

Abstract
The N-terminal SH2 domain from the p85α subunit of phosphatidylinositol 3` kinase is cleaved specifically into 9- and 5-kD fragments by limited proteolytic digestion with trypsin. The noncovalent SH2 domain complex and its constituent tryptic peptides have been investigated using high-resolution heteronuclear magnetic resonance (NMR). These studies have established the viability of the SH2 domain as a fragment complementation system. The individual peptide fragments are predominantly unstructured in solution. In contrast, the noncovalent 9-kD + 5-kD complex shows a native-like 1H-15N HSQC spectrum, demonstrating that the two fragments fold into a native-like structure on binding. Chemical shift analysis of the noncovalent complex compared to the native SH2 domain reveals that the highest degree of perturbation in the structure occurs at the cleavage site within a flexible loop and along the hydrophobic interface between the two peptide fragments. Mapping of these chemical shift changes on the structure of the domain reveals changes consistent with the reduction in affinity for the target peptide ligand observed in the noncovalent complex relative to the intact protein. The 5-kD fragment of the homologous Src protein is incapable of structurally complementing the p85 9-kD fragment, either in complex formation or in the context of the full-length protein. These high-resolution structural studies of the SH2 domain fragment complementation features establish the suitability of the system for further protein-folding and design studies.
Keywords: Fragment complementation, fragment reconstitution, protein–protein interactions, protein folding, nuclear magnetic resonance spectroscopy, limited proteolysis
Certain single-domain proteins show native-like structure and function as noncovalent complexes of their constituent peptide fragments. This complementation of domain fragments has been used to study protein–protein interactions in folding/binding events (Eustance and Schleif 1996; Neira et al. 1996; Pecorari et al. 1996; Vainshtein et al. 1996; Chaffotte et al. 1997; Gegg et al. 1997; Rietveld and Ferreira 1998; Chakshusmathi et al. 1999; Georgescu et al. 1999; Goldberg and Baldwin 1999; Honda et al. 1999; Kobayashi et al. 1999; Tsuji et al. 1999; Jourdan and Searle 2000; Yu et al. 2000; for review, see de Prat-Gay 1996) as a method to screen for protein interactions in vivo (Johnsson and Varshavsky 1994; Rossi et al. 1997; Pelletier et al. 1998; Remy and Michnick 1999) and for developing semisynthetic techniques by religation of the fragments (Homandberg and Laskowski 1979; Vogel and Chmielewski 1994; Vogel et al. 1996; for review, see Wallace 1995). Residual structure within the individual peptides of these noncovalent complexes has been investigated as a model for initiation sites of protein folding (Oas and Kim 1988; Wright et al. 1988; Dyson et al. 1992a, 1992b; for review, see de Prat-Gay 1996). Fragment complementation systems offer an opportunity to scrutinize the recognition elements necessary for peptides to associate and fold into a stable native-like protein domain complex. Whereas several peptide complexes have been investigated using structural and biophysical techniques, detailed high-resolution structural information is still limited (Kim et al. 1992; de Prat-Gay et al. 1994; Neira et al. 1996; Ladurner et al. 1997; Tasayco et al. 2000; Yu et al. 2000). High-resolution structural data on these complementation systems are needed to provide sufficiently detailed characterization of the molecular interactions necessary to understand the recognition processes that facilitate folding. Here, we present high-resolution NMR studies of the SH2 domain establishing this m as a suitable fragment complementation system for structural studies.
