

Abstract
We recently described two protein G variants (NuG1 and NuG2) with redesigned first hairpins that were almost twice as stable, folded 100-fold faster, and had a switched folding mechanism relative to the wild-type protein. To test the structural accuracy of our design algorithm and to provide insights to the dramatic changes in the kinetics and thermodynamics of folding, we have now determined the crystal structures of NuG1 and NuG2 to 1.8 Å and 1.85 Å, respectively. We find that they adopt hairpin structures that are closer to the computational models than to wild-type protein G; the RMSD of the NuG1 hairpin to the design model and the wild-type structure are 1.7 Å and 5.1 Å, respectively. The crystallographic B factor in the redesigned first hairpin of NuG1 is systematically higher than the second hairpin, suggesting that the redesigned region is somewhat less rigid. A second round of structure-based design yielded new variants of NuG1 and NuG2, which are further stabilized by 0.5 kcal/mole and 0.9 kcal/mole.
Keywords: Computational protein design, β-hairpin design, protein folding, protein G, crystal structure of protein G
Protein design is a challenging and exciting problem because it tests our understanding of the basic principles governing protein folding and opens up the possibility of creating proteins with novel structures and functions. The early approach toward protein redesign mainly involved looking at a protein structure and making an educated guess as to the mutations necessary to achieve a certain goal, for example, increasing the stability of the protein (Cordes et al. 1996). More recently, efforts have been made to streamline this procedure by incorporating current knowledge of protein energetics into protein design algorithms (Desjarlais and Handel 1995; Dahiyat and Mayo 1996; Kuhlman and Baker 2000). The general approach is to specify a fixed target backbone and search for sequences with low free energies based on a potential energy function (Gordon et al. 1999). Mayo and coworkers have successfully redesigned a zinc finger and the B1 domain of protein G (protein G) using this approach (Dahiyat and Mayo 1997; Malakauskas and Mayo 1998). There has been less success in generating structures with new backbone conformations, with the notable exception of the design of a novel right-handed coiled-coil (Harbury et al. 1998) by using parametric equations to describe the protein backbone. Although elegant, this approach is currently limited to proteins with symmetric backbone structures, such as the coiled-coil and Triosephosphate Isomerage (TIM) barrels.
An alternative to a complete parameterization of the backbone is to replace the wild-type backbone with, for example, a structural element from the Protein Data Bank (PDB). We used this approach to redesign the first hairpin of protein G (Nauli et al. 2001). The design energy function simulates the physical interactions stabilizing protein structures and is dominated by a Lennard-Jones packing term and an implicit solvation term (Kuhlman and Baker 2000). NuG1 and NuG2 are 4 kcal/mole more stable and fold 100-fold faster when compared to wild-type protein G (Nauli et al. 2001). Furthermore, the folding pathways of the two proteins are opposite that of wild-type protein G. In wild-type protein G, the second β-hairpin is formed and the first disrupted in the folding transition state, whereas in NuG1 and NuG2, the first hairpin is formed and the second is disrupted.
In this article, we investigate the accuracy of structural prediction made by our design program by comparing the computational models with the crystal structures of NuG1 and NuG2. The root-mean-square deviation (RMSD) of the backbone of the redesigned turn between the crystal structure and the computational model is 1.7 Å for NuG1 and 3.4 Å for NuG2. In contrast, the RMSD between the crystal structure and the structure of wild-type protein G is 5.1 Å for NuG1 and 7.2 Å for NuG2. We also seek to further stabilize both NuG1 and NuG2 by incorporating low energy sequence changes suggested by the design algorithm and the new crystal structure. The resulting variants of both NuG1 and NuG2 are either more stable than their respective wild-type proteins or maintain wild-type stability.
Results
Structural analyses of NuG1 and NuG2 redesigned hairpin
The design process for NuG1 and NuG2 has been described previously (Nauli et al. 2001). Briefly, we replaced an 11-residue region of the first hairpin of protein G (Table 1) with 322 new backbone conformations from the PDB. For each backbone, sequences with low energy were identified with our protein design algorithm and the lowest-scoring sequence/structure combinations predicted to have a type I` turn (NuG2) and a type II` turn (NuG1) were selected for experimental study. These turn types are commonly seen in β-hairpins in naturally occurring proteins (Sibanda and Thornton 1985).
Table 1.
Amino acid sequence of redesigned proteins
| WT | 6-ILNGKTLKGET-16 |
| NuG1 | 6-FIVIGDRVVVV-16 |
| NuG2 | 6-VIVLNGTTFTY-16 |
Turn residues in bold.
