

Human flap endonuclease 1 complexed with nicked DNA has been crystallized. A diffraction data set was collected to a resolution of 2.75 Å.
Keywords: DNA–protein complexes, DNA repair, DNA replication, nucleases
Abstract
Flap endonuclease 1 (FEN1) is a structure-specific nuclease that removes the RNA/DNA primer associated with Okazaki fragments in DNA replication. Here, crystals of the complex between the catalytic domain of human FEN1 and a DNA product have been obtained. For efficient crystallization screening, a DNA–protein complex crystallization screening (DPCS) kit was designed based on commercial crystallization kits. The crystal was found to belong to space group P21, with unit-cell parameters a = 61.0, b = 101.3, c = 106.4 Å, β = 106.4°. The asymmetric unit is predicted to contain two complexes in the crystallographic asymmetric unit. A diffraction data set was collected to a resolution of 2.75 Å.
1. Introduction
In lagging-strand DNA synthesis, DNA polymerase δ (pol δ) displaces the 5′-terminal RNA/DNA primers associated with Okazaki fragments into single-stranded flaps (Harrington & Lieber, 1994 ▶). Similarly, during long-patch base-excision repair, DNA fragments containing damaged nucleotides are also displaced into a flap (Murante et al., 1995 ▶). These 5′-flap DNA strands are removed by flap endonuclease 1 (FEN1). This nuclease activity is specific to 5′-flaps but is independent of flap-strand length (Harrington & Lieber, 1994 ▶) and sequence (Kaiser et al., 1999 ▶). FEN1 is not capable of cleaving 3′-flap strands (Harrington & Lieber, 1994 ▶). The cleavage provides nicked substrates for ligation by DNA ligase I. In mice, knockout of FEN1 genes leads to embryonic lethality (Kucherlapati et al., 2002 ▶): blastocysts fail to enter the S phase to carry out normal DNA synthesis and are arrested in the endocycle (Larsen et al., 2003 ▶). Thus, FEN1 is a structure-specific 5′-endonuclease that plays an essential role in maintaining genome stability (Liu et al., 2004 ▶).
FEN1 belongs to the FEN1/XPG family of nucleases (Lieber, 1997 ▶). The crystal structures of FEN1/XPG-family nucleases from eukaryotes (Sakurai et al., 2005 ▶), archaea (Chapados et al., 2004 ▶; Hosfield et al., 1998 ▶; Hwang et al., 1998 ▶), prokaryotes (Kim et al., 1995 ▶) and bacteriophages (Ceska et al., 1996 ▶; Mueser et al., 1996 ▶; Devos et al., 2007 ▶) have been reported. All of these nucleases show cleavage preference for a double-flap DNA substrate containing 5′-flap nucleotides with a single-nucleotide 3′-flap (Fig. 1 ▶), suggesting that their catalytic mechanism is conserved (Kaiser et al., 1999 ▶; Kao et al., 2002 ▶; Harrington & Lieber, 1995 ▶; Storici et al., 2002 ▶). Double-flap DNA structures are proposed to be created in vivo by a transient flap-migration equilibration following strand displacement by pol δ (Kao et al., 2002 ▶; Reynaldo et al., 2000 ▶; Fig. 1 ▶). Of the flap isomers produced in the equilibrium, FEN1 recognizes the double-flap structure that contains 5′-flap nucleotides with a one-nucleotide 3′-flap (Harrington & Lieber, 1995 ▶). Double-flap DNA is cleaved accurately one site into the downstream region by FEN1 and this cleaved product is a ligatable nick (Kao et al., 2002 ▶). The requirement of double-flap structures for maximum FEN1 activity was discovered from in vitro FEN1 activity measurements using artificial double-flap substrates containing 5′-flap and 3′-flap strands that are noncomplementary to the template strand in order to prevent branch-migration equilibration (Harrington & Lieber, 1995 ▶; Xie et al., 2001 ▶; Kao et al., 2002 ▶; Kaiser et al., 1999 ▶; Storici et al., 2002 ▶). The products of these double-flap substrates remain one-nucleotide 3′-flaps.
Figure 1.

