

Abstract
Replication and related processes in eukaryotic cells require replication factor C (RFC) to load a molecular clamp for DNA polymerase in an ATP-driven process, involving multiple molecular interactions. The detailed understanding of this mechanism is hindered by the lack of data regarding structure, mutual arrangement, and dynamics of the players involved. In this study, we analyzed interactions that take place during loading onto DNA of either the PCNA clamp or the Rad9-Rad1-Hus1 checkpoint complex, using computationally derived molecular models. Combining the modeled structures for each RFC subunit with known structural, biochemical, and genetic data, we propose detailed models of how two of the RFC subunits, RFC1 and RFC3, interact with the C-terminal regions of PCNA. RFC1 is predicted to bind PCNA similarly to the p21-PCNA interaction, while the RFC3-PCNA binding is proposed to be similar to the E. coli δ-β interaction. Additional sequence and structure analysis, supported by experimental data, suggests that RFC5 might be the third clamp loader subunit to bind the equivalent PCNA region. We discuss functional implications stemming from the proposed model of the RFC1-PCNA interaction and compare putative clamp-interacting regions in RFC1 and its paralogs, Rad17 and Ctf18. Based on the individual intermolecular interactions, we propose RFC and PCNA arrangement that places three RFC subunits in association with each of the three C-terminal regions in PCNA. The two other RFC subunits are positioned at the two PCNA interfaces, with the third PCNA interface left unobstructed. In addition, we map interactions at the level of individual subunits between the alternative clamp loader/clamp system, Rad17-RFC2–5/Rad9-Rad1-Hus1. The proposed models of interaction between two clamp/clamp loader pairs provide both structural framework for interpretation of existing experimental data and a number of specific findings that can be subjected to direct experimental testing.
Keywords: Comparative modeling, AAA+ proteins, DNA replication, clamp loader, pentameric complex, Rad17, Ctf18, 9-1-1 checkpoint complex
Replication factor C (RFC) is an essential heteropentameric protein complex that functions in DNA replication, DNA repair, and cell cycle checkpoints. During DNA replication, the RFC complex together with proliferating cell nuclear antigen (PCNA) function as accessory proteins of the eukaryotic DNA polymerases. RFC binds to primed DNA and uses ATP to drive the loading of PCNA onto DNA. PCNA is a ring-shaped homotrimeric protein that encircles DNA, forming a freely sliding clamp that tethers DNA polymerase to the DNA template for processive synthesis.
Eukaryotic RFC complexes are composed of one large (95–140 kD) and four small (36–40 kD) subunits (Mossi and Hubscher 1998). Genes encoding each of the subunits are essential, and all five subunits are required for efficient PCNA loading onto DNA. Clamp loading complexes, functionally corresponding to eukaryotic RFC, are present in all kingdoms of life, with the closest RFC relative found in Archaea. However, the archaeal clamp loading complex is composed of only two types of subunits, RFCL and RFCS (large and small, respectively), indicating that the clamp loading function itself does not necessitate four smaller subunits to be unique. Most likely, in eukaryotes distinct subunits evolved to allow RFC complex to interact with an extended set of partners and to perform other functions in addition to PCNA loading. An example of eukaryotic RFC versatility was revealed in the recent identification of two new alternative RFC complexes. In one of these complexes, the large RFC subunit is replaced by cell cycle checkpoint protein Rad17 (Rad24 in S. cerevisiae; Green et al. 2000; Lindsey-Boltz et al. 2001). In the second complex, the large subunit is replaced by Ctf18/Chl12 protein (Hanna et al. 2001; Mayer et al. 2001; Naiki et al. 2001), required for high-fidelity chromosome segregation. The Rad17-RFC2–5 complex is predicted to load a different DNA sliding clamp (Venclovas and Thelen 2000), composed of Rad checkpoint proteins (Kostrub et al. 1998; Kondo et al. 1999; St. Onge et al. 1999; Volkmer and Karnitz 1999; Caspari et al. 2000; Hang and Lieberman 2000; Burtelow et al. 2001) and recently termed the 9-1-1 complex (Burtelow et al. 2001), whereas the exact biochemical role of Ctf18-RFC complex has yet to be identified.
Interestingly, while all five RFC subunits are distinct, they all display significant sequence similarity with each other as well as with Rad17 and Ctf18 (Kouprina et al. 1994; Cullmann et al. 1995; Griffiths et al. 1995; Venclovas and Thelen 2000), proteins that can replace the large subunit within the RFC complex. The highest homology between RFC subunits is concentrated within a region containing seven conserved sequences motifs referred to as RFC boxes II–VIII (Cullmann et al. 1995). This high homology region links all RFC subunits, Rad17, and Ctf18 protein families into a highly abundant and functionally diverse group of proteins referred to as the AAA+ class (ATPases associated with various cellular activities; Neuwald et al. 1999).
Detailed three-dimensional (3D) structures are not available for the RFC complex or for any of its five subunits. However, recently solved crystal structures of the pentameric E. coli clamp loader (Jeruzalmi et al. 2001a) and the small subunit of the archaeal clamp loader (Oyama et al. 2001) provide strong clues for the structure of individual RFC subunits as well as the entire RFC complex. Moreover, a crystal structure of E. coli clamp loader subunit δ complexed with the monomer of β clamp (Jeruzalmi et al. 2001b) provides significant insight into the mechanism of bacterial clamp loading. Because the E. coli clamp loader subunits γ, δ, and δ′, all have similar structure, and are related to the RFC subunits, Rad17, and Ctf18 proteins, much of this new data can be used to facilitate our understanding of the structure and functional mechanism of "regular" and alternative eukaryotic clamp loaders.
Despite these recent advances, the clamp loading process is still poorly understood. A number of questions remain, including the structural effect of ATP binding on individual subunits as well as on the overall structure of a clamp loader; the role of each clamp loader subunit in interaction with a clamp; where the DNA binds within the clamp loader; and how the binding of the clamp and DNA triggers ATP hydrolysis. In eukaryotes, these questions are complicated by the fact that "regular" and alternative clamp loaders act on at least two different clamps (PCNA and 9-1-1 complex), and that each version of the eukaryotic clamp loader is a pentamer composed of related yet distinct proteins.
In this study, we integrated advanced sequence comparison with molecular modeling to generate 3D models for individual RFC subunits, and to analyze both their similarities and differences. Combining the resulting models with known experimental data on RFC-PCNA interactions, we propose models of how two of the subunits, RFC1 and RFC3, bind the C-terminal regions of PCNA, and we discuss the functional importance of the RFC1-PCNA interaction. Based on sequence and structure analysis, we also propose that RFC5 is likely to bind the remaining third such region in PCNA, a possibility that is supported, albeit indirectly, by experimental data. In addition, we analyzed the putative clamp-interacting regions in Rad17 and Ctf18. In the case of Rad17-substituted RFC, we mapped its interaction with the 9-1-1 checkpoint complex at the level of individual subunits, offering several novel predictions. We conclude with a summary of common structural steps in the clamp loading mechanism for "regular" and Rad17-substituted RFC complexes.
Results and Discussion
Models for individual subunits of the RFC complex
The resulting sequence-structure alignment of RFC subunits with known structures of subunits of clamp loaders from both Archaea and E. coli is given in Figure 1A ▶. In this figure, the alignment of related E. coli sequences was derived from the corresponding structure superpositions with archaeal RFCS. With the exception of the C-terminal region of RFC5, the alignment reported here mostly agrees with the published structure-based alignment of human RFC subunits and archaeal RFCS (Oyama et al. 2001).
Fig. 1.