Crystals of single point mutants of NuG1 and NuG2 were grown because these proteins could be expressed at high levels. The mutations T49A for NuG1 and D46A for NuG2 correspond to sites in the second hairpin of protein G, far removed from the redesigned hairpin. In the crystal structures, the backbone of the second β hairpin superimposes very well with that of wild-type protein G, suggesting that these mutations have only local effects and do not affect the analyses of the first β hairpin. From here on, the crystal structures will simply be referred to as NuG1 and NuG2. Data collection and refinement statistics for both proteins are given in Table 2.
Table 2.
Data collection and refinement statistics
| Dataset | NuG1 | NuG2 |
| Data collection and processing | ||
| Wavelength | 0.97 | 1.5418 |
| Resolution | 1.8 | 1.85 |
| Space group | I422 | C2 |
| Cell dimensions | ||
| a(Å) | 49.46 | 47.33 |
| b(Å) | 49.46 | 73.79 |
| c(Å) | 103.02 | 39.18 |
| beta(deg) | n/a | 96 |
| Number of unique reflections | 6383 | 10905 |
| Rsym | 11.6 | 6.7 |
| Completeness | 95.4 | 97.3 |
| I/sigma I | 8.7 | 14 |
| Refinement | ||
| Resolution range | 22.3–1.8 | 15–1.85 |
| Number of reflections | 5900 | 9780 |
| R/Rfree (%) | 21.2/22.5 | 26.0/26.5 |
| Number of refined atoms | ||
| Protein | 521 | 956 |
| Water | 114 | 110 |
| Average B-factors (Å-squared) | 21.3 | 37.3 |
| RMSD | ||
| Bonds | 0.006 | 0.02 |
| Angles | 1.3 | 3.8 |
Figure 1 ▶ shows a comparison of the NuG1 structure with wild-type protein G and the computational model. The redesigned hairpin is clearly in a different conformation than that of the wild-type protein and forms a type II` turn as designed (Fig. 1 ▶). To more precisely determine the differences between the three structures they were superimposed using residues from the helix and the second hairpin (23–55) that are the least likely to be affected by the mutations. With this superposition the RMSD for the first turn residues (9–12) is 1.7 Å between NuG1 and the model, and a considerably larger 5.1 Å between NuG1 and wild-type protein G.
Fig. 1.

Comparison of the backbone (A) and side chain (B) of the redesigned hairpin in NuG1. The crystal structure of wild-type protein G (1PGA) is shown in green; the computational model, in blue; and the crystal structure of NuG1, in red.
The agreement between the model and the crystal structure is not as good for the side chain atoms. Five of the 10 redesigned residues were modeled into the correct chi 1 rotamer (F6, V8, V13, V14, and V15) (Fig. 1 ▶). Ile7 is observed in two separate side chain conformations in the crystal, neither of which match with the design model. It is interesting that Ile7 can assume multiple conformations because it is located in the center of the hydrophobic core and makes most of the interactions between the first hairpin and the helix. This implies that the interface between the hairpin and the helix is not uniquely packed, but rather flexible. This conclusion is further supported by an examination of crystallographic B factors in NuG1 (Fig. 2 ▶). Unlike wild-type protein G that has a uniform distribution of B factors below 30, most of the atoms in the hairpin of NuG1 have B factors between 30 and 50. Another important difference is at position 14 where in the wild-type protein Gly14 has dihedral angles of (φ = 169°,ξ = −161°), which is unfavorable for the β-strand conformation. In NuG1, Val14 adopts dihedral angles (−115°, 131°), which is regularly observed for β-strands (Table 3).
Fig. 2.
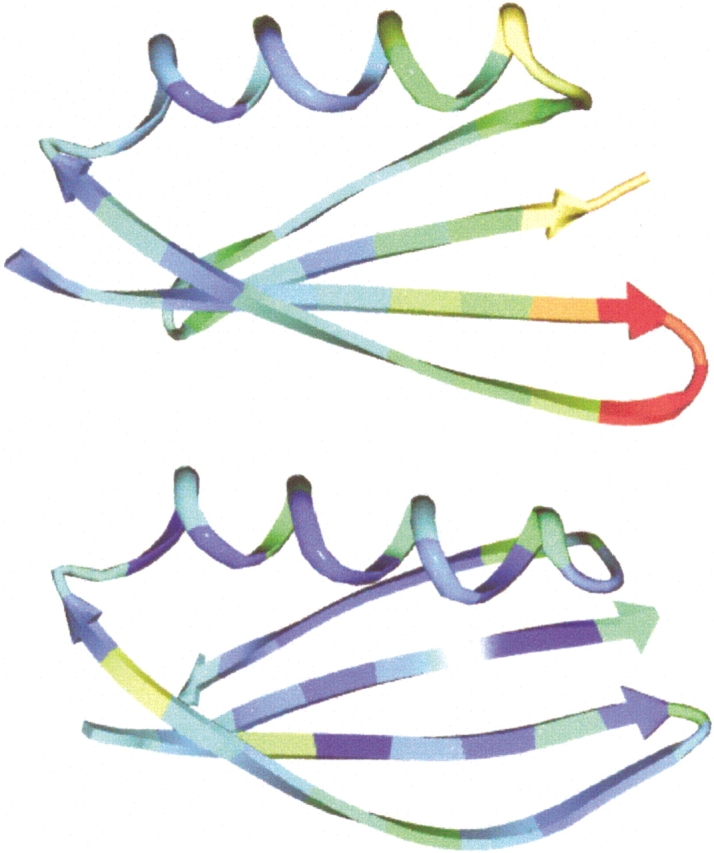
Crystallographic B factors of NuG1 (top) and wild-type protein G (bottom). The B factor is colored from high (red) to low (blue).