SH2 domains display many critical features that make them good candidates for complementation studies. SH2 domains are small and monomeric, contain no disulfide bonds, and do not require a ligand to stabilize the overall native α/β fold. SH2 domains are also highly conserved as a family in sequence, structure, and function. The domains are mediators of cellular-signaling pathways and bind peptides containing phosphorylated tyrosines in a sequence-dependent context. The human Src SH2 domain and the human N-terminal SH2 domain from the p85α subunit of phosphatidylinositol 3` kinase form 5- and 9-kD peptide fragments on trypsin cleavage at an exposed lysine in the flexible BC loop (Fig. 1 ▶). These two peptides have been shown to be capable of fragment complementation (Williams and Shoelson 1993). CD spectropolarimetry showed that the noncovalent SH2 domain complex displayed secondary structure similar to the native domain but was less stable to temperature denaturation. The noncovalent complexes retain limited biological activity and can bind their target peptide, albeit with an order of magnitude reduced affinity when compared to the native protein. It was proposed by Williams and Shoelson (1993) that the reduced binding affinity of the SH2 domain for its specific phosphopeptide was because of ineffective recognition of the phosphate by the cleaved BC-binding loop rather than to inefficient complex formation.
Fig. 1.

Backbone ribbon structure of the bovine p85α N-terminal SH2 domain (PDB 2PNB). Limited tryptic digestion cleaves the domain at an exposed lysine (K52) within the flexible BC loop between the βB and βC strands of the domain. The resulting 5- and 9-kD fragments are colored purple and red, respectively.
The 5- and 9-kD SH2 domain fragments divide the native domain structure along a large β-sheet structure within the interior of the protein. Thus, the two fragments associate along a large hydrophobic surface area that traverses the core of the domain. This presents an intriguing protein–protein recognition problem in which the recognition elements necessary for the two fragments to come together in solution also stabilize the hydrophobic core of the protein. A detailed understanding of these interactions will provide insight into the role of hydrophobic cores in stabilizing protein structure. In addition to these favorable physical characteristics, a wealth of sequence information is available for the domain family as well as structural information that could facilitate analysis of the sequence dependence of complex formation. Therefore, fragment reconstitution of SH2 domains is a promising model system for probing the molecular interactions governing folding interactions and protein stability. However, high-resolution structural data on the noncovalent complex of an SH2 domain are needed before a detailed understanding of the complementation process can be obtained.
In this study, heteronuclear NMR spectroscopy is used to characterize the intact protein, peptide fragments, and noncovalent complex of the p85 SH2 domain. Although the fragments are found to be highly disordered in solution, the noncovalent complex of these peptides shows a native-like structure. This is unusual, as it is more commonly found that residual structure exists in one of the complementation competent fragments, and few well-characterized systems have shown fragment complementation from completely disordered states (Neira et al. 1996; Chaffotte et al. 1997; Tasayco et al. 2000). Evaluation of the 1H-15N HSQC spectra of fragment complexes is a valuable analysis tool for determining which specific interactions between these fragments are perturbed in the noncovalent complex. Analysis of chemical shift changes between the native protein and noncovalent complex reveals only small differences, indicating that the two have similar three-dimensional structures. The differences rationalize the reduced phosphopeptide binding affinity observed in the complex. To evaluate the sequence specificity of complex formation, the 5-kD fragment of p85 was replaced by the homologous Src protein 5-kD fragment. It was found that this related sequence is incapable of structurally complementing the p85 9-kD fragment, either in bimolecular complex formation or in the context of a full-length chimeric protein.
Results
Characterization of the native p85 SH2 domain
Structural features of the native p85 SH2 domain were studied using NMR spectroscopy. The 1H-15N HSQC spectrum of the native p85 SH2 domain, seen in Figure 2A ▶, shows the peak dispersion and linewidths indicative of a stably folded protein. The structure of a slightly different construct of the p85 bovine SH2 domain has been solved previously using NMR (Booker et al. 1992). The bovine SH2 domain differs from the human p85α N-terminal SH2 domain at only three residues: D19N, N88S, and L119K. The backbone resonance assignments were obtained independently at our experimental conditions using standard heteronuclear NMR experiments on 13C, 15N uniformly labeled protein. Assignments are available as supplementary material and residues are numbered according to the bovine construct as given by Hensmann et al. (1994a). Positive 1H-15N heteronuclear NOEs were measured for all amide crosspeaks (residues S16–D123) except those assigned to the N and C termini and sidechains (Fig. 4A ▶) (Farrow et al. 1994). The positive NOEs are indicative of a stably folded macromolecule tumbling with an overall correlation time (τc) in the nanosecond time regime.
