Clark and Georgiou 1991), protein purification (Przybycien 1998), and formulation design (Cleland et al. 1993). Biomedical research has also been directed toward protein stability because aggregation in vivo is a critical step in various human conditions such as Alzheimer's disease and the formation of cataracts (Tan and Pepys 1994; Serpell et al. 1997).
The formation of insoluble aggregates is generally thought to proceed via a two-step process in which the protein first unfolds and subsequently precipitates (Lumry and Eyring 1954; Lumry and Biltonen 1969). Unfolded protein molecules have low solubilities due to exposed hydrophobic groups normally buried in the native state. For a given total protein concentration, avoiding aggregation via this pathway could involve either stabilizing the native state of the protein or improving the solubility of the unfolded state. Although there is generally a correlation between native-state stability and aggregation propensity, protein engineering studies have shown that this is not universal (Wetzel et al. 1988; Mulkerrin and Wetzel 1989; Chrunyk and Wetzel 1993). Discrepancies in the correlation were attributed to the effect of the mutational or chemical modification of the protein on the intermolecular interactions that stabilize the aggregated state. In some applications, such as protein refolding, mild concentrations of denaturants such as urea or guanidine hydrochloride (GdnHCl) are used to discourage precipitation of folding intermediates, despite their destabilizing effect on the native structure (De Bernardez-Clark and Georgiou 1991). In any strategy for avoiding aggregation, it is of interest to understand the conformational changes that occur and how the unfolded proteins interact to form aggregates. Such structural information would aid in process design or protein engineering efforts intended to stabilize the protein or disrupt intermolecular interactions.
However, the aggregation prone conformations in these systems are difficult to investigate due to their inherent lack of stability and low solubilities. Both partially (London et al. 1974; DeFelippis et al. 1993; Speed et al. 1996) and fully (De Young et al. 1993; Stigter and Dill 1993) unfolded states of the protein have been implicated as the aggregation prone conformation. More structural information has been obtained for the aggregated protein, primarily with the use of infrared spectroscopy (IR). IR measurements have revealed nonnative secondary structure for various protein aggregates (Wetzel 1994; Fink 1998; Kendrick et al. 1998b) and for amyloid fibrils (Booth et al. 1997). Often, losses in α-helical content with increases in both intermolecular and intramolecular β-sheet are observed, suggesting a general aggregation mechanism for a number of proteins under a variety of conditions. However, amorphous protein aggregates are not well suited for examination by high-resolution techniques, such as nuclear magnetic resonance (NMR) or X-ray diffraction. Consequently, less is known about the tertiary structure of the aggregated protein, both in terms of which native-like contacts remain and which residues may have rearranged to form intermolecular contacts.
In complex processing applications not amenable for direct measures of tertiary structure, hydrogen-deuterium exchange (HX) can be used as an indirect probe for protein conformation. For example, HX experiments have been applied to study protein structure in salt-induced precipitation (Chang and Fernandez 1998; Tobler et al. 2001), reversed phase chromatography (McNay and Fernandez 1999), and lyophilized solids (Desai et al. 1994). To obtain residue level information about HX, nuclear magnetic resonance (NMR) has been used extensively in protein folding studies in the last few decades (Englander and Kallenbach 1984; Roder et al. 1988; Englander et al. 1997). Alternatively, mass spectrometry (MS) has proven useful in distinguishing different conformations that existed during the HX experiment (Miranker et al. 1993; Chung et al. 1997; Tobler et al. 2001). Further, proteolytic digestion prior to MS analysis can provide structural information with close to the resolution that NMR affords. This technique has been largely pioneered by Smith and coworkers for kinetic, thermodynamic, and structural studies of protein folding (Zhang and Smith 1993; Smith et al. 1997; Deng and Smith 1999).
We have chosen recombinant human interferon-γ (IFN-γ) as a model protein to study aggregation using HX and MS methods. This 32-kD homodimer consists of primarily α-helical secondary structure with no β-sheet or disulfide bonds. IFN-γ aggregates under a variety of denaturing conditions, such as high temperature, low pH, and in the presence of chaotropes or the formulation preservative benzyl alcohol (Hsu and Arakawa 1985; Mulkerrin and Wetzel 1989; Lam et al. 1997; Kendrick et al. 1998b). IR measurements of the insoluble aggregates induced by heat, acid, and chaotropic salts indicated very similar secondary structures that had lost some but not all of their native-like characteristics, suggesting that IFN-γ may aggregate through a common pathway under various denaturing conditions (Kendrick et al. 1998b). Using HX, proteolysis, and electrospray ionization MS, we have probed the tertiary structure of IFN-γ in aggregates induced by two chaotropic salts, 1 M GdnHCl and 0.3 M KSCN. Patterns of protection from HX illustrate which parts of the native structure remained intact in the aggregated phase and which parts had unfolded or rearranged. A general correlation between regions of the protein most stable in the native state and those that remained intact in the aggregated state was observed. The solution-phase HX behavior of IFN-γ in the same chaotrope conditions was also investigated to gain insight into the aggregation pathway, which was found to be different in each of the two salt conditions.
Results
A primary goal of this study was to obtain HX rates for small segments of IFN-γ under various solution and precipitated phase conditions. These HX rates reflect solvent accessibility, thus providing insight into the tertiary structure of the protein. Typically, concentrated IFN-γ in H2O was diluted into D2O and the condition of interest to initiate exchange. At various times aliquots of the protein were digested using pepsin, and the resulting digest peptides were separated on a reversed-phase chromatography column and mass analyzed to create a HX curve for each peptide. Before the HX data can be interpreted, the peptic fragments had to be identified, and the back exchange during the analysis steps had to be determined (see Materials and Methods).
Figure 1 ▶ shows the 18 peptides analyzed in this study. Although >50 different peptides were detected, these were chosen based on the confidence identifying the peptide, MS signal intensity, size, and contribution to primary sequence coverage. The peptides numbered 1–14 provide excellent sequential coverage of the primary sequence, with little gaps or overlap. The extra overlapping peptides, indicated with either a single or a double quotation mark, were included because of their high MS signal intensity or usefulness in providing HX information and could be used occasionally as a test for internal consistency. In each peptide, the N-terminal amide and side-chain sites exchange at least an order of magnitude more rapidly than backbone amides during the analysis steps (Wüthrich 1986), so that only the remaining amide sites were included in the measured deuterium contents (Thevenon-Emeric et al. 1992). Although the slowest exchanging side chains (e.g., Asn, Gln, and Arg) may contribute somewhat to measured masses, we have not included them in calculating the maximum number of exchangeable hydrogens in reporter peptides. This digest therefore affords HX information for ∼89% of the backbone amide sites at a spatial resolution ranging from 3 to 20 residues.
Fig. 1.

Primary sequence of the recombinant human IFN-γ monomer. The numbered arrows indicate the peptic peptides identified and analyzed in this study, and the dashed lines indicate the locations of the six helices labeled A–F (Ealick et al. 1991).
HX data for four example peptides (2, 4, 7, and 12) from IFN-γ exchanging in 5 mM succinate, pD 5, are shown in Figure 2 ▶. Rate data were obtained for the time range from 3 min to 24 h for this native-solution case, although typically HX was monitored for only 10 h. Figure 2A ▶ shows the uncorrected relative mass changes for the four peptides, where m is the measured mass, mp is the measured mass in 1hydrogen-containing buffer, and N is the total number of backbone amide sites. The maximum degree of labeling that could be achieved for fully solvent accessible peptides were determined as previously described (Zhang and Smith 1993) and is indicated as m100% on Figure 2A ▶. The corrected deuterium content, D/N (see Materials and Methods) is shown in Figure 2B ▶.
