

Abstract
Serum retinol binding protein (RBP) is a member of the lipocalin family, proteins with up-and-down β-barrel folds, low levels of sequence identity, and diverse functions. Although tryptophan 24 of RBP is highly conserved among lipocalins, it does not play a direct role in activity. To determine if Trp24 and other conserved residues have roles in stability and/or folding, we investigated the effects of conservative substitutions for the four tryptophans and some adjacent residues on the structure, stability, and spectroscopic properties of apo-RBP. Crystal structures of recombinant human apo-RBP and of a mutant with substitutions for tryptophans 67 and 91 at 1.7 Å and 2.0 Å resolution, respectively, as well as stability measurements, indicate that these relatively exposed tryptophans have little influence on structure or stability. Although Trp105 is largely buried in the wall of the β-barrel, it can be replaced with minor effects on stability to thermal and chemical unfolding. In contrast, substitutions of three different amino acids for Trp24 or replacement of Arg139, a conserved residue that interacts with Trp24, lead to similar large losses in stability and lower yields of native protein generated by in vitro folding. The results and the coordinated nature of natural substitutions at these sites support the idea that conserved residues in functionally divergent homologs have roles in stabilizing the native relative to misfolded structures. They also establish conditions for studies of the kinetics of folding and unfolding by ideying spectroscopic signals for monitoring the formation of different substructures.
Keywords: Protein stability; mutagenesis; evolution; conserved residues; lipocalins; protein folding, serum retinol-binding protein; crystal structures
Serum retinol binding protein (RBP) is a monomeric protein of molecular weight 21,000 that transports vitamin A in the circulation; most circulating RBP is in the form of a holoprotein complexed with transthyretin (Fex et al. 1979; Monaco et al. 1995; Naylor and Newcomer 1999). RBP is a member of the extensive lipocalin superfamily that includes >35 distinct proteins and domains distributed in bacteria, animals, molds, and plants (Pervaiz and Brew 1985Pervaiz and Brew 1987; Flower et al. 1993; Bishop and Weiner 1996; Toh et al. 1996; Ganfornina et al. 2000). The main feature of crystallographic structures of holo and apo human and bovine RBPs (Zanotti et al. 1993c; Cowan et al. 1990) is an eight-stranded up-and-down β-barrel accompanied by a short α-helix, formed by a region close to the C terminus (Fig. 1 ▶). Eleven other structurally characterized lipocalins are similar in structure (Huber et al. 1987; Bocskei et al. 1992; Newcomer 1993; Brownlow et al. 1997; Weichsel et al. 1998; Coles et al. 1999; Lascombe et al. 2000). However, the helix is absent in triabin, a salivary thrombin inhibitor from the triatomine bug Triatoma pallidipennis (Fuentes-Prior et al. 1997), and in bovine odorant binding protein engages in dimer stabilization through domain swapping with the cognate subunit (Bianchet et al. 1996; Tegoni et al. 1996).
Fig. 1.
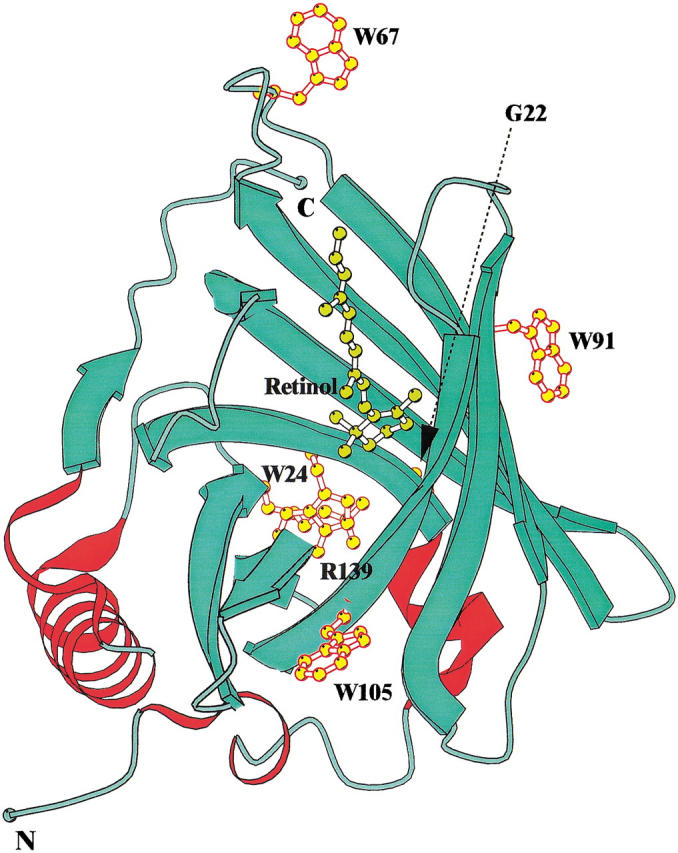
Structure of retinol binding protein showing the position of the four tryptophan residues and retinol molecule in the holo-protein. The figure was prepared using the program BOBSCRIPT (Esnouf 1997).
The lipocalins are particularly interesting because of their wide range of functions and high levels of sequence divergence with closely similar folds. RBP is part of a major subgroup of lipocalins that bind nonpolar ligands within the solvent-accessible barrel but use different amino acid side-chains, even when the ligands are closely similar (Newcomer 1995). Proteins in this group bind a wide range of ligands that includes retinoids, heme, pheromones, odorants, biliverdin, astaxanthine, prostaglandins, cholesterol, lipids, and progesterone. Other lipocalins bind proteins; triabin (Fuentes-Prior et al. 1997) and a lipocalin found in Von Ebner's gland and tears (Van't Hof et al. 1997) are protease inhibitors, whereas neutrophil gelatinase–associated lipocalin (NGAL) forms a noninhibitory complex with progelatinase B and biologically active peptides (Kjeldsen et al. 1993; Coles et al. 1999). Brain prostaglandin D synthase is a catalytically active lipocalin (Nagata et al. 1991) and a domain of the enzyme violoxanthine de-epoxidase is also a lipocalin (Bugos and Yamamoto 1996). Biological processes mediated by lipocalins include blood coagulation, complement fixation, regulation of inflammatory processes, crustacean and insect coloration, prostaglandin synthesis, immune system modulation, regulation of cell homeostasis, and neural system development, and several are major allergens (Flower 1996; Gregoire et al. 1996; Fuentes-Prior et al. 1997; Mantyjarvi et al. 1997; van't Hof et al. 1997). The functional versatility of the lipocalin fold indicates that it can provide a useful framework for engineering proteins with novel activities (Beste et al. 1999). The ideication of residues that are important for molecular stability is vital for such studies.
The folding mechanisms of β-sheet proteins and, in particular, β-barrel proteins are less understood than those of other structural classes. In particular, the conversion of an unstructured polypeptide to form an open barrel would require major structural reorganization after hydrophobic collapse (Clark et al. 1996). The structures of partially folded states may be relevant to this process, and in RBP a molten globule (MG) state (Ohgushi and Wada 1983) is stabilized at low pH (Bychkova et al. 1992); the MG of another lipocalin, β-lactoglobulin, is reported to be identical with an early intermediate in the refolding process (Hamada et al. 1996). In β-lactoglobulin it appears that nonnative structures form during folding because the MG has higher helix content than the native structure (Ragona et al. 1997). Despite their low global levels of sequence identity, different lipocalins show remarkable conservation of five patches of sequence that are associated with a structurally conserved mixed nonpolar/polar core at the base of the β-barrel. This region appears to be a key structural element of the lipocalin fold and has been suggested to play a pivotal role in folding (Greene and Brew 1995; Brew and Greene 1997; Greene 1998).
We describe here initial studies of the role of the four tryptophans of RBP and two additional residues that are adjacent to the highly conserved Trp24 (see Fig. 1 ▶) in the structure and stability of the apo-protein. Expression systems have been previously reported for apo-forms of human and porcine rRBP (Muller and Skerra 1993; Sivaprasadarao and Findlay 1993; Wang et al. 1993), but the recombinant proteins were not subject to detailed structural and biophysical characterization. We present the high-resolution crystal structure of recombinant RBP, folded from inclusion bodies by a new process, and a variant with substitutions for the surface tryptophans 67 and 91. Spectroscopic and stability measurements are reported for human apo-rRBP and 10 mutants. The properties of these proteins idey unique contributions made by different tryptophans to the CD and fluorescence spectrum of the native protein, indicating approaches and spectroscopic signals to investigate the development and loss of structure during folding and unfolding processes. The effects of mutations underscore the uniquely important role of interactions involving Trp24 in molecular stability.
Results
High-resolution structures of rRBP and rRBP67L/91H
Human RBP was selected as a model lipocalin for investigating the roles of conserved residues in folding because it has a well-characterized function, is devoid of cis-prolyl residues and structural cofactors, and is monomeric. A high-yield expression system, in vitro refolding protocol, and purification scheme were developed to produce native functional protein (see Materials and Methods). The high-resolution crystal structure of this recombinant protein confirms that it has a structure that is closely similar to those of the apo- and holo-proteins isolated from human serum.
The structures of rRBP and rRBP67L/91H were determined at 1.7 Å and 2.0 Å resolution, respectively. Although the complete amino acid sequence of RBP comprises 182 amino acids (Rask et al. 1979), clear electron density was visible only for 174 out of 182 residues in both structures. The overall structures of the two proteins are very similar (root mean square deviation, 0.32 Å) apart from the flexible loop formed by residues 62–68 and minor conformational changes of some surface residues (Fig. 2 ▶).
Fig. 2.
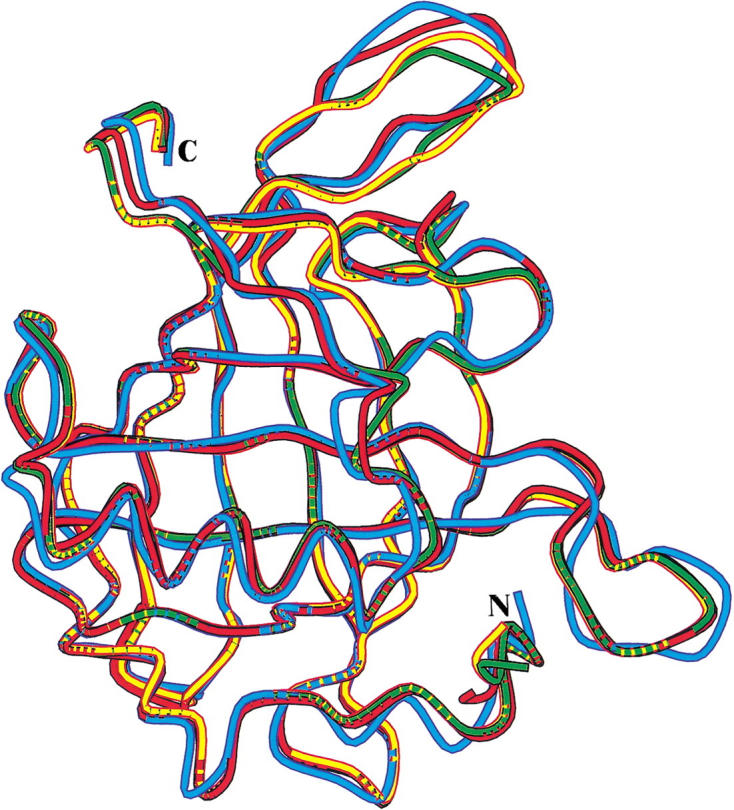
Superposition of the overall structure (only Cα atoms included) of recombinant human retinol-binding protein (RBP), recombinant RBP67L/91H, human serum RBP with retinol bound (Cowan et al. 1990), and human serum RBP when in complex with TTR (Naylor and Newcomer 1999) are shown in yellow, green, red, and cyan, respectively. N and C indicate the locations of the N-terminal and C-terminal residues.