Fig. 2.

1H-15N HSQC spectra collected at 500 MHz of (A) 730 μM native p85, (B) 220 μM 5-kD peptide, and (C) 220 μM 9-kD peptide. All NMR samples were prepared in 50 mM potassium phosphate at pH 5.8, 50 mM NaCl, 0.02% NaN3, and 10% D2O. Crosspeaks were assigned in the native spectrum. The boxed region (A) has been enlarged and shown as an inset in the lower right-hand corner. Resonances arising from residues D83 and Q124 and residues D38 and I94 overlap.
Fig. 4.
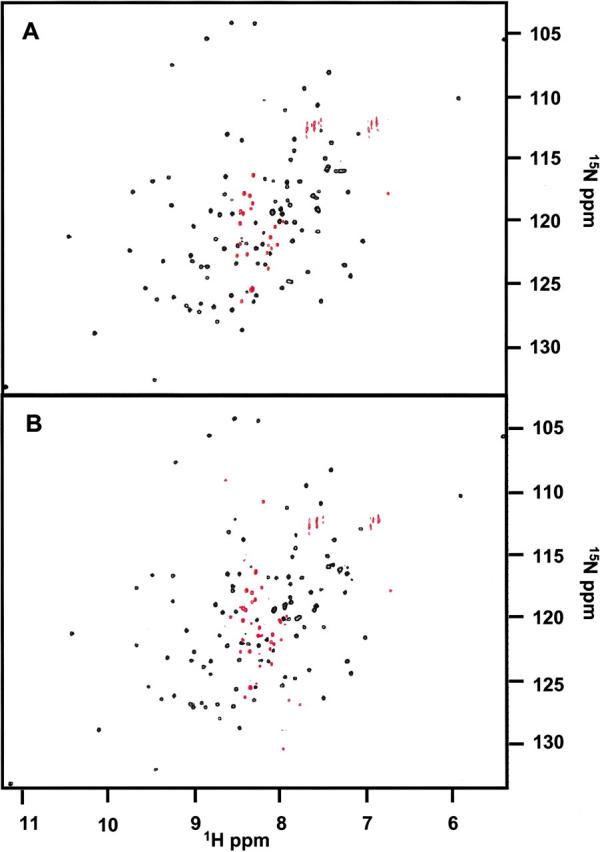
Heteronuclear NOE experiments for the native His-tagged p85 domain and complex. Positive peaks are shown in black and negative peaks in red for (A) a 240 μM sample of 15N-labeled native p85 SH2 domain and (B) a 100 μM noncovalent complex.
Characterization of the 5-kD p85 SH2 domain fragment
The ideication of residual structures in individual peptide fragments could provide insight to the folding and complementation pathway of the domain. To determine if residual structure is present, each isotopically labeled fragment was studied individually. The 1H-15N HSQC spectrum of 15N-labeled 5-kD fragment of p85, shown in Figure 2B ▶, differs dramatically from that of the native protein and strongly suggests that the peptide is unfolded in solution. The amide proton resonances show little chemical shift dispersion and fall between 7.5 and 8.5 ppm, consistent with random coil assignments for the 20 amino acids (Bundi and Wüthrich 1979; Braun et al. 1994; Wishart et al. 1995). The majority of the proton amide resonances expected for the 5-kD fragment are observed (40 out of 43), indicating that there are not multiple conformations exchanging slowly on the NMR timescale. The 1H-15N heteronuclear NOEs are uniformly negative throughout the sequence, which differs from what is observed in the intact p85 SH2 domain, indicating global motions on a subnanosecond time scale consistent with an unfolded peptide. No evidence for the presence of residual structure could be detected from a temperature denaturation study; no significant changes in the 1H-15N HSQC spectrum of the 5-kD fragment were observed over a temperature range of 25° to 50°C. Samples prepared in 5 M GndHCl also did not show any significant changes in the 1H-15N HSQC spectrum. These data are consistent with an unstructured peptide that is rapidly sampling many conformational states on the NMR timescale.