Flap migration and double-flap generation. The preferred substrate of FEN1/XPG-family nucleases is a double-flap DNA. Because single-stranded flap DNA is complementary to the template region, the 3′-end of the upstream strand and the 5′-end of the downstream strand form various flap structures following displacement synthesis by pol δ. During the transient flap-migration equilibration, FEN1 specifically recognizes the double-flap structure containing 5′-flap nucleotides with a one-nucleotide 3′-flap. A displaced 5′-flap DNA (red) was produced by DNA polymerase from the upstream strand. The flap is capable of migrating back and forth and forms various flap structures based on sequence complementarity between the template (black) and the upstream/downstream (blue) strands. FEN1 removes the 5′-flap by cleaving the double-flap substrate with one-nucleotide 3′-flap after the first base pair preceding the 5′-flap DNA to generate a nicked DNA product for the next DNA-ligase reaction.
FEN1 activity is enhanced by direct binding to proliferation cell nuclear antigen (PCNA), which acts as a DNA sliding clamp (Li et al., 1995 ▶; Wu et al., 1996 ▶; Tom et al., 2000 ▶). The structure of human FEN1 bound to PCNA has recently been solved and the conformational switch of FEN1 proposed, highlighting changes in the orientation and position of the FEN1 core domain with respect to the PCNA ring from an inactive OFF state to an active ON state (Sakurai et al., 2005 ▶).
Despite the accumulated three-dimensional structures, the catalytic mechanism by which FEN1 produces a precise nick remains unknown. We have undertaken a crystallographic study of human FEN1 in complex with flap DNA in an effort to elucidate the structural basis of the mechanism pertaining to FEN1 structure-specific activity. We have crystallized the catalytic core domain of human FEN1 in complex with nicked DNA, a product of the double-flap DNA. Elucidation of the three-dimensional structure of the FEN1–DNA complex is expected to advance our understanding of the structure-specific recognition and catalytic action of this biologically and medically important enzyme.
2. Materials and methods
2.1. Construction and expression of human FEN1
Human FEN1 (residues 1–380) consists of a catalytic core domain (1–336) and a C-terminal tail (337–380) containing the PCNA-interacting peptide (PIP) box (337–344) followed by a basic region. In the PCNA-free state, the C-terminal tail should be flexible and may prevent crystallization. Therefore, we prepared C-terminal tail-truncated FEN1 as well as the full-length protein for crystallization. The construct for the catalytic domain of human FEN1 (1–336) was amplified from the plasmid pT7-hFEN-1 (Sakurai et al., 2003 ▶) using the polymerase chain reaction (PCR) and then subcloned into the NdeI/BamHI site of the pET22b vector (Novagen), which contains a C-terminal His6 tag after the thrombin cleavage site. To suppress the nuclease activity of FEN1, mutations of two catalytic residues (Shen et al., 1996 ▶), D86A and D181A, were introduced into both full-length and C-terminal truncated FEN1 using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The subcloning and mutagenesis were confirmed by DNA sequencing.
The expression and purification of full-length FEN1 were performed as described previously (Sakurai et al., 2003 ▶). The catalytic core domain of FEN1 was expressed in Escherichia coli strain BL21-CodonPlus (DE3)-RIL (Stratagene) cells cultured in LB media containing 100 µg ml−1 ampicillin and 50 µg ml−1 chloramphenicol. Cells were grown at 310 K to an OD660nm of ∼0.8 and protein expression was induced following the addition of 0.5 mM IPTG. After 5 h, cells were harvested by centrifugation (6000g, 15 min at 277 K) and pellets were frozen at 193 K. Cell pellets were resuspended in lysis buffer (50 mM K HEPES pH 7.5, 100 mM KCl, 5 mM MgCl2 and 2 mM DTT) and lysed by sonication. The lysate was then ultracentrifuged (140 000g, 1 h at 277 K) and the resultant supernatant was loaded onto a heparin Sepharose column (Amersham Biosciences) equilibrated with lysis buffer and then eluted with a KCl gradient ranging from 0.1 to 0.6 M. Fractions containing FEN1 were pooled and then loaded onto Ni2+ Sepharose (Amersham Biosciences) equilibrated with lysis buffer, washed with lysis buffer containing 20 mM imidazole and finally eluted with an imidazole gradient ranging from 20 to 300 mM. Following removal of the His tag by thrombin digestion, FEN1s were further purified by passage through Superdex 75 HR 26/60 (Amersham Biosciences). N-terminal sequence analysis (M492, Applied Biosystems), matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI–TOF MS; PerSpective) and polyacrylamide gel electrophoresis in the presence of 0.1%(w/v) sodium dodecyl sulfate (SDS–PAGE) were performed to validate the purity of the resulting samples.