(Continued on facing page)
Comparative modeling of RFC subunits. (A) Alignment of human and yeast sequences with the structures of archaeal small RFC subunit (RFCS) and subunits of the E. coli clamp loader (γ, δ′, and δ). Mapped above the alignment is the RFCS secondary structure, with different coloring representing individual structural domains. Some of the conserved sequence motifs (RFC-boxes) are also indicated. The structure of the RFC5 subunits within the region, enclosed in the red frame, is expected to deviate from that in RFCS (and RFC2–4), contributing to increased rigidity. Red background indicates motifs, known (δ) or predicted (RFC1 and RFC3) to mediate interaction with corresponding clamps. The alignment was generated with the structure of δ taken from E. coli γ-complex. In the complex with the β clamp, δ undergoes structural changes such that the clamp-interacting motif shifts by four residues and is aligned with that indicated for RFC3 (see also Fig.2B). (B) Ribbon diagram of the archaeal RFCS crystal structure and the GRASP (Nicholls et al. 1991) representation of the molecular surface electrostatic potential for human RFC3 and RFC5. Structures are shown in the same orientation; color coding and labeling for RFCS corresponds with that used in A. GRASP molecular surface regions of positive charge are colored blue, and those having negative charge are red. For the calculation of the surface electrostatic potential, we used dielectric constants of 2.0 for the protein interior and 80.0 for solvent at ionic strength equivalent to 150 mM NaCl. This and other structural figures were prepared using Molscript (Kraulis 1991) and Raster3D (Merritt and Bacon 1997).
RFC2, RFC3, and RFC4
As expected from sufficiently high sequence similarity, the structure of the clamp loader small subunit (RFCS) from Archaea Pyrococcus furiosus turns out to be a very close approximation for the structural models of the three small RFC subunits: RFC2, RFC3, and RFC4. To estimate the quality of the constructed models we used ProsaII, a program designed to detect errors in 3D protein structures with the help of an empirically derived energy function (Sippl 1993). The more the model properties correspond to those from experimentally observed structures, the lower is the energy (and the Z-score) assigned to the model. Both ProsaII position-dependent profiles and Z-scores for the models of these subunits are comparable with those for P. furiosus RFCS (Z-scores for RFCS: −11.7, and for RFC2–4: [−11.0; −11.8]), indicating that sequences for these RFC subunits can be accommodated within the framework of archaeal RFCS structure without any significant perturbation of main chain conformation.
RFC5
Although similar to the other small subunits (Z-score for RFC5 models: [−10.0; −10.6]), RFC5 is distinct in several respects. Perhaps the most significant difference is that the RFC5 ATP-binding site contains a modified P-loop (Walker A) and DEXX (Walker B) motifs. The modification of the DEXX motif includes a substitution of the first acidic residue with a neutral one (Thr or Asn). Since the acidic nature of the first residue is known to be critical for catalytic activity (e.g., Pause and Sonenberg 1992), RFC5 is apparently inactive in ATP hydrolysis. The functional consequences of the departure of the RFC5 sequence from the canonical P-loop motif are less clear; however, one feature stands out compared to the models of other RFC subunits, which is a markedly increased positive electrostatic potential of the altered RFC5 nucleotide binding site (Fig. 1B ▶). In fact, the appearance of an additional conserved Lys residue (Lys-49 in human protein) instead of Thr in the modified P-loop (GXXXXGKK) is only partly responsible for the increased positive electrostatic potential. The reduction of the negative charges in the Walker B and Sensor-2 motifs also contributes to the increase of the positive potential within the area of the nucleotide binding site. In general, all of the residue substitutions in and around the nucleotide binding site in RFC5 seem to alter catalytic properties and surface electrostatic properties, but not the 3D structure of this region in any significant way. Note that this is in sharp contrast to δ′, a subunit of the prokaryotic clamp loader, for which a modified nucleotide binding site was the basis for establishing the correspondence with RFC5 (O’Donnell et al. 2001). In the δ′ subunit, this site has a negative electrostatic potential (Guenther et al. 1997). Moreover, access to the nucleotide binding site of δ′ is completely blocked by bulky hydrophobic residues (Guenther et al. 1997). On the other hand, in RFC5 there are no apparent factors that would prevent nucleotide binding, unless the domain orientation is significantly different from that in the structure of RFCS, which served as a structural template for models of all RFC subunits. Although mutations of one of the Lys residues within the RFC5 P-loop motif were found to have little effect on RFC-dependent DNA replication in vitro (Cai et al. 1998; Podust et al. 1998), yeast RFC5 mutant rfc5–1, which has another residue substituted in the P-loop (G43E), is defective in DNA replication and also impaired in S-phase checkpoint (Sugimoto et al. 1996). One possible explanation for this is that normally ATP can bind to RFC5, but the binding plays only a stabilizing role. Alternatively, this positively charged surface patch in RFC5 could play some role in helping to direct DNA into the RFC complex, and into the open PCNA clamp during loading, but the effect of the P-loop mutation may only be noticeable in vivo.
In addition to the specific change of the electrostatic properties within the altered nucleotide binding site, the lack of convergence of the alignments generated with PSI-BLAST-ISS suggests that the polypeptide chain region, connecting the second and third domains of RFC5, has a different local conformation from that in RFCS (or RFC2, RFC3, and RFC4) (Fig. 1 ▶). Assessment of the RFC5 models indicated that using an unmodified template backbone in this region results in structural flaws, including poor residue packing and unfavorable distribution of hydrophilic/hydrophobic residues on the surface. Forcing the extension of the N-terminal end of the first helix of the third domain reduces these defects, suggesting that this helix in RFC5 indeed is longer than in the other small RFC subunits. In this respect, RFC5 closely resembles the δ′ subunit, which is the only E. coli clamp loader subunit that has a long rigid helix as a connector between domains two and three. One additional prominent difference between RFC5 and all of the other RFC subunits is the insertion within the N-terminal domain of a short subdomain (23 aa). Interestingly, its position coincides with that of a zinc-binding subdomain in the E. coli δ′ and γ structures; however, its sequence lacks the characteristic pattern of zinc-coordinating Cys residues. Furthermore, the sequence of this subdomain seems to lack any signature associated with known structural motifs, prompting us to exclude it from modeling. RFC5 comparison with other RFC subunits suggests that this subdomain is not critical for the structural integrity, but whether it has functional importance is yet to be determined.
RFC1
Eukaryotic RFC1, the largest RFC subunit, has an N-terminal extension with some similarity to DNA ligases (Cullmann et al. 1995). However, this region is not needed for the main RFC function—clamp loading onto DNA (Uhlmann et al. 1997). Indeed, archaeal RFCL, corresponding to RFC1, does not possess this region at all. RFC1 has a region of similarity (RFC boxes II–VIII) common to all subunits (also referred to as the AAA+ module; Neuwald et al. 1999) which spans two of the three structural domains in the small RFC subunits. The RFC1 C-terminal region, which extends beyond the common AAA+ module, is very divergent and is also considerably longer than the region encompassing the third (C-terminal) domain in small RFC subunits of eukaryotes and archaea.
For RFC1 we constructed 3D models that include only the AAA+ module. Although this module in RFC1 is clearly homologous to that of the other RFC subunits, it is the most divergent of these five subunits. One of the notable differences is the lack of SRC-motif that is absolutely conserved in all small RFC subunits. The RFC1 subunit also features two long loops that are not characteristic of the small subunits. The positions of these two loops, although distal in sequence, are proximal in space. We excluded them from modeling because loops of such length are impossible to predict accurately with existing methods (Tramontano et al. 2001). Moreover, the exact conformation of these loops is likely to depend on whether RFC1 is isolated, in the complex with other RFC subunits, or interacts with PCNA, the latter being most pertinent to this study (see the section below entitled "Models of RFC subunit interactions with PCNA"). The poorer ProsaII Z-scores for the RFC1 models (RFC1: [−6.9; −8.7], corresponding structural region of RFCS: −9.9) reflect larger structural differences between RFC1 and the RFCS compared to those for the RFC2–5 subunits.