The final rRBP and rRBP67L/91H structures include 205 and 218 water molecules, respectively. This is considerably more than in RBP structures reported previously because of the higher resolution of the data. Visual inspection of the rRBP electron density map revealed the presence of small but continuous pieces of density in the core of the β-barrel and on the surface of the molecule that could not be accounted for by addition of water molecules. Simulated annealing omit maps indicated the presence of six possible glycerol molecules (used as cryoprotectant during data collection) bound to the protein. Five putative glycerol molecules were ideied in the rRBP67L/91H structure, bound in the core of the barrel and on the surface of the molecule. Three of these are located in identical positions in rRBP and rRBP67L/91H in the interior of the β-barrel that forms the binding site for retinol in holo-RBP (Fig. 3 ▶).
Fig. 3.
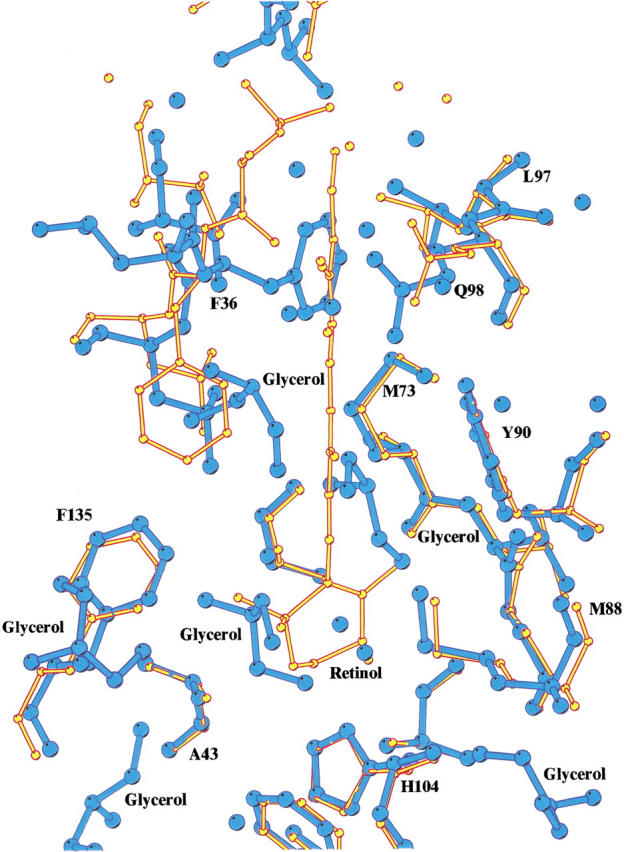
Superposition of the structures of rRBP and the retinol bound form of human retinol-binding protein (RBP; Cowan et al. 1990), showing the conformational change of the side-chain of Phe 36 and the binding of glycerol molecules in the place of retinol in the interior of the β-barrel. Recombinant RBP is depicted in blue; holo-RBP, in yellow.
Superposition of the rRBP structure onto apo and holo human serum RBP structures determined previously by Zanotti et al. (1993c) and Cowan et al. (1990) indicates that there is good agreement between them, with root mean square deviation of 0.5 Å and 0.6 Å, respectively.
Flexible loop (residues 62–68)
The loop region 62–68 in the rRBP structure was highly disordered, and the incorporation of the excluded residues, which were represented as alanines in the model, was possible only after applying the automated refinement procedure of Lamzin and Wilson (1993). Subsequent cycles of refinement considerably improved the quality of the electron density map, and the amino acids forming the flexible loop were introduced into the model, according to the sequence, only toward the final stage of refinement. The rRBP67L/91H structure had features similar to rRBP, except the loop region is better defined in the electron density.
This flexible loop and, in particular, Trp 67 is known to be involved in molecular packing interactions at the interface of the human RBP-TTR (transthyretin) complex (Monaco et al. 1995; Naylor and Newcomer 1999). In the two structures reported for this complex, Trp67 and Trp91 have critical roles in heterodimer stabilization, but a more detailed examination of these particular residues is not possible because of the low resolution (3.1 Å). Thus, when the rRBP67L/91H structure is compared with the structure of RBP in the complex, only gross structural differences can be ideied. The most profound differences between the recombinant apo-RBP and RBP-TTR complex are those around residue 62 and at the C terminus (root mean square, 1.05 Å; Fig. 2 ▶); both regions are implicated in interactions with TTR in the complex. In the rRBP structure, Trp67 is disordered, as are the rest of the residues that form the flexible loop, whereas in the rRBP67L/91H structure, the effect of the sequence substitution at position 67 was not investigated in detail because of the poor electron density in this region.
β-Barrel structure
The hydrophobic β-barrel structure is very similar to those in previously reported RBP structures (Figs. 1, 2 ▶ ▶). The structural similarity of RBP and β-lactoglobulin, which is isolated from milk as an apo-protein (Sawyer et al. 1985), also shows that the absence of a ligand does not cause a collapse of the β-barrel. The structures of rRBP and rRBP67L/91H and the apo-form of RBP obtained by depleting the natural holo-protein of retinol (Zanotti et al. 1993c) confirm this suggestion. The barrel of RBP accommodates one retinol molecule (Cowan et al. 1990), located in the interior with the β-ionone ring pointing to the deepest region of the barrel and the isoprene tail stretching almost to the barrel entrance. The apo-form of RBP derived from protein obtained from human plasma was determined at 2.5 Å resolution by Zanotti et al. (1993c), who observed that in the absence of retinol, the internal cavity contained significant portions of Fo–Fc electron density. Molecules responsible for this density could not be ideied because of insufficient resolution of the X-ray data, and it was suggested that the unexplained density might be attributed to solvent molecules. The structure of rRBP at the higher resolution (1.7 Å) presented here clarifies the tentative conclusions of Zanotti and coworkers by revealing the presence of both water and what appear to be glycerol molecules in the β-barrel in the rRBP and rRBP67L/91H structures. Glycerol molecules were incorporated in the model only during the final stage of refinement and are well positioned in the density, being stabilized by hydrogen bonds and van der Waals interaction with residues lining the interior of the β-barrel.
The hydrophobic nature of the interior of the β-barrel is evident from the presence of a large number of aromatic rings located within the lumen of the barrel. Phe36, which is located at the entrance to the interior, appears to block the opening of the barrel in the apo-RBP structure (Zanotti et al. 1993c) but adopts a different conformation when retinol is bound that opens the entrance of the barrel (Cowan et al. 1990; Naylor and Newcomer 1999). Thus, the side-chain of Phe36 seems to act like a tollgate for retinol binding at the entrance of the barrel. Although a similar feature is observed in the structures of rRBP and rRBP67L/91H (Fig. 3 ▶), Phe36 does not prevent the binding of small organic molecules such as glycerol in the barrel. Interestingly, the position of two glycerol molecules in the barrel in the rRBP and the variant structures appear to mimic the interactions made by the retinol molecule in holo-RBP structure.
Environments of tryptophans within the three-dimensional structure and conservation in sequence
The four tryptophans vary in their degree of conservation in a comparison with 32 members of the lipocalin superfamily (Fig. 4 ▶), with Trp24 and the proximal Gly22 (numbering in accordance with recombinant human RBP) being the most highly conserved residues. Lipocalins differ in function and structure-function relationships, and the conservation of Trp24 does not reflect a conserved role in function because it does not directly interact with bound ligands in currently known structures. Two other tryptophans, Trp67 and Trp91, are not conserved but Trp105 is conserved in all 12 known RBP sequences (data not shown).
Fig. 4.