2.2. Preparation of DNA oligonucleotides
For DNA–protein cocrystallization, utilization of a DNA fragment with precise length suitable for crystal packing is critical since the DNA mostly packs end-to-end and can form a pseudo-continuous helix (Joachimiak & Sigler, 1991 ▶). We designed three different-length double-flap DNA substrates (Fig. 2 ▶) by model-building studies based on previous structural and biochemical studies concerning FEN1–DNA interactions (Allawi et al., 2003 ▶; Chapados et al., 2004 ▶; Sakurai et al., 2005 ▶). These studies suggested that 12 base pairs of the downstream duplex and 6–10 base pairs of the upstream duplex could be fitted to the FEN1 catalytic domain. The 3-nucleotide 5′-flap was also chosen based on the model-building studies: 5′-flap strands longer than three nucleotides seemed to stick out from the catalytic domain. The protruding longer flap strands may be flexible and prevent crystallization. The sequences of the double-flap DNA were designed based on the single-flap DNA previously studied (Harrington & Lieber, 1994 ▶) and a cytosine was used as the one base of the 3′-flap in this crystallization study.
Figure 2.

Flap DNA sequences used for cocrystallization. The cleaved site in the crystal of the FEN1–DNA complex is shown with an arrow.
DNA oligonucleotides were purchased from Hokkaido System Science Co. Ltd. After each oligonucleotide had been dissolved in annealing buffer (10 mM K HEPES, 100 mM KCl and 0.5 mM EDTA), three equimolar oligonucleotides, representing an upstream primer containing a 3′ single flap, a template strand and a downstream primer containing a 5′-flap, were mixed to a final concentration of 1 mM. The oligonucleotide mixture was heated to 343 K and then cooled gradually to 277 K.
2.3. Preparation of the DNA–protein complexes
To survey the optimal combination of FEN1 construct and DNA length for crystallization of the FEN1–DNA complex, all combinations of the wild-type and two mutant (D86A and D181A) FEN1 proteins of the two constructs, full-length and the C-terminally truncated as described above, were tested in our initial crystallization screening with three different-length double-flap DNA substrates (Fig. 2 ▶). The FEN1–DNA complexes were formed by mixing 300 µM FEN1 and 320 µM double-flap DNA and dialyzed in equilibration buffer (10 mM HEPES pH 7.5, 50 mM KCl, 5 mM MgCl2 and 1 mM DTT) for 12 h at 277 K. The final concentration of the FEN1–DNA complexes was 0.15 mM.
2.4. DNA–protein complex crystallization screening kit
Polyethylene glycols (PEGs) represent the most successful precipitants for protein–DNA cocrystallization (Joachimiak & Sigler, 1991 ▶). Thus, we designed a DNA–protein complex crystallization screening (DPCS) kit for the efficient crystallization screening of the FEN1–DNA complex. Solutions containing PEGs as precipitants were obtained from commercial crystallization screening kits: Crystal Screen I, Crystal Screen II, Crystal Screen Lite, Index, Natrix, Grid Screen PEG (Hampton Research), JBScreen 1–6 (Jena Bioscience), Wizard I, II and III (Emerald BioStructures) and PEGs (Nextal Biotechnologies). Furthermore, Grid Screen MPD (Hampton Research) was added to the above collection since DNA–protein complexes have occasionally been crystallized using MPD as a precipitant. Solutions that contained similar or overlapping compositions were then excluded from the collection. Solutions containing acidic buffers (pH < 5) or divalent ions (zinc, copper and cadmium) were also excluded since FEN1 tends to denature or aggregate under these conditions. Altogether, 338 solutions were selected for the DPCS kit (see supplementary material1).