One might expect that since RFC1 is similar to the small RFC subunits, its divergent C-terminal region would also include a domain similar to the C-terminal α-helical domain in the small RFC subunits. The architecture of the E. coli clamp loader also argues for that. As the C-terminal region of RFC1 is significantly longer than in the small subunits, it clearly should contain additional structural domain(s), responsible for the observed stable DNA binding by the RFC1 subunit that lacks the N-terminal ligase-like domain (Uhlmann et al. 1997). However, making assignments of structural domains within the divergent C-terminal region of RFC1 proved to be difficult. For example, PSI-BLAST searches using complete RFC1 sequences as queries did align the C-terminal region with the third (C-terminal) structural domain of the small RFC subunits. However, in such cases the C-terminal region of RFC1 was also aligned with the C-terminal domain of RuvB helicases, a domain that has an entirely different fold. If the divergent RFC1 C-terminal region alone was used as a query, PSI-BLAST failed to detect statistically significant similarity to the small RFC subunits. Exploring this further, we generated an extended sequence profile representing C-terminal domains of all known structures for the clamp loading subunits (RFCS, γ, δ, and δ′) and their homologs. However, independently of whether this profile was used to initiate PSI-BLAST or Hidden Markov Model searches, we failed to detect a statistically significant match between any of these C-terminal domains and the RFC1 C-terminal region. Fold recognition methods also were unable to find a significant match between RFC1 and any known structures, including bacterial and archaeal clamp loader subunits, when the RFC1 C-terminal region alone was used as a probe. These negative results do not necessarily mean that RFC1 lacks an equivalent structural domain. Most likely, the corresponding domain in RFC1 has diverged beyond the sensitivity limits of the methods we used. Nevertheless, we emphasize that the absence of the 3D model for the C-terminal region of RFC1 does not significantly hinder addressing the issues central to this study.
Models of RFC subunit interactions with PCNA
The RFC interaction with PCNA is one of the key steps in the loading of PCNA onto DNA. Two subunits, RFC1 and RFC3, have been shown using several methods to physically interact with the C-terminal region of PCNA (Mossi et al. 1997). Interaction of these subunits with PCNA is also substantiated by genetics, in that several mutant RFC subunits, including RFC1 and RFC3, can suppress PCNA mutations, mapping close to the C-terminus (Amin et al. 1999). We investigated possible modes of RFC1 and RFC3 interaction with PCNA using an analogy to the E. coli clamp loader δ subunit interaction with a monomer of β, the prokaryotic sliding clamp (Jeruzalmi et al. 2001b). For this, we superimposed PCNA and a model of either RFC1 or RFC3 onto the structures of β monomer and δ subunit respectively, mimicking the arrangement of the β-δ complex (Jeruzalmi et al. 2001b). In the δ subunit, two hydrophobic residues (L73 and F74) from the C-terminus of the helix α4 upon clamp binding become part of a short loop and provide major points of interaction with β (Jeruzalmi et al. 2001b). All RFC subunits have loops of different length following the helix corresponding to α4 in δ, with the loop of RFC1 being the longest (Fig. 1A ▶). Jeruzalmi et al. (2001b) suggested that two aromatic residues (F701 and Y702 in human RFC1) within this long RFC1 loop might interact with PCNA in a similar way as L73 and F74 in δ interact with β. In contrast, we found different residues, one hydrophobic (I698) and one charged (K699), corresponding to the clamp-interacting residue pair of the δ subunit. It appears that in human RFC1, residues I698 and K699 as well as F701 and Y702 are part of a larger motif which is almost identical to the one mediating cell-cycle checkpoint protein p21 binding to PCNA (Fig. 2A ▶; Gulbis et al. 1996). Although the p21 and δ subunit clamp-interacting motifs partially overlap (Jeruzalmi et al. 2001b), the p21 residues corresponding to F701 and Y702 of human RFC1 belong to a 310 helix that extends beyond the common substructure. In p21, these two aromatic and one aliphatic residue (equivalent to I698 in human RFC1) make a critical contribution towards hydrophobic interaction with PCNA (Gulbis et al. 1996). Corresponding RFC1 residues are highly conserved not only in mammal but also in yeast sequences. Human p21 serine (S146), known to be important for stabilizing the 310 helical conformation in PCNA-bound p21 fragment (Gulbis et al. 1996), is also absolutely conserved in the analyzed RFC1 subunits. In addition to the analogy of p21-PCNA complex, the structural model of human RFC1 interaction with PCNA (Fig. 2A ▶) is strongly corroborated by yeast genetic data. In budding yeast, it was found that the PCNA mutation K253E (pol30-100), responsible for its temperature-sensitive phenotype, could be suppressed by the RFC1 mutation D397H (RFC1-19; Amin et al. 1999). It can be seen in Figure 2A ▶ that in human PCNA and RFC1, the side chains of corresponding residues (K254 and N694, indicated with red arrows) are capable of direct interaction with each other. In wild-type yeast, PCNA Lys-253 could form a salt bridge with Asp-397; however in the K253E PCNA mutant, not only would this salt bridge be absent, but two proximal negative charges would create a repulsion, weakening the PCNA-RFC1 interaction and presumably thereby giving rise to the observed mutant phenotype. It follows that the RFC1 D397H mutation suppresses the PCNA K253E phenotype by restoring a favorable electrostatic interaction.
Fig. 2.


Interactions of the RFC subunits with PCNA. (A) Comparison of the crystal structure for the PCNA-p21 complex with a model for the PCNA-RFC1 interaction. Three conserved hydrophobic residues important for the p21 interaction with PCNA and the corresponding residues in RFC1 are indicated with red labels in the structures and stars above the alignment. "p"s indicate known phosphorylation sites in p21. For comparison, structure-based alignment of the corresponding region in the E. coli δ subunit from δ-β complex is also included. Yeast RFC mutation D397H can suppress the PCNA K253E mutant. Corresponding residues in human proteins are indicated with labels and red arrows. (B) Comparison of the δ-β interaction in the crystal structure with a modeled RFC3-PCNA interaction. Labels and stars indicate residues, important for δ interaction with β.
In the case of RFC3, it is obvious even from the sequence alignment (Fig. 1A ▶) that the binding of this subunit to PCNA must be different from that of the p21-like mode of interaction predicted for RFC1. The corresponding loop in RFC3 is too short to be able to form a helical turn characteristic of the p21-like PCNA interacting motif. On the other hand, if RFC3 is superimposed with the structure of δ in complex with β, two hydrophobic residues (L73 and F74) of δ that are critical for this association line up with two highly conserved hydrophobic residues of RFC3 (I114 and F115 in the case of human RFC3; Fig. 2B ▶). To test whether these two hydrophobic residues in RFC3 could interact with PCNA similarly to how δ residues interact with β, we modeled the putative clamp-interacting loop of human RFC3 after the corresponding region of δ. Both hydrophobic residues could be accommodated with adjustments of only a few side-chain rotamers within PCNA in the vicinity of contact with the bulky side chain of Phe-115 from human RFC3. Some remaining minor steric clashes with Phe-115 could be easily removed by shifting the flexible PCNA interdomain linker by fractions of an Ångstrom. Thus, the model of δ-like RFC3 interaction with PCNA is consistent with the predicted structure and residue interactions.