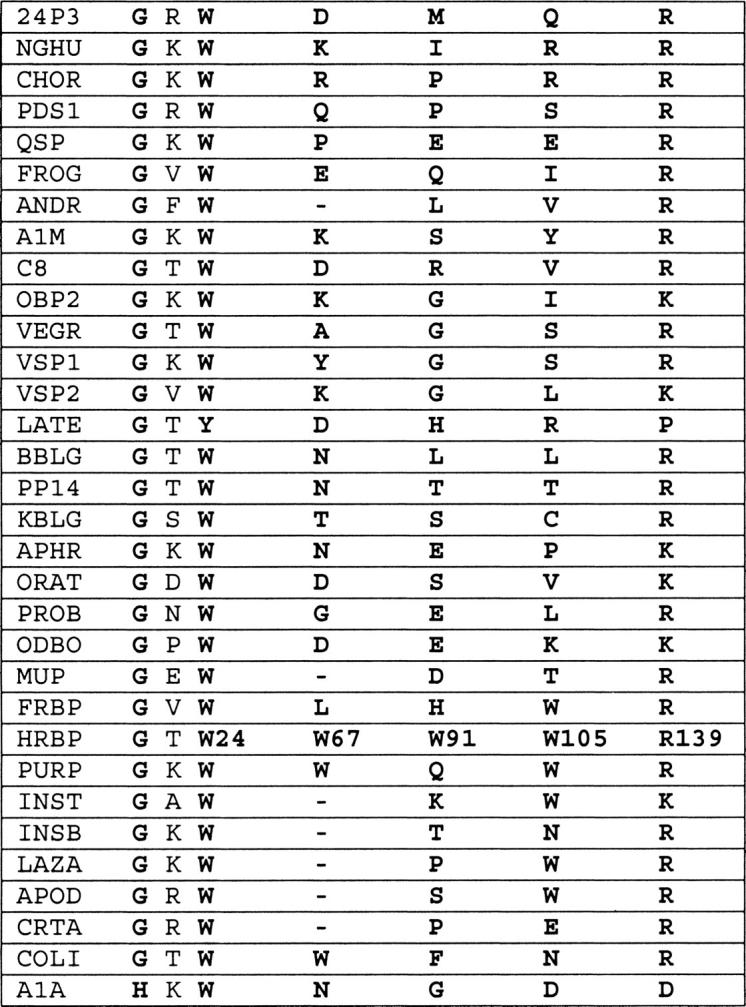
High conservation of Trp24 and structurally associated residues Gly22 and Arg139 of retinol-binding protein (RBP) relative to tryptophans 67, 91, and 105 in divergent lipocalins. This is based on an alignment of the proteins listed below together with their abbreviations and database accession codes: 24P3, mouse 24P3 (PIR: S07397); NGHU, human neutrophil gelatinase-associated lipocalin (P25; Swiss-Prot: NGAL HUMAN); CHOR, giant toad choriod plexus lipocalin (Swiss-Prot: LIPO BUFMA); PDS1, human brain prostaglandin D synthase-1 (PIR: A39400); QSP, chicken quiescence-related protein (PIR: A30230); FROG, Northern leopard frog odorant-binding protein (PIR: OVFGP); A1M, pig α1-microglobulin or protein HC (PIR: S13493); C8, human complement component 8 γ-chain (PIR: C8HUG); OBP2, rat odorant-binding protein II (PIR: A40464); VEGR, rat Von Ebner's gland protein or tear prealbumin (Swiss-Prot: VEG RAT); VSP1, mouse vomeronasal secretory protein I (PIR: D38580); VSP2, mouse vomeronasal secretory protein II (PIR: D38581); LATE, Tammar wallaby late lactation protein (PIR: A33672); ANDR, rat androgen-dependent epididymal retinoic acid–binding protein (PIR: SQRTAD); BBLG, bovine β-lactoglobulin (PIR: S05505); PP14, human placental protein 14 (PIR: A31242); KBLG, Eastern gray kangaroo β-lactoglobulin (PIR: A29699); APHR, hamster aphrodisin (PIR: A31243); ORAT, rat odorant-binding protein I (PIR: A28713); PROB, rat probasin (PIR: A32602); ODBO, bovine odorant-binding protein (Swiss-Prot: OBP BOVIN); MUP, mouse major urinary protein or α2u-globulin (PIR: UAMS); FRBP, African-clawed frog retinol-binding protein (PIR: S30013); HRBP, human serum tetinol-binding protein (PIR: VAHU); PURP, chicken purpurin (Swiss-Prot: PURP CHICK); INST, tobacco hornworm insecticyanin (PIR: CUWOI); INSB, white cabbage butterfly insecticyanin B(PIR: S00819); APOD, human apolipoprotein D (PIR: A26958); LAZA, South Americana lazarillo lipocalin (Genpept: U15656 1); CRTA, lobster crusticyanin A (Swiss-Prot: CRA2 HOMGA); COLI, bacterial lipocalin from Escherichia coli (Swiss-Prot: YJEL ECOLI); and A1A, rabbit α-1-acid glycoprotein (PIR: JH0617).
Trp24 is a component of the first (A) of the strands that form the β-barrel, whereas another highly conserved residue, Arg139, is located at the end of the final H strand. The interactions between these two residues contribute significantly to the formation of the barrel cylinder and closing of its base (Tables 1, 2). In some lipocalins, Arg139 is replaced by lysine, a change that does not disrupt the electrostatic interaction between these residues. In late lactation proteins from marsupials, Trp24 is uniquely replaced by tyrosine (Collet et al. 1989; Beg and Shaw 1994), a change that is invariably linked with the replacement of the residue corresponding to Arg139 by proline. These coupled natural substitutions seem likely to reflect the conservation of a tertiary interaction between these sites by substitution of a Tyr-Pro stacking interaction (Thornton 1992) for the Trp-Arg aromatic-amine interaction. Based on sequence conservations in the other lipocalins, similar interactions involving the conserved tryptophan are likely to be maintained throughout the entire superfamily (Fig. 4 ▶).
Table 1.
Hydrogen-bond interactions of tryptophan residues in rRBP
| Trp 24 | Trp 91 | Trp 105 | |||||||
| Atom | H-Bonds Distance < 3.3 Å | Distance (Å) | Angle (°) | H-bonds Distance < 3.3 Å | Distance (Å) | Angle (°) | H-Bonds Distance < 3.3 Å | Distance (Å) | Angle (°) |
| Ala43 O | 2.84 | 154.9 | Val74 O | 2.95 | 157.4 | Tyr118 O | 2.71 | 165.5 | |
| Nɛ1 | Phe20 O | 2.76 | 145.2 | Wat | 3.17 | Wat | 2.87 | ||
| O | Ala43 N | 2.87 | 162.3 | Val74 N | 2.73 | 160.3 | Tyr118 N | 2.82 | 142.0 |
| Val74 O | 3.20 | 112.9 | |||||||
| Environment of trypotophan residues in rRBP | |||||||||
| Residue | van der Waals Interactions/ No. of contacts | Accessible protein surface, (Å2) | Temperature factor (Å2) | Comments | |||||
| van der Waals interactions were calculated using CCP4 (CCP4 1994). van der Waals distances are the maximum allowed values of C-C, 4.1 Å; C-N, 3.8 Å; C-O, 3.7 Å; O-O, 3.3 Å; O-N, 3.4 Å; and N-N, 3.4 Å. Accessible protein surface per atom for each tryptophan residue was calculated using DSSP (Kabsch and Sanders 1983). | |||||||||
| Trp24 | Phe20/15, Thr23/13, | 0.0 | 19.5 | well defined in the electron density map | |||||
| Tyr25/12, Ala43/1, Phe45/1, | |||||||||
| Thr109/1, Tyr111/6, | |||||||||
| Thr113/1, Tyr114/6, | |||||||||
| Ala115/5, Phe137/1, | |||||||||
| Ser138/3, Arg139/16 | |||||||||
| Trp67 | — | — | — | disordered well defined in the electron density map | |||||
| Trp91 | Met73/3, Val74/9, Thr76/2, | 77.0 | 28.3 | ||||||
| Lys89/5, Tyr90/18, | |||||||||
| Gly92/14, Lys99/2 | |||||||||
| Trp105 | Val6/5, Phe9/1, Val11/1, | 21.0 | 18.6 | well defined in the electron density map | |||||
| Ala84/2, Lys85/18, | |||||||||
| His104/17, Ile106/11, | |||||||||
| Val107/3, Gly117/4, | |||||||||
| Tyr118/5, Wat/6 | |||||||||
Table 2.
Conserved aromatic-amine interaction and indole–hydrogen bonds in known lipocalin structures
| 3D Structures | Aromatic-amine interaction | Hydrogen bond (atom) |
| Retinol-binding protein (current) | Try24–Arg139 | Trp24 (NE1)–Phe20 (O) |
| β-Lactoglobulin | Trp19–Arg124 | Trp19 (NE1)–Val15 (O) |
| Bilin-binding protein | Trp27–Arg137 | Trp27 (NE1)–His24 (O) |
| Major urinary protein | Trp23–Arg126 | Trp23 (NE1)–Ile19 (O) |
| Odorant-binding protein | Trp17–Lys121 | Trp17 (NE1)–Leu13 (O) |
| Androgen-dependent epididymal retinoic acid–binding protein | Trp15–Arg119 | Trp15 (NE1)–Phe11 (O) |
| Allergen Bos D2 | Trp16–Lys119 | Trp16 (NE1)–Ile12 (O) |
| Triabin | Trp25–Arg129 | Trp25 (NE1)–Phe20 (O) |
Table 1 gives details of the environments of the tryptophan residues in the three-dimensional structure of rRBP. The four tryptophans differ in the number of contacts with other residues, solvent exposure, and mobility (based on temperature factors). Tryptophans 24 and 105 are the least accessible and most rigid, and tryptophans 67 and 91 are more exposed. Trp67 is highly flexible, being in the C-D loop between β-strands, and is flanked on one side by Asn65. Trp91 is located in the E-F loop and is close to Val74, Lys89, and Lys99. Trp105 is located on strand F (Fig. 1 ▶), with its side-chain pointing out from the barrel wall toward solvent, and is surrounded by Lys85, Asp103, and Tyr118. The ɛN of Lys85 in the present rRBP structure is ∼3.5 Å from the center of the indole ring of Trp105 (measured using Hyperchem), reflecting their involvement in an amine-aromatic electrostatic interaction.
Design and characterization of mutants
Ten single and multisite mutants were expressed and characterized to investigate the contributions of the different tryptophans to structure, stability, and spectroscopic properties of RBP. Therefore, the substitutions were designed to introduce minimal changes in structure and stability using, where possible, amino acids at corresponding sites in closely related lipocalins or to introduce a structurally similar amino acid. Substitutions of Leu for Trp67 and of His for Trp91 reflect a sequence change in Xenopus RBP, and Phe for Trp105 was based on structural conservation. Trp24 was replaced by Tyr, based on the previously discussed substitution in late lactation proteins from marsupials (Collet et al. 1989) and by Phe and Leu to introduce a weakly fluorescent or nonfluorescent residue. These changes will also eliminate the H-bond between the indole nitrogen and the carbonyl oxygen of Phe20. Gln was substituted for Arg139 as an alternative method of disrupting the Trp24-Arg139 interaction (Table 2). A mutant with substitutions of Phe for Trp24 and of Ala for the highly conserved Gly22 was also produced by a polymerase chain reaction (PCR)-derived mutation during the construction of the Trp24-to-Phe mutant. Designed multisite mutants were rRBP67L/91H, rRBP67L/91H/105F, and rRBP24Y/67L/91H. All rRBP mutants gave single bands of similar size to wild-type RBP on sodium dodecyl sulfat (SDS) polyacrylamide gel electrophoresis. Retinol-binding studies with selected tryptophan mutants indicated that the mutations did not eliminate the ability of the protein to bind all-trans retinol (data not shown). After folding and purification, the mutants were isolated in yields of ∼20 mg/L, except for those with substitutions for Trp24 or Arg139, in which the yields were 4- to 20-fold lower. To obtain small quantities of pure forms of rRBP24L and rRBP139Q, it was necessary to perform an additional separation by fast protein liquid anion exchange chromatography.
Contributions of tryptophans to the CD spectrum
Near-ultraviolet (UV) CD spectra of proteins contain features that reflect the fixed asymmetric environments of aromatic amino acids and cystines in the tertiary structure (Strickland 1974). Far-UV CD spectra largely arise from secondary structure, but aromatic residues can also contribute to this region (Woody 1995). Figure 5A and 5D ▶ ▶ shows near- and far-UV CD spectra of rRBP under native conditions (pH 7.4 and 20°C) and in acid-denatured (pH 2.0) and solvent-denatured (6 M guanidine hydrochloride [GndHCl]) states. The near- and far-UV CD spectra of rRBP in the native state are very similar to those previously reported by Bychkova et al. (1992) for human serum apo-RBP. rRBP unfolds at low pH and elevated temperatures to produce partially denatured conformers that have no near-UV CD spectrum and characteristic far-UV CD spectra that differ from the spectrum of the fully denatured protein. Both acid-denatured and thermally denatured rRBP (spectrum not shown) have greater negative ellipticity between 210 and 240 nm than the native protein, and the spectrum at pH 2.0 is similar to that previously described as a MG by Bychkova et al. (1992).