Characterization of the 9-kD p85 SH2 domain fragment
The 1H-15N HSQC spectrum of the 9-kD fragment shown in Figure 2C ▶, like the 5-kD fragment, shows little peak dispersion, suggesting that this fragment is also unstructured. However, unlike the 5-kD spectrum, there are variations in peak intensities and larger linewidths in the 1H-15N HSQC spectrum leading to a great degree of peak overlap, thus preventing accurate quantitation of the number of resonances observed. Although difficult to quantitate, the number of crosspeaks observed clearly exceeds the predicted number for the 9-kD fragment. The variation in linewidth and number of crosspeaks may be because of the presence of several conformers interconverting slowly on the NMR chemical shift time scale, which would be consistent with the presence of residual structure, or may be due to the presence of oligomers. The 1H-15N heteronuclear NOEs are negative for all of the amides in the 9-kD fragment, consistent with the majority of the fragment population being not highly structured in solution.
To explore the possibility of two populations in intermediate-to-slow exchange between a partially folded and unfolded state, denaturation studies of the 9-kD fragment with temperature and chaotropic agent were performed. 1-D and 2-D spectra were collected at 5°, 25°, and 45°C and examined for changes in chemical shifts and linewidths of the resonances. No significant changes were observed as a function of temperature. In addition, no significant changes in the spectrum were observed in either chemical shift or linewidth in the 1 H-15N HSQC spectrum in the presence of 5 M GndHCl. Denaturations monitored by CD spectropolarimetry also indicated that no cooperatively formed residual structure was present. From these data it seems unlikely that there is any cooperatively formed residual structure present within the 9-kD p85 fragment. Instead, these data suggest that the 9-kD fragment is nonspecifically self-associating into higher order structures.
DLS measurements were performed on the 9-kD fragment and compared to the native p85 SH2 domain to determine its aggregation state in solution. Cumulants analysis (Koppel 1972) of the native p85 domain gave an effective diameter of 4.73 ± 0.06 nm and a polydispersity of 0.06 ± 0.03. The expected diameter, as calculated from the solution structure (Booker et al. 1992), is 3–4 nm, which is in good agreement with the DLS results. In contrast, the effective diameter of the 9-kD fragment is 73 ± 4 nm, with a polydispersity of 0.21 ± 0.02. This is indicative of a solution containing multiple populations of higher order species in which the average oligomeric state consists of several 9-kD peptide molecules. The presence of aggregated states, however, is not surprising given the hydrophobic stretches within the 9-kD fragment, and that self association of peptide fragments have been observed in other complementation systems (Tasayco and Carey 1992; Kanaya and Kanaya 1995; Ladurner et al. 1997; Georgescu et al. 1999; Neira and Fersht 1999a). Addition of 5 M GndHCl to the 9-kD DLS sample resulted in a decreased effective diameter of 47 ± 2 nm and an increased polydispersity of 0.33 ± 0.02. Although the denaturant succeeded in lowering the average effective diameter by breaking up some aggregates, the number of populated aggregated states actually increased. This may explain the lack of significant changes in the 1H-15N HSQC spectrum of the 9-kD fragment that were observed with addition of denaturant.
The degree of self association may be exacerbated by the high concentrations (> 1 mM) necessary to do DLS analysis, but even at the lower concentrations used for NMR (100 μM), aggregation is likely to be an important contribution to the variation in NMR linewidths. However, the monomeric state must still be present in the population, because the 9-kD peptide resonances are readily observable by NMR. The presence of multiple states within the 9-kD sample, because of higher order oligomers, should not preclude high-resolution studies of the noncovalent peptide complexes, because the aggregated state(s) are apparently in equilibrium with the monomeric form. As shown below, formation of a specific structured complex between the 9- and 5-kD fragments is thermodynamically more stable than the 9-kD conformational ensemble.