Table 3.
Backbone φ, ξ angles in the redesigned region of wild-type protein G (WT), NuG1 and NuG2
| Residue number | WT | NuG1 | NuG2 |
| 6 | −105,117 | −100,108 | −122,129 |
| 7 | −101,117 | −115,99 | −121,112 |
| 8 | −118,70 | −83,88 | −103,124 |
| 9 | −92,−177 | −108,118 | −111,129 |
| 10 | −77,−35 | −59,−104 | −101,89 |
| 11 | −103,−41 | −102,−33 | −72,−4 |
| 12 | −112,129 | −103,138 | −116,149 |
| 13 | −138,138 | −102,122 | −131,133 |
| 14 | 169,−161 | −115,131 | −111,140 |
| 15 | −146,135 | −114,117 | −129,152 |
| 16 | −140,173 | −111,128 | −140,128 |
NuG2 crystallizes as a dimer with the second β strand of each monomer forming the dimer interface, giving rise to an eight-stranded β sheet. The dimer may be formed due to the pairing of surface Thr residues on both molecules, which is favorable for β sheet formation (Smith and Regan 1995). As a caution, we note that the structure could only be refined to an R factor of 26% (see Materials and Methods). For clarity in differentiating among the structures, we do refer to this model here as the crystal structure of NuG2. When the structure of NuG2 is superimposed with the computational model and wild-type protein G, the RMSD of turn residues (9–12) is 3.4 Å and 7.2 Å, respectively. The comparatively large RMSD to the computational model results from movement of the hairpin backbone away from the helix (Fig. 3 ▶). This deviation may be due to the partner molecule in the dimer, which forms hydrogen bonds with the second β strand. Because the R factor is high for NuG2, it is not appropriate to make detailed comparisons between side chain atoms and torsion angles in the design model and the crystal structure.
Fig. 3.

Comparison of the backbone of the redesigned hairpin in NuG2. The hairpin of the crystal structure of wild-type protein G (1PGA) is shown in green; the computational model, in blue; and the crystal structure of NuG2, in red.
To independently asses the rigidity of NuG1 and NuG2, one-dimensional nuclear magnetic resonance (NMR) spectra were collected (Fig. 4 ▶). Unlike many designed proteins, the spectra exhibit the sharp and well-dispersed peaks typical of native proteins.
Fig. 4.
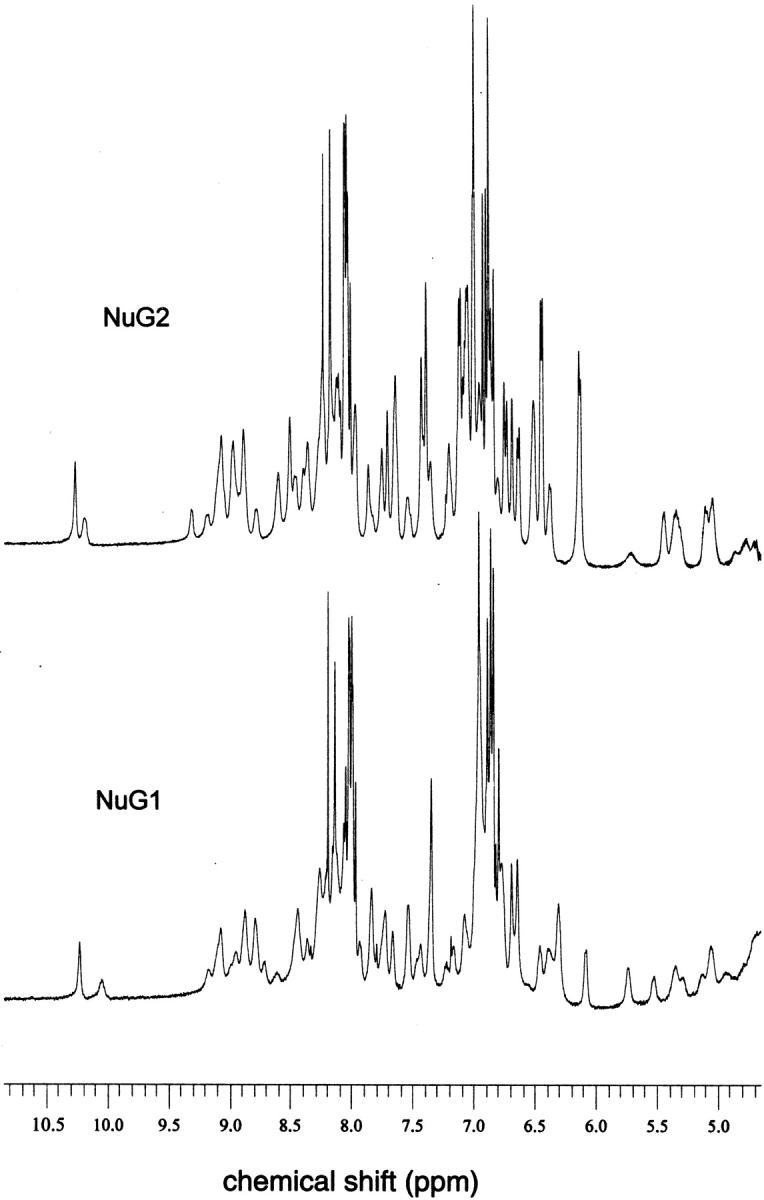
One-dimensional 500-MHz 1H NMR spectra of NuG1 (0.7 mM) and NuG2 (1.2 mM) at 300 K.