Fig. 5.

Near-ultraviolet (UV; 240 to 320 nm) and far-UV (200 to 240nm) CD spectra of recombinant retinol-binding protein (RBP) and variants. (A) Wild-type RBP: Native state at pH 7.4 and 20°C (thick solid line); unfolded state, 6 M GndHCl at pH 7.0 (broken line); and molten globule state at pH 2.0 (dotted line). (B) Mutants with substitutions for W67, W91, and W105: rRBP67L/91H/105F (– • •), rRBP67L/91H (– – –), rRBP91H (– •), and rRBP105F (dotted line). (C) Mutants with substitutions for G22, W24, and/or R139: rRBP24Y (broken line), rRBP24L (– • •), and rRBP139Q (thin solid line). The spectrum of native wild-type RBP is shown as a bold continuous line in all plots. Near-UV spectra were measured using a pathlength of 1 cm; far-UV spectra, with a pathlength of 0.1 cm.
Analysis of the far-UV CD spectrum of native RBP using the k2d neural network program (Andrade et al. 1993) gave proportions of α-helix and β-sheet (Table 3) that are in good agreement with those present in the crystallographic structure, although agreement of the fitted spectrum with experimental data is poor. This discrepancy appears to reflect contributions from tertiary structure to the far-UV CD spectrum, particularly from aromatic side-chains, because the fitted spectra of the acid and thermally unfolded states in which tertiary structure interactions are disrupted, are in better agreement with the experimentally determined spectra. In a comparison of different programs for analyzing far-UV CD spectra, k2d was found to give a very good estimate of β-structure contents of proteins, even though the calculated curves do not always match well with experimental data (Greenfield 1996). These analyses indicate that the thermally denatured protein has a secondary structure content closely similar to that of the native protein, but the pH 2.0 state has a higher helix content than the native structure (24% versus 8%) and a lower content of β-sheet (40% vs 45%). This is consistent with previous findings with β-lactoglobulin, in which the CD spectrum of the acid MG state indicates a higher helical content than the native protein (Hamada et al. 1996). The enhanced helicity of the MG of β-lactoglobulin was proposed to result from the greater propensity for α-helix formation in the primary structure in comparison with the helix content present in the native structure determined by X-ray crystallography. However, the helix content of RBP predicted using several sequence-based secondary structure prediction algorithms is in good agreement with the secondary structure content of the native structure (data not shown), indicating that the formation of MG with enhanced helix content may be a feature of the folding process in both lipocalins.
Table 3.
Secondary structure content estimated from far UV CD spectra of rRBP and rRBP mutants
| Protein | Temperature (°C) | pH | % α-Helix | % β-Sheet | Random | Quality of fit |
| rRBP (structure) | 9 | 45 | 47 | — | ||
| rRBP | 20 | 7.4 | 8 | 45 | 47 | pa |
| rRBP | 20 | 2.0 | 24 | 40 | 36 | g |
| rRBP | 80 | 7.4 | 8 | 41 | 51 | g |
| rRBP24Y | 20 | 7.4 | 14 | 34 | 52 | p |
| rRBP22A/24F | 20 | 7.4 | 14 | 34 | 52 | p |
| rRBP91H | 20 | 7.4 | 9 | 47 | 44 | p |
| rRBP67L/91H | 20 | 7.4 | 8 | 44 | 48 | p |
| rRBP105F | 20 | 7.4 | 11 | 40 | 49 | p |
| rRBP67L/91H/105F | 20 | 7.4 | 15 | 35 | 51 | p |
a (p) a poor fit of the calculated spectrum to experimentally determined spectrum (maximum error > 0.227); (g) a good fit (maximum error < 0.227).
The analyses were conducted using the k2d program (Andrade et al. 1993).
Near- and far-UV CD spectra of rRBP mutants are shown in comparison to the wild-type protein in Figure 5 ▶. Substitutions for tryptophans 67, 91, and/or 105 result in a progressive loss of negative ellipticity in broad trough centered at ∼280 nm (Fig. 5B ▶), whereas substitutions of Leu, Tyr, or Phe for Trp24 have increased negative ellipticity at ∼275 nm (Fig. 5C ▶). In contrast, the mutation of Arg139 to Gln has little effect on the near-UV CD spectrum (Fig. 5C ▶). All substitutions for Trp24 also change the far-UV CD spectrum as reflected in increased negative ellipticity at ∼228 nm (Fig. 5F ▶). This change is similar to one that has been attributed, in other proteins, to an interaction between the tryptophan indole ring system and a basic residue (Clark et al. 1996). The Arg139-to-Gln mutation will eliminate this particular interaction, but the spectrum of this mutant is closely similar to that of the wild-type protein, indicating that this feature in the CD spectrum does not arise from the electrostatic Arg139-Trp24 interaction in RBP. rRBP105F also shows that Trp105 contributes to negative ellipticity at ∼228 nm (Fig. 5E,F). This region of the far-UV CD spectra is only affected by substitutions for Trp24 or Trp105, and the ellipticity at 228 nm is therefore a signal that can be used to monitor the tertiary structure interactions of Trp24 and Trp105. Substitutions for Trp24 do not affect the trough at ∼208 nm (Fig. 5F ▶), but rRBP91H, rRBP67L/91H, and rRBP67L/91H/105F show a progressive loss in negative ellipticity at 208 nm (Fig. 5E ▶).
Roles of different tryptophans in fluorescence spectra of native and unfolded rRBP
RBP contains 4 tryptophan, 8 tyrosine, and 10 phenylalanine residues. The fluorescence emission spectra of rRBP and mutants missing tryptophan 24 (rRBP24F), tryptophan 105 (rRBP105F), or tryptophans 67, 91, and 105 (rRBP67L/91H/105F) in native and unfolded states when excited at 295 nm are shown in Figure 6 ▶. Wild-type rRBP shows a red shift in the emission maximum from 345 to 355 nm and a 20% decrease in fluorescence intensity on unfolding with 6 M GndHCl. Emission maxima for the denatured states of most mutants were similar (352 to 357 nm), but those of mutants with substitutions of Trp24 by Phe or Tyr (data not shown) were shifted to lower wavelength (351 and 349 nm). The blue-shifted contributions of Trp 24 and Trp 105 mutants in the native state fluorescence spectra compared with the denatured state (Fig. 6 ▶) are consistent with their low levels of solvent-accessibility (Table 1) and location in a hydrophobic environment (Table 1). The tryptophan fluorescence spectra indicate that Trp 24 makes the largest contribution to the decrease of fluorescence on unfolding, which may be partly attributable to a change in polarity of its environment on folding (Burnstein et al. 1973). However, this tryptophan is also part of a cluster of aromatic residues, its indole ring being 6 to 7 Å from the side-chains of tyrosines 25, 111, and 114 in the structure of rRBP, and it is also possible that the enhanced fluorescence on folding associated with Trp24 arises from fluorescence energy transfer from the adjacent tyrosine side-chains. These results indicate that Trp24 would serve as a good fluorescent probe for monitoring the formation of the core during studies of folding and unfolding kinetics.
Fig. 6.

Fluorescence spectra of recombinant retinol-binding protein (RBP) and selected mutants with substitutions for different tryptophans. Wild-type protein (solid line), rRBP24F (– • •), rRBP105F (– •), and rRBP67L/91H/105F (broken line). Spectra for the native states are denoted by thick lines; denatured states, by thin lines.
Effects of mutations on stability
Thermal unfolding in RBP monitored by CD ellipticity at 250 nm is reversible, the native UV CD spectrum being restored when the protein was cooled to 20°C after heating to 80°C. The Tm for recombinant human apo-RBP (68.9°C ± 0.04; Table 4a) is closely similar to the Tm of 70.3°C calculated for human apo-RBP by Muccio et al. (1992). Substitutions for individual or combinations of tryptophans 67, 91, and 105 reduce the Tm by 0.5°C to 2.7°C, reflecting relatively minor reductions in thermal stability of 0.1 to 0.5 kcal/mole (Table 4a; Fig. 7A ▶). However, all substitutions for tryptophan 24 (Phe, Tyr, or Leu) result in large losses in stability; the reductions in Tm of 11°C to 12.4°C reflect a decrease in stability of 2.2 to 2.6 kcal/mole (Table 4a). There is a similar loss of thermal stability in the Arg139 to Gln variant. rRBP22A/24F, which has substitutions for two highly conserved residues, Gly22 and Trp24, was the least stable mutant, with a decrease in Tm of 16.7°C, reflecting a reduction in stability of 3.8 kcal/mole (Table 4a).
Table 4a.
Effects of mutations on the thermal stability of rRBP
| Protein | Tm (°C) | Δ%m(WT−mutant) | δΔG(WT−mutant) (kcal/mole) |
| rRBP | 68.9 ± 0.04 | — | — |
| rRBP24Y | 57.9 ± 0.06 | 11.0 ± 0.10 | −2.20 ± 0.10 |
| rRBP24F | 56.9 ± 0.11 | 12.0 ± 0.12 | −2.52 ± 0.12 |
| rRBP24L | 56.6 ± 0.08 | 12.4 ± 0.09 | −2.60 ± 0.06 |
| rRBP22A/24F | 52.2 ± 0.07 | 16.7 ± 0.15 | −3.80 ± 0.12 |
| rRBP91H | 68.4 ± 0.06 | 0.5 ± 0.07 | −0.10 ± 0.01 |
| rRBP105F | 66.7 ± 0.03 | 2.2 ± 0.05 | −0.44 ± 0.10 |
| rRBP67L/91H | 66.2 ± 0.06 | 2.7 ± 0.07 | −0.50 ± 0.10 |
| rRBP67L/91H/105F | 66.7 ± 0.04 | 2.2 ± 0.06 | −0.50 ± 0.02 |
| rRBP24Y/67L/105F | 55.0 ± 0.10 | 13.9 ± 0.11 | −2.92 ± 0.07 |
| rRBP139Q | 56.5 ± 0.14 | 12.4 ± 0.15 | −2.60 ± 0.07 |
Fig. 7.

Stability studies of the recombinant retinol-binding protein (rRBP) and variants. (A) Thermal unfolding for rRBP and mutants measured by CD ellipticity at 250 nm. All proteins were dissolved in 5 mM phosphate buffer (pH 7.4). Protein samples were heated from 20°C to 80°C at a rate of 50°C/h. The pathlength was 1.0 cm. (B) Solvent-induced equilibrium denaturation and refolding of rRBP monitored by tryptophan fluorescence: (closed circles) Unfolding of rRBP; (open circles) refolding. Protein samples were incubated in 5 mM NaPO4 buffer and a range of GndHCl concentrations (0 to 6 M) for 24 h at 23°C. Fluorescence emission was measured from 329 to 331 nm using an excitation wavelength of 290 nm. Each spectrum is an average of nine scans. (C) Plot of ΔG versus GndHCl concentration for rRBP and mutants. Chemical unfolding was conducted with varying concentrations of GndHCl: rRBP (circles), rRBP24Y (triangles), rRBP22A/24F (hexagon), rRBP67L/91H/105F (squares), rRBP91H (open hexagons), rRBP105F (open circles), and rRBP67L/91H (open triangles).