Fig. 2.


Hydrogen-deuterium exchange (HX) curves for four peptic peptides from IFN-γ undergoing HX in 5 mM succinate, pD 5. (A) Uncorrected mass measurements; (B) corrected deuterium content, D/N, calculated as explained in the text. The data for peptides 2, 7, and 12 were fit using equation 2 (peptide 2, As = 0.8, Am = 5.4, km = 0.04 min-1; peptide 7, As = 2.6, Am = 2.4, km = 0.001; peptide 12, As = 1.6, Am = 6, km = 0.002 min−1). The line through data for peptide 4 is the line D/N = 0 (As = 1, Af = Am = 0). Parallel samples created and analyzed separately for each condition are shown in open and solid symbols.
Each peptide has N amide sites that will exchange at different rates, so that these curves can be described by the sum of N first-order rate equations. We simplify our data analysis by assuming that the amides on each peptide can be divided into three HX groups, so that
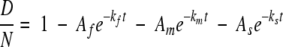 |
(1) |
where Af, Am, and As are the fraction of N amides that belong to the fast-, medium-, and slow-exchanging groups (Af + Am + As = 1), respectively, and kf, km, and ks are the corresponding HX rate constants. Because it would be very difficult to reliably resolve multiple exponential contributions from the exchange curve of each peptide, we simplify the expression further by defining the fast HX group to be those amides that exchange too quickly to be significant in these experiments (e−kft<0.1 ∼ 0 after 3 min). Indeed, a value of e−kft equal to 0.1 would indicate that kf > 0.8 min−1, which is approximately equal to the intrinsic HX rate at pD 5 and 20°C (Bai et al. 1993). Further, we define the slow-exchanging group to be those that remain protected during the HX experiments (e−kst > 0.9 ∼ 1 after 10 h). For the amides assigned to the slow HX group, their ks values would be <1.8 × 10−4 min−1. This is equivalent to protection factors of ∼103 or better, which would be expected for at least a few amides in a protein with a stability of ∼δGunf > 4 kcal/mole (Bai et al. 1994). Equation 1 then simplifies further to
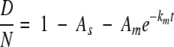 |
(2) |
The curve fits using equation 2 are shown for the data in Figure 2B ▶ and lead to the determination of Af, Am, As, and km.
Hydrogen exchange of native IFN-γ
To probe the HX behavior of the native IFN-γ dimer, exchange was initiated in 5 mM succinate, pD 5, and allowed to proceed for 24 h. The protein in this condition exhibits native secondary structure based on spectroscopic measurements (Lam et al. 1997; Kendrick et al. 1998b) and a stability of δGunf ∼ 7 kcal/mole based on calorimetry data under similar conditions (Beldarrain et al. 1999). Figure 3 ▶ summarizes the HX data from this experiment by showing the number of amides assigned to the three HX groups from each peptide. This graph indicates that the last four helices (C–F) possess the most slowly exchanging amides. Of these, helix C appears to exhibit the highest amount of stability because there are ∼12 consecutive amide sites that exchange slowly (peptides 3", 4, and 5). The graph also indicates that peptide 3" contains approximately the same amount of slow exchangers as peptide 3, revealing that all of the strong protection observed in peptide 3 is located in helix C and not in helix B. Helix C in each monomer forms much of the dimer interface area and consequently becomes the most buried of the helices in the core of the native dimer structure (Ealick et al. 1991; Grzesiek et al. 1992). Of course, many peptides will cover areas of varying local stability, which is reflected in the relative amounts of amides in each HX group. This can be seen in helices D, E, and F (peptides 7, 8, 9, 10, 12, and 13), in which the helix structures are more closely exposed to the solvent in the crystal structure (Ealick et al. 1991). The data also indicates regions of the protein almost completely exposed to the solvent, such as helices A and B (peptides 1, 2, and part of 3) and the loop regions (peptides 6, 11, and 14). Helices A and B have been reported to be relatively flexible (Grzesiek et al. 1992; Waschutza et al. 1996), perhaps to facilitate the conformational change that the loop region in between the two helices undergoes in the receptor-bound form of the active protein (Walter et al. 1995). The pattern of protection observed using MS agrees closely with the semiquantitative HX-NMR data reported by Grzesiek et al. (1992) under similar conditions. In general, the amides represented in the slow HX group for each peptide will serve as reporter groups for structural disruptions that may occur in more denaturing environments.
Fig. 3.

Number of fast-, medium-, and slow-exchanging amide sites in each peptic peptide from IFN-γ undergoing hydrogen exchange in 5 mM succinate, pD 5. Based on the standard errors of the fit parameters (Ai), error bars for each fraction are generally ±0.5 amides.
The overlapping peptides indicate an internal consistency to the HX methods and analysis used here. For example, based on the results for peptide 4, peptide 3′ should have four extra amides in the slow HX group relative to peptide 3 and the same number of amides in the other two groups. These expectations are confirmed in Figure 3 ▶, which indicates that peptide 3′ contains approximately five more slow-exchanging amides than peptide 3. In addition, the number of amides in the three groups from peptide 6` should be a subset of those in peptide 6, and the sum of those in peptides 10 and 11 should include all but one of the amides in peptide 10′. The data in Figure 3 ▶ are again in agreement with both of these statements. In the other experiments in this study, the data for these overlapping peptides provided similar consistency tests but for simplicity are not presented here.
Hydrogen exchange in the aggregated phase
To probe the tertiary structure of aggregated IFN-γ, HX experiments were then performed on insoluble aggregates induced in chaotropic salt solutions. Previous HX studies on protein crystals (Pederson et al. 1991) and protein precipitates (Chang and Fernandez 1998; Tobler et al. 2001) showed that HX rates for protein in the solid phase could be comparable or only slightly attenuated relative to rates for solution-phase protein, as long as the protein maintained a native conformation. The slight attenuation in HX was attributed to a reduction in structural fluctuations experienced by solid-phase protein (Pederson et al. 1991). A denatured protein precipitate, however, can still experience enhanced HX in regions where the tight native structure is lost (Chang and Fernandez 1998; Tobler et al. 2001).
To label insoluble IFN-γ aggregates, the protein was first incubated in H2O (15 mg/mL protein, 5 mM succinate, pH 5), with either 1 M GdnHCl or 0.3 M KSCN. After 24 h, the slurry was centrifuged and the supernatant was removed and replaced with an appropriate D2O buffer containing the same chaotrope concentration. We observed that the solubility of IFN-γ was <0.1 mg/mL in these solution conditions after 24 h and do not believe any significant amount of IFN-γ redissolved into the new supernatant. The aggregate was then incubated for a specified HX time, redissolved quickly with 6 M GdnDCl, and digested and analyzed. Although the redissolution in 6 M GdnDCl was performed at conditions meant to quench HX, the harsh denaturing condition needed to quickly redissolve the aggregate might also unfold the protein and jeopardize the isotope label. To determine how much exchange occurs during this step, a control was performed in which the D2O replacement supernatant and GdnDCl redissolution buffer were added simultaneously. HX for each of the peptides was generally between 15% and 20% of full exchange after the calculation for back exchange during the digest and chromatography steps (data not shown). This enhanced exchange caused by the redissolution condition may preclude quantitative detection of small reductions in HX protection observed in the aggregates relative to the solution phase native state. However, the relative protection patterns in the two aggregates can be productively compared.