Our proposed models of the RFC1 and RFC3 interactions with PCNA differ in the size of the respective interfaces. RFC1 has a significantly more extensive network of interactions with PCNA than RFC3, suggesting that RFC1 binds PCNA with a higher affinity. These different modes of interaction agree with the experimental findings that although both RFC1 and RFC3 protect a modified PCNA C-terminus from phosphorylation, protection by RFC1 is more effective (Mossi et al. 1997).
An obvious next question is whether the other RFC subunits bind to PCNA. Considering that PCNA has three equivalent C-terminal regions, it is highly probable that another RFC subunit, in addition to RFC1 and RFC3, binds to the unoccupied third C-terminal region of PCNA. Based on our analysis, RFC5 seems to be the best candidate as the third binder. The position corresponding to the first of the two hydrophobic residues, which are responsible for δ binding to the β clamp and also predicted here to be critical for RFC3 binding to PCNA, is invariably occupied by aliphatic residues (L121 in human RFC5) in all currently available RFC5 sequences, including ESTs (data not shown). RFC2 sequences specifically lack hydrophobic residues in the positions corresponding to the pair of clamp-binding residues in δ, strongly suggesting that RFC2 is unable to bind the C-terminal region of PCNA. There also are arguments against the ability of RFC4 to bind the same PCNA region. In the case of RFC4, the first of the two corresponding positions is mostly occupied by small hydrophilic residues, in contrast to RFC1, RFC3, and RFC5, where the hydrophobic character of this position is very strongly conserved. Comparison of the p21 and δ complexed with the respective clamps also reveals that the first of the two structurally equivalent residue pairs in both p21 and δ is similarly buried in the hydrophobic pocket of the clamps, but the second residue (T148 in human p21) is not necessarily buried. These arguments speak against the ability of RFC4 to bind the C-terminal region of PCNA.
What do experiments reveal about the clamp-binding properties of RFC2, RFC4, or RFC5? To our knowledge, there is no experimental evidence of RFC2 binding to PCNA, but RFC5 binding indeed has been reported (Cai et al. 1998; Mossi and Hubscher 1998). Reports regarding RFC4 binding to PCNA are conflicting. One of the earlier studies found human RFC4 to weakly interact with PCNA (Pan et al. 1993), but in a more recent study no significant binding could be detected (Mossi et al. 1997). Importantly, the latter study demonstrated that RFC4, unlike RFC1 and RFC3, could not protect the C-terminal region of PCNA from phosphorylation, suggesting that even if RFC4 does have affinity to PCNA, it does not bind the C-terminal region.
Taken together, our analysis and the experimental data support the notion that RFC5 is likely to bind the C-terminal region of one of the PCNA monomers in a fashion similar to that of RFC3. However, because only one of the two hydrophobic residues implicated in clamp-binding is present in RFC5, we predict that this interaction is weaker than that of RFC3. Although lacking direct evidence, RFC5 binding to one of the three equivalent PCNA C-terminal regions would also be consistent with the most likely arrangement of RFC subunits within the RFC complex (see the section below entitled "Mapping the topography of RFC/PCNA and Rad17-RFC2–5/9-1-1 interactions“).
Functional implications of the model of RFC1 interaction with PCNA
Considering all possible interactions of PCNA with the RFC subunits, the interaction with RFC1 appears to be the most intriguing in two ways: First, the similarity of the PCNA-interacting region of RFC1 with that of p21 suggests parallels for the regulation of these interactions with PCNA. It has been shown that the PCNA-interacting region of human p21 is subject to AKT-dependent phosphorylation at either Thr-145 or Ser-146 (see Fig. 2A ▶), and that the phosphorylation of either residue inhibits p21 binding to PCNA (Scott et al. 2000; Li et al. 2001; Rossig et al. 2001). All RFC1 subunits have a highly conserved Ser residue, and vertebrate RFC1 subunits have a conserved Thr, in positions equivalent to Ser-146 and Thr-145, respectively, of human p21. Moreover, it was demonstrated (Maga et al. 1997) that human RFC1 can be phosphorylated by the Ca2+/calmodulin-dependent protein kinase II (CaMKII), an enzyme required for cell-cycle progression in eukaryotic cells. Phosphorylation by CaMKII reduces RFC1 binding to PCNA; however, once bound to PCNA, RFC1 is protected from phosphorylation (Maga et al. 1997). These findings coupled with our model of the RFC1-PCNA interaction strongly suggest that Thr and Ser residues within the RFC1 PCNA-binding motif (Thr-696 and Ser-697 in human RFC1) are the most likely phosphorylation targets. Phosphorylation at these sites may not only inhibit RFC1 (and therefore the RFC complex) binding to PCNA, but perhaps also influence the interaction of RFC1 with the rest of the RFC complex. This speculation is based on the crystallographic study of the E. coli clamp loader (Jeruzalmi et al. 2001a). The modeling in that study suggested that in the absence of ATP, the α4 helix harboring clamp-interacting residues of the δ subunit is sequestered by the N-terminal domain of δ′ through hydrophobic interactions. RFC1 and RFC5 are thought to be the RFC equivalents of the E. coli clamp loader subunits δ and δ′, respectively (Jeruzalmi et al. 2001b; O’Donnell et al. 2001). Accordingly, in the absence of ATP, the RFC1 clamp-interacting region might be at least partially shielded by its interaction with RFC5. In this case, phosphorylation of the PCNA-interacting region would then interfere with the RFC1-RFC5 interface and could potentially facilitate exchange of RFC1 with other RFC1-related proteins such as Rad17 or Ctf18, known to form modified RFC complexes by substituting RFC1 (Green et al. 2000; Hanna et al. 2001; Lindsey-Boltz et al. 2001; Mayer et al. 2001; Naiki et al. 2001).
Secondly, the predicted similarity of the PCNA-interacting region in RFC1 to that of p21, along with the proposed role of RFC1 as the ring opener equivalent to the E. coli δ subunit (Jeruzalmi et al. 2001b; O’Donnell et al. 2001), raises an interesting question. In the crystal structure of the δ-β complex, the conformation of the β monomer is incompatible with the closed ring structure, indicating that the interaction with δ causes β-ring to open (Jeruzalmi et al. 2001b). Likewise, the RFC1 interaction with PCNA is expected to facilitate opening of the PCNA ring. However, the complex of the p21 clamp-interacting region with PCNA maintains an intact trimeric PCNA ring. What makes the RFC1 interaction with PCNA different from that of p21? Comparison of the p21-PCNA structure and the model of the RFC1-PCNA interaction reveals one substantial difference. The C-terminal residues, following 310 helix in the p21 peptide, form a β-strand that runs antiparallel and is paired to the β-strand formed by a portion of the PCNA interdomain connector loop (Fig. 2A ▶; Gulbis et al. 1996). Our analysis indicates that the RFC1 clamp-interacting loop would be too short to form the analogous secondary structure (β-strand–β-strand) interaction. The crystal structure of the δ-β complex (Jeruzalmi et al. 2001b) indicates that the reduced curvature of the β monomer results from the rotation of individual domains as rigid bodies at the domain boundaries affecting the interdomain linkers. Therefore it is reasonable to expect that in the case of p21 binding to PCNA, the rigidity of the resulting two-stranded β-sheet prevents the PCNA ring from opening, whereas in the case of the RFC1-PCNA interaction, the same region of the PCNA interdomain linker would not be similarly constrained, allowing PCNA to open.