RBP folds and unfolds reversibly on solvent denaturation, a necessary criterion for measurements of stability and folding/unfolding kinetics (Fig. 7B ▶). The kinetics of the GndHCl-induced unfolding of rRBP is biphasic and has a very slow second phase (Greene 1998). Therefore, in stability measurements by solvent unfolding, rRBP and mutants were incubated at different concentrations of guanidine HCl for 24 h to ensure that the process reached equilibrium. Denatured RBP was prepared by 24-h incubation in 6 M guanidine HCl and subsequently refolded at lower denaturant concentrations to equilibrium also by incubation for 24 h (see also Muller and Skerra 1993). Data were analyzed to determine the guanidine HCl concentration at the midpoint of the transition (Gnd50) and the m value. These were used to calculate the free energy of stability in the absence of denaturant (ΔGH2O) and at Gnd50 (Table 4b). Because solvent denaturation requires much more protein than thermal unfolding, only six mutants were characterized by this method (Fig. 7C ▶). However, the results obtained are in good qualitative agreement with thermal stability data. The mutant with His for Trp91 shows a small and insignificant increase in stability. The losses in stability of both rRBP105F and rRBP67L/91H/105F are significant, indicating that Trp105 has a role in molecular stability in RBP, but the change is far less than in all mutants with substitutions for Trp24.
Table 4b.
Effect of mutations on the stability of rRBP as measured by chemical unfolding
| Protein | Δf(H2O) (kcal/mole) | m (kcal/mole/M) | Gnd50 | δΔG(WT−mutant) (water) | δΔG(WT−mutant) (Gnd50) |
| rRBP | −4.76 ± 0.16 | 2.67 ± 0.10 | 1.79 ± 0.01 | — | — |
| rRBP24Y | −2.99 ± 0.17 | 2.49 ± 0.13 | 1.22 ± 0.03 | −1.77 ± 0.23 | −1.47 ± 0.09 |
| rRBP24F | −3.36 ± 0.33 | 2.46 ± 0.24 | 0.37 ± 0.03 | −1.40 ± 0.22 | −1.09 ± 0.04 |
| rRBP22A/24F | −2.78 ± 0.08 | 2.86 ± 0.09 | 0.97 ± 0.01 | −1.98 ± 0.18 | −2.26 ± 0.06 |
| rRBP91H | −4.77 ± 0.23 | 2.92 ± 0.14 | 1.77 ± 0.03 | 0.01 ± 0.28 | 0.06 ± 0.08 |
| rRBP105F | −4.44 ± 0.15 | 2.92 ± 0.10 | 1.52 ± 0.06 | −0.32 ± 0.22 | −0.43 ± 0.10 |
| rRBP67L/91H | −4.13 ± 0.26 | 2.61 ± 0.16 | 1.65 ± 0.01 | −0.45 ± 0.31 | −0.37 ± 0.04 |
| rRBP67L/91H/105F | −4.61 ± 0.17 | 3.16 ± 0.11 | 1.42 ± 0.01 | −0.15 ± 0.23 | −0.96 ± 0.02 |
ΔG values are the free energy difference between the unfolded and folded states of a particlular protein.
m values represent the change in stability with GndHCl.
[GND]50 = is the midpoint of the unfolding transition.
δΔGmut = ΔGwt − ΔGmut
δΔGGND50 = ([GND]50wt − [GND]50mut) * (mwt + mmut)/2, the free energy of stability at the midpoint of the unfolding transition.
Discussion
Conserved residues are important for native state stability and for preventing misfolding
The present study highlights the unique role of Trp24 and its local and long-range interactions in the stability of apo-RBP. Previously, the substitution of Tyr for the residue corresponding to RBP Trp24 in dimeric bovine β-lactoglobulin (Trp19) was found to be highly destabilizing but did not eliminate retinol binding activity (Katakura et al. 1994). RBP and β-lactoglobulin are distantly related homologs, differing by 76% in amino acid sequence so that the importance of this tryptophan for molecular stability is likely to be a general feature of lipocalins. In RBP, a similar destabilization is obtained by introducing Tyr, Phe, or Leu and also by replacing Arg139 by Gln. The double-mutant rRBP22A/24F is further reduced in stability, indicating additive or synergistic effects of substitutions for multiple conserved residues in this region of structure. In β-lactoglobulin, changes of the equivalent residue, Trp 19, to Phe or Ala resulted in insignificant yields compared with those of wild-type protein or substitution by Tyr (Katakura et al. 1994). In that study, recombinant β-lactoglobulin was expressed using a yeast secretion system in contrast to the bacterial system used here for rRBP, in which in vitro folding is used to generate the native protein.
Although all rRBP mutants were expressed in similar yields in Escherichia coli as inclusion bodies, after in vitro folding the native forms of all Trp24 mutants were isolated in lower yields (∼25%) compared with those of the wild-type protein and other mutants; in the case of the Trp24Leu and Arg139Gln variants, the yields were about fourfold lower. This suggests that these mutations increase the stability of misfolded forms relative to the native structure so that a larger proportion of protein molecules fold to nonnative structures. This view is reinforced by the lack of secretion of the Trp19 to Ala and Phe mutants of β-lactoglobulin by the eukaryotic expression system because such systems tend to degrade wrongly folded proteins. These findings are conceptually significant because they link mutations in conserved residues to protein misfolding, which is the basis of many diseases (Dobson 2001).
The folding behavior of RBP has a specific biological interest because only the holo form of the protein is secreted after biosynthesis in mammalian and other cells; RBP molecules that do not acquire a retinol ligand within the cell are retained in the endoplasmic reticulum (Melhus et al. 1992) and appear not to be fully folded (Kaji and Lodish 1993). Previously determined structures for human and bovine apo-RBP show close similarity to the holo-protein. These apo-proteins were prepared from natural holo-protein after extraction with ethyl ether to remove the bound ligand (Zanotti et al. 1993a). Here we find that recombinant human apo-RBP produced by in vitro folding of material extracted from inclusion bodies has a structure and spectroscopic properties that are closely similar to those of apo and holo forms of natural human holo-RBP (Cowan et al. 1990), holo and apo bovine RBP (Zanotti et al. 1993a,c), and RBP in complexes with different retinoids (Zanotti et al. 1993b). Because our preparations of apo-rRBP have never bound a retinol ligand, we can conclude that although retinol may enhance folding yields, the ligand is not necessary for an irreversible maturation step in folding. Thus, the degradation of apo-RBP in vivo must be linked to some specific structural feature or property of the apo- versus holo-protein.
The results described here are necessary for the design and interpretation of unfolding and folding kinetics of rRBP because they establish conditions in which the folded and unfolded conformers are most populated. They also allow comparisons of the effects of mutations on the stability of the transition states for folding and unfolding with those of the native and unfolded states. The spectroscopic properties of the mutants indicate signals that provide information about the structure formation in different parts of the RBP molecule during folding processes. A major focus of our work is to determine if there is a relationship between folding and sequence conservation in functionally divergent paralogous proteins. This is a concept that is recently receiving attention from experimentalists (Martinez and Serrano 1999; Hamill et al. 2000; Nishimura et al. 2000; Plaxco et al. 2000). rRBP, as an experimental model for the large lipocalin superfamily, represents an ideal system to further our understanding between evolution and folding. Toward this end, kinetic and additional X-ray crystallographic studies are in progress with recombinant RBP and mutants.
Materials and methods
Preparation of rRBP
The cDNA encoding human serum RBP was amplified from a mp9 phage vector by PCR in a Perkin Elmer Cetus DNA Thermal Cycler using two primers designed specifically for directional cloning into the pET-3a vector: 5`-d(GCACTGATACTACATAT GGAGCGCGACTGCCGA)-3`, containing the NdeI cloning site and Met-1, and 5`-d(GCAGGTACTGACGGATCCCTACAAAA GGTTTC)-3`, containing the BamHI cloning site and stop codon. Twelve extra bases were included outside each restriction site to allow for efficient digestion. Amplifications by PCR were performed using a Perkin Elmer/Cetus thermocycler using the following reaction conditions: 25-pmole primers, 1.5 units Vent DNA polymerase, 1× buffer (supplied by the manufacturer), 50 mM dNTP mixture, and 1.5 mM MgCl2, template empirically determined, 100 μL total volume. The first cycle was 2 min at 94°C; the next 20 cycles, 1 min at 94°C, 2 min at 50°C, and 1.5 min at 74°C; and the final cycle, 10 min at 74°C and cooling to 4°C. The product was purified by electrophoresis, extracted with phenol, precipitated with ethanol, digested with NdeI and BamHI, and cloned into a preparation of the expression vector (pET3a) that had been previously digested with the same restriction enzymes. Recombinant plasmid (pRBP) was amplified for characterization by sequencing and for use as templates in PCR mutagenesis experiments using E. coli strain DH5α and transformed into BL21(DE3) cells for expression.
DNA sequencing of a selected clone showed that it is identical to that published by Colantuoni and coworkers (1983) apart from two silent substitutions, TCC for TCG in the codon for serine 46 and GTG for GTA in the codon for valine 116.
Construction of RBP mutants
Mutations were introduced using the "megaprimer" method (Sarkar and Sommer 1990). The megaprimer was generated by amplification with T7 promoter primer or T7 terminator primer together with an appropriate mutagenic primer (Grobler et al. 1994). The megaprimer was purified by agarose gel electrophoresis and used in the second amplification with the same template and the cognate T7 primer. The final amplification product was purified and cloned into pET3a as described for RBP. The complete sequences of the inserts in the recombinant vectors were determined by automated DNA sequencing using both the T7 promoter and T7 terminator primers. Oligonucleotide synthesis and automated DNA sequencing were performed in the DNA Core Facility, University of Miami School of Medicine.
Protein expression and initial purification of rRBP
Cultures of E. coli strain BL21(DE3) transformed with the recombinant vector (pRBP) or variant were grown at 37°C with 100 mg/mL ampicillin, induced with 0.4 mM isopropylthiogalactopyranoside (IPTG) when the optical density reached 0.6, and harvested after 3 h. Cells were suspended in lysis buffer (0.05 M Tris, 0.1 M NaCl, and 1 mM EDTA at pH 8.3) and lysed by treatment with lysozyme and deoxycholic acid and treated with DNAse (Sambrook et al. 1989). Inclusion bodies containing recombinant RBP (rRBP) were isolated by centrifugation and washed initially by resuspension in lysis buffer and then with double distilled water. The protein was extracted with 8 M urea containing 20 mM Tris base and 20 mM dithiothreitol (∼50 mL per 6 L of culture), centrifuged at 15,000 rpm for 10 min, and loaded on a column (2 × 10 cm) of Macroprep 50Q equilibrated with 20 mM Tris-HCl (pH 8.5) containing 8 M urea. The column was washed with equilibration buffer until the absorbance at 280 nm was nearly to baseline, and the protein was eluted with a linear gradient from 0 to 0.5 M NaCl in 20 mM Tris-HCl (pH 8.5) containing 8 M urea at a flow rate of 2 mL/min. Fractions of 6.0 mL were collected and monitored by absorbance at 280 nm. SDS gel electrophoresis of aliquots of different fractions indicated that the major UV-absorbing peak, which eluted at ∼25 min, contained rRBP (molecular weight 21,000) in a relatively pure form.