Figure 4 ▶ compares the HX patterns for both the GdnHCl- and KSCN-induced aggregates. These two precipitates were reported to have very similar secondary structure contents, even though GdnHCl induced the formation of a translucent gel, whereas the KSCN aggregates were white precipitates (Kendrick et al. 1998b). We also observed these different macroscopic morphologies, and Figure 4 ▶ shows that they also have similar HX behavior. One common feature to both aggregates was the much smaller fraction of intermediate exchangers, Am, for all peptides. For the GdnHCl-induced aggregate, the HX data had almost no curvature, so that a confident curve fit to equation 2 could not be obtained. Instead, an average value of the points was used for each peptide, essentially separating the fast exchangers from the slow exchangers, with no medium group. A reasonable explanation for this observation is that in a precipitated phase, the protein structure will not fluctuate as it does in solution. It is this structural breathing on both local and global levels that gives rise to a continuous range of exchange rates for a protein in solution. However, the intermolecular interactions in the precipitated phase can reduce these fluctuations, thereby `locking' the amide sites into either open (exchange competent) or closed (exchange incompetent) positions. Another observation common to both aggregates was that each peptide exhibited single, tight-mass distributions, suggesting that the structure of aggregated IFN-γ was homogeneous. If the precipitated-phase protein contained a heterogeneous mixture of varying structures, then a broad or bimodal distribution of masses would be expected (Tobler et al. 2001).
Fig. 4.

Number of fast-, medium-, and slow-exchanging amide sites in each peptic peptide from aggregated IFN-γ. Aggregation was induced in the presence of either 0.3 M KSCN (left-hand bar in each pair) or 1 M GdnHCl (right-hand bar) in 5 mM succinate, pH 5, for 24 h prior to hydrogen-deuterium-exchange labeling in the analogous D2O buffer.
For both aggregates, most of the slowly exchanging amides were located in peptides 4 and 5, which are both in helix C. Peptide 3 also contains ∼6 strongly protected amide sites, which are most likely the sites also contained in helix C, based on the native state data in Figure 3 ▶ (in these experiments, the MS signal intensity for peptide 3" was not sufficient to obtain confident molecular weight values). These results suggest that the aggregate maintained a relatively stable, native-like helix C. Helices D, E, and F, which were also well protected in the native state, appear more disrupted in the aggregates. In both aggregates, peptides 9 and 10 from Helix E retain no slowly exchanging amides. The main differences between the two aggregates are seen in helices D and F. Although peptides 7, 8, 12, and 13 from the KSCN-induced aggregate each contain close to the number of slow-exchange groups as was observed in the native protein, the GdnHCl-induced aggregate exhibits almost no protection in those regions.
Hydrogen exchange in chaotropic salt solutions and low protein concentration
To gain insight into the aggregation pathway for the protein in the presence of these two chaotropic salts, HX experiments were performed at low protein concentrations in the solution phase. IFN-γ at 20 mg/mL (H2O) was diluted 10× into either 0.3 M KSCN or 1 M GdnDCl (both in D2O, 5mM succinate, pD 5) to initiate exchange. GdnDCl was not used for redissolution of aggregates in these experiments, and thus any precipitated protein was most likely not digested and mass analyzed. Less than ∼15% of the total protein precipitated during the 10-h HX experiments, based on the UV chromatograms obtained during the peptide separation. This indicates that the apparent solubility during these shorter experiments was greater than 0.1 mg/mL after 24 h of incubation, as estimated earlier.
Consequently, we used high-performance size-exclusion chromatography (HPSEC) to characterize the aggregation state of the protein as shown in Figure 5 ▶. For the native condition, a single peak was detected, indicating that no small molecular weight oligomers were present. Under these conditions, no visible precipitate was observed upon centrifugation. In the presence of 0.3 M KSCN, a native-like chromatogram was obtained at 1 h. After 10 h, the peak area decreased ∼30%, indicating that some of the protein had aggregated and been removed by centrifugation and/or filtration. This indicates some aggregation, but there are no intermediate molecular weight oligomers. Thus, in the presence of 0.3 M KSCN, the protein is mostly homodimer, and our structural analysis can be taken to reflect the effect of KSCN on the dimeric state. GdnDCl also gave a single peak, indicating no intermediate molecular weight oligomers. However, the reduced UV peak area showed that a significant portion of the protein was removed prior to SEC as high molecular weight aggregates. Although these aggregates may not have dissolved during proteolytic fragmentation, the structural analysis cannot be taken to reflect only the dimeric state.
Fig. 5.

Size-exclusion chromatography analysis of 2 mg/mL IFN-γ under native conditions (upper trace), with added KSCN (middle traces), and with added GdnHCl (lower traces). For the KSCN and GdnHCl conditions, two chromatograms are shown, obtained at 1 h and 10 h.
HX patterns for the 14 reporter peptides in these chaotropic salt solutions are summarized in Figure 6 ▶. IFN-γ exhibits native-like HX patterns in 0.3 M KSCN. Figure 6 ▶ indicates that each peptide contained at least as many slow-exchanging amide sites as observed in the native case. Peptides 7 and 12 (and peptide 3, to a smaller extent) actually show stronger protection from HX with the added KSCN, implying a greater local stability in those areas of the protein. It is difficult to determine whether there are also stability differences for peptides 4, 5, and 8 because all amide sites in these peptides are assigned as slow exchangers in both cases and a decrease in HX would not be measured in the timescale of these experiments. This apparent increase in stability was surprising because KSCN is considered a chaotrope and is able to bind aggressively to peptides and proteins, thus disrupting their structure (Arakawa and Timasheff 1982; 0000Watanabe and Saito 1985).
Fig. 6.

Number of fast-, medium-, and slow-exchanging amide sites in each peptic peptide from IFN-γ in a 0.3 M KSCN (left-hand bar in each pair) or 1 M GdnDCl (right-hand bar) solution (5 mM succinate, pD 5, 2 mg/mL protein).
Because IFN-γ at 2 mg/mL in 0.3 M KSCN exhibited some aggregation over the time of these experiments (see Fig. 5 ▶), it is possible that selective precipitation of unfolded protein may have occurred. Filtration of the samples would remove that protein from digestion and mass analysis here, leading to an overestimation of stability. Thus, similar experiments were performed at a total IFN-γ concentration of 0.01 mg/mL, in which no precipitation could be visually observed after at least 5 h. In these experiments, mass spectra for the intact protein were obtained so that the overall HX behavior could be observed for solution conditions with and without KSCN (Fig. 7 ▶). In any HX experiment, the protein quickly loses the majority of its labile protons, which represent the fast-exchanging amide and side-chain sites. The HX curve then levels because the remaining well-protected protons are slow to exchange. In comparison to the HX curve for IFN-γ in buffer only, the HX curve for IFN-γ in the presence of KSCN shifts downward, suggesting that some exchangers have been converted from the fast- or medium-exchange group to the slow group, as was indicated by the fragmentation experiment. This experiment shows that the addition of KSCN does in fact increase the stability of IFN-γ slightly, at least in the local regions indicated highlighted by differences between Figures 3 and 6 ▶ ▶. A third HX curve is included in Figure 7 ▶ for IFN-γ in the presence of 0.3 M KCl, which shows a behavior similar to that in 0.3 M KSCN. Thus, the stabilizing effect is most likely a nonspecific electrostatic effect of increasing the ionic strength.