Analysis of putative clamp-interaction regions in Rad17 and Ctf18
As mentioned above, two RFC1 paralogs, Rad17 and more recently Ctf18 were found to substitute RFC1 in alternative RFC complexes (Green et al. 2000; Hanna et al. 2001; Lindsey-Boltz et al. 2001; Mayer et al. 2001; Naiki et al. 2001). Using molecular modeling, in a previous study we predicted that the Rad17-RFC2–5 complex loads the PCNA-like Rad9-Hus1-Rad1 (9-1-1) complex onto processed DNA lesions (Venclovas and Thelen 2000). Experimental evidence for this proposed mechanism continues to grow (Caspari et al. 2000; Hang and Lieberman 2000; Burtelow et al. 2001; Kaur et al. 2001; Lindsey-Boltz et al. 2001; Zou et al. 2002). Hence it appears that the large subunit of the RFC complex, either RFC1 or Rad17, is the sole determinant of the type of interacting clamp, either PCNA or the 9-1-1 complex, respectively. Although the biochemical role of the Ctf18-substituted RFC complex is not yet clear, the analogy with both "regular" and Rad17-substituted RFC suggests that this complex also may interact with either PCNA, the 9-1-1 checkpoint complex, or an as yet unidentified clamp-like structure.
Since our model holds that the RFC1 interaction with PCNA is mediated through a universal p21-like motif, we examined corresponding regions in Rad17 and Ctf18 to see whether they too possess some identifiable conserved motifs.
Sequences within the Rad17 family are much more divergent both in terms of insertions/deletions and residue conservation in comparison to those within the RFC1 or Ctf18 families. That is also true for the region, which is expected to harbor putative clamp-interacting residues. Despite high divergence, we were able to identify a conserved hydrophobic motif in the Rad17 sequences in vertebrates, plants, and yeasts. The generalized form of this motif is F-X-X-F-[L/V/M], where X denotes a nonconserved position (185-FKEFL-189 in human Rad17; GenBank gi:4102916). Incidentally, the spacing between the hydrophobic positions in this motif is identical to that in the p21-like motif, but the chemical nature of the residues in the conserved positions is altered (see Fig. 2A ▶). It is interesting to note that according to secondary structure prediction, this sequence motif shows strong preference for α-helical conformation, also characteristic of p21 motif. However, due to the lack of sequence conservation between Rad17 and RFC1 in this region, it remains uncertain whether this Rad17 motif is in the position equivalent to the p21-like motif of RFC1, or whether it is embedded in the helix that corresponds to the one preceding the RFC1 p21-like motif (Fig. 2A ▶).
The results of our analysis of the putative clamp-binding region in Ctf18 family turned out to be both more definite and more intriguing. Because the Ctf18 proteins are more closely related to the RFC1 family than are the Rad17 proteins, generating the alignment between the RFC1 and Ctf18 families was straightforward. The putative clamp-interacting region in the Ctf18 family is considerably shorter than that in the RFC1 family, clearly too short to assume the p21-like structural motif. Instead, closer inspection revealed that this region is similar to the corresponding regions of RFC3 and the evolutionarily related large subunit (RFCL) of the archaeal clamp loader (Fig. 3 ▶). In particular, both the Ctf18 and RFCL families have predominantly hydrophobic residues in two positions that also line up with the pair of conserved hydrophobic residues in the predicted RFC3 clamp-binding motif, suggesting that the Ctf18-substituted RFC complex might interact at least with PCNA. This idea is also supported by the fact that eukaryotic and archaeal PCNA have very similar 3D structures (Matsumiya et al. 2001). In addition, biochemical experiments demonstrate that human RFC can load archaeal PCNA (Ishino et al. 2001), further emphasizing that the respective clamp structures are similar enough to allow, albeit inefficient, a functional molecular substitution.
Fig. 3.
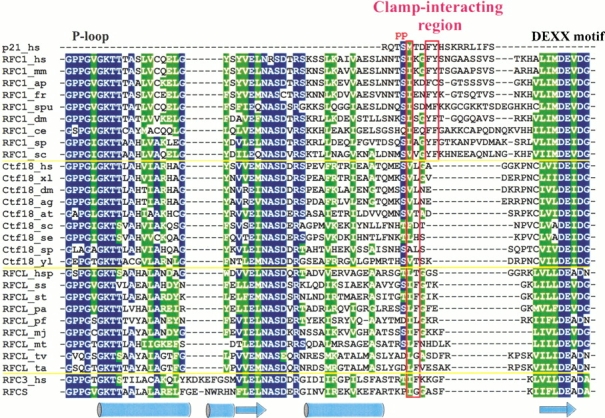
Multiple sequence alignment of the putative clamp-interaction regions in RFC1, Ctf18, and archaeal RFCL families. Sequences of known structures (p21 and RFCS) and RFC3 are included for reference. Secondary structure of the RFCS subunit is also shown. Red frames enclose positions that correspond to residues either known (for p21) or predicted here (for RFC3) to be important for interaction with PCNA. "p" indicates known sites of phosphorylation in human p21.
Archaeal RFCL sequences share homology beyond the AAA+ module with all three eukaryotic RFC1 paralogs (RFC1, Rad17, and Ctf18). However, the detected similarity of the putative clamp-interacting regions raises a possibility that Ctf18, and not RFC1, is the direct descendant of RFCL. Ctf18 might have been reduced to nonessential functions later in evolution after RFC1 acquired a p21-like motif within its clamp-interacting region. The acquisition of this motif could have significantly improved the efficiency of the RFC1-based clamp loading complex, making its presence essential for DNA replication and related processes.
Mapping the topography of RFC/PCNA and Rad17-RFC2–5 / 9-1-1 interactions
Both the analogy to the E. coli clamp loader and available experimental data suggest (O’Donnell et al. 2001) that the five subunits of RFC are arranged in a circular fashion such that their C-terminal regions mediate major intersubunit contacts whereas the face formed by the N-terminal domains interacts with PCNA. Parallels with the E. coli clamp loader also infer specific mechanical roles for the RFC subunits, with RFC1 acting as the "wrench," RFC5 as the "stator," and RFC2–4 performing a "motor" function (Jeruzalmi et al. 2001a,b; O’Donnell et al. 2001). Our modeling results indicate that of all the RFC subunits, RFC1 has the most elaborate PCNA-interacting motif, thus supporting the proposed "wrench" function of RFC1. Likewise, additional structural parallels determined in this study between RFC5 and the E. coli δ′ subunit reinforce the notion of RFC5 acting as the "stator". As part of the RFC complex, RFC1, RFC3, and likely RFC5, would bind to the three equivalent C-terminal regions of PCNA (Fig. 4 ▶). These particular interactions between the three RFC subunits and PCNA are consistent with the subunit arrangement within the RFC complex advocated in the recent review (O’Donnell et al. 2001) on the basis of analogy with the E. coli clamp loader and pairwise interactions detected in biochemical studies. In this scenario, the N-terminal domains of RFC2 and RFC4 subunits would have to be positioned at two of the three PCNA interfaces. Perhaps these subunits could even stabilize the two interfaces between the PCNA monomers, but their modes of interaction would be different from those proposed here for RFC1, RFC3, or RFC5. It is noteworthy that a single PCNA interface unobstructed by any other RFC subunit would be positioned between the "wrench" and the "stator" (RFC1 and RFC5, respectively), exactly where the opening of the E. coli clamp is proposed to occur (Jeruzalmi et al. 2001a).
Fig. 4.

Interactions between clamp loaders and clamps (A) Topography of interactions between subunits of “regular” or Rad17-substituted RFC complexes and PCNA or Rad9-Rad1-Hus1 (9-1-1) clamps, respectively. Colored ellipses indicate the three C-terminal regions of the clamp (PCNA/9-1-1) predicted to interact with the specified clamp loader subunits. The size of the ellipses represents the extent of the interactions between the clamp loader subunits and the clamp. The interface predicted to open up during clamp loading onto DNA is indicated with red arrows. (B) Schematic arrangement of the individual subunits within the two clamp loaders. The reader is facing the side formed by the C-terminal domains. The coloring of the subunits is the same as in A.