Characterization of the noncovalent p85 SH2 domain complex
A dramatic change in the NMR spectrum is observed when the individual 5- and 9-kD fragments are combined. The 15N-labeled complex of the p85 SH2 domain 5-kD fragment with the 9-kD fragment shows an 1H-15N HSQC spectrum that is remarkably similar to the spectrum of the intact protein. The superposition of these two spectra is shown in Figure 3A ▶. The close overall match of the chemical shifts of the noncovalent complex with the intact protein indicates that their structures are very similar. In addition, the 1H-15N heteronuclear NOEs are positive for the same crosspeaks as in the native domain, indicating the presence of a stably folded protein complex (Fig. 4 ▶). These observations show that native-like structure is essentially completely recovered on complementation, because the 1H-15N HSQC spectrum is exquisitely sensitive to the precise details of the molecular environment of each amide proton and nitrogen.
Fig. 3.


(A) Superposition of the 1H-15N HSQC spectra for the noncovalent complex of His-tagged p85, titrated to a 1:1 complex at 100 μM (red), and native His-tagged p85 at 240 μΜ (black). Most of the native crosspeaks are completely recovered in the complex spectrum; however, some small chemical shift differences are readily apparent. The His-tagged 9-kD fragment allowed for a better 1:1 titration of peptide fragments as it was slightly less susceptible to aggregation. Titrations using 9-kD fragments without the His-tag also produced native-like spectra and these were used for further chemical shift analysis. (B) Temperature denaturation of the p85 His-tagged noncovalent complex. One-dimensional proton spectra at 25°, 30°, 35°, 40°, 45°, and 50°C are shown for a noncovalent complex of p85 at 100 μM peptide fragments. Native peaks disappear and denatured peptide peaks arise as the temperature is raised.
The titration of 5-kD peptide with 9-kD peptide showed the complex to be in slow exchange with the fragments on the NMR chemical shift timescale. NMR spectra obtained on serial dilutions of the complex down to a 5 μM sample still displayed peaks consistent with a native-like HSQC spectrum, indicating that the upper limit for the Kd of the complex is in the low to sub μM range, consistent with values obtained on other fragment complementation systems (Sancho and Fersht 1992; Wu et al. 1993; de Prat-Gay and Fersht 1994; Kobayashi et al. 1995, 1999; Georgescu et al. 1999). To test the thermal stability of the complex, one-dimensional 1H spectra were collected at temperatures of 25°, 30°, 35°, 40°, 45°, and 50°C. These data, shown in Figure 3B ▶, indicate a reduction in thermal stability relative to the intact protein. Signals from the folded state melted out between 40° and 45°C, whereas the intact protein was stable to 65°C, consistent with reported thermal denaturations monitored by CD spectropolarimetry (Williams and Shoelson 1993).
Strikingly, nearly all of the native 1H-15N crosspeaks observed in the native p85 SH2 domain (Fig. 3A ▶) are recovered in the p85 complex with only some slight chemical shift changes. Native structure is clearly recovered on fragment complementation. Therefore, the reduction in stability of the complex is not because of any significant change in structure compared to the native domain. The 15N chemical shift changes (Fig. 5A ▶) were mapped onto the structure of the p85 SH2 domain to localize the regions of structural difference between the intact and complexed protein (Fig. 5B ▶). The most dramatic differences occur in the loop residues containing the trypsin cleavage site (K52) that separates the 5- and 9-kD fragments. Smaller chemical shift changes were observed at the interface between fragments near the cleavage site, and some additional changes were observed for the N terminus of the α-helix in the 5-kD peptide, which were likely because of fraying of the helix at that terminus. Thus, when the two unfolded peptide fragments of p85 are added back to each other they are able to fold spontaneously into a stable structure. This folded structure is essentially structurally identical to the native domain and shows only a moderate change in melting temperature.
Fig. 5.


Chemical shift changes observed in 1