Fig. 7.

Hydrogen-deuterium exchange (HX) curves for intact IFN-γ undergoing HX in 5 mM succinate, pD 5, and with the addition of either 0.3 M KSCN or 0.3 M KCl. Masses reported were not corrected for further exchange occurring during the chromatography step prior to mass spectrometry analysis.
The presence of 1 M GdnDCl severely disrupted the structure of IFN-γ as measured by hydrogen exchange. This was expected because this GdnDCl concentration is near the midpoint for equilibrium denaturation (∼1.3 M GdnHCl) as measured by fluorescence at either pH 7 (Waschutza et al. 1996) or pH 5 (data not shown). The HX data indicates that the protein had almost no amides in the slow-exchanging group but instead exhibited a larger fraction of medium-exchanging amides relative to the native-state HX experiments. The mass distributions for a few of the peptides in 1 M GdnDCl were clearly bimodal (peptides 3, 4, and 12; see Fig. 8 ▶), as were mass spectra for the intact protein under identical conditions (data not shown). These bimodal mass distributions arise when HX occurs through the classic EX1 condition (Hvidt and Nielson 1966), in which refolding is sufficiently slow that full exchange of newly exposed sites occurs with every opening event (for a more detailed description of HX and MS, see Arrington et al. 1999 and Deng et al. 1999). The observed HX rate under this condition is equivalent to the kinetic unfolding rate. Although not all of the peptide mass spectra exhibited obvious bimodal patterns, the km values determined in 1 M GdnDCl for peptides well protected in the folded conformation (i.e., slow exchangers in the native condition) indicate the unfolding rate for local regions of the protein structure. In contrast, refolding rates are faster under HX for the protein under other solution conditions in this study, and HX is believed to occur primarily through the EX2 condition, in which the observed-rate constant is related to the stability of each specific region of the protein. An alternative explanation for the bimodal distributions would be slow dissolution of oligomers present in the original 20 mg/mL stock solution. However, no oligomers were observed in the HPSEC of the native IFN-γ (see Fig. 5 ▶).
Fig. 8.

Mass spectra for peptide 4 (left) and peptide 12 (right) from IFN-γ undergoing hydrogen-deuterium exchange in 5mM succinate, pD 5, with (solid lines) and without (dashed lines) 1 M GdnDCl.
In 1 M GdnDCl, the values of km for many of the effective reporter peptides of unfolding (peptides 7, 8, 9, 10, 12, and 13) were found to be between 0.013 and 0.023 min−1, with standard errors of the parameters corresponding to ∼10%–20% of the fit value. These consistent values suggest that much of the protein (involving helices D, E, and F) unfolds at a rate of ∼0.02 min−1. First-order aggregation kinetics for IFN-γ in 1 M GdnHCl reported by Kendrick et al. (1998b) suggested that aggregation was rate limited by an unfolding step with a rate constant of 0.017 min−1, consistent with our result using HX and MS. However, values for km obtained for peptides 4 and 5 in helix C were significantly smaller (0.006 and 0.007 min−1 for peptides 4 and 5, respectively). The mass spectra for peptide 4 included in Figure 8 ▶ display this difference in HX relative to peptide 12. Slower unfolding in this region suggests that IFN-γ first unfolds to an intermediate state, which maintains appreciable secondary and/or tertiary structure in the helix C region of the native structure. Similar experimental measurements have previously been applied in the determination of folding intermediates of rabbit aldolase (Deng and Smith 1998).
It should be noted that because the protein aggregated extensively over 10 h, we cannot ascribe this behavior to the soluble, dimeric state alone. Nonetheless, the mixture of the soluble dimer and any aggregate digested with pepsin were markedly more solvent accessible (see Fig. 6 ▶) than the aggregates formed at higher protein concentrations (see Fig. 4 ▶). Unfortunately, sensitivity limits of current techniques preclude further reducing protein concentration to isolate soluble protein in the presence of GdnDCl. Improved MS techniques might ultimately allow such an analysis.
Structural rearrangement in a KSCN-induced precipitate
Because IFN-γ was not destabilized in a KSCN solution but was denatured in a KSCN-induced aggregate, the possibility of structural rearrangements occurring in the solid phase was explored. The protein was precipitated with KSCN in H2O as before except that the precipitation time before centrifugation varied from 1 h to the previous 24 h. The supernatant was then replaced with D2O and exchange was allowed to proceed for only 30 min for each precipitation time. Redissolution of the insoluble aggregate was performed as before except that no pepsin was included and the mass of the intact protein was obtained to determine overall extent of exchange. UV chromatograms obtained in parallel with the MS analysis of the protein eluting from the column did not indicate any losses in total protein analyzed in the early time points. This suggests that the precipitation process was nearly complete after the first hour, because a significant amount of any supernatant protein at that time would have been removed when the supernatant H2O buffer was replaced with fresh D2O buffer. Figure 9 ▶ shows the measured masses for several aggregation times. Because each sample was labeled for the same amount of time, the difference in HX observed implies that the structure of the aggregated protein was changing during the incubation time as a precipitated solid. The difference of ∼7 daltons from 1 to 14 h translates to a difference of >10 slowly exchanging sites after correcting for the 75% D content in the labeling step and the estimated exchange that occurs during desalting. This number of amides roughly corresponds to the difference in number of slowly exchanging amides observed in the low protein concentration (Fig. 6 ▶) and aggregated-phase experiments (Fig. 4 ▶). It appears that KSCN precipitates the native form of IFN-γ but then works to unravel some of the strongly protected native structure (primarily helix E and portions of helices D and F) in the precipitated protein phase.
Fig. 9.

Mass of intact IFN-γ after undergoing hydrogen-deuterium exchange (HX) in the precipitated state. The aggregation time indicates how long the protein was incubated in 0.3 M KSCN at 15 mg/mL before initiating HX. Masses reported were not rescaled to account for the 75% D content in the labeling solution and were not corrected for further exchange occurring during the chromatography step prior to mass spectrometry analysis.
Discussion
The results in Figures 4 and 6 ▶ ▶ show that two chaotropic salts that have markedly different effects on IFN-γ at low protein concentrations can induce the formation of insoluble aggregates with similar tertiary structures. Similar secondary structures of IFN-γ under these and other aggregation conditions were previously reported by Kendrick et al. (1998b). IR measurements indicated that 1 M GdnHCl- and 0.3 M NaSCN-induced aggregates contain ∼20% and 29% α-helical content, respectively, compared to the 70% α-helical content measured for the native state. In contrast to the information afforded by IR spectroscopy, the HX experiments used here provide a probe for tertiary structure by indicating which regions of the protein are protected from the surrounding solvent. It is therefore difficult to directly compare the HX results to the spectroscopic measurements (for example, helices A and B contribute to the helical content in the native state but do not provide substantial protection from HX), but the trends from both studies are in agreement. Both HX and IR reveal a partial loss of native structure upon aggregation, in which the −SCN-induced aggregate retained measurably more native-like structure than the GdnHCl-induced aggregate. More specifically, the HX data indicates that the helix C region of the protein remains tightly structured in both aggregates and that the −SCN-induced aggregate also remains protected in helices D and F. The results in Figure 4 ▶ suggest that helices are lost cooperatively because helices D and F were either reasonably well retained or completely lost.