The model for the mutual arrangement between RFC subunits and PCNA can be extrapolated to map the interactions between the Rad17-substituted RFC complex and the 9-1-1 checkpoint complex. This extrapolation infers that Rad17 and RFC3 (and likely RFC5) bind the 9-1-1 complex at the C-terminal regions of the individual proteins (Rad9, Rad1, Hus1). The important difference from PCNA, however, is that all three C-terminal regions in 9-1-1 are distinct. Therefore, even with the similarity to the RFC-PCNA complex in mind, one is left with the three unique mutual arrangements between the Rad17-RFC2–5 and 9-1-1 complexes, where, for example, Rad17 could interact with any of the three subunits (Rad9, Rad1, or Hus1) of the 9-1-1 ring. Fortunately, there are several lines of evidence indicating that the Rad17 interaction with the 9-1-1 complex is mediated through Rad1 (Parker et al. 1998; Caspari et al. 2000; Rauen et al. 2000). Furthermore, a Rad1 mutant has been constructed that efficiently associated with Rad9 and Hus1 but failed to interact with Rad17 (Rauen et al. 2000). Given that at least some of the mutated residues were in the vicinity of the Rad1 C-terminal region, these findings provide additional support for the homology-based assumption that the RFC1-PCNA and the Rad17-Rad1 interactions involve topologically similar regions. Taking into account that our earlier prediction (Venclovas and Thelen 2000) of the subunit order within the 9-1-1 complex has been confirmed experimentally (Burtelow et al. 2001), these data allow for only one particular mutual arrangement of the two complexes (shown in Fig. 4 ▶). Besides the experimentally identified Rad17-Rad1 interaction, the proposed arrangement of the two complexes leads to the prediction that RFC3 interacts with Hus1, and that RFC5 is proximal to and likely interacts with Rad9. Moreover, based on this arrangement, during its loading onto DNA by the Rad17-substituted complex, the 9-1-1 complex must be opened at least at the interface formed by Rad1 and Rad9. Conversely, if the interface between Rad1 and Rad9 is sealed (e.g., by chemical crosslinking or protein engineering), the loading of the 9-1-1 complex onto DNA should not occur.
Collectively, analyses of the individual intermolecular interactions lead to a topological model of the clamp loading process by either the "regular" or Rad17-substituted RFC complex that is summarized in Figure 5 ▶. Upon ATP binding, the clamp loading complex undergoes an initial conformational change exposing the clamp-interacting regions of the individual subunits, where the number of subunits that bind ATP could be a critical factor in establishing the initial clamp/clamp loader complex (Gomes et al. 2001). The large subunit (either RFC1 or Rad17) binds to the C-terminal region of the PCNA monomer or the Rad1 protein, destabilizing the adjacent interface of the clamp. Binding of the other subunits, particularly of RFC3 and possibly RFC5, to the C-terminal regions of the other two clamp monomers temporarily fixes the clamp in the state with the open interface positioned between RFC1(Rad17) and RFC5. This ATP-bound quasistable structure of the complex between the clamp loader and the clamp has a high affinity to the double-stranded/single-stranded DNA structure. When DNA binds, its single-stranded portion triggers ATP hydrolysis with subsequent ADP dissociation. That resets the conformation of the clamp loader, causing the release of the clamp, which then spontaneously closes, trapping DNA inside. This summary, of course, includes only the major landmarks in the eukaryotic clamp-loading process. Differences in the finer details are likely to occur between the two clamp-loading processes, and, perhaps, even between different organisms in the loading of the same type of clamp.
Fig. 5.

Schematic view of the RFC-dependent loading of PCNA or the 9-1-1 complex onto DNA.
Computational Methods
Sequence-structure alignments and model building
Models of the RFC subunits were derived using a crystal structure of the small subunit of archaeal RFC (PDB code: 1IQP) (Oyama et al. 2001). In a few cases, short fragments from X-ray structures of the E. coli clamp loader subunits (Jeruzalmi et al. 2001a,b) and other AAA+ proteins were used to better represent local conformation.
In high homology cases, such as for the small RFC subunits RFC2, RFC3, and RFC4, constructing sequence alignments with archaeal RFC subunit was straightforward, with only a few deletions or insertions required. For the more distantly related RFC subunits, RFC5 and RFC1, alignments were derived using the intermediate sequence search procedure PSI-BLAST-ISS, which is described in more detail elsewhere (Venclovas 2001). Briefly, a set of sequences that are homologous to both the query and the sequence to which the query will be aligned (in this case, the structural template), are used as seeds to generate corresponding PSI-BLAST (Altschul et al. 1997) profiles. Using the SEALS package (Walker and Koonin 1997) and in-house Perl scripts, query sequence alignments with the template are extracted from the resulting individual PSI-BLAST profiles and compared. The convergence of the query-template alignments in a particular region is then used as an indicator of the alignment reliability in this region. If several alternative alignments are present, they are all investigated further by building and assessing the corresponding 3D models. Secondary structure predictions by Psipred (Jones 1999b) were also used to guide sequence-structure alignments in questionable regions.
To more rigorously assess the alignments in the questionable regions, models were built for orthologous proteins from at least three organisms: human, budding yeast, and fission yeast. Evaluation of the 3D models was done by visual inspection with emphasis on significant structural flaws, such as buried uncompensated charges or hydrogen donors/acceptors and severe steric clashes as detected with the structure verification module of WHATIF (Vriend 1990). In parallel, ProsaII (Sippl 1993) energy profiles and Z-scores were used to estimate the energy of local regions and the overall modeled structures, respectively.
Three-dimensional models were generated automatically from the constructed alignments with MODELLER (Šali and Blundell 1993). The amino acid side chains for the resulting models were positioned using a backbone-dependent rotamer library implemented in SCWRL (Bower et al. 1997). If after this step there were remaining severe side-chain clashes, they were resolved by manual rotamer positioning.
Possible interactions of RFC subunits with PCNA were studied by comparison with the structures of different clamps complexed with interacting peptides or proteins. Both types of analysis were based on structure superpositions generated either with DALI (Holm and Sander 1993) or with LGA, a locally written program available on the WWW (http://PredictionCenter.llnl.gov/local/lga/).
Analysis using structure-based sequence profiles and fold recognition
Structure-based sequence profiles were constructed as follows. Initially, multiple sequence alignments were derived from structure superposition of all clamp loader subunits that have known crystal structures. Next, these alignments were extended to include additional homologous proteins from respective families. The extended structure-based alignments were then used either as starting profiles for iterative PSI-BLAST searches or as models for Hidden Markov Model searches with HMMer (Eddy 1998).
For fold recognition, we used three methods in parallel: mGenThreader (Jones 1999a), FFAS (Rychlewski et al. 2000), and 3D-PSSM (Kelley et al. 2000).
Database ids of sequences presented in the alignments
RFC1 sequences are from human (hs, GenBank id: gi3334456), mouse (mm, gi6755306), duck (ap, gi1399917), fish (fr, JGI_14941), sea urchin (spu, gi5713366), fly (dm, gi17737765), worm (ce, gi17563226), and yeasts (sp, gi7491516; sc, gi6324791). Ctf18 sequences are from human (hs, gi13623351), frog (xl, gi13567707), fly (dm, gi15617460), mosquito (ag, gi19612274), plant (at, gi15219798), and yeasts (sc, gi6323724; se, gi12175545; sp, gi6522992; yl, gi12178242). RFCL family includes the following archaeal sequences: hsp, gi15790579; ss, gi15897671; st, gi15920689; pa, gi18312141; pf, gi18976464; mj, gi15669074; mt, gi15678268; tv, gi13541365; ta, gi16082282.