2.5. Crystallization
Initial crystallization conditions were screened by the sitting-drop vapour-diffusion method using a Hydra II-Plus-One crystallization robot (Matrix Technology) with our DPCS kit. Each droplet, prepared by mixing 0.2 µl of the FEN1–DNA complex solution with an equal volume of each reservoir solution, was equilibrated against 100 µl reservoir solution at 277 K using 96-well crystallization plates (Hampton Research). The drops were observed 2 d, one week and one month after setting up the crystallization plates. Conditions yielding crystals were further optimized by varying the PEG concentration and the pH. For the optimization, 2 µl FEN1–DNA complex solution was mixed with an equal volume of the reservoir solution and then equilibrated against 200 µl reservoir solution. Crystals were mounted in a nylon loop (Hampton Research) and then flash-cooled in liquid nitrogen. Initial diffraction tests were performed using a home-source X-ray generator (Rigaku FR-E) equipped with a Rigaku R-AXIS IV detector at 100 K. The presence of both protein and DNA in the crystals was confirmed by MALDI–TOF MS and SDS–PAGE. The crystals were washed with the precipitant solution and dissolved in equilibration buffer prior to these analyses.
2.6. Data collection
Diffraction data sets were collected using the BL41XU beamline at SPring-8 (Hyogo, Japan). For data collection, the crystals were cryoprotected by soaking in mother liquor with 16%(v/v) glycerol and then flash-cooled in liquid nitrogen. A complete data set was collected to 2.75 Å, integrated and scaled using DENZO and SCALEPACK as implemented in HKL-2000 (Otwinowski & Minor, 1997 ▶). Calculation of the self-rotation function was carried out using the program POLARRFN (Collaborative Computational Project, Number 4, 1994 ▶) with structure-factor amplitudes to a maximum resolution of 3.0 Å.
3. Results and discussion
In the initial crystallization screening, crystals of the FEN1–DNA complex were obtained under many conditions using the 18 bp double-flap DNA substrate, whereas no crystals were obtained using the 20 and 22 bp flap DNA substrates (Table 1 ▶). On the other hand, only three conditions yielded crystals using the catalytic core domains of wild-type and D181A FEN1 without DNA. These results indicate that our DPCS kit is effective for DNA–protein cocrystallization, especially in light of the comprehensive survey using various DNA lengths and protein constructs. Although most crystals diffracted poorly (to less than 7 Å resolution), a well diffracting crystal (∼3 Å resolution) was found under crystallization conditions using the 18 bp double-flap DNA substrate and the catalytic domain of D181A FEN1. Following optimization of the crystallization conditions, crystals grew within one week to dimensions of approximately 0.05 × 0.1 × 0.2 mm by mixing 2 µl FEN1–DNA complex solution with an equal volume of reservoir solution (10% PEG 6K, 50 mM KCl, 5 mM MgCl2 and 1 mM DTT) and equilibrating against 200 µl reservoir solution (Fig. 3 ▶ a). These crystals contained both FEN1 and DNA, as determined from SDS–PAGE (12.5%) and native PAGE (15%) gel analyses (Figs. 3 ▶ b and 3 ▶ c). Since the DNA band from the dissolved crystal solution was shifted slightly from that of the original DNA sample, we doubted that the double-flap DNA substrate had been digested by the enzyme. Using MALDI–TOF MS, we found a peak of 3500.1 for the digested 11-mer downstream primer DNA (calculated mass 3500.3), but no peak for the intact 15-mer downstream primer DNA (calculated mass 4632.1). We also observed peaks of 2041.1 for the 7-mer upstream primer (calculated mass 2041.4) and 5462.3 for the 18-mer template strand (calculated mass 5459.6) as expected, but no peak for the 4-mer fragment. It is probable that the small cleaved fragment has been lost from the crystals. Thus, we concluded that the 15-mer downstream primer was cleaved in the crystal during crystallization at the cleavage site shown in Fig. 2 ▶.
Table 1. Number of conditions yielding FEN1–DNA complex crystals using the six FEN1 constructs in combination with three double-flap DNA substrates.
The number of conditions yielding large-size crystals (>50 µm) is shown in parentheses. ND, no data.
| Full-length (1–380) | Catalytic domain (1–336) | |||||
|---|---|---|---|---|---|---|
| Wild type | D86A | D181A | Wild type | D86A | D181A | |
| No DNA | 0 (0) | ND | 0 (0) | 3 (3) | ND | 3 (1) |
| 18 bp DNA | 0 (0) | 14 (1) | 136 (61) | 2 (0) | 36 (9) | 76 (18) |
| 20 bp DNA | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| 22 bp DNA | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Figure 3.