In vitro folding and purification of folded rRBP
Fractions containing rRBP or a mutant were pooled, and the concentration of protein was estimated from the absorbance at 280 nm; the extinction coefficients for RBP and tryptophan mutants were calculated from their contents of tryptophan, tyrosine, and cysteine (Mach et al. 1992). E280nm0.1% values are as follows: wild-type rRBP and rRBP139Q, 1.61; rRBP24Y, 1.44; rRBP24F, rRBP24L, rRBP91H, rRBP105F, and rRBP22A/24F, 1.34; rRBP67L/91H, 1.06; rRBP67L/91H/105F, 0.79; and rRBP24Y/67L/105F, 0.80. Native protein was generated by adding the denatured material in 8 M urea drop-wise to folding buffer (20 mM Tris HCl at pH 8.5 containing 10 mM β-mercaptoethanol, 1 mM 2-hydroxyethyldisulfide, and 1% glycerol) at a rate of ∼30 drops/min. The final concentrations of protein and urea were 50 to 100 μg/mL and 0.3 M, respectively. Folding was allowed to proceed for 16 h at 4 °C, and the solution was then concentrated using an Amicon Spiral Filter Ultrafiltration System. The protein was loaded on a column (2 × 10 cm) of DE52 DEAE cellulose equilibrated with 20 mM Tris-HCl (pH 8.5) at 22°C. After washing with 20 mM Tris HCl (pH 8.5), the sample was eluted with a linear gradient from zero to 0.5 M NaCl in the same buffer over 85 min at a flow rate of 2 mL/min. A single peak containing folded protein eluted at ∼0.3 M NaCl and was dialyzed against water at 4°C and lyophilized. Further purification was performed by gel filtration with a column of Superdex G75 (7 × 21cm) equilibrated with 20 mM Tris-HCl (pH 7.4) and 0.2 M NaCl at a flow rate of 1.5 mL/min. CD spectroscopy showed that the second, larger protein peak contained native protein.
Folded rRBP was obtained by this method in a yield of 20 mg/L of bacterial culture. The purified protein was homogeneous on SDS polyacrylamide gel electrophoresis. Fourteen cycles of Edman degradation using an Applied Biosystems model 470A Protein/Peptide Sequencer gave a single N-terminal sequence identical to that of human RBP, except for the presence of an N-terminal methionyl extension that reflects its intracellular expression in E. coli. The ligand-binding properties of RBP were measured using retinoic acid, the interaction of which with RBP quenches tryptophan fluorescence. The calculated dissociation constant (Kd) for rRBP was 1.9 ± 0.2 × 10−7 M (data not shown), which is within experimental error of the value of 2.1 × 10−7 M reported for natural human apo-RBP (Cogan et al. 1976).
X-ray crystallographic analysis
Crystallization rRBP
Large needle shaped crystals of native recombinant human RBP were grown by the vapor diffusion method at 16°C. Equal volumes (2.0 μL) of protein (∼12 mg/mL in 10 mM sodium cacodylate and 3.9 M sodium chloride at pH 6.8) and reservoir solution (20 mM sodium cacodylate and 4.5 M sodium chloride at pH 6.8) were mixed on siliconized coverslips and left to equilibrate against the reservoir solution.
rRBP67L/91H variant
rRBP double mutant was crystallized by the vapor diffusion method at 16°C. Single crystals were obtained by applying the seeding technique from crystals of the native recombinant protein, and their size was optimized by addition of 0.05% β-octyl-glucoside. Equal volumes (2.0 μL) of protein (∼12 mg/mL) and reservoir solution (4.5 M sodium chloride, 50 mM Tris-HCl at pH 9.1, and 0.05% β-octyl-glucoside) were used.
Diffraction data collection-rRBP
Data were collected to 1.7 Å resolution from one crystal at the Synchrotron Radiation Source, Daresbury on station PX 9.5 (oscillation range, 2.0°; λ = 0.9 Å) under cryogenic conditions (100 K) using a 30-cm MAR Research image plate. Before data collection, crystals were transferred to buffer solution (same as the reservoir solution) containing 25% (v/v) glycerol (cryoprotectant) for ∼30 sec. The data set was indexed in rhombohedral spacegroup R3, with one molecule in the asymmetric unit and solvent content of 68.3% (Matthews coefficient of ∼3.9 Å3/Da). Data integration and reduction were performed with the programs DENZO and SCALEPACK (Otwinowski and Minor 1997). The data collection statistics are presented in Table 5.
Table 5.
Data collection and refinement statistics of rRBP and rRBP67L/91H variant
| Spacegroup details | ||
| Cell dimensions (a, b, c) (Å) | 1, b, 101.34, c, 71.98 | 1, b, 101.76, c, 72.72 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 |
| Space group | R3 | R3 |
| No. of molecules per asymmetric unit | 1 | 1 |
| Data collection and processing statistics | ||
| Station (Synchrotron) | Daresbury PX9.5 | EMBL-Hamburg X31 |
| Image plate | Mar30 | Mar18 |
| Wavelength (Å)0.90 | 1.09 | |
| No. of observations | 67,359 | 109,525 |
| No. of unique reflections | 28,189 | 18,890 |
| Maximum resolution (Å) | 1.7 | 2.0 |
| Completeness (outermost shell) (%)a | 90.5 (91.0) | 98.0 (92.7) |
| Rsym (I) (outermost shell) (%)b | 4.7 (50.5) | 10.2 (37.0) |
| <I/σ (I) > (outermost shell) | 7.68 (2.59) | 7.53 (3.19) |
| Outermost shell (Å) | 1.72–1.70 | 2.07–2.00 |
| Refinement statistics and model quality | ||
| Resolution range (Å) | 40.0–1.7 | 40.0–2.0 |
| No. of reflections27,458 | 18,877 | |
| No. of protein atoms | 1,407 | 1,398 |
| No. solvent molecules | 205 | 218 |
| No. of glycerol molecules | 6 | 5 |
| Rfree (%)a | 27.0 | 22.9 |
| Rconv (%)b | 24.8 | 20.5 |
| RMS deviations in | ||
| bonds (Å) | 0.007 | 0.006 |
| angles (°) | 1.469 | 1.379 |
| Temperature factors (Å2) | ||
| overall | 26.3 | 22.6 |
| average main-chain | 24.1 | 20.1 |
| average side-chain | 24.7 | 21.6 |
| from Wilson plot | 23.1 | 18.8 |
aCompleteness in the range ∞-resmax, where resmax is the maximum resolution to which data were collected. Rfree = ∑hkl⊂τ||Fobs-k|Fcalc||/∑hdl⊂τ|Fobs|×100, where hkl|Ndτ represents the test set (5% of the diffraction data).
bRconv=∑hkl||Fobs-k|Fcalc||/∑hkl|Fobs|×100.
Deviations from ideal values (Engh and Humber 1991).
rRBP67L/91H mutant
Data from a single crystal were collected to 2.0 Å resolution on station X31 at EMBL-Hamburg Synchrotron Radiation Source, using an 18-cm MAR Research image plate (Table 5; oscillation range per image, 1.2°; λ = 1.09 Å). The data set was collected under cryogenic conditions (100 K) using 20% (v/v) glycerol as cryoprotectant. Both DENZO and SCALEPACK programs were used for data processing (Otwinowski and Minor 1997). The double variant was crystallized in rhombohedral spacegroup R3, like the native protein, with one molecule in the asymmetric unit and solvent content of 69.5% (Matthews coefficient of ∼4.0 Å3/Da; Table 5).
Structure determination rRBP
The structure of human serum RBP (hRBP) determined previously at 2.0 Å resolution (Cowan et al. 1990) was used for molecular replacement with the program AMoRe (Navaza 1994). Superposition of hRBP with rat androgen-dependent epididymal retinoic acid–binding protein (E-RABP; Newcomer 1995) indicated that residues 14–30, 70, 80, 84–86, 106–115, 135–139, 146–151, 158–160, 165, 172, and 174 are conserved in the RBP structure; hence, only these were included in the search model. Residues that were part of flexible loops or other disordered regions were excluded from the starting model.
rRBP67L/91H variant
The structure of the double variant at 2.0 Å resolution was determined by molecular replacement with X-PLOR (Brünger 1992b) using the rRBP structure determined at 1.7 Å resolution as a search model.
Structure refinement
The resultant models of rRBP and rRBP67L/91H were first subjected to restrained refinement with maximum likelihood method using the program REFMAC (CCP4 1994) followed by the automated refinement procedure (ARP; Lamzin and Wilson 1993) and to rigid body refinement using the program X-PLOR, respectively. The combination of REFMAC and ARP refinement protocol, applied to the starting model of rRBP, significantly improved the quality of the electron density map and enabled the building of the residues that had been initially excluded from the molecular replacement solution (only conserved residues). Cycles of refinement were performed using the slowcool protocol, at first with the program X-PLOR (Brünger 1992b) and during the final stages of refinement with CNS (Brünger et al. 1998), for both structures. The progress of refinement was monitored through both free and conventional R-factors (Brünger 1992a). Alternate cycles of manual rebuilding with the graphics program O (Jones et al. 1991) and refinement cycles using the standard protocol improved the quality of the model. Simulated annealing omit maps calculated using either X-PLOR or CNS for the two structures indicated density in the core of the β-barrel and on the surface sufficient to accommodate one or more glycerol molecules. Six glycerol molecules were incorporated into the rRBP model (five in the rRBP67L/91H model) at the very final stage of refinement. In each case, water molecules were inserted in the model only if there were peaks in the Fo–Fc electron density maps with heights >3σ and they were at hydrogen bond–forming distances from appropriate atoms. 2Fo–Fc electron density maps were also used to check the consistency in peaks. The final refinement statistics and model quality are summarized in Table 5. Analysis of the Ramachandran plot, calculated with the program PROCHECK (Laskowski et al. 1993), showed that all residues lay in the allowed regions except Tyr 111 in both the rRBP and rRBP67L/91H structures, for which the ϕ and ξ values are ϕ = 69.1°, ξ = `39.8° and ϕ = 64.6°, ξ = −36.7°, respectively. The details of refinement statistics and model accuracy are listed in Table 5. Structural superpositions were performed with the program MAPS (Lu 2000) and the accessibility of the protein surface was calculated with the program DSSP (Kabsch and Sanders 1983). The atomic coordinates have been deposited at the RCSB Protein Data Bank (accession codes 1JYD for apo-rRBP and 1JYJ for rRBP67L/91H).