Less apparent from the HX-MS data presented here is the newly formed structure involved in the intermolecular interactions in the aggregate. The aforementioned IR measurements also indicated the formation of both intermolecular and intramolecular β-sheet. The HX-MS measurements do not indicate any obvious increases in protection that were thought to arise from such newly formed structure, although peptides 2, 3, and 6 do show some increase in the number of slowly exchanging amides after aggregation. Although HX rates can be affected by the involvement in hydrogen bonding, tightly packed tertiary structure that can effectively exclude water is also required. Intermolecular β-sheet structures in aggregated states may well have insufficiently tight packing to exclude water molecules from exchangeable amide protons.
We also found that the two chaotropic salts had very different effects on IFN-γ stability at lower protein concentrations. 1 M GdnHCl was shown to destabilize the protein and produce aggregates even at lower protein concentrations. Helix C, which was highly protected from HX in the native state, unfolded more slowly than the rest of the native structure in 1 M GdnHCl, suggesting the existence of either an unfolding intermediate or a denatured state with residual, albeit transient, native-like structure. Previous studies support the view that IFN-γ does not follow a simple two-state unfolding process (from a native to a fully denatured state) during aggregation in a denaturing solution condition. Several observations, including biphasic unfolding kinetics, have implicated a folding intermediate in the aggregation pathway for thermally denatured IFN-γ (Mulkerrin and Wetzel 1989). Also, it has been observed that the addition of sucrose can slow the rate of aggregation of IFN-γ in 0.9 M GdnHCl (Kendrick et al. 1998a). In that study, a model that linked aggregation kinetics and conformational thermodynamics was used to show that a slight and transient expansion of the native state preceded further unfolding and subsequent aggregation. Further, IFN-γ in 3 M GdnHCl maintains ∼30% α-helical content, which indicates that the denatured state is not a fully random chain configuration (Kendrick et al. 1998b). Attempts to elucidate the structure in this solution condition using HX-MS failed because no protection from HX could be detected in 3 M GdnDCl, even at short labeling times (data not shown). Thus, the residual structure measured by IR and circular dichroism is not persistent enough in 3 M GdnHCl to provide significant protection from HX, as has been reported for some, but not all, compact denatured states for other proteins such as hen lysozyme (Buck et al. 1994).
Unlike 1 M GdnHCl, the addition of 0.3 M KSCN to the succinate buffer does not destabilize IFN-γ in solution, but rather appears to stabilize the structure, at least in peptides 7 and 12. A similar stabilization was observed with 0.3 M KCl, and is thus a more general effect of increasing ionic strength. IFN-γ at acidic pH is highly positively charged, and it is possible that the added ionic strength effectively shields the intramolecular charge repulsions that decrease stability of the native structure (Stigter et al. 1991). For IFN-γ in 0.3 M KSCN, the effect of ionic strength might overcome the denaturing effect that −SCN is generally known to have on protein structure (Arakawa and Timasheff 1982; Watanabe and Saito 1985; Tobler et al. 2001).
Instead of unfolding the protein in solution to an unfolded state with a low solubility, KSCN first precipitates the folded form of IFN-γ and then works further to unfold the structure in the solid phase. KSCN has been previously shown to act effectively as a precipitant for the native state of lysozyme, also a positively charged protein (Ries-Kautt and Ducruix 1989). This was attributed to the ability of the anion to form ion bridges between the positive charges on the protein surface (Ries-Kautt and Ducruix 1991). In contrast to this study, the previous study of lysozyme precipitated in 0.2 and 1 M KSCN indicated that unfolding occurred in the solution phase, but was inhibited if the protein was precipitated in the native state (Tobler et al. 2001). Such behavior suggested that protein molecules packed in a precipitated phase have less opportunity to undergo cooperative structural changes because of steric limitations imposed by surrounding molecules. We speculate that this conclusion may depend on subtleties in the native-state topology that would dictate the kinetically accessible conformational alternatives for a protein packed in a precipitated phase. Lysozyme contains a stable α-helical domain, a β-sheet/loop domain, and four disulfide bonds and generally follows a cooperative two-state transition during unfolding (Privalov 1992). On the other hand, IFN-γ is a non-disulfide-bonded α-helical bundle with helices that may have the ability to unravel from the rest of the structure without forcing the whole protein to completely unfold. Bovine growth hormone is another helical bundle that has exhibited such behavior, with a specific helix being implicated in a domain-swapping-like mechanism (Brems 1988). With that view, it is imaginable that not only does the precipitated IFN-γ have the ability to structurally rearrange, but that the surrounding protein molecules may even encourage unfolding by acting as a surface or ligand to stabilize the newly formed, nonnative structures (Miller and Dill 1997). This would account for the unfolding of the solid-phase protein in the KSCN-induced precipitate, even though the protein in a KSCN solution was not destabilized relative to the native-solution condition in this study.
These results show that helix C, which forms the hydrophobic core of the IFN-γ dimer, is highly protected from HX under native conditions, is more stable in 1 M GdnHCl than the rest of the protein, and appears to remain intact in both GdnHCl- and KSCN-induced aggregates. In addition to the unfolding intermediates and denatured states discussed above, residual secondary structure measured by circular dichroism has also been observed in acid- (Hsu and Arakawa 1985) and benzyl alcohol- (Lam et al. 1997) denatured IFN-γ. It is unclear how related these various partially unfolded states may be, but it is tempting to suppose that they also share structural features, particularly an intact helix C. In detailed studies for a few proteins, HX has been used to show common structural features of folding intermediates, molten globule states, and rare but detectable high energy conformations under native conditions (Jennings and Wright 1993; Arai and Kuwajima 1996; Chamberlain and Marqusee 1997). This suggests that native-state HX patterns may also presage regions of the protein susceptible to unfolding during aggregation, consistent with our observations.
Determining regions of the native state of a protein that either remain intact or become unfolded can aid in rational efforts to avoid aggregation because such information could provide insight into the aggregation pathway or implicate any specific regions of the protein that may induce the aggregation. For example, in this study there are two classes of local stability behavior that might warrant closer scrutiny. The native state experiment indicated that helices A and B and the AB loop region connecting these helices were loosely structured under stable conditions, consistent with previous reports (Ealick et al. 1991; Grzesiek et al. 1992). A previous engineering effort has already targeted this region, adding a disulfide bond, which stabilized the protein against irreversible temperature- and GdnHCl-induced denaturation (Waschutza et al. 1996). The second class of stability includes helices D, E, and F. Although these helices showed reasonable protection from HX in stable conditions, they were notably denatured in the aggregated phase and residues from these regions are likely involved in the intermolecular interactions in the aggregated state. In fact, it has already been shown that histidines H19 (AB loop region) and/or H111 (helix F) play a critical and specific role in the aggregation propensity of IFN-γ (Mulkerrin and Wetzel 1989; Beldarrain et al. 1999).