Crystal of the obtained FEN1–DNA complex and PAGE analyses. (a) Crystal of the catalytic domain of D181A FEN1 in complex with nicked DNA. (b) A 12.5% SDS–PAGE gel stained with Coomassie Brilliant Blue; lane 1, molecular-weight markers (kDa); lane 2, purified catalytic domain of D181A FEN1; lane 3, dissolved crystal. (c) A 15% native PAGE gel stained with SYBER Green I (Molecular Probes); lane 1, base-pair markers; lane 2, 18 bp double-flap DNA; lane 3, dissolved crystal.
The crystal of the FEN1–DNA complex belongs to space group P21, with unit-cell parameters a = 61.0, b = 101.3, c = 106.4 Å, β = 106.4°. The data-collection statistics are summarized in Table 2 ▶. The crystal of the FEN1–DNA complex diffracted X-rays to beyond 3 Å resolution, although the diffraction was highly anisotropic (2.6 × 3.2 Å resolution). The low value of the completeness in the highest resolution shell is a consequence of this anisotropy. Assuming the presence of two FEN1–DNA complexes in the asymmetric unit, the calculated value of the Matthews coefficient is 3.3 Å3 Da−1, corresponding to a solvent content of 62%. The self-rotation function calculated in polar coordinates with the rotation angle (κ) set to 180° is shown in Fig. 4 ▶. These results indicate that two FEN1–DNA complexes are present in the asymmetric unit and are related by a twofold noncrystallographic axis. Initial structure determination of the FEN1–DNA complex via molecular replacement is currently being undertaken using the catalytic domain of the human FEN1 structure (PDB code 1ul1) as a search model.
Table 2. Data-collection and processing statistics for the FEN1–DNA complex.
Values in parentheses are for the outer resolution shell.
| Beamline | SPring-8/BL41XU |
| Detector | ADSC Quantum 315 |
| Wavelength (Å) | 1.00 |
| Temperature (K) | 100 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 61.0, b = 101.3, c = 106.4, β = 106.4 |
| Resolution (Å) | 50.0–2.75 (2.85–2.75) |
| No. of reflections (total/unique) | 117875/29915 |
| Completeness (%) | 93.0 (70.2) |
| Mosaicity | 0.3–0.5 |
| 〈I/σ(I)〉 | 10.1 (3.5) |
| Rmerge† (%) | 7.9 (37.0) |
R
merge = 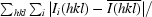
 , where Ii(hkl) is the intensity of the ith measurement of reflection h and
, where Ii(hkl) is the intensity of the ith measurement of reflection h and 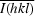 is the mean value of Ii(hkl) for all i measurements.
is the mean value of Ii(hkl) for all i measurements.
Figure 4.

Self-rotation function for κ = 180° section calculated using POLARRFN in the resolution range 20–3.0 Å; the integration radius was 20 Å.
Supplementary Material
Supplementary material file. DOI: 10.1107/S1744309107065372/fw5149sup1.pdf
Acknowledgments
We would like to thank J. Tsukamoto for technical support in performing the MALDI–TOF MS analysis. This work was supported in part by a Protein 3000 project on Signal Transduction from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (to TH). The work was also supported by the CREST project from Japan Science and Technology Corporation to TH. We also acknowledge Drs N. Shimizu, M. Kawamoto and M. Yamamoto at SPring-8 for help during data collection at the synchrotron beamline BL41XU.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: FW5149).