Increasing the stability of NuG1 and NuG2
The stabilities of the protein G variants were measured using guanidine denaturation experiments monitored by circular dichroism. Previously, we showed that both NuG1 and NuG2 are about twice as stable as wild-type protein G at 22°C (Fig. 5A ▶). Because both variants are too stable at 22°C to give an accurate unfolded baseline in a guanidine unfolding curve, we measured stabilities at 50°C instead (Fig. 5B ▶). NuG1 (5.2 kcal/mole) and NuG2 (4.9 kcal/mole) have free energies of unfolding at 50°C, which are almost twice as large as wild-type protein G (2.8 kcal/mole) (Malakauskas and Mayo 1998).
Fig. 5.
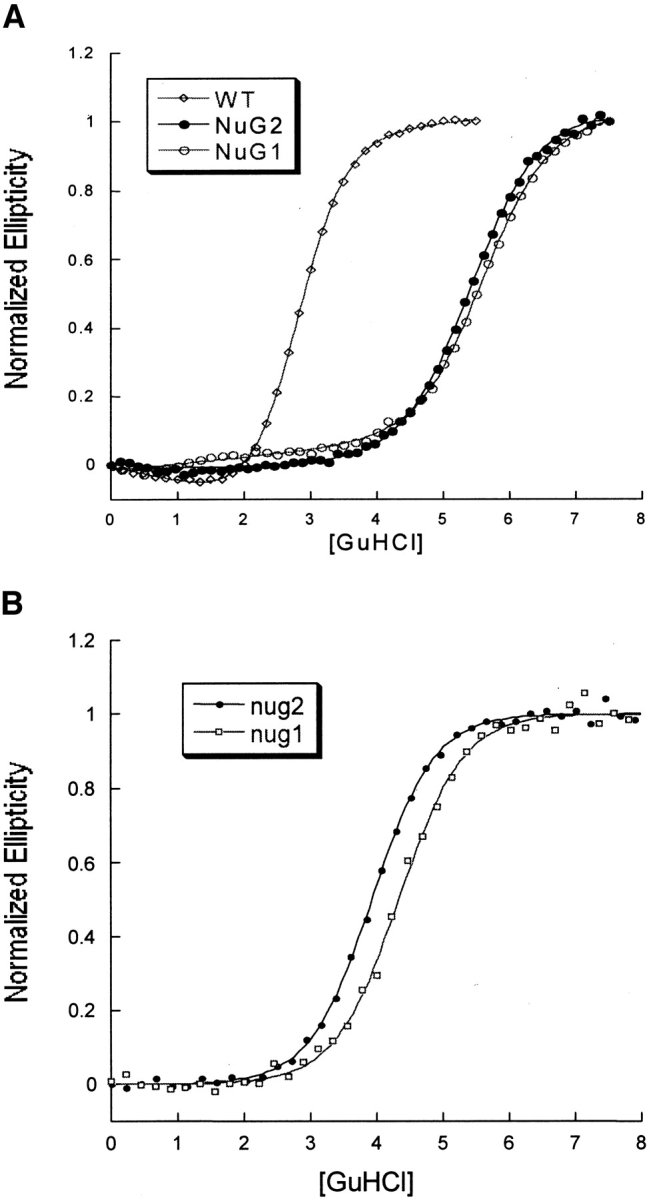
Stabilities of wild-type protein G, NuG1, and NuG2 at 25°C (A) and at 50°C (B).
We used the design program to identify mutations that would further stabilize NuG1 and NuG2 (see Materials and Methods). In the case of NuG2, we ran the computer simulations before the crystal structure was solved, and therefore, restricted our search to residues outside of the redesigned hairpin. The design program was used to compile a list of five mutations predicted to be stabilizing (Fig. 6B ▶; Table 4). Three of the substitutions—Y3F, T25E, D47A—have been shown previously to stabilize wild-type protein G (Minor and Kim 1994; Smith et al. 1994; Smith and Regan 1995; McCallister et al. 2000). T53V was shown to be destabilizing in the context of wild-type protein G, whereas V21A has not been studied previously.
Fig. 6.

Location of rationally designed mutations on NuG1 (A) and NuG2 (B). The redesigned hairpin is colored in yellow, and the mutated residues are colored in red and labeled.
Table 4.
Thermodynamic stability of NuG1 and NuG2 mutants
| Protein | Cm (m) | ΔG (kcal/mol) | ΔΔG (kcal/mol) | ΔΔE (computed) |
| NuG1 | 4.3 | −5.2 | — | — |
| V39I/V54I/NuG1 | 4.7 | −5.7 | −0.5 | −2.0 |
| I9K/V39I/V54I/NuG1 | 4.4 | −5.3 | −0.1 | −4.1 |
| NuG2 | 3.9 | −4.9 | — | — |
| Y3F/NuG2 | 3.7 | −4.7 | 0.2 | −0.5 |
| V21A/NuG2 | 4.0 | −5.0 | −0.1 | −1.8 |
| T25E/NuG2 | 4.3 | −5.3 | −0.4 | −1.7 |
| D47A/NuG2 | 4.4 | −5.5 | −0.6 | −1.8 |
| T53V/NuG2 | 4.0 | −5.0 | −0.1 | −1.1 |
| Y3F/V21A/T25E/D47A/NuG2 | 4.6 | −5.8 | −0.9 | −6.2 |
Cm is the [GuHCl] where half of the protein is unfolded ΔΔG = mWT ΔCm where ΔCm = Cmredesigned − CmWT. The wild-type protein is either NuG1 or NuG2.
Figure 7B ▶ shows the results of denaturation experiments on NuG2 variants. Y3F slightly destabilizes NuG2, whereas both V21A and T53V maintain wild-type stability. Both T25E and D47A stabilize NuG2 and a mutant containing four mutations (Y3F/V21A/T25E/D47A) is more stable than NuG2 by 0.9 kcal/mole. There is only a weak correlation between the computed energies and the actual changes in free energy (Table 4), but it is encouraging that none of the mutations were significantly destabilizing.
Fig. 7.
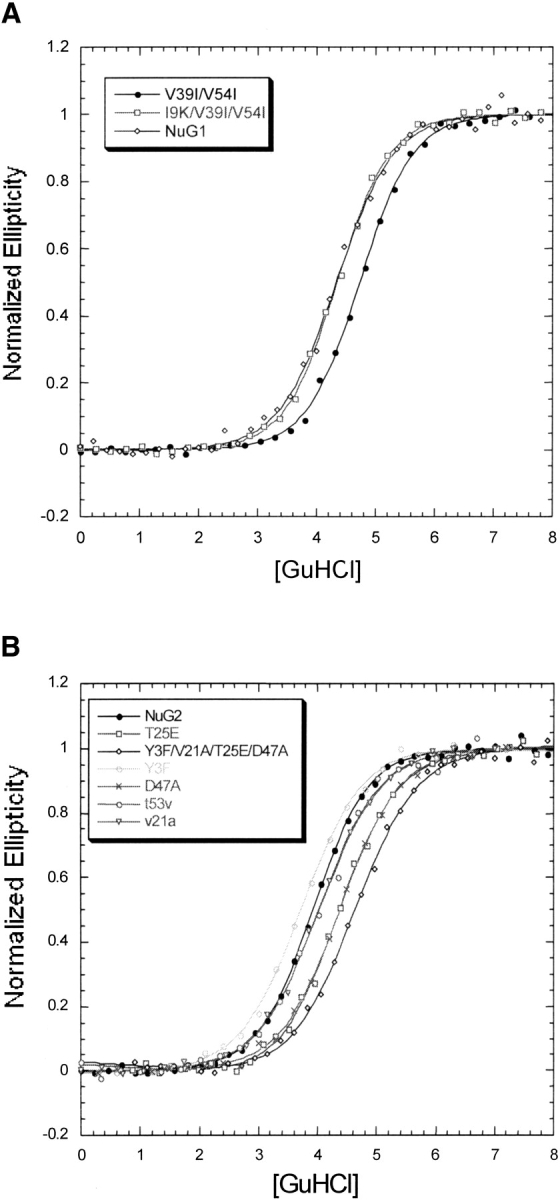
Guanidine-induced denaturation of NuG1 (A) and NuG2 (B) and their mutants at 50°C followed by CD.
In the case of NuG1, we took an iterative design approach. We used the crystal structure of NuG1 as the template for the computational modeling. For this reason we restricted our simulations to the redesigned hairpin and the residues that contact it. In contrast to the strategy for selecting NuG2 mutants, the design program was allowed to make multiple substitutions simultaneously. V39I/V54I and I9K/V39I/V54I were experimentally characterized and the double mutant is stabilized relative to NuG1, whereas the triple mutant is as stable as NuG1 (Fig. 7A ▶).
Discussion
Crystal structures of NuG1 and NuG2
Previously, we demonstrated that NuG1 and NuG2 are more stable, fold 100-fold faster, and have a switched folding pathway relative to wild-type protein G (Nauli et al. 2001). Here, we show that the redesigned hairpins have the intended canonical turn types and that the final structures more closely resemble the computationally predicted design models than wild-type protein G. The NuG1 redesign was particularly successful with an RMSD of only 1.7 Å between the redesigned hairpin and the design model.
Given the overall success of the designs, it is interesting that the central core residues, Ile7, can populate multiple conformations and that the B factors are significantly higher for this region of the protein. Increased entropy in the folded state may be one of the reasons that NuG1 is significantly more stable than wild-type protein G. We recently redesigned a hairpin in protein L using methods identical to those used in the protein G redesigns. Unlike NuG1 and NuG2, the protein L variants were not more stable than wild-type protein, but the crystal structure of the redesigned protein matched the design model almost identically and the B factors were not higher in the redesigned hairpin (Kuhlman et al. 2002). The differences between the protein L and protein G results suggest a tradeoff between specificity and stability similar to what has been observed in the design of helical bundles (Bryson et al. 1995).
Alternatively, both the increased stability and B factors may be consequences of the significant increase in the hydrophobicity of the redesigned hairpin (Table 1), which may allow for greater burial of hydrophobic surface with less constraint on maintaining interactions among solvating polar residues. Designed proteins have frequently been observed to be more molten than their naturally occurring counterparts. The NuG1 structure shows that this can extend to redesigned substructure within a single protein; the B factors are much higher in the first hairpin than the second.
Given their switched folding pathways relative to wild-type protein G, we anticipate that the NuG1 and NuG2 crystal structures will be useful for the testing of computational models that try to predict folding mechanisms. The crystal structures of NuG1, NuG2, and wild-type protein G have been used by two theoretical models of folding to examine the importance of the two β-hairpins. Both models correctly predict that the first hairpin turn is formed before the second hairpin turn in NuG1 and NuG2, but the exact opposite in wild-type protein G (E. Alm and Y. Zhou, pers. comm.).
Rationally designed mutations that increase stability
As a test of the design algorithm, we used the program to identify mutations that would stabilize NuG1 and NuG2. Guanidine unfolding experiments showed that some of the mutations were stabilizing and that none of them were significantly destabilizing, but there was only a weak correlation between the actual and computed changes in free energy. Some of the discrepancy probably arises because the energy function has been optimized to reproduce native-like sequences, and therefore it tries to maintain the correct balance of hydrophobic and polar amino acids so as to maintain solubility as well as stability. For example, two of the mutations that were computationally favorable, but experimentally neutral, were I9K in NuG1 and V21A in NuG2. Both of these mutations are highly solvent exposed and less hydrophobic residues are favored at these positions by the Lazaridis-Karplus solvation model used in the design algorithm.
In the cases where the mutations were stabilizing different features seem to be optimized in each case. In NuG2 Glu 25 forms a hydrogen bond with the backbone nitrogen from residue 21 that lowers the energy by 1.4 kcal/mole. In NuG1 Ile 39 and Ile 54 bury more hydrophobic surface area and make more favorable van der Waals contacts. The computed Lennard-Jones score is −271.1 kcal/mole for the double mutant as opposed to −268.5 kcal/mole for the wild-type structure.
The results described in this paper highlight our ability to make rational changes in a protein sequence, predict the resulting structure, and also make rational mutations that would increase protein stability. Most mutants of both NuG1 and NuG2 are more stable or maintain wild-type stability. Where the algorithm fails, the problem seems to be in its current inability to allow backbone changes. To deal with this problem we are currently combining our ab initio structural prediction method Rosetta (Bonneau and Baker 2001) with the design algorithm (B.Kuhlman, unpubl.) to allow simultaneous searching in sequence and structure space.
Materials and methods
Sample preparation
Both proteins are purified using published methods (Nauli et al. 2001). The protein is dialyzed into 10 mM Tris buffer at pH 7.5 for NuG1, and 10 mM HEPES buffer at pH 7.5 for NuG2 for crystallization. NuG1 could be crystallized in 8% n-propanol, 3.6 M Na formate. Single crystals of NuG2 were observed in 1.6 M ammonium sulfate, 0.1 M Tris at pH 8.0. Both proteins are crystallized using the hanging drop method with Q plates from Hampton Research.
Crystal structures
Diffraction data for NuG1 were collected at the Stanford Synchotron Radiation Laboratory (SSRL) at Beamline 9–1. For NuG2, we use an in-house Cu-K-alpha X-ray source with a Rigaku R-Axis IIC detector and an RU200 rotating anode generator. Data processing was with Denzo and Scalepack. As NuG2 was readily available, it was crystallized first and a dataset was collected for it. A molecular replacement (MR) solution was found using the wild-type protein G coordinates (1PGA; Gallagher et al. 1994) as the search model in the MR program EPMR (Kissinger et al. 1999). The NuG2 model could not be refined to values of R and Rfree that are normally expected for a dataset extending to 1.85 Å resolution. We tried various space groups for the data; however, the best model still refined only to an R factor of 26%. Because of this reason, we report here only a model that fits the diffraction data. Coordinates and structure factors have been deposited in the PDB for both NuG1 and NuG2.
NuG1 crystals were then grown and a dataset was collected for them at the SSRL. An MR solution was found with Amore (Navaza 2001) using a model of NuG2 where the redesigned hairpin was converted to polyalanine. Refinement of both structures uses the following programs: CCP4 suite, XtalView (McRee 1999) and CNS (Brunger et al. 1998).
Although the data set for NuG2 extends to 1.85 Å resolution, the model can only be refined to 26% R factor. We note that the dataset has a relatively large Wilson B factor. Considering how the stability of this protein, a mobile structure is unlikely to be the cause of the high Wilson B factor. A more likely reason is the high degree of static disorder in the crystal due to each protein molecule packing in a slightly different orientation relative to its neighbors.
Guanidine denaturation
Guanidine denaturations were monitored using an Aviv 16A DS CD machine. Proteins were equilibrated at 50°C for 2 min and each data point is an average of 1 min of CD signal at 220 nm. Protein concentrations are 10 μM. The data are an average of two measurements.
Modeling
The design program and energy function used to identify stabilizing mutations have been described in more detail previously (Kuhlman and Baker 2000). The energy function used to rank structures is a linear combination of the following terms: (1) a 12–6 Lennard-Jones potential truncated at E = 0; (2) a linear repulsive term below E = 0 that ramps up to E = 10 at no separation; (3) the Lazaridis-Karplus implicit solvation model; (4) an empirically based hydrogen bonding potential derived from the PDB database (T. Kortemme and D. Baker, pers. comm.); (5) side chain internal free energies derived from PDB statistics (Dunbrack and Cohen 1997); (6) an approximation to electrostatic interactions in protein based on PDB statistics (Simons et al. 1999); and (7) reference values for each of the 20 amino acids. The desired hydrophobicity of a given residue is largely determined by the combination of the Lennard-Jones term, the Lazaridis-Karplus term, and the reference energies.
For NuG1, the newly solved crystal structure was used as the template for modeling and only positions in the redesigned hairpin were allowed to vary during the simulations: 5, 7, 9, 14, 16, 33, 34, 39, and 54. All other side chains were held fixed in positions observed in the NuG1 crystal structure. The two variants selected for experimental study, V39I/V54I and I9K/V39I/V54I, were the lowest scoring sequences with two and three mutations, respectively. A third sequence containing five mutations was also identified (I7V/I9K/Y33W/A34L/V39Y) but has not been studied experimentally.
In the case of NuG2, mutations were restricted to regions outside of the redesigned hairpin and therefore, the crystal structure of wild-type protein G was used as the template for modeling. To identify a set of proteins that varied from one to five mutations, the energy function was modified so that the Monte Carlo search of sequence space was biased toward a desired number of mutations. Adding the following quadratic energy term to the total energy proved suitable for creating structure with the desired number of mutations: (number of mutations − desired number of mutations)2. In the simulations all residues and rotamers outside of the redesigned hairpin were allowed to vary and in the first run the desired number of mutations was set to 1. This mutation was then fixed and the desired number of mutations was set to 2. This procedure was followed for up to five mutations and the following variants were picked out: V21A, V21A/Y3F, V21A/Y3F/T25E, V21A/Y3F/T25E/T53V, and V21A/Y3F/T25E/T53V/D47A. At this point it was noticed that these mutations are not located near each other in the structure; therefore we decided make each of them independent and combine the stabilizing mutations in one final variant. These sequences are shown in Table 4 with their respective experimental and computed energies.
Acknowledgments
We thank Craig Behnke for help in data collection for NuG2. We also thank Eric Alm and Yaoqi Zhou for sharing their data with us before publication. S.N. thanks Salem Faham for helpful discussion during refinement of NuG1 and NuG2. B.K. thanks Rachel Klevit, Lynne Spencer, Peter Brzovic, Ponni Rajagopal, and Jennifer Keefe for aid in acquiring the NMR spectra. S.N. was supported by the Hall-Ammerer-Washington Research Foundation fellowship. B.K. was supported by the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation. This work was partially supported by grants to D.B. from the National Institutes of Health, the National Science Foundation, and Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
GyHCl
guanidine hydrochloride
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0216902
References
- Bonneau, R. and Baker, D. 2001. Ab initio protein structure prediction: Progress and prospects. Annu. Rev. Biophys. Biomol. Struct. 30 173–189. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Bryson, J.W., Betz, S.F., Lu, H.S., Suich, D.J., Zhou, H.X., O'Neil, K.T., and DeGrado, W.F. 1995. Protein design: A hierarchic approach. Science 270 935–941. [DOI] [PubMed] [Google Scholar]
- Cordes, M.H., Davidson, A.R., and Sauer, R.T. 1996. Sequence space, folding and protein design. Curr. Opin. Struct. Biol. 6 3–10. [DOI] [PubMed] [Google Scholar]
- Dahiyat, B.I. and Mayo, S.L. 1996. Protein design automation. Protein Sci. 5 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997. De novo protein design: Fully automated sequence selection. Science 278 82–87. [DOI] [PubMed] [Google Scholar]
- Desjarlais, J.R. and Handel, T.M. 1995. New strategies in protein design. Curr. Opin. Biotechnol. 6 460–466. [DOI] [PubMed] [Google Scholar]
- Dunbrack, Jr., R.L. and Cohen, F.E. 1997. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6 1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher, T., Alexander, P., Bryan, P., and Gilliland, G.L. 1994. Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochemistry 33 4721–4729. [PubMed] [Google Scholar]
- Gordon, D.B., Marshall, S.A., and Mayo, S.L. 1999. Energy functions for protein design. Curr. Opin. Struct. Biol. 9 509–513. [DOI] [PubMed] [Google Scholar]
- Harbury, P.B., Plecs, J.J., Tidor, B., Alber, T., and Kim, P.S. 1998. High-resolution protein design with backbone freedom. Science 282 1462–1467. [DOI] [PubMed] [Google Scholar]
- Kissinger, C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol. Crystallogr. 55 484–491. [DOI] [PubMed] [Google Scholar]
- Kuhlman, B. and Baker, D. 2000. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. 97 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman, B., O'Neill, J.W., Kim, D.E., Zhang, K.Y., and Baker, D. 2002. Accurate computer-based design of a new backbone conformation in the second turn of protein L. J. Mol. Biol. 315 471–477. [DOI] [PubMed] [Google Scholar]
- Lazaridis, T. and Karplus, M. 1999. Effective energy function for proteins in solution. Proteins. 35 133–152. [DOI] [PubMed] [Google Scholar]
- Malakauskas, S.M. and Mayo, S.L. 1998. Design, structure and stability of a hyperthermophilic protein variant. Nat. Struct. Biol. 5 470–475. [DOI] [PubMed] [Google Scholar]
- McCallister, E.L., Alm, E., and Baker, D. 2000. Critical role of β-hairpin formation in protein G folding. Nat. Struct. Biol. 7 669–673. [DOI] [PubMed] [Google Scholar]
- McRee, D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125 156–165. [DOI] [PubMed] [Google Scholar]
- Minor, Jr., D.L. and Kim, P.S. 1994. Measurement of the β-sheet-forming propensities of amino acids. Nature 367 660–663. [DOI] [PubMed] [Google Scholar]
- Nauli, S., Kuhlman, B., and Baker, D. 2001. Computer-based redesign of a protein folding pathway. Nat. Struct. Biol. 8 602–605. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276A 307–326. [DOI] [PubMed] [Google Scholar]
- Sibanda, B.L. and Thornton, J.M. 1985. β-hairpin families in globular proteins. Nature 316 170–174. [DOI] [PubMed] [Google Scholar]
- Simons, K.T., Ruczinski, I., Kooperberg, C., Fox, B.A., Bystroff, C., and Baker, D. 1999. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins 34 82–95. [DOI] [PubMed] [Google Scholar]
- Smith, C.K. and Regan, L. 1995. Guidelines for protein design: The energetics of β sheet side chain interactions. Science 270 980–982. [DOI] [PubMed] [Google Scholar]
- Smith, C.K., Withka, J.M., and Regan, L. 1994. A thermodynamic scale for the β-sheet forming tendencies of the amino acids. Biochemistry 33 5510–5517. [DOI] [PubMed] [Google Scholar]