RFC2 sequences: human (hs, gi1703052), yeast (sc, gi6322528); RFC3: human (hs, gi728777) and yeasts (sc, gi6324039; sp, gi13431787); RFC4: human (hs, gi2507300) and yeast (sc, gi6324478); RFC5: human (hs, gi3915601) and yeast (sc, gi586518); bacterial δ subunits: vc, gi15640969; hi, gi1170333); mouse p21 (mm, gi1705726). Sequences of known 3D structures were derived from respective PDB files.
Acknowledgments
This research was partly funded by a DOE research grant to M.T., and was performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Livermore National Laboratory under contract W-7405-ENG-48. We thank Dorota Sawicka for critically reading the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Note Using electron microscopy, Griffith et al. (2002) found that the hRad9, hHus1, and hRad1 proteins make a trimeric ring structure (checkpoint 9-1-1 complex) reminiscent of the PCNA ring. Similarly they found that hRad17 makes a heteropentameric complex with the four RFC small subunits (hRad17-RFC) with a deep groove or cleft and is similar to the RFC clamp loader.
Abbreviations
AAA+, ATPases associated with various cellular activities
PCNA, proliferating cell nuclear antigen
RFC, replication factor C
RFCL, archaeal RFC large subunit
RFCS, archaeal RFC small subunit
9-1-1, Rad9-Hus1-Rad1 checkpoint protein complex
The nomenclature for the RFC subunits was developed independently for human and budding yeast, and therefore sometimes causes confusion as to which RFC subunit in yeast corresponds to that in human. When appropriate, we use the yeast nomenclature of RFC subunits. The correspondence between the yeast/human nomenclatures is the following: RFC1/p140, RFC2/p37, RFC3/p36, RFC4/p40, and RFC5/p38. Proteins that are homologous to the large RFC subunit and are known to be able to replace it in the RFC complex, to date, consist of two families: Rad17 family, including Rad17 in humans and other eukaryotes except budding yeast, where the corresponding protein is Rad24; and Ctf18 family, including Ctf18/Chl12 protein in budding yeast, and putative orthologs in fission yeast and other eukaryotes. Rad1, Rad9, and Hus1 denote respective protein families in all eukaryotes, except budding yeast, where corresponding proteins are termed Rad17, Ddc1, and Mec3, respectively.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0214302.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs.Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, N.S., Tuffo, K.M., and Holm, C. 1999. Dominant mutations in three different subunits of replication factor C suppress replication defects in yeast PCNA mutants.Genetics 153 1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower, M.J., Cohen, F.E., and Dunbrack Jr., R.L. 1997. Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: A new homology modeling tool.J. Mol. Biol. 267 1268–1282. [DOI] [PubMed] [Google Scholar]
- Burtelow, M.A., Roos-Mattjus, P.M., Rauen, M., Babendure, J.R., and Karnitz, L.M. 2001. Reconstitution and molecular analysis of the hRad9-hHus1-hRad1 (9–1–1) DNA damage responsive checkpoint complex.J. Biol. Chem. 276 25903–25909. [DOI] [PubMed] [Google Scholar]
- Cai, J., Yao, N., Gibbs, E., Finkelstein, J., Phillips, B., O’Donnell, M., and Hurwitz, J. 1998. ATP hydrolysis catalyzed by human replication factor C requires participation of multiple subunits.Proc. Natl. Acad. Sci. 95 11607–11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari, T., Dahlen, M., Kanter-Smoler, G., Lindsay, H.D., Hofmann, K., Papadimitriou, K., Sunnerhagen, P., and Carr, A.M. 2000. Characterization of Schizosaccharomyces pombe Hus1: A PCNA-related protein that associates with Rad1 and Rad9.Mol. Cell Biol. 20 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullmann, G., Fien, K., Kobayashi, R., and Stillman, B. 1995. Characterization of the five replication factor C genes of Saccharomyces cerevisiae.Mol. Cell Biol. 15 4661–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy, S.R. 1998. Profile hidden Markov models.Bioinformatics 14 755–763. [DOI] [PubMed] [Google Scholar]
- Gomes, X.V., Schmidt, S.L., and Burgers, P.M. 2001. ATP utilization by yeast replication factor C. II. Multiple stepwise ATP binding events are required to load proliferating cell nuclear antigen onto primed DNA.J. Biol. Chem. 276 34776–34783. [DOI] [PubMed] [Google Scholar]
- Green, C.M., Erdjument-Bromage, H., Tempst, P., and Lowndes, N.F. 2000. A novel Rad24 checkpoint protein complex closely related to replication factor C.Curr. Biol. 10 39–42. [DOI] [PubMed] [Google Scholar]
- Griffith, J.D., Lindsey-Boltz, L.A., and Sancar, A. 2002. Structures of the human Rad17-replication factor C and checkpoint Rad 9-1-1 complexes visualized by glycerol spray/low voltage microscopy.J. Biol. Chem. 277 15233–15236. [DOI] [PubMed] [Google Scholar]
- Griffiths, D.J., Barbet, N.C., McCready, S., Lehmann, A.R., and Carr, A.M. 1995. Fission yeast rad17: A homologue of budding yeast RAD24 that shares regions of sequence similarity with DNA polymerase accessory proteins.EMBO J. 14 5812–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther, B., Onrust, R., Sali, A., O’Donnell, M., and Kuriyan, J. 1997. Crystal structure of the δ′ subunit of the clamp-loader complex of E. coli DNA polymerase III.Cell 91 335–345. [DOI] [PubMed] [Google Scholar]
- Gulbis, J.M., Kelman, Z., Hurwitz, J., O’Donnell, M., and Kuriyan, J. 1996. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA.Cell 87 297–306. [DOI] [PubMed] [Google Scholar]
- Hang, H. and Lieberman, H.B. 2000. Physical interactions among human checkpoint control proteins HUS1p, RAD1p, and RAD9p, and implications for the regulation of cell cycle progression.Genomics 65 24–33. [DOI] [PubMed] [Google Scholar]
- Hanna, J.S., Kroll, E.S., Lundblad, V., and Spencer, F.A. 2001. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion.Mol. Cell Biol. 21 3144–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices.J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Ishino, Y., Tsurimoto, T., Ishino, S., and Cann, I.K. 2001. Functional interactions of an archaeal sliding clamp with mammalian clamp loader and DNA polymerase delta.Genes Cells 6 699–706. [DOI] [PubMed] [Google Scholar]
- Jeruzalmi, D., O’Donnell, M., and Kuriyan, J. 2001a. Crystal structure of the processivity clamp loader γ (gamma) complex of E. coli DNA polymerase III.Cell 106 429–441. [DOI] [PubMed] [Google Scholar]
- Jeruzalmi, D., Yurieva, O., Zhao, Y., Young, M., Stewart, J., Hingorani, M., O’Donnell, M., and Kuriyan, J. 2001b. Mechanism of processivity clamp opening by the δ subunit wrench of the clamp loader complex of E. coli DNA polymerase III.Cell 106 417–428. [PubMed] [Google Scholar]
- Jones, D.T. 1999a. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences.J. Mol. Biol. 287 797–815. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. 1999b. Protein secondary structure prediction based on position-specific scoring matrices.J. Mol. Biol. 292 195–202. [DOI] [PubMed] [Google Scholar]