Conclusions
Hydrogen-deuterium exchange coupled with mass spectrometry remains a relatively young technique for probing protein conformation. This study shows its use in elucidating tertiary structure in amorphous protein aggregates, as well as ascertaining stability under conditions in which protein solubility is very low. It has become increasingly evident in the literature that partially unfolded states of proteins are critical in aggregation and that native-state HX patterns may reflect the structural features of those states. The HX and MS approaches employed here could serve as a rapid and sensitive technique to guide buffer selection or protein engineering efforts to increase pharmaceutical protein stability during purification and delivery, as well as other applications in which aggregation plays a crucial role.
Materials and methods
IFN-γ and D2O buffer preparation
IFN-γ was a gracious gift provided by Genentech as a liquid formulation. To remove polysorbate and other formulation additives, the protein was loaded onto a cation exchange column (CM650 S media; TosoHaas), washed, and eluted with a step change from 0 to 1 M NaCl (50 mM Tris buffer at pH 8). Peak fractions were pooled and desalted into 5 mM succinate (pH 5) using a HiTrap desalting column (Amersham Pharmacia Biotech). The protein was concentrated (typically to ∼20 mg/mL) using either a Microcon-10 or Centriprep-10 centrifugal filtration device (Millipore) and stored at 4°C. Deuterium oxide (D2O, 99.9% atom D) was purchased from Cambridge Isotope Laboratories, Inc. The pD of buffers made in D2O were read and adjusted using pD = pHread + 0.4 (Glasoe and Long 1960). Deuterated GdnDCl was prepared by dissolving GdnHCl in D2O, freezing and lyophilizing the solution, and repeating the process two times more. The same concentrated stock solution was then used to make all the solutions of GdnDCl used in this study.
Identifying peptic digest peptides
Digests were run for 5 min at a concentration of 0.5 mg/mL for both IFN-γ and pepsin in a ∼0.1 M phosphate buffer (pH 2.5, 0°C). The digest mix was loaded into a 100 μL injection loop using a prechilled syringe and syringe filter (Millex-GV, 0.2 μm; Millipore) and injected onto a stainless steel HPLC column containing Source RPC media (2.1 × 30 mm, packed and donated by Amersham Pharmacia Biotech). The injection loop and RPC column were kept in an ice bath. The solvents used here were 0.05% triflouroacetic acid in H2O (solvent A) and 0.05% triflouroacetic acid in acetonitrile (solvent B). The peptic peptides were eluted from the column using a 30-min gradient from 5% to 55% B. The flow rate through the column was set to 0.25 mL/min, and ∼25 μL/min was split off and directed into an LCQ Duo ion trap mass spectrometer (Finnigan). The rest of the flow was directed to a UV detector. The LCQ Duo was set to acquire mass spectra in the triple-play mode (full scan, zoom scan, MS/MS scan), and the SEQUEST software provided by Finnigan was used to identify peptides against possible matches found in the IFN-γ primary sequence.
MS analysis of peptide-level HX in IFN-γ
In HX experiments, the digest conditions described above were used except that extra salts (e.g., GdnHCl and KSCN) were often included in the digest as the HX experiments were diluted into pepsin solutions. To shorten the reversed phase chromatography step, the HPLC flow rate was increased to 0.5 mL/min, and a 13-min gradient was used to elute the peptides after a 2-min desalting step with 5% B. Full-scan mass spectra were acquired in the mass range from 400 to 1400 m/z. Typically, 5–10 scans were averaged for each peptide, and the centroid of the isotope envelope was used as the average molecular weight for the peptide.
In the solution-phase HX experiments, IFN-γ (20 mg/mL in 5 mM succinate at pH 5) was diluted 10× into a D2O buffer (5 mM succinate, pD 5) and an appropriate amount of GdnDCl or KSCN. The solutions were kept at 20°C. At various time points, 125 μL aliquots of the solution were taken and added to 375 μL of a pepsin solution in 0.1 M phosphate buffer (pH 2.5) on ice to initiate the digest. For the aggregate-phase HX experiments, the aggregates were first formed in H2O solutions at a total protein concentration of 15 mg/mL. The protein, at a starting concentration of ∼25 mg/mL, was mixed with a succinate buffer (pH 5) containing an appropriate amount of GdnHCl or KSCN to a total volume of 50 μL in 1.5 mL centrifuge tubes. After 24 h at ambient room temperature (∼25°C), the precipitated and supernatant phases were separated by centrifugation at 14,000×g for 25 min. Labeling was initiated by replacing 25 μL of the supernatant with 75 μL of D2O with 5 mM succinate (pD 5) and the appropriate chaotrope concentration. The slurry was briefly agitated with a syringe needle and was kept at 20°C during the HX time. For each aggregate sample, the tube was chilled on ice at the end of the HX time. Then 300 μL of ice-cold 8 M GdnDCl (pD 2.8) was added to quickly redissolve the aggregate for 1 min. One minute was sufficient to completely dissolve GdnDCl aggregates visually. KSCN aggregates dissolved even more rapidly. Further, 6 M GdnDCl was twice the concentration previously found to be effective for dissolving similar IFN-γ aggregates (Kendrick et al. 1998b). Then 267 μL of the resulting solution was added to 733 μL of pepsin in 0.1 M phosphate buffer (pH 2.5) to start the digest. Several of these aliquots were analyzed with variable HX times to create a HX curve for the aggregated protein.
The measured deuterium content in each peptide was corrected for back exchange that occurs during the digest and chromatography steps using the equation presented by Zhang and Smith (1993):
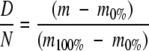 |
(3) |
where D is the corrected deuterium content, N is the total number of exchangeable sites, m is the measured mass of the peptide in a labeling experiment, and m0% and m100% are the measured peptide masses from 0% and 100% deuteration controls, respectively. The 0% deuteration control was obtained by mixing concentrated IFN-γ (H2O) with appropriate amounts of premixed and prechilled succinate buffer (D2O) and pepsin solution to initiate the digest. Four different HX conditions were tested to obtain m100%. IFN-γ was diluted 10× in D2O with succinate buffer and allowed to exchange at pD 2 for (a) 1 d and (b) 2 d, and at pD 5 and 55°C for 2 h (c) with and (d) without the addition of GdnDCl during the digest. For each peptide, an average value from the four 100% deuteration controls (a)–(d) was used to determine m100%. The standard deviations of these four values for each peptide corresponded to ∼5% of the total possible exchange. The consistency in HX between the various denaturing conditions supports the assertion that full exchange would occur in any one of them and shows a robustness in the amount of back exchange occurring during digests from variable experimental conditions (GdnDCl concentration, pD, etc.). These controls were performed in a 90% D solution, as in the solution-phase experiments in this study. HX labeling in the aggregate phases was performed in 75% D solutions, and so for these experiments the same correction was applied but the value was then rescaled by multiplying by (90/75) to correct for this difference. Curve fits described in Results were performed using SigmaPlot (SPSS, Inc.).
MS analysis of HX for intact IFN-γ
MS analysis of HX was performed on the intact protein in some instances. For the whole-protein HX curves in succinate buffer, 0.3 M KSCN, and 0.3 M KCl, IFN-γ (8 mg/mL) was diluted to 0.01 mg/mL in an appropriate D2O buffer and kept at 20°C. Aliquots were removed at various time points and loaded into a 1-mL injection loop through a 0.2 μm syringe filter. The protein was then injected onto a Vydac C4 guard column (2.1 × 10 mm). As before, the injection loop and RPC column were kept in an ice bath. The HPLC solvents used here were 0.5% acetic acid in D2O (solvent A) and 0.5% acetic acid in acetonitrile (solvent B). The initial flow condition was 10% B at 1 mL/min. At 2 min, the flow rate was slowed to 0.5 mL/min and the protein was eluted off the column with 60% solvent B. The flow rate was slowed once again to 0.2 mL/min as the protein eluted off the column, and ∼20 scans (500–1900 m/z) of the protein could be acquired. As before, the total flow was split between the mass spectrometer and the UV detector. The average mass of the protein used to determine HX curves under these conditions represents the average of mass values at the top of the peak and the center of the peak at half height (which were always within 1 dalton of each other).