References
- Allawi, H. T., Kaiser, M. W., Onufriev, A. V., Ma, W. P., Brogaard, A. E., Case, D. A., Neri, B. P. & Lyamichev, V. I. (2003). J. Mol. Biol.328, 537–554. [DOI] [PubMed]
- Ceska, T. A., Sayers, J. R., Stier, G. & Suck, D. (1996). Nature (London), 382, 90–93. [DOI] [PubMed]
- Chapados, B. R., Hosfield, D. J., Han, S., Qiu, J., Yelent, B., Shen, B. & Tainer, J. A. (2004). Cell, 116, 39–50. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Devos, J. M., Tomanicek, S. J., Jones, C. E., Nossal, N. G. & Mueser, T. C. (2007). J. Biol. Chem.282, 31713–31724. [DOI] [PubMed]
- Harrington, J. J. & Lieber, M. R. (1994). EMBO J.13, 1235–1246. [DOI] [PMC free article] [PubMed]
- Harrington, J. J. & Lieber, M. R. (1995). J. Biol. Chem.270, 4503–4508. [DOI] [PubMed]
- Hosfield, D. J., Mol, C. D., Shen, B. & Tainer, J. A. (1998). Cell, 95, 135–146. [DOI] [PubMed]
- Hwang, K. Y., Baek, K., Kim, H. Y. & Cho, Y. (1998). Nature Struct. Biol.5, 707–713. [DOI] [PubMed]
- Joachimiak, A. & Sigler, P. B. (1991). Methods Enzymol.208, 82–99. [DOI] [PubMed]
- Kaiser, M. W., Lyamicheva, N., Ma, W., Miller, C., Neri, B., Fors, L. & Lyamichev, V. I. (1999). J. Biol. Chem.274, 21387–21394. [DOI] [PubMed]
- Kao, H. I., Henricksen, L. A., Liu, Y. & Bambara, R. A. (2002). J. Biol. Chem.277, 14379–14389. [DOI] [PubMed]
- Kim, Y., Eom, S. H., Wang, J., Lee, D. S., Suh, S. W. & Steitz, T. A. (1995). Nature (London), 376, 612–616. [DOI] [PubMed]
- Kucherlapati, M., Yang, K., Kuraguchi, M., Zhao, J., Lia, M., Heyer, J., Kane, M. F., Fan, K., Russell, R., Brown, A. M., Kneitz, B., Edelmann, W., Kolodner, R. D., Lipkin, M. & Kucherlapati, R. (2002). Proc. Natl Acad. Sci. USA, 99, 9924–9929. [DOI] [PMC free article] [PubMed]
- Larsen, E., Gran, C., Saether, B. E., Seeberg, E. & Klungland, A. (2003). Mol. Cell. Biol.23, 5346–5353. [DOI] [PMC free article] [PubMed]
- Li, X., Li, J., Harrington, J., Lieber, M. R. & Burgers, P. M. (1995). J. Biol. Chem.270, 22109–22112. [DOI] [PubMed]
- Lieber, M. R. (1997). Bioessays, 19, 233–240. [DOI] [PubMed]
- Liu, Y., Kao, H. I. & Bambara, R. A. (2004). Annu. Rev. Biochem.73, 589–615. [DOI] [PubMed]
- Mueser, T. C., Nossal, N. G. & Hyde, C. C. (1996). Cell, 85, 1101–1112. [DOI] [PubMed]
- Murante, R. S., Rust, L. & Bambara, R. A. (1995). J. Biol. Chem.270, 30377–30383. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Reynaldo, L. P., Vologodskii, A. V., Neri, B. P. & Lyamichev, V. I. (2000). J. Mol. Biol.297, 511–520. [DOI] [PubMed]
- Sakurai, S., Kitano, K., Okada, K., Hamada, K., Morioka, H. & Hakoshima, T. (2003). Acta Cryst. D59, 933–935. [DOI] [PubMed]
- Sakurai, S., Kitano, K., Yamaguchi, H., Hamada, K., Okada, K., Fukuda, K., Uchida, M., Ohtsuka, E., Morioka, H. & Hakoshima, T. (2005). EMBO J.24, 683–693. [DOI] [PMC free article] [PubMed]
- Shen, B., Nolan, J. P., Sklar, L. A. & Park, M. S. (1996). J. Biol. Chem.271, 9173–9176. [DOI] [PubMed]
- Storici, F., Henneke, G., Ferrari, E., Gordenin, D. A., Hübscher, U. & Resnick, M. A. (2002). EMBO J.21, 5930–5942. [DOI] [PMC free article] [PubMed]
- Tom, S., Henricksen, L. A. & Bambara, R. A. (2000). J. Biol. Chem.275, 10498–10505. [DOI] [PubMed]
- Wu, X., Li, J., Li, X., Hsieh, C. L., Burgers, P. M. & Lieber, M. R. (1996). Nucleic Acids Res.24, 2036–2043. [DOI] [PMC free article] [PubMed]
- Xie, Y., Liu, Y., Argueso, J. L., Henricksen, L. A., Kao, H. I., Bambara, R. A. & Alani, E. (2001). Mol. Cell. Biol.21, 4889–4899. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material file. DOI: 10.1107/S1744309107065372/fw5149sup1.pdf