Circular dichroism
Near- and far-UV CD spectra of rRBP and mutants were determined with a Jasco J-710/720 spectropolarimeter. Twelve spectra were scanned for each sample at a speed of 50 nm/min and were averaged. Near-UV CD spectra (240 to 320 nm) were determined using a cell with a path length of 1.0 cm; far-UV spectra (200 to 240 nm), using a path length of 0.1 cm. Protein samples were dissolved in 5 mM sodium phosphate buffer (pH 7.4) at concentrations of 0.2 to 0.5 mg/mL. A constant temperature of 20°C was maintained with a circulating NESLab RTE-111 water bath. MG spectra were determined with protein samples dissolved in 50 mM phosphate buffer (pH 2.0). Secondary structure analysis of far-UV CD spectra was performed using the k2d neural network program (Andrande et al. 1993).
Thermal unfolding
Thermal unfolding data were analyzed by fitting to the following equation:
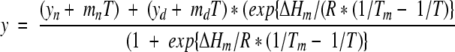 |
where y is the ellipticity at 250 nm, T is the temperature in Kelvin, R is the gas constant, ΔHm is the enthalpy of unfolding, Tm is the midpoint of the unfolding transition, yn is the ellipticity of the native protein and yd is the ellipticity of the unfolded protein (both extrapolated to 0 K), and mn and md are the baseline slopes for the native and denatured states, respectively. The approximate effects of mutations on stability were calculated from the changes in Tm (ΔTm) produced by the following mutation:
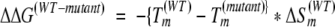 |
where ΔΔG(WT-mutant) is the stability difference between wild-type protein and mutant, Tm(WT) and Tm(mutant) are the mid-points of the unfolding transition of the wild-type protein and mutant, and ΔSm(WT) is entropy of unfolding of the wild-type protein. ΔSm is calculated from the relationship ΔSm = ΔHm/Tm.
Chemical-induced equilibrium unfolding
Fluorescence spectra of native and unfolded forms of protein and equilibrium GndHCl unfolding were determined using a Perkin Elmer Luminescence LS-50 Fluorimeter at 23°C. Protein samples (50 to 130 μg/mL) in 5 mM phosphate buffer (pH 7.0) containing 0 to 6 M GndHCl were preincubated for 24 h at 23°C, and fluorescence spectra were measured from 300 to 400 nm using an excitation wavelength of 295 nm. For unfolding measurements at different denaturant concentrations, samples were excited at 290 nm, and the average fluorescence emission between 329 and 331 nm was determined. An excitation wavelength of 290 nm was used in the latter experiments to maximize the fluorescence yield and difference between native and unfolded states. Each data point is the average of nine scans. The concentrations of GndHCl in sample solutions were determined by refractive index at 25°C (Nozaki 1972) using a Milton Roy ABBE-3L refractometer. In all unfolding studies, solutions were prepared using Hamilton syringes to ensure accuracy.
The fluorescence spectra were analyzed by fitting to appropriate equations using the curve fitter program Sigma Plot, version 3 (Jandel Scieic). Baseline data from the linear native and unfolded regions of the curve (ideied from initial plots of fluorescence against denaturant) were analyzed by linear regression to obtain baseline corrections reflecting the response of the native and unfolded states to denaturant. Subtraction of these across the unfolding curve allowed the calculation of the fraction of folded and unfolded protein across the transition (Pace 1986). Free energies of stability were calculatedas ΔG = −RTln(Kf), where Kf = fraction folded/fraction unfolded. Linear plots of ΔG[denaturant] versus [denaturant] allowed the calculation of ΔG(water) and [denaturant]50%—the free energy of stability in the absence of denaturant and the denaturant concentration at the midpoint of the unfolding transition, respectively—together with their standard errors.
Acknowledgments
We thank Vittorio Collantuoni for generously providing the hRBP clone for this work and V. Ondricek for the sequence determination of rRBP. We are grateful to the staff at the Synchrotron Radiation Sources at Daresbury, United Kingdom, and EMBL-Hamburg, Germany, for their assistance during X-ray data collection and also to the members of the Brew laboratory for helpful discussions. Parts of this work were supported by a European Community Marie Curie postgraduate training fellowship to E.D. Chrysina and by National Institutes of Health grant GM21363.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RBP, retinol-binding protein
rRBP, recombinant human retinol-binding protein
rRBPmX, single site mutants (indicating the substitution of amino acid X for residue number m)
rRBPmX/nY, multi-site mutants (indicating the substitution of X for residue m and Y for residue n)
MG, molten globule state
PCR, polymerase chain reaction
GndHCl, guanidine hydrochloride
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.22901.
References
- Andrade, M.A., Chacon, P., Merelo, J.J., and Moran, F. 1993. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6 383–390. [DOI] [PubMed] [Google Scholar]
- Beg, O.U. and Shaw, D.C. 1994. The complete primary structure of late lactation protein from quokka (Setonix brachyurus). J. Protein Chem. 13 513–516. [DOI] [PubMed] [Google Scholar]
- Beste, G., Schmidt, F.S., Stibora, T., and Skerra, A. 1999. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc. Natl. Acad. Sci. 96 1898–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchet, M.A., Bains, G., Pelosi, P., Pevsner, J., Snyder, S.H., Monaco, H.L., and Amzel, L.M. 1996. The three-dimensional structure of bovine odorant binding protein and its mechanism of odor recognition. Nat. Struct. Biol. 3 934–939. [DOI] [PubMed] [Google Scholar]
- Bishop, R.E. and Weiner, J.H. 1996. Outlier lipocalins more than peripheral. Trends Biochem. Sci. 21 127. [PubMed] [Google Scholar]
- Bocskei, Z., Groom, C.R., Flower, D.R., Wright, C.E., Phillips, E.V., Cavaggioni, A., Findlay, J.B.C., and North, A.C.T. 1992. Pheromone binding to two rodent urinary proteins revealed by X-ray crystallography. Nature 360 186–188. [DOI] [PubMed] [Google Scholar]
- Brew, K. and Greene, L. 1997. Evolution, folding and flexibility. Protein Eng. 10 S44. [Google Scholar]
- Brownlow, S., Morais Cabral, J.H., Cooper, R., Flower, D.R., Yewdall, S.J., Polikarpov, I., North, A.C.T., and Sawyer, L. 1997. Bovine β-lactoglobulin at 1.8 Å resolution: Still an enigmatic lipocalin. Structure 5 481–495. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992a. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475 [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992b. X-PLOR Version 3.1 Manual: A system for X-ray Crystallography and NMR, Yale University Press, New Haven.
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., GrosseKunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Bugos, R.C. and Yamamoto, H.Y. 1996. Molecular cloning of violaxanthin de-epoxidase from romaine lettuce and expression in Eschericia coli. Proc. Natl. Acad. Sci. 93 6320–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstein, E.A., Vedenkina, N.S., and Ivkova, M.N. 1973. Fluorescence and the location of tryptophan residues in protein molecules. Photochem. and Photobiol. 18 263–279. [DOI] [PubMed] [Google Scholar]
- Bychkova, V.E., Berni, R., Rossi, G.L., Kutyshenko, V.P., and Ptitsyn, O.B. 1992. Retinol-binding protein is in the molten globule state at low pH. Biochemistry 31 7566–7571. [DOI] [PubMed] [Google Scholar]
- CCP4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Clark, P.L., Liu, Z., Zhang, J., and Gierasch, L.M. 1996. Intrinsic tryptophans of CRABPI as probes of structure and folding. Protein Sci. 5 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogan, U., Kopelman, M., Mokady, S., and Shinitsky, M. 1976. Binding affinities of retinol and related proteins to retinol binding proteins. Eur. J. Biochem. 65 71–78. [DOI] [PubMed] [Google Scholar]
- Colantuoni, V., Romano, V., Bensi, G., Santoro, C., Costanzo, F., Raugei, G., and Cortese, R. 1983. Cloning and sequencing of a full-length cDNA for human retinol-binding protein. Nucl. Acids Res. 11 7769–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles, M., Diercks, T., Muehlenweg, B., Bartsch, S., Zolzer, V., Tschesche, H., and Kessler, H. 1999. The solution structure and dynamics of human neutrophil gelatinase-associated lipocalin. J. Mol. Biol. 289 139–157. [DOI] [PubMed] [Google Scholar]
- Collet, C., Joseph, R., and Nicholas, K. 1989. Molecular cloning and characterization of a novel marsupial milk protein gene. Biochem. Biophys. Res. Commun. 164 1380–1383. [DOI] [PubMed] [Google Scholar]
- Cowan S.W., Newcomer, M.E., and Jones, T.A. 1990. Crystallographic refinement of human serum retinol-binding protein at 2 Å resolution. Proteins 8 44–61. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M. 2001. The structural basis of protein folding and its links with human disease. Phil. Trans. R. Soc. B 356 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engh, R.A. and Huber, R. 1991. Accurate bond and angle parameters for X-ray structure refinement. Acta Crystallogr. A47 100–119. [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of Molscript that includes greatly enhanced coloring capabilities. J. Mol. Graphics and Modelling 15 132–134. [DOI] [PubMed] [Google Scholar]
- Fex, G., Albertsson, P., and Hansson, B. 1979. Interaction between prealbumin and retinol-binding protein studied by affinity chromatography, gel filtration and two-phase partition. Eur. J. Biochem. 99 353–360. [DOI] [PubMed] [Google Scholar]
- Flower, D.R. 1996. The lipocalin protein family: Structure and function. Biochem. J. 318 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower, D.R., North, A.C.T., and Attwood, T.K. 1993. Structure and sequence relationships in the lipocalins and related proteins. Protein Sci. 2 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Prior, P., Noeske-Jungblut, C., Donner, P., Schleuning, W., Huber, R., and Bode, W. 1997. Structure of the thrombin complex with triabin, a lipocalin-like exosite-binding inhibitor derived from the triatomine bug. Proc. Natl. Acad. Sci. 94 11845–11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina, M.D., Gutierrez, G., Bastiana, M., and Sanchez, D. 2000. A phylogenetic analysis of the lipocalin protein family. Mol. Biol. Evol. 17 114–126. [DOI] [PubMed] [Google Scholar]
- Greene, L. 1998. "Investigation into the relationship between sequence, structure and folding in a model lipocalin, human serum retinal-binding protein." Ph.D. thesis, University of Miami, Florida.