Intact protein MS analysis of HX for KSCN-induced IFN-γ aggregates was also performed, using a procedure similar to the one described for the fragmentation experiments. The procedure was scaled down 5× because less sample was necessary for whole-protein MS analysis. After a 30-sec redissolution step with GdnDCl, the protein was further diluted in 0.5% acetic acid (D2O, 0°C), loaded into a 1-mL injection loop, and analyzed via the HPLC and MS method described above.
Size-exclusion chromatography
High-performance size-exclusion chromatography (HPSEC) was used to characterize the aggregation state of IFN-γ under native and low protein concentrations. Samples were centrifuged (14,000×g) for 20 sec filtered (Millex-GV, 0.22 μm; Millipore) and 20 μL samples were run on a TSK-GEL G3000SWXL column (Tosoh Biosep). The elution buffer contained 5 mM succinate (pH 5), 0.05% NaN3, and any salts included in the sample (i.e., KSCN or GdnDCl).
Acknowledgments
We thank Mary Cromwell for helpful discussions on this project and Xiaolin Fan for assistance with HPSEC analysis. This work has been supported by the National Science Foundation through a Career Award (BES-9501909) to E.J.F., the Thomas F. and Kate Miller Jeffress Memorial Trust, and Genentech, Inc.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3770102.
References
- Arai, M. and Kuwajima, K. 1996. Rapid formation of a molten globule intermediate in refolding of α- lactalbumin. Fold. Des. 1 275–287. [DOI] [PubMed] [Google Scholar]
- Arakawa, T. and Timasheff, S.N. 1982. Preferential interactions of proteins with salts in concentrated solutions. Biochemistry 21 6545–6552. [DOI] [PubMed] [Google Scholar]
- Arrington, C.B., Teesch, L.M., and Robertson, A.D. 1999. Defining protein ensembles with native-state NH exchange: Kinetics of interconversion and cooperative units from combined NMR and MS analysis. J. Mol. Biol. 285 1265–1275. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Milne, J.S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Prot. Struct. Funct. Genet. 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———.1994. Protein stability parameters measured by hydrogen exchange. Prot. Struct. Funct. Genet. 20 4–14. [DOI] [PubMed] [Google Scholar]
- Beldarrain, A., Lopez-Lacomba, J.L., Furrazola, G., Barberia, D., and Cortijo, M. 1999. Thermal denaturation of human γ-interferon. A calorimetric and spectroscopic study. Biochemistry 38 7865–7873. [DOI] [PubMed] [Google Scholar]
- Booth, D.R., Sunde, M., Bellotti, V., Robinson, C.V., Hutchinson, W.L., Fraser, P.E., Hawkins, P.N., Dobson, C.M., Radford, S.E., Blake, C.C., and Pepys, M.B. 1997. Instability, unfolding, and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 385 787–793. [DOI] [PubMed] [Google Scholar]
- Brems, D.N. 1998. Solubility of different folding conformers of bovine growth hormone. Biochemistry 27 4541–4546. [Google Scholar]
- Buck, M., Radford, S.E., and Dobson, C.M. 1994. Amide hydrogen exchange in a highly denatured state. J. Mol. Biol. 237 247–254. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K. and Marqusee, S. 1997. Touring the landscapes: Partially folded proteins examined by hydrogen exchange. Structure 5 859–863. [DOI] [PubMed] [Google Scholar]
- Chang, S.T. and Fernandez, E.J. 1998. Probing residue-level unfolding during lysozyme precipitation. Biotechnol. Bioeng. 59 144–155. [DOI] [PubMed] [Google Scholar]
- Chrunyk, B.A. and Wetzel, R. 1993. Breakdown in the relationship between thermal and thermodynamic stability in an interleukin-1-β point mutant modified in a surface loop. Protein Eng. 6 733–738. [DOI] [PubMed] [Google Scholar]
- Chung, E.W., Nettleton, E.J., Morgen, C.J., Grob, M., Miranker, A., Radford, S.E., Dobson, C.M., and Robinson, C.V. 1997. Hydrogen exchange properties in native and denatured states monitored by mass spectrometry and NMR. Protein Sci. 6 1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland, J.L., Powell, M.F., and Shire, S.J. 1993. The development of stable protein formulations : A close look at protein aggregation, deamidation, and oxidation. Crit. Rev. Ther. Drug Carrier Syst. 10 307–377. [PubMed] [Google Scholar]
- De Bernardez-Clark, E. and Georgiou, G. 1991. Inclusion bodies and recovery of proteins from the aggregated state. In Protein refolding (eds. G. Georgiou and E. De Bernardez-Clark), pp. 1–20. American Chemical Society, Washington, DC.