- Kaur, R., Kostrub, C.F., and Enoch, T. 2001. Structure-function analysis of fission yeast hus1-rad1-rad9 checkpoint complex.Mol. Biol. Cell 12 3744–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM.J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Kondo, T., Matsumoto, K., and Sugimoto, K. 1999. Role of a complex containing Rad17, Mec3, and Ddc1 in the yeast DNA damage checkpoint pathway.Mol. Cell Biol. 19 1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrub, C.F., Knudsen, K., Subramani, S., and Enoch, T. 1998. Hus1p, a conserved fission yeast checkpoint protein, interacts with Rad1p and is phosphorylated in response to DNA damage.EMBO J. 17 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N., Kroll, E., Kirillov, A., Bannikov, V., Zakharyev, V., and Larionov, V. 1994. CHL12, a gene essential for the fidelity of chromosome transmission in the yeast Saccharomyces cerevisiae.Genetics 138 1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. Molscript—A program to produce both detailed and schematic plots of protein structures.J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Li, Y., Dowbenko, D., and Lasky, L.A. 2002. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival.J. Biol. Chem. 277 11352–11361. [DOI] [PubMed] [Google Scholar]
- Lindsey-Boltz, L.A., Bermudez, V.P., Hurwitz, J., and Sancar, A. 2001. Purification and characterization of human DNA damage checkpoint Rad complexes.Proc. Natl. Acad. Sci. 98 11236–11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maga, G., Mossi, R., Fischer, R., Berchtold, M.W., and Hubscher, U. 1997. Phosphorylation of the PCNA binding domain of the large subunit of replication factor C by Ca2+/calmodulin-dependent protein kinase II inhibits DNA synthesis.Biochemistry 36 5300–5310. [DOI] [PubMed] [Google Scholar]
- Matsumiya, S., Ishino, Y., and Morikawa, K. 2001. Crystal structure of an archaeal DNA sliding clamp: Proliferating cell nuclear antigen from Pyrococcus furiosus.Protein Sci. 10 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, M.L., Gygi, S.P., Aebersold, R., and Hieter, P. 2001. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): An alternative RFC complex required for sister chromatid cohesion in S. cerevisiae.Mol. Cell 7 959–970. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A., and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics.Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Mossi, R. and Hubscher, U. 1998. Clamping down on clamps and clamp loaders—The eukaryotic replication factor C.Eur. J. Biochem. 254 209–216. [PubMed] [Google Scholar]
- Mossi, R., Jonsson, Z.O., Allen, B.L., Hardin, S.H., and Hubscher, U. 1997. Replication factor C interacts with the C-terminal side of proliferating cell nuclear antigen.J. Biol. Chem. 272 1769–1776. [DOI] [PubMed] [Google Scholar]
- Naiki, T., Kondo, T., Nakada, D., Matsumoto, K., and Sugimoto, K. 2001. Chl12 (ctf18) forms a novel replication factor c-related complex and functions redundantly with rad24 in the DNA replication checkpoint pathway.Mol. Cell Biol. 21 5838–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald, A.F., Aravind, L., Spouge, J.L., and Koonin, E.V. 1999. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes.Genome Res. 9 27–43. [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons.Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- O’Donnell, M., Jeruzalmi, D., and Kuriyan, J. 2001. Clamp loader structure predicts the architecture of DNA polymerase III holoenzyme and RFC.Curr. Biol. 11 R935–946. [DOI] [PubMed] [Google Scholar]
- Oyama, T., Ishino, Y., Cann, I.K., Ishino, S., and Morikawa, K. 2001. Atomic structure of the clamp loader small subunit from Pyrococcus furiosus.Mol. Cell 8 455–463. [DOI] [PubMed] [Google Scholar]
- Pan, Z.Q., Chen, M., and Hurwitz, J. 1993. The subunits of activator 1 (replication factor C) carry out multiple functions essential for proliferating-cell nuclear antigen-dependent DNA synthesis.Proc. Natl. Acad. Sci. 90 6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, A.E., Van de Weyer, I., Laus, M.C., Verhasselt, P., and Luyten, W.H. 1998. Identification of a human homologue of the Schizosaccharomyces pombe rad17+ checkpoint gene.J. Biol. Chem. 273 18340–18346. [DOI] [PubMed] [Google Scholar]
- Pause, A. and Sonenberg, N. 1992. Mutational analysis of a DEAD box RNA helicase: The mammalian translation initiation factor eIF-4A.EMBO J. 11 2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podust, V.N., Tiwari, N., Ott, R., and Fanning, E. 1998. Functional interactions among the subunits of replication factor C potentiate and modulate its ATPase activity.J. Biol. Chem. 273 12935–12942. [DOI] [PubMed] [Google Scholar]
- Rauen, M., Burtelow, M.A., Dufault, V.M., and Karnitz, L.M. 2000. The human checkpoint protein hRad17 interacts with the PCNA-like proteins hRad1, hHus1, and hRad9.J. Biol. Chem. 275 29767–29771. [DOI] [PubMed] [Google Scholar]
- Rossig, L., Jadidi, A.S., Urbich, C., Badorff, C., Zeiher, A.M., and Dimmeler, S. 2001. Akt-dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells.Mol. Cell Biol. 21 5644–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychlewski, L., Jaroszewski, L., Li, W., and Godzik, A. 2000. Comparison of sequence profiles. Strategies for structural predictions using sequence information.Protein Sci. 9 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šali, A. and Blundell, T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints.J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Scott, M.T., Morrice, N., and Ball, K.L. 2000. Reversible phosphorylation at the C-terminal regulatory domain of p21(Waf1/Cip1) modulates proliferating cell nuclear antigen binding.J. Biol. Chem. 275 11529–11537. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J. 1993. Recognition of errors in three-dimensional structures of proteins.Proteins 17 355–362. [DOI] [PubMed] [Google Scholar]
- St. Onge, R.P., Udell, C.M., Casselman, R., and Davey, S. 1999. The human G2 checkpoint control protein hRAD9 is a nuclear phosphoprotein that forms complexes with hRAD1 and hHUS1.Mol. Biol. Cell 10 1985–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto, K., Shimomura, T., Hashimoto, K., Araki, H., Sugino, A., and Matsumoto, K. 1996. Rfc5, a small subunit of replication factor C complex, couples DNA replication and mitosis in budding yeast.Proc. Natl. Acad. Sci. 93 7048–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano, A., Morea, V., and Leplae, R. 2001. Analysis and assessment of comparative modeling predictions in CASP4.Proteins Suppl. 5 22–38. [DOI] [PubMed] [Google Scholar]
- Uhlmann, F., Cai, J., Gibbs, E., O’Donnell, M., and Hurwitz, J. 1997. Deletion analysis of the large subunit p140 in human replication factor C reveals regions required for complex formation and replication activities.J. Biol. Chem. 272 10058–10064. [DOI] [PubMed] [Google Scholar]
- Venclovas, Č. 2001. Comparative modeling of CASP4 target proteins: Combining results of sequence search with three-dimensional structure assessment.Proteins Suppl. 5 47–54. [DOI] [PubMed] [Google Scholar]
- Venclovas, Č. and Thelen, M.P. 2000. Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes.Nucleic Acids Res. 28 2481–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmer, E. and Karnitz, L.M. 1999. Human homologs of Schizosaccharomyces pombe rad1, hus1, and rad9 form a DNA damage-responsive protein complex.J. Biol. Chem. 274 567–570. [DOI] [PubMed] [Google Scholar]
- Vriend, G. 1990. WHAT IF: A molecular modeling and drug design program.J. Mol. Graph. 8 52–56. [DOI] [PubMed] [Google Scholar]
- Walker, D.R. and Koonin, E.V. 1997. SEALS: A system for easy analysis of lots of sequences.Proc. Int. Conf. Intell. Syst. Mol. Biol. 5 333–339. [PubMed] [Google Scholar]
- Zou, L., Cortez, D., and Elledge, S.J. 2002. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin.Genes & Dev. 16 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]