- Greene, L. and Brew, K. 1995. Conserved residues in the lipocalins suggest a folding m. Protein Eng. 8 S100. [Google Scholar]
- Greenfield, N.J. 1996. Methods to estimate the conformation of proteins and polypeptides from circular dichroism data. Anal. Biochem. 235 1–10. [DOI] [PubMed] [Google Scholar]
- Gregoire, C., Rosinski-Chupin, I., Rabillon, J., Alzari, P.M., David, B., and Dandeu, J. 1996. cDNA cloning and sequencing reveal the major horse allergen Equ c1 to be a glycoprotein member of the lipocalin superfamily. J. Biol. Chem. 271 32951–32959. [DOI] [PubMed] [Google Scholar]
- Grobler, J.A., Wang, M., Pike, A., and Brew, K. 1994. Study by mutagenesis of the roles of two aromatic clusters of α-lactalbumin in aspects of its action in the lactose synthase system. J. Biol. Chem. 269 5106–5114. [PubMed] [Google Scholar]
- Hamada, D., Segawa, S., and Goto, Y. 1996. Non-native α-helical intermediate in the refolding of β-lactoglobulin, a predominantly β-sheet protein. Nat. Struct.Biol. 3 868–873. [DOI] [PubMed] [Google Scholar]
- Hamill, S.J., Cota, E., Chothia, C., and Clarke, J. 2000. Conservation of folding and stability within a protein family: The tyrosine corner as an evolutionary cul-de-sac. J. Mol. Biol. 295 641–649. [DOI] [PubMed] [Google Scholar]
- Huber, R., Schneider, M., Mayr, I., Muller, R., Deutzmann, R., Suter, F., Zuber, H., Falk, H., and Kayser, H. 1987. Molecular structure of the bilin binding protein (BBP) from Pieris brassicae after refinement at 2.0 Å resolution. J. Mol. Biol. 198 499–513. [DOI] [PubMed] [Google Scholar]
- Johnson, W.C., Jr. 1988. Secondary structure of proteins through circular dichroism spectroscopy. Ann. Rev. Biophys. Chem. 17 145–166. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 22 No 12, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kaji, E.H. and Lodish, H.F. 1993. Unfolding of newly made retinol-binding protein by dithiothreitol: Sensitivity to retinoids. J. Biol. Chem. 268 22188–22194. [PubMed] [Google Scholar]
- Katakura, Y., Totsuka, M., Ametani, A., and Kaminogawa, S. 1994. Tryptophan-19 of β-lactoglobulin, the only residue completely conserved in the lipocalin superfamily, is not essential for binding retinol, but relevant to stabilizing bound retinol and maintaining its structure. Biochim. Biophys. Acta 1207 58–67. [DOI] [PubMed] [Google Scholar]
- Kjeldsen, L., Johnsen, A.H., Sengelov, H., and Borregaard, N. 1993. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 268 10425–10432. [PubMed] [Google Scholar]
- Lamzin, V.S. and Wilson, K.S. 1993. Automated refinement of protein models. Acta Crystallogr. D 49 129–147. [DOI] [PubMed] [Google Scholar]
- Lascombe, M.B., Gregoire, C., Poncet, P., Tavares, G.A., Rosinski-Chupin, I., Rabillon, J., Goubran-Botros, H., Mazie, J.C., David, B., and Alzari, P.M. 2000. Crystal structure of the allergen Equ c 1: A dimeric lipocalin with restricted IgE-reactive epitopes. J. Biol. Chem. 275 21572–21577. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Lu, G. 2000. TOP: A new method for protein structure comparisons and similarity searches. J. Appl. Cryst. 33 176–183. [Google Scholar]
- Mach, H., Middaugh, C.R., and Lewis, R.V. 1992. Statistical determination of the average values of the extinction coefficients of tryptophan and tyrosine in native proteins. Anal. Biochem. 200 74–80. [DOI] [PubMed] [Google Scholar]
- Mantyjarvi, R., Parkkinen, S., Rytkonen, M., Pentikainen, J., Pelkonen, J., Rautiainen, J., Zeiler, T., and Virtanen, T. 1997. Complimentary DNA cloning of the predominant allergen of bovine dander: A new member in the lipocalin family. J. Allergy Clin. Immunol. 97 1297–1303. [DOI] [PubMed] [Google Scholar]
- Martinez, J.C. and Serrano, L. 1999. The folding transition state between SH3 domains as conformationally restricted and evolutionarily conserved. Nat. Struct. Biol. 6 1010–1016. [DOI] [PubMed] [Google Scholar]
- Melhus, H., Laurent, B., Rask, L., and Peterson, P.A. 1992. Ligand-dependent secretion of rat retinol-binding protein expressed in HeLa cells. J. Biol. Chem. 267 12036–12041. [PubMed] [Google Scholar]
- Monaco, H.L., Rizzi, M., and Coda, A. 1995. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 268 1039–1041. [DOI] [PubMed] [Google Scholar]
- Muccio, D.D., Waterhous, D.V., Fish, F., and Brouillette, C.G. 1992. Analysis of the two state behavior of the thermal unfolding of serum retinol-binding protein containing a single retinol ligand. Biochemistry 31 5560–5567. [DOI] [PubMed] [Google Scholar]
- Muller, H.N. and Skerra, A. 1993. Functional expression of the uncomplexed serum retinol-binding protein in Eschericha coli. J. Mol. Biol. 230 725–732. [DOI] [PubMed] [Google Scholar]
- Nagata, A., Suzuki, Y., Igarashi, M., Eguchi, N., Toh, H., Urade, Y., and Hayaishi, O. 1991. Human brain prostaglandin D synthase has been evolutionarily differentiated from lipophilic-ligand carrier proteins. Proc. Natl. Acad. Sci. 88 4020–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor, H.M. and Newcomer, M.E. 1999. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry 38 2647–2653. [DOI] [PubMed] [Google Scholar]
- Newcomer, M.E. 1993. Structure of the epididymal retinoic acid binding protein at 2.1Å resolution. Structure 1 7–18. [DOI] [PubMed] [Google Scholar]
- ————. 1995. Retinoid-binding proteins: Structural determinants important for function. FASEB J. 9 229–239. [DOI] [PubMed] [Google Scholar]
- Nishimura, C., Prytulla, S., Dyson, H.J., and Wright, P.E. 2000. Conservation of folding pathways in evolutionarily distant globin sequences. Nat. Struct. Biol. 7 679–686. [DOI] [PubMed] [Google Scholar]
- Nozaki, Y. 1972. The preparation of guanidine hydrochloride. Methods Enzymol. 26 43–50. [DOI] [PubMed] [Google Scholar]
- Ohgushi, M. and Wada, A. 1983. "Molten-globule state": A compact form of globular proteins with mobile side-chains. FEBS. Lett. 164 21–24. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Pervaiz, S. and Brew, K. 1985. Homology of β-lactoglobulin, serum retinol-binding protein and protein HC. Science 228 335–337. [DOI] [PubMed] [Google Scholar]
- ———. 1987. Homology and structure-function correlations between α1-acid glycoprotein and serum retinol-binding protein and its relatives. FASEB J. 1 209–214. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Larson, S., Ruczinski, I., Riddle, D.S., Thayer, E.C., Buchwitz, B., Davidson, A.R., and Baker, D. 2000. Evolutionary conservation in protein folding kinetics. J. Mol. Biol. 298 303–312. [DOI] [PubMed] [Google Scholar]
- Ragona, L., Pusterla, F., Zetta, L., Moaco, H.L., and Molinari, H. 1997. Ideication of a conserved hydrophobic cluster in partially folded bovine β-lactoglobulin at pH2. Folding and Design 2 281–290. [DOI] [PubMed] [Google Scholar]
- Rask, L., Anundi, H., and Peterson, P.A. 1979. The primary structure of the human retinol-binding protein. FEBS Lett. 104 55–58. [DOI] [PubMed] [Google Scholar]
- Read, R.J. 1986. Improved fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A42 140–149. [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, pp. 17.38–17.39.
- Sarkar, G. and Sommer, S.S. 1990. The "megaprimer" method of site-directed mutagenesis. Biotechniques 8 404–407. [PubMed] [Google Scholar]
- Sawyer, L., Papiz, M.Z., North, A.C.T., and Eliopoulos, E.E. 1985. Structure and function of bovine β-lactoglobulin. Biochem. Soc. Trans. 13 265–266. [Google Scholar]
- Sivaprasadarao, A. and Findlay, J.B.C. 1993. Expression of functional human retinol-binding protein in Eschericha coli using a secretion vector. Biochem. J. 296 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland, E.H. 1974. Aromatic contributions to circular dichroism spectra of proteins. CRC Crit. Rev. Biochem. 2 113–175. [DOI] [PubMed] [Google Scholar]
- Tegoni, M., Ramoni, R., Bignetti, E., Spinelli, S., and Cambillau, C. 1996. Domain swapping creates a third putative combining site in bovine odorant binding protein dimer. Nat. Struct. Biol. 3 863–867. [DOI] [PubMed] [Google Scholar]
- Thornton, J.M. 1992. Protein structures: The end point of the folding pathway. In Protein folding (ed. T.E. Creighton), pp 59–81. W.H. Freeman and Company, New York.
- Toh, H., Kubodera, H., Nakajima, N., Sekiya, T., Eguchi, N., Tanaka, T., Urade, Y., and Hayaishi, O. 1996. Glutathione-independent prostaglandin D synthase as a lead molecule for designing new functional proteins. Protein Eng. 9 1067–1082. [DOI] [PubMed] [Google Scholar]
- Van't Hof, W., Blankenvoorde, M.F.J., Veerman, E.C.I., and Amerongen, A.V.N. 1997. The salivary lipocalin Von Ebner's gland is a cysteine proteinase inhibitor. J. Biol. Chem. 272 1837–1841. [DOI] [PubMed] [Google Scholar]
- Wang, T.T.Y., Lewis, K.C., and Phang, J.M. 1993. Production of human plasma retinol-binding protein in Escherichia coli. Gene 133 291–294. [DOI] [PubMed] [Google Scholar]
- Weichsel, A., Andersen, J.F., Champagne, D.E., Walker, F.A., and Montfort, W.R. 1998. Crystal structures of a nitric oxide transport protein from a blood-sucking insect. Nat. Struct. Biol. 5 304–309. [DOI] [PubMed] [Google Scholar]
- Woody, R.W. 1995. Circular dichroism. Methods Enzymol. 246 34–71. [DOI] [PubMed] [Google Scholar]
- Zanotti, G., Berni, R., and Monaco, H.L. 1993a. Crystal structure of liganded and unliganded forms of bovine plasma retinol-binding protein. J. Biol. Chem. 268 10728–10738 [PubMed] [Google Scholar]
- Zanotti, G., Malpeli, G., and Berni, R. 1993b. The interaction of N-ethyl retinamide with plasma retinol-binding protein (RBP) and the crystal structure of the retinoid-RBP complex at 1.9-Å resolution. J. Biol. Chem. 268 24873–24379. [DOI] [PubMed] [Google Scholar]
- Zanotti, G., Ottonello, S., Berni, R., and Monaco, H.L. 1993c. Crystal structure of the trigonal form of human plasma retinol-binding protein at 2.5Å resolution. J. Mol. Biol. 230 613–624. [DOI] [PubMed] [Google Scholar]