- De Young, L.R., Dill, K.A., and Fink, A.L. 1993. Aggregation and denaturation of apomyoglobin in aqueous urea solutions. Biochemistry 32 3877–3886. [DOI] [PubMed] [Google Scholar]
- DeFelippis, M.R., Alter, L.A., Pekar, A.H., Havel, H.A., and Brems, D.N. 1993. Evidence for a self-associating equilibrium intermediate during folding of human growth hormone. Biochemistry 32 1555–1562. [DOI] [PubMed] [Google Scholar]
- Deng, Y. and Smith, D.L. 1998. Identification of unfolding domains in large proteins by their unfolding rates. Biochemistry 37 6256–6262. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Rate and equilibrium constants for protein unfolding and refolding determined by hydrogen exchange-mass spectrometry. Anal. Biochem. 276 150–160. [DOI] [PubMed] [Google Scholar]
- Deng, Y.Z., Zhang, Z.Q., and Smith, D.L. 1999. Comparison of continuous and pulsed labeling amide hydrogen exchange/mass spectrometry for studies of protein dynamics. J. Amer. Soc. Mass. Spectrom. 10 675–684. [DOI] [PubMed] [Google Scholar]
- Desai, U.R., Osterhout, J.J., and Klibanov, A.M. 1994. Protein structure in the lyophilized state: A hydrogen isotope exchange/NMR study with bovine pancreatic trypsin inhibitor. J. Am. Chem. Soc. 116 9420–9422. [Google Scholar]
- Ealick, S.E., Cook, W.J., Vijay-Kumar, S., Carson, M., Nagabhusan, T.L., Trotta, P.P., and Bugg, C.E. 1991. Three-dimensional structure of recombinant human interferon-γ. Science 252 698–702. [DOI] [PubMed] [Google Scholar]
- Englander, S.W. and Kallenbach, N.R. 1984. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16 521–655. [DOI] [PubMed] [Google Scholar]
- Englander, S.W., Mayne, L., Bai, Y., and Sosnick, T.R. 1997. Hydrogen exchange: The modern legacy of Linderstrom-Lang. Protein Sci. 6 1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink, A.L. 1998. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des. 3 R9–R23. [DOI] [PubMed] [Google Scholar]
- Glasoe, P.K. and Long, F.A. 1960. Use of glass electrodes to measure acidities in D2O. J. Phys. Chem. 64 188–193. [Google Scholar]
- Grzesiek, S., Dobeli, H., Gentz, R., Garotta, G., Labhardt, A.M., and Bax, A. 1992. 1H, 13C, and 15N NMR backbone assignments and secondary structure of human interferon-γ. Biochemistry 31 8180–8190. [DOI] [PubMed] [Google Scholar]
- Hsu, Y.-R. and Arakawa, T. 1985. Structural studies on acid unfolding and refolding of recombinant human interferon γ. Biochemistry 24 7959–7963. [DOI] [PubMed] [Google Scholar]
- Hvidt, A. and Nielson, S.O. 1966. Hydrogen exchange in proteins. Adv. Protein Chem. 21 287–386. [DOI] [PubMed] [Google Scholar]
- Jennings, P.A. and Wright, P.E. 1993. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science 262 892–896. [DOI] [PubMed] [Google Scholar]
- Kendrick, B.S., Carpenter, J.F., Cleland, J.L., and Randolph, T.W. 1998a. A transient expansion of the native state precedes aggregation of recombinant human interferon-γ. Proc. Natl. Acad. Sci. 95 14142–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick, B.S., Cleland, J.L., Lam, X., Nguyen, T., Randolph, T.W., Manning, M.C., and Carpenter, J.F. 1998b. Aggregation of recombinant human interferon γ: Kinetics and structural transitions. J. Pharm. Sci. 87 1069–1076. [DOI] [PubMed] [Google Scholar]
- Lam, X.M., Patapoff, T.W., and Nguyen, T.H. 1997. The effect of benzyl alcohol on recombinant human interferon-γ. Pharm. Res. 14 725–729. [DOI] [PubMed] [Google Scholar]
- London, J., Skrzynia, C., and Goldberg, M.E. 1974. Renaturation of Escherichia coli tryptophanase after exposure to 8 m urea: Evidence for the existence of nucleation centers. Eur. J. Biochem. 47 409–415. [DOI] [PubMed] [Google Scholar]
- Lumry, R. and Biltonen, R. 1969. Thermodynamic and kinetic aspects of protein conformations in relation to physiological function. In Structure and stability of biological macromolecules (eds. S.N. Timasheff and G.D. Fasman), pp. 65–212. Marcel Dekker, Inc., New York.
- Lumry, R. and Eyring, H. 1954. Conformation changes in proteins. J. Phys. Chem. 58 110–120. [Google Scholar]
- McNay, J.L. and Fernandez, E.J. 1999. How does a protein unfold on a reversed-phase liquid chromatography surface? J. Chromatogr. A 849 135–148. [Google Scholar]
- Miller, D.W. and Dill, K.A. 1997. Ligand binding to proteins: The binding landscape model. Protein Sci. 6 2166–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranker, A., Robinson, C.V., Radford, S.E., Aplin, R.T., and Dobson, C.M. 1993. Detection of transient protein folding populations by mass spectrometry. Science 262 896–900. [DOI] [PubMed] [Google Scholar]
- Mulkerrin, M.G. and Wetzel, R. 1989. Ph dependence of the reversible and irreversible thermal denaturation of γ interferons. Biochemistry 28 6556–6561. [DOI] [PubMed] [Google Scholar]
- Pederson, T.G., Sigurskjold, B.W., Anderson, K.V., Kjaer, M., Poulsen, F.M., Dobson, C.M., and Redfield, C. 1991. A nuclear magnetic resonance study of the hydrogen-exchange behaviour of lysozyme in crystals and solution. J. Mol. Biol. 218 413–426. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. 1992. Physical basis of the stability of the folded conformations of proteins. In Protein folding (ed. T.E. Creighton), pp. 83–126. W. H. Freeman and Company, New York.
- Przybycien, T.M. 1998. Protein–protein interactions as a means of purification. Curr. Opin. Biotechnol. 9 164–170. [DOI] [PubMed] [Google Scholar]
- Ries-Kautt, M.M. and Ducruix, A.F. 1989. Relative effectiveness of various ions on the solubility and crystal growth of lysozyme. J. Biol. Chem. 264 745–748. [PubMed] [Google Scholar]
- ———.1991. Crystallization of basic proteins by ion pairing. J. Cryst. Growth 110 20–25. [Google Scholar]
- Roder, H., Elove, G.A., and Englander, S.W. 1988. Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature 335 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell, L.C., Sunde, M., and Blake, C.C.F. 1997. The molecular basis of amyloidosis. Cell. Mol. Life Sci. 53 871–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D.L., Yuzhong, D., and Zhang, Z. 1997. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J. Mass Spectrom. 32 135–146. [DOI] [PubMed] [Google Scholar]
- Speed, M.A., Wang, D.I.C., and King, J. 1996. Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition. Nat. Biotechnol. 14 1283–1287. [DOI] [PubMed] [Google Scholar]
- Stigter, D. and Dill, K.A. 1993. Theory for protein solubilities. Fluid Phase Equilib. 82 237–249. [Google Scholar]
- Stigter, D., Alonso, D.O., and Dill, K.A. 1991. Protein stability: Electrostatics and compact denatured states. Proc. Natl. Acad. Sci. 88 4176–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, S.Y. and Pepys, M.B. 1994. Amyloidosis. Histopathology 25 403–414. [DOI] [PubMed] [Google Scholar]
- Thevenon-Emeric, G., Kozlowski, J., Zhang, Z., and Smith, D.L. 1992. Determination of amide hydrogen exchange rates in peptides by mass spectrometry. Anal. Chem. 64 2456–2458. [DOI] [PubMed] [Google Scholar]
- Tobler, S.A., Sherman, N.E., and Fernandez, E.J. 2001. Tracking lysozyme unfolding kinetics during salt-induced precipitation with hydrogen exchange and mass spectrometry. Biotech. Bioeng. 71 194–207. [DOI] [PubMed] [Google Scholar]
- Walter, M.R., Windsor, W.T., Nagabhushan, T.L., Lundell, D.J., Lunn, C.A., Zauodny, P.J., and Narula, S.K. 1995. Crystal structure of a complex between interferon-γ and its soluble high-affinity receptor. Nature 376 230–235. [DOI] [PubMed] [Google Scholar]
- Waschutza, G., Li, V., Schafer, T., Schomburg, D., Villmann, C., Zakaria, H., and Otto, B. 1996. Engineered disulfide bonds in recombinant human interferon-γ: The impact of the N-terminal helix A and the AB-loop on protein stability. Protein Eng. 9 905–912. [DOI] [PubMed] [Google Scholar]
- Watanabe, S. and Saito, T. 1985. Circular dichrosim study of nascn effect on conformation of poly-l-lysine. J. Peptide Protein Res. 26 439–447. [DOI] [PubMed] [Google Scholar]
- Wetzel, R. 1994. Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 12 193–198. [DOI] [PubMed] [Google Scholar]
- Wetzel, R., Perry, L.J., Baase, W.A., and Becktel, W.J. 1988. Disulfide bonds and thermal-stability in T4 lysozyme. Proc. Natl. Acad. Sci. 85 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. In NMR of proteins and nucleic acids, pp. 24–25. John Wiley and Sons, New York.
- Zhang, Z. and Smith, D.L. 1993. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 2 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]