

Abstract
Modeling the inherent flexibility of the protein backbone as part of computational protein design is necessary to capture the behavior of real proteins and is a prerequisite for the accurate exploration of protein sequence space. We present the results of a broad exploration of sequence space, with backbone flexibility, through a novel approach: large-scale protein design to structural ensembles. A distributed computing architecture has allowed us to generate hundreds of thousands of diverse sequences for a set of 253 naturally occurring proteins, allowing exciting insights into the nature of protein sequence space. Designing to a structural ensemble produces a much greater diversity of sequences than previous studies have reported, and homology searches using profiles derived from the designed sequences against the Protein Data Bank show that the relevance and quality of the sequences is not diminished. The designed sequences have greater overall diversity than corresponding natural sequence alignments, and no direct correlations are seen between the diversity of natural sequence alignments and the diversity of the corresponding designed sequences. For structures in the same fold, the sequence entropies of the designed sequences cluster together tightly. This tight clustering of sequence entropies within a fold and the separation of sequence entropy distributions for different folds suggest that the diversity of designed sequences is primarily determined by a structure's overall fold, and that the designability principle postulated from studies of simple models holds in real proteins. This has important implications for experimental protein design and engineering, as well as providing insight into protein evolution.
Keywords: Protein design, sequence space, designability, backbone flexibility, distributed computing
The aim of protein design is to find amino acid sequences that are compatible with specific protein structures. Screening of sequences for compatibility with a protein structure was introduced in the early 1980s, with the definition of the inverse folding problem (Pabo 1983). Whereas protein folding involves finding the native three-dimensional structure for a particular amino acid sequence, the inverse folding problem seeks to define the entire set of sequences that can specifically form a stable protein with some target structure. Protein design, whether experimental, computational, or some hybrid approach, provides important clues towards a solution of the inverse protein folding problem by sampling the sequence space of known protein structures (Pande et al. 1997).
An important practical use of protein design is in the stabilization of known protein folds (Dahiyat 1999). The optimization schemes used in most protein design algorithms are written to find local or globally optimized sequences, with the lowest or near-lowest free energy of folding for an existing target structure; much recent work has addressed this topic (Desjarlais and Clarke 1998; Shakhnovich 1998; Koehl and Levitt 1999a; Voigt et al. 2000; Wernisch et al. 2000; Pokala and Handel 2001). Finding sequences that will form a given structure often results in sequences with increased stability over the wild type (Malakauskas and Mayo 1998). An exciting potential direction for protein design lies in creating totally novel protein structures. The successful design of a family of right-handed coiled coils demonstrated the capability of computational protein design to create novel structures, and highlighted the importance of allowing for backbone flexibility in the design process (Harbury et al. 1998).
Experimental techniques for protein design have also enjoyed much success in the last decade. Rational design, based on structural analysis and site-directed mutagenesis, has been used extensively in redesigning enzymes for increased stability and/or altered function (Cedrone et al. 2000; Kazlauskas 2000). The most successful experimental methods for protein design involve directed protein evolution, using genetic recombination of natural diversity and in vitro functional assays to explore sequence space (Tobin et al. 2000; Bornscheuer and Pohl 2001). Directed protein evolution generates a diversity of functional sequences through iterations of mutation and recombination, allowing the exploration of areas of sequence space that are not accessible using rational design or random mutagenesis techniques. However, because current methods for in vitro protein evolution are limited to searching spaces in the range of 103–106 sequences, computational techniques for reducing the search space for experimental protein design are of great importance and current relevance (Kono and Saven 2001; Voigt et al. 2001). By computationally designing large libraries of viable sequences, favorable and unfavorable regions of sequence space could be identified and combinatorial libraries could be greatly constrained by tailoring the range of diversity allowed at each position of the protein.
Most computational studies to date have produced designed sequences that tend to resemble the native sequence of the protein structure (Koehl and Levitt 1999b , 2002a; Kuhlman and Baker 2000; Raha et al. 2000). This result has generally been attributed to the constraints imposed by using fixed backbones. Backbone flexibility in the target structure is desirable when computationally designing amino acid sequences, because it is well known that natural proteins use small backbone adjustments to accommodate disruptive mutations (Eriksson et al. 1992; Baldwin et al. 1993). Indeed, when designing sequences to a structure, one does not expect these sequences to fold to exactly the target structure with zero deviation, but rather some ensemble of highly similar structures. Incorporating backbone flexibility into computational protein design more realistically models real proteins, and is a critical prerequisite for de novo protein design, where the exact structure of the resulting protein cannot be known (Desjarlais and Handel 1999).
Some recent studies have described methods incorporating some form of backbone flexibility, with excellent success in designing sequences that stably fold to the target structure (Su and Mayo 1997; Harbury et al. 1998; Desjarlais and Handel 1999). However, due to the extreme computational demands of including backbone flexibility in the design process, previous work has been limited to coarse-grained variation of backbone structure parameters (e.g., relative arrangement of secondary or supersecondary structure element; Su and Mayo 1997; Harbury et al. 1998; or designing only a subset of residues in the target protein; Desjarlais and Handel 1999). In all cases, only a small number of minimum-energy sequences for several proteins of interest were identified. Some recent work of note (Zou and Saven 2000; Kono and Saven 2001) has developed a generally applicable statistical theory for exploring protein sequence space, analogous to other mean-field methods used in protein design (Koehl and Delarue 1994; Lee 1994; Koehl and Levitt 1999a), which does not require the explicit articulation of minimum-energy sequences. Instead, this approach estimates amino acid probabilities at each residue position, which are energetically consistent with a given protein structure. In designing sequence profiles for protein L, backbone flexibility was incorporated by considering those sequence properties that were robust with respect to 21 backbone variants in an NMR ensemble (Kono and Saven 2001).
In this study, using a distributed computing network (Shirts and Pande 2000) of over 3000 processors has allowed us to design hundreds of minimum-energy sequences per structure, with the incorporation of fine-grain backbone variability, for the set of all protein structures in the Protein Data Bank (Berman et al. 2000) of length less than 100 residues, solved by X-ray crystallography: 253 structures in total. Designing to an ensemble of slight structural variants of the target structure produces a large diversity of high-quality sequences, allowing for the exploration of a much broader range of sequence space than previous studies, and leading to novel insights into the determinants of protein sequence space.
Results
The Genome@home distributed computing architecture allowed for the collection of a very large data set (Table 1). For each of the 253 structures in our data set, roughly 750 unique sequences were amassed over a period of 2 months. With a total of almost 200,000 processor days (∼3000 active processors over the 62-day course of data collection), almost 200,000 distinct sequences were returned. These overall figures agree well with tests that show a protein of 100 amino acids requiring roughly 24 h for completion of one full sequence design on a 500-MHz Celeron workstation.
Table 1.
Summary of the Genome@home data set
| Total number of structures | 253 |
| Total number of backbone variants used for design | 25,300 |
| Total time of data collection | 62 days |
| Processors available | 3000 |
| Total number of distinct sequences generated | 187,342 |
Generation of meaningful sequence diversity and the incorporation of backbone flexibility are two challenges facing modern protein design. By designing sequences for an ensemble of slightly varying three-dimensional structures, we achieve these two goals simultaneously. To create structural ensembles for each of the 253 structures studied, 100 structural variants were generated for each protein by gently perturbing the dihedral angles of the protein backbone (see Materials and Methods). The RMSD (Cα RMSD, specifically, is used throughout this study) of each variant is no more than 1 Å from the native target structure. For example, Figure 1 ▶ shows 10 structural variants of the SH3 domain from Abl tyrosine kinase (Musacchio et al. 1994) superimposed on the native crystal structure.
Fig. 1.

Ten representative backbone traces from the structural ensemble used in designing sequences for 1abo, the SH3 domain from Abl tyrosine kinase. All structures are within 1 Å RMSD of each other.
For clarity, let us briefly define some terms used in this paper: “Residue entropy”, S(i), refers to the informational entropy of the set of amino acids that appear at any one position, or column, in a sequence alignment, and is reported in its exponentiated form, exp(S(i)) (see Materials and Methods). This statistic, which ranges from 1 to 20, gives a rough measure of how many different amino acids appear at a sequence position. “Sequence entropy” refers to the mean residue entropy over an entire set of amino acid sequences. The sequence entropy quoted for any one structure in this study refers to the entropy of the entire set of sequences designed from the 100 variants of that structure.
Increased sequence diversity with structural ensembles
To assess the amount of diversity generated by our method, the entropy of the designed sequences for each structure was calculated (Shenkin et al. 1991). Figure 2a ▶ displays the distribution of residue entropies for each position in the total set of 253 structures. The residue entropies range from 1.0 to 14.4, with a mean of 6.6. As a control, between 70 and 100 sequences were designed for the fixed native backbone of each of the 253 target structures (i.e., no structural ensembles were used). The residue entropies of these sequences range from 1.0 to 3.3, with a mean of 2.4.
Fig. 2.
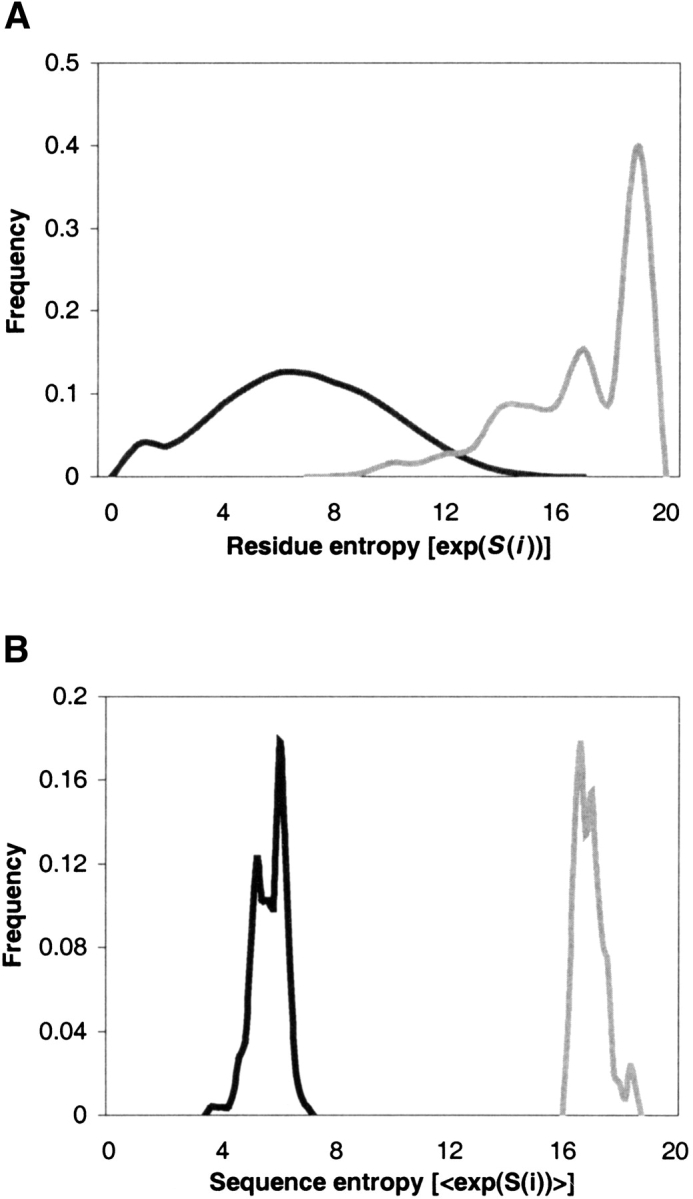
Entropy distributions of designed and sterically allowed residues and sequences. (A) Residue entropies of all designed positions are plotted in black. As well, the set of all sterically allowed rotamers at each position of each structure was calculated. The distribution of residue entropies for this set is plotted in gray. (B) The sequence entropy (mean residue entropy) for each structure was calculated. The distribution of sequence entropies for the designed sequences is plotted in black, with the sequence entropy from the allowed rotamers in gray.
To put the designed sequence sets into context within sequence space, some relevant baseline is needed for comparison. A trivial space against which to make comparisons is the set of all possible amino acid sequences of length L. This space is of size 20L, with a residue entropy at each position of exp(S(i)) = 20. A more realistic upper bound for sequence diversity was calculated for each structure as follows. At each position, each rotamer from the rotamer library used by the sequence prediction algorithm (Raha et al. 2000) was tested for steric clashes with the atoms of the peptide backbone. Any rotamers that clashed with the backbone were ignored, and the remaining set of all sterically allowed amino acids at each position was used to calculate sequence entropy. This set encompasses “what we have to work with” for each structure: the set of all amino acid side chains that could fit onto the protein structure at each position. The mean of the residue entropies over all positions in a protein, using the amino acid frequencies given by this process of identifying all sterically allowed rotamers, serves as an upper bound on the size of the structure’s sequence space.
Figure 2a ▶ shows that the residue entropy distribution of the sterically allowed set of rotamers is more sharply peaked and shifted higher than the residue entropy distribution of the final designed sequences. This shows the effects of the other terms in the energy function, such as hydrogen bonding and solvation, in constraining the sequence space of a protein structure. Figure 2b ▶ plots the distribution of sequence entropies (i.e., mean residue entropy over an entire sequence) for the 253 structures. The sequence entropies for the designed sequences have a more sharply peaked distribution than the overall pool of designed residue entropies, and the separation between the designed and sterically allowed distributions is even greater than in the case of residue entropy.
Previous studies have reported designed sequences retaining a high degree of similarity with the native sequence of the target structure (Koehl and Levitt 1999b; Kuhlman and Baker 2000; Raha et al. 2000). In a study on a set of 108 proteins, Kuhlman and Baker found that 51% of the core residues in designed sequences and 27% of all residues matched those found in the native sequence of the target structure. Koehl and Levitt found a 36% average identity to the native sequence over ten independent designs of 1ctf, but only a 16% average identity to native in 13 designed TIM sequences. In a study using a very slightly modified version of the design algorithm used here, Desjarlais and colleagues (Raha et al. 2000) found a 24%–28% identity to the native target structure. The results of applying our method to single, fixed, native backbones agree well with results such as these. When only the native fixed backbone is used for design (as described above), average identity to the native sequence of the target structure ranges from 1% to 40%, with a mean of 24% (Fig. 3 ▶). For buried positions, this value ranges from 0% to 75%, with an average of 43%. These distributions, both in mean and range, are strikingly similar to those produced by Kuhlman and Baker. When structural ensembles of 100 structural variants are used as design targets, the average identity of the resulting sequences to the native sequence drops to 17%, and the average pairwise identity of the sequences is 29%. The distributions of identity to the native sequence for both full sequences and core positions alone also narrow dramatically when structural ensembles are used. This suggests that the inclusion of backbone flexibility, even in the fairly simple manner used here, allows for the design of a much greater diversity of sequences compatible with the target structure.
Fig. 3.

Distribution of average amino acid identity of the designed sequences to the native target sequence for 253 structures. Identity to the native target sequence was calculated first for the set of sequences designed using only a single fixed target backbone as a target template (all residues: black dashed line; buried residues: gray dashed line). Using structural ensembles of 100 structural variants as target templates narrows and lowers the distribution of identity to the target native sequence (all residues: black solid line; buried residues: gray solid line).
How large a structural ensemble is needed to generate substantial sequence diversity by this method? In Figure 4 ▶, we see that an ensemble of just 30 structural variants results in a sequence set with “maximum” entropy. Adding more structural variants will not increase the diversity of the sequence set, as measured by sequence entropy. It is tempting to suggest that the sequence space for a structure has an entropy close to that which is reached by the method of designing to a structural ensemble. However, adding more structural variants does result in additional unique sequences, even though the entropy of the sequence set does not increase, showing that we have sampled only a fraction of sequence space. Ideally, this means that our sampling of sequence space, although sparse, is well-distributed throughout all the regions of that space. The maximum entropy, asymptotically approached as the structural ensemble grows, is noticeably distinct for each structure, varying evenly over a range from 3.2 to 6.8. There is a weak correlation between sequence entropy (i.e., mean residue entropy) and sequence length, but this disappears altogether once sequences are longer than 40 residues.
Fig. 4.

Sequence entropy increases with the size of the structural ensemble used for design. The traces represent the sequence entropy of the designed sequences obtained when using increasing numbers of structural variants as targets for design. The black traces represent the two structures that produced sequence sets with the highest and lowest average sequence entropy. The gray traces are for 100 different structures randomly picked from the remaining 251 proteins.
In any computational design study, it is important that a substantial fraction of the designed sequences are reasonable, that they would indeed take on a stable native structure closely resembling that of the target protein. The design algorithm used in this study has been shown to produce viable sequences in previous work (Desjarlais and Handel 1995; Raha et al. 2000). A number of recent papers have assessed the efficacy of computational design methods by using sequence profiles to compare the designed sequences to the native structure (Koehl and Levitt 1999b, 2002a; Raha et al. 2000; Kono and Saven 2001). Using a sequence profile generated from a set of designed sequences to scan a natural sequence database should be able to identify true homologues of the target structure. To assess the quality of our designed sequences, we performed such a test. PSI-BLAST(Altschul et al. 1997) searches against the PDB using our designed sequence sets as input profiles produced significant hits (E < 1) against the target structure and/or its structural homologues for 75% of the structures, with very few false positives (i.e., very few significant hits against nonhomologs). Figure 5 ▶ shows the distribution of PSI-BLAST E-values (a measure of sequence matching significance) for the 241 of 253 sequence profiles that produced any hits whatsoever against the PDB. At a significance level of E < 1.0, 74% (186 of 253) of the sequence profiles produced hits to putative structural homologues; 92% (172 of 186) of these are indeed hits to true structural homologs. In fact, half of the 241 profiles identify the native target structure itself as a significant match. True homologs are found even by sequence profiles with very low sequence identity to the target structure (10%–20%). As expected, the significance of hits generally increases slightly as the sequence profiles become more native-like (Fig. 5 ▶).
Fig. 5.

Results of PSI-BLAST searches against the Protein Data Bank using sequence profiles generated from the designed sequences. Two hundred forty-one of the 253 structures (those that gave hits) are represented here, ranked along the x-axis by the E-value of the most significant hit obtained from that structure’s designed sequence profile. Dark columns represent sequence profiles that gave hits against true structural homologues (true positives). Light columns identify sequence profiles that produced hits to nonhomologs (false positives). A threshold of E < 1.0 gives an accuracy of 92% (176 of 186) for 74% (186 of 253) of all sequence profile searches. The gray points plot the average amino acid identity of each sequence profile to the native target sequence.
Sequence diversity within folds
The 253 structures in our data set were clustered, based on structural similarity, using the VAST structure alignment algorithm (see Materials and Methods). These groups represent sets of structurally similar proteins, or folds. Our data set included six folds that contained at least 10 proteins each (Table 2). All structures in a fold are within 3 Å RMSD of each other. Figure 6 ▶ shows the distribution of sequence entropies of the designed sequence sets for the six folds. Although the average identity of designed sequences within a fold is quite low (26%–33%; Table 2), the entropies of the designed sequence sets for each structure within a fold cluster together quite tightly. The sequence entropy distributions for each fold peak around a mean (see Table 2), with relatively little variation around that mean. Interestingly, the residue entropies across corresponding positions in sequences within a fold do not show significant correlations (data not shown). In other words, the range of allowed amino acids at any one position seems to vary from structure to structure within a fold, but the fold itself tightly defines the overall diversity of the allowed sequence space.
Table 2.
Six folds and their corresponding PFAM families
| PFAM family name | Toxin | Copper-bind | Rubredoxin | Kunitz_BPTI | Antifreezea | Phage_DNA_binda | |
| Structures included in fold classification | 1ctx | 1ag6 | 1be7 | 1bpi | 1ame | 1ae3 | |
| 1fas | 1bxu | 1bq8 | 1bpt | 1gzi | 1gkh | ||
| 1fsc | 1bxv | 1bq9 | 1bti | 1msi | 1mho | ||
| 1ntn | 1byp | 1brf | 1dtx | 1ops | 1vqa | ||
| 1nxb | 1iuz | 1caa | 1fan | 2msi | 1vqc | ||
| 1qke | 1pnd | 1cad | 1knt | 3msi | 1vqd | ||
| 1qm7 | 2pcy | 1irn | 1nag | 4msi | 1vqe | ||
| 2ctx | 3pcy | 1iro | 2knt | 5msi | 1vqf | ||
| 2era | 4pcy | 1rb9 | 4pti | 6msi | 1vqg | ||
| 3ebx | 5pcy | 4rxn | 5pti | 7msi | 1vqh | ||
| 5ebx | 6pcy | 5rxn | 7pti | 1vqi | |||
| 7pcy | 7rxn | 8pti | 1vqj | ||||
| 8rxn | 9pti | 1yhb | |||||
| Natural sequences | Average % ID | 39% | 40% | 44% | 68% | 37% | 47% |
| Mean entropy | 3.2 | 3.5 | 3.3 | 1.6 | 4.0 | 1.9 | |
| Designed sequences | Average % ID | 30% | 27% | 33% | 26% | 30% | 27% |
| Mean entropy | 5.7 | 6.2 | 5.1 | 6.0 | 5.5 | 6.1 | |
The structures were grouped together according to structural similarity and are identified by the names assigned to their corresponding PFAM sequence families.
a These two PFAM alignments contain less than 30 sequences. Reliable conclusions about the “natural” sequence space of these folds cannot be drawn from such small samples.
Fig. 6.

Sequence entropy distributions of designed sequences, grouped by structure into folds. The six folds are identified by the names corresponding to their PFAM sequence families. The frequencies for each fold are normalized to unity. The sequence entropy distribution for all 253 structures is also shown.
It is tempting to suggest that there might be some structural characteristic, shared within a fold and different between folds, that determines sequence entropy. Definition of such a metric would not only allow some fundamental insight into the sequence–structure relationship, but could serve as an empirical tool for prediction of sequence/structure properties, akin to the utility of contact order in predicting rates of folding kinetics (Plaxco et al. 1998). Several such characteristics were tested: sequence length, alpha helix character, beta sheet character, and contact order (see Materials and Methods). None of these structural metrics showed significant correlations with sequence entropy (all regressions produced correlation coefficients below R2 = 0.4), suggesting that the structural determinants of a fold’s sequence space are somewhat more complex.
All six groups of structures, defined initially by structural similarity, corresponded to PFAM families of natural sequences (Bateman et al. 2000), which are defined solely by sequence similarity. The full alignment of natural sequences for each fold was obtained from PFAM. To reduce the inherent biases in natural sequence alignments, the alignments were reduced to 90% sequence redundancy, and were weighted according to the Henikoff algorithm (Henikoff and Henikoff 1994). These measures are critical in compensating for the artefactually low diversity of natural sequence alignments arising from the evolutionary relatedness of natural sequences (Larson et al. 2000). This weighting is unnecessary for designed sequences because each is completely independent of the others; the sampling of sequence space is not biased by an evolutionary constraints. Summary statistics for the designed and natural sequence sets for each of the six folds are tabulated in Table 2. In all cases, the designed sequence sets had greater overall sequence entropy than the natural sequence alignments. Surprisingly, there seems to be no correlation between the diversity (as measured by sequence entropy) of natural sequence alignments and the diversity of corresponding sets of designed sequences, perhaps stemming from the aforementioned sampling biases of natural sequence diversity.
Discussion
Defining sequence space
It is generally thought that the set of unique sequences that can stably fold into a specified three-dimensional protein structure must be enormous. Even just considering those protein sequences we have found in nature, many natural sequence alignments contain thousands of distinct sequences. In this work, we initially defined the sequence space of a structure as all those sequences that will fold to a low free-energy structure anywhere within 1.0 Å RMSD of the original protein backbone. Within this definition, we see that the sequence space for most structures is diverse and likely quite large. A very striking observation is that there is no general trend in overall sequence entropy for the structures studied. On the contrary, a broad range of sequence entropies is seen across the 253 structures, in agreement with another recent study using a different design algorithm (Koehl and Levitt 2002b). However, by looking at several proteins within the same fold, across several folds, we also see here that structures sharing an overall fold do tend to have quite similar sequence entropies.
The use of protein folds, as opposed to individual structures, as landmarks in sequence space facilitates meaningful comparisons between experimental or computational explorations of sequence space and those regions of sequence space known to be inhabited by natural protein sequences. As computational protein design has become more tractable, a number of recent studies have sought to compare sets of designed sequences to their natural counterparts, by looking at the identity of designed sequences to the native sequence of the target structure. Instead of comparing designed sequences to the native sequence alone, it is more meaningful to make comparisons against the natural sequence alignment of structural homologs (see, e.g., Koehl and Levitt 2002b). Natural sequence alignments are a reliable, albeit small, sample of sequence space, to which we can compare larger computationally predicted samples of the same sequence space. By broadening the boundaries of sequence space to encompass larger ensembles of similar structures, meaningful comparisons to natural sequences and structures can be made, while taking into account the known plasticity of proteins.
Backbone flexibility in protein design
Incorporating backbone flexibility is of general importance to computational protein design, and is certainly a prerequisite for de novo structure design, where the exact structure of the target is not known. Although computational protein design does not seek to directly simulate a physical process, it is highly desirable to build the realistic behavior of proteins (i.e., backbone relaxation to accommodate mutations) into design algorithms. Previous studies incorporating backbone flexibility, although quite successful, have been hindered by the increased computational complexity of annealing in conformation space on top of annealing in sequence space (Su and Mayo 1997; Harbury et al. 1998; Desjarlais and Handel 1999). By utilizing a distributed computing architecture, we have been able to incorporate fine-grained backbone flexibility in a large-scale protein design effort.
Designing to a structural ensemble is a fairly simple way of incorporating backbone flexibility, but we see that it allows for a much broader search of sequence space than fixed-backbone methods. Designing to a single, fixed backbone produces results very similar to other recently published studies. Designing to a structural ensemble, however, produces a much greater diversity of sequences, and allows movement away from the region of sequence space immediately surrounding the native sequence. Homology searches against natural sequence databases (a method used by a number of recent studies to confirm relevance of their designed sequences) show that the quality of these sequences is not diminished. In fact, the increased diversity of the sequence set improves the utility of designed sequence libraries in fold recognition for structural and functional genomics (S.M. Larson, A. Garg, J.R. Desjarlais, V.S. Pande, in prep.).
Designability
The concept of designability (Li et al. 1996, 1998; Helling et al. 2001) has been proposed as an explanation for the oft-noted observation that certain protein structures or folds are more commonly seen in nature than others (Chothia 1992; Orengo et al. 1994; Murzin et al. 1995; Brenner et al. 1997). Designability is defined simply as the number of sequences that can fold into a specific structure. Numerous theoretical studies have investigated this property through complete enumeration of sequences and structures of lattice (Buchler and Goldstein 1999, and references therein) and off-lattice models (Miller et al. 2002). In this study, we can estimate designability of real protein structures by comparing the sequence entropies of large sets of diverse designed sequences for different folds (see Fig. 6 ▶; Table 2). Recall that the entropies of designed sequences for structures within a fold cluster together tightly. The range of allowed amino acids at any one position varies from structure to structure within a fold, but the overall diversity of the allowed sequence space seems to be defined by the structural properties of the fold. The relatively tight clustering of sequence entropies within a fold and the separation of sequence entropy distributions for different folds suggests (a) that the diversity of the designed sequences for a structure is primarily determined by some structural characteristics of its overall fold, and (b) that the designability principle postulated from studies of simple models may hold in real proteins.
The results of this study are, of course, based on our particular model of the protein sequence–structure relationship, and it would be of great interest to see how the results of other theoretical and/or experimental protein design studies of a similar scale might compare. Most importantly, further theoretical and experimental work is needed to identify the specific structural characteristics that determine a fold’s sequence space.
Materials and methods
Genome@home distributed computing cluster
Assessing the diversity of sequence space requires the design of hundreds of thousands of protein sequences, an extremely demanding computational task. To allow for a study of this scope, a distributed computing project (Shirts and Pande 2000), dubbed `Genome@home' , was created (see http://genomeathome.stanford.edu). During the course of this study, the global cluster of available computers exceeded 3000 processors. The Genome@home server sends out “work units,” a set of protein backbone coordinates and design parameters, which are downloaded to the Genome@home client running on a user’s computer. The client verifies the work unit and runs the sequence prediction algorithm (Raha et al. 2000), summarized below. Work units of the size used in this study require a few hours to a day on a 500-MHz Intel Celeron processor. Upon completion of the sequence design, the results are verified by the client and sent back to the server, where the data is again verified, stored, and processed. At the time of this writing, the Genome@home global cluster produces over ten thousand new sequences daily.
Protein sequence design
Sequences were designed using SPA (Raha et al. 2000). Briefly, protein structures are created by modeling the placement of amino acid side-chain rotamers onto a fixed target backbone. Models are scored using a combination of the Amber potential function (Weiner et al. 1984) with OPLS nonbonded parameters (Jorgensen and Tirado-Rives 1988), a surface-area term that accounts implicitly for solvation effects (Eisenberg and McLachlan 1986), and a set of amino acid baseline corrections, which are critical for maintaining reasonable amino acid compositions. The models are optimized by a sequence selection process that involves initial filtering of rotamers, and a genetic algorithm for finding an optimal sequence for the target structure. A diversity of sequences can be designed for the same target backbone, as the initial population of 300 models is randomly assigned from a filtered rotamer library, analogous to starting in a random point of sequence space. Two hundred rounds of model building and evaluation, selective recombination, and a small amount of random mutagenesis are performed, and the entire cycle is repeated 30 times.
To create an ensemble of 100 target backbones for each structure, a Monte Carlo expansion and contraction algorithm was used to gently perturb the dihedral angles of the target backbone. The algorithm works by creating random perturbations of up to 5 degrees to the dihedral angles of the target structure, followed by simple Monte Carlo with smaller random perturbations until the target RMSD from the native structure is reached. In this study, the perturbation was constrained such that no two backbones in the ensemble differ by more than 1.0 Å RMSD. Studying an ensemble of such slightly varying structures is justified by the fact that structure determination techniques, NMR and X-ray crystallography, are generally accurate to about the 1.0-Å level. Each work unit of sequence design is done against a fixed backbone (i.e., one of the 100 variants of the target structure), and the designed sequences for all 100 variants are included in the resulting overall sequence set for the target structure.
Structure and sequence analyses
The set of protein structures used for this study consisted of all records in the Protein Data Bank (Berman et al. 2000) that contained only one chain, less than 100 amino acids long, solved by X-ray crystallography; a total of 292 structures. A sufficient amount of data was returned to complete the described analyses for 253 of these structures. The complete set of designed sequences for each structure can be obtained at http://gah.stanford.edu/cgi-bin/results/SqlCgi.pl.
Residue entropy was calculated according to the standard formulation:
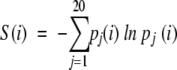 |
where pj(i) is the frequency of residue type j at position i in the alignment. To get a rough sense of how many amino acids appear at a specific position, we display the residue entropy in its exponentiated form, exp(S(i)), which ranges from 1 to 20. Sequence entropy is the mean residue entropy over all residues i from 1 through L in the alignment, where L is the length of the sequence.
Structures were grouped into folds using VAST (Madej et al. 1995); all 253 structures were clustered into their assigned structural groupings from MMDB (Wang et al. 2000). Natural sequence alignments corresponding to the VAST structural groupings were obtained from PFAM (Bateman et al. 2000). To reduce sequence bias and increase the relative diversity of the natural sequence sets, the alignments were reduced to 90% redundancy and weighted according to the Henikoff algorithm (Henikoff and Henikoff 1994). α-Helix and β-sheet character for each structure was defined as the fraction of residues assigned to the corresponding secondary structure by DSSP (Kabsch and Sander 1983). DSSP was also used to automate the identification of buried residues (i.e., less than 10% exposed side-chain surface area). Contact order was calculated as described by Plaxco and colleagues (1998).
Acknowledgments
This work would not have been possible without the enthusiastic participation of thousands of Genome@home users around the world. The authors are greatly indebted to everyone who contributed processor time to this study. A full list of users is available at http://gah.stanford.edu/userstats.txt. The authors thank members of the Pande group for insightful discussion throughout the course of this work. Special thanks to Amit Garg for his help in generating the data for Figure 6 ▶, and to Patrice Koehl, Chris Snow, and Bojan Zagrovic for critical reading of the manuscript. S.M.L. is a James Clark Fellow of the SGF program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RMSD, root-mean-square deviation
PDB, Protein Data Bank
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0203902.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, E.P., Hajiseyedjavadi, O., Baase, W.A., and Matthews, B.W. 1993. The role of backbone flexibility in the accommodation of variants that repack the core of T4 lysozyme. Science 262 1715–1718. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Durbin, R., Eddy, S.R., Howe, K.L., and Sonnhammer, E.L. 2000. The Pfam protein families database. Nucleic Acids Res. 28 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornscheuer, U.T. and Pohl, M. 2001. Improved biocatalysts by directed evolution and rational protein design. Curr. Opin. Chem. Biol. 5 137–143. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E., Chothia, C., and Hubbard, T.J. 1997. Population statistics of protein structures: Lessons from structural classifications. Curr. Opin. Struct. Biol. 7 369–376. [DOI] [PubMed] [Google Scholar]
- Buchler, N.E. and Goldstein, R.A. 1999. Effect of alphabet size and foldability requirements on protein structure designability. Proteins 34 113–124. [PubMed] [Google Scholar]
- Cedrone, F., Menez, A., and Quemeneur, E. 2000. Tailoring new enzyme functions by rational redesign. Curr. Opin. Struct. Biol. 10 405–410. [DOI] [PubMed] [Google Scholar]
- Chothia, C. 1992. Proteins. One thousand families for the molecular biologist. Nature 357 543–544. [DOI] [PubMed] [Google Scholar]
- Dahiyat, B.I. 1999. In silico design for protein stabilization. Curr. Opin. Biotechnol. 10 387–390. [DOI] [PubMed] [Google Scholar]
- Desjarlais, J.R. and Clarke, N.D. 1998. Computer search algorithms in protein modification and design. Curr. Opin. Struct. Biol. 8 471–475. [DOI] [PubMed] [Google Scholar]
- Desjarlais, J.R. and Handel, T.M. 1995. De novo design of the hydrophobic cores of proteins. Protein Sci. 4 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1999. Side-chain and backbone flexibility in protein core design. J. Mol. Biol. 290 305–318. [DOI] [PubMed] [Google Scholar]
- Eisenberg, D. and McLachlan, A.D. 1986. Solvation energy in protein folding and binding. Nature 319 199–203. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., Zhang, X.J., Heinz, D.W., Blaber, M., Baldwin, E.P., and Matthews, B.W. 1992. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255 178–183. [DOI] [PubMed] [Google Scholar]
- Harbury, P.B., Plecs, J.J., Tidor, B., Alber, T., and Kim, P.S. 1998. High-resolution protein design with backbone freedom. Science 282 1462–1467. [DOI] [PubMed] [Google Scholar]
- Helling, R., Li, H., Melin, R., Miller, J., Wingreen, N., Zeng, C., and Tang, C. 2001. The designability of protein structures. J. Mol. Graph. Model 19 157–167. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1994. Protein family classification based on searching a database of blocks. Genomics 19 97–107. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L. and Tirado-Rives, J. 1988. The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110 1657–1666. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kazlauskas, R.J. 2000. Molecular modeling and biocatalysis: Explanations, predictions, limitations, and opportunities. Curr. Opin. Chem. Biol. 4 81–88. [DOI] [PubMed] [Google Scholar]
- Koehl, P. and Delarue, M. 1994. Application of a self-consistent mean field theory to predict protein side-chains conformation and estimate their conformational entropy. J. Mol. Biol. 239 249–275. [DOI] [PubMed] [Google Scholar]
- Koehl, P. and Levitt, M. 1999a. De novo protein design. I. In search of stability and specificity. J. Mol. Biol. 293 1161–1181. [DOI] [PubMed] [Google Scholar]
- ———. 1999b. De novo protein design. II. Plasticity in sequence space. J. Mol. Biol. 293 1183–1193. [DOI] [PubMed] [Google Scholar]
- ———. 2002a. Improved recognition of native-like protein structures using a family of designed sequences. Proc. Natl. Acad. Sci. 99 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2002b. Protein topology and stability define the space of allowed sequences. Proc. Natl. Acad. Sci. 99 1280–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono, H. and Saven, J.G. 2001. Statistical theory for protein combinatorial libraries. Packing interactions, backbone flexibility, and the sequence variability of a main-chain structure. J. Mol. Biol. 306 607–628. [DOI] [PubMed] [Google Scholar]
- Kuhlman, B. and Baker, D. 2000. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. 97 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, S.M., Di Nardo, A.A., and Davidson, A.R. 2000. Analysis of covariation in an SH3 domain sequence alignment: Applications in tertiary contact prediction and the design of compensating hydrophobic core substitutions. J. Mol. Biol. 303 433–446. [DOI] [PubMed] [Google Scholar]
- Lee, C. 1994. Predicting protein mutant energetics by self-consistent ensemble optimization. J. Mol. Biol. 236 918–939. [DOI] [PubMed] [Google Scholar]
- Li, H., Helling, R., Tang, C., and Wingreen, N. 1996. Emergence of preferred structures in a simple model of protein folding. Science 273 666–669. [DOI] [PubMed] [Google Scholar]
- Li, H., Tang, C., and Wingreen, N.S. 1998. Are protein folds atypical? Proc. Natl. Acad. Sci. 95 4987–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej, T., Gibrat, J.F., and Bryant, S.H. 1995. Threading a database of protein cores. Proteins 23 356–369. [DOI] [PubMed] [Google Scholar]
- Malakauskas, S.M. and Mayo, S.L. 1998. Design, structure and stability of a hyperthermophilic protein variant. Nat. Struct. Biol. 5 470–475. [DOI] [PubMed] [Google Scholar]
- Miller, J., Zeng, C., Wingreen, N.S., and Tang, C. 2002. Emergence of highly designable protein–backbone conformations in an off-lattice model. Proteins. 47 506–512. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Musacchio, A., Saraste, M., and Wilmanns, M. 1994. High-resolution crystal structures of tyrosine kinase SH3 domains complexed with proline-rich peptides. Nat. Struct. Biol. 1 546–551. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Jones, D.T., and Thornton, J.M. 1994. Protein superfamilies and domain superfolds. Nature 372 631–634. [DOI] [PubMed] [Google Scholar]
- Pabo, C. 1983. Molecular technology. Designing proteins and peptides. Nature 301 200. [DOI] [PubMed] [Google Scholar]
- Pande, V.S., Grosberg, A.Y., and Tanaka, T. 1997. Statistical mechanics of simple models of protein folding and design. Biophys J. 73 3192–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., and Baker, D. 1998. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277 985–994. [DOI] [PubMed] [Google Scholar]
- Pokala, N. and Handel, T.M. 2001. Review: Protein design—Where we were, where we are, where we're going. J. Struct. Biol. 134 269–281. [DOI] [PubMed] [Google Scholar]
- Raha, K., Wollacott, A.M., Italia, M.J., and Desjarlais, J.R. 2000. Prediction of amino acid sequence from structure. Protein Sci. 9 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhnovich, E.I. 1998. Protein design: A perspective from simple tractable models. Fold. Des. 3 R45–R58. [PubMed] [Google Scholar]
- Shenkin, P.S., Erman, B., and Mastrandrea, L.D. 1991. Information-theoretical entropy as a measure of sequence variability. Proteins 11 297–313. [DOI] [PubMed] [Google Scholar]
- Shirts, M. and Pande, V.S. 2000. COMPUTING: Screen savers of the world unite! Science 290 1903–1904. [DOI] [PubMed] [Google Scholar]
- Su, A. and Mayo, S.L. 1997. Coupling backbone flexibility and amino acid sequence selection in protein design. Protein Sci. 6 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin, M.B., Gustafsson, C., and Huisman, G.W. 2000. Directed evolution: The "rational" basis for "irrational" design. Curr. Opin. Struct. Biol. 10 421–427. [DOI] [PubMed] [Google Scholar]
- Voigt, C.A., Gordon, D.B., and Mayo, S.L. 2000. Trading accuracy for speed: A quantitative comparison of search algorithms in protein sequence design. J. Mol. Biol. 299 789–803. [DOI] [PubMed] [Google Scholar]
- Voigt, C.A., Mayo, S.L., Arnold, F.H., and Wang, Z.G. 2001. Computational method to reduce the search space for directed protein evolution. Proc. Natl. Acad. Sci. 98 3778–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Addess, K.J., Geer, L., Madej, T., Marchler-Bauer, A., Zimmerman, D., and Bryant, S.H. 2000. MMDB: 3D structure data in Entrez. Nucleic Acids Res. 28 243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, S.J., Kollman, P.A., Case, D.A., Singh, U.C., Ghio, C., Alagona, G., Profeta, S., and Weiner, P. 1984. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 106 765–784. [Google Scholar]
- Wernisch, L., Hery S., and Wodak, S.J. 2000. Automatic protein design with all atom force-fields by exact and heuristic optimization. J. Mol. Biol. 301 713–736. [DOI] [PubMed] [Google Scholar]
- Zou, J. and Saven, J.G. 2000. Statistical theory of combinatorial libraries of folding proteins: Energetic discrimination of a target structure. J. Mol. Biol. 296 281–294. [DOI] [PubMed] [Google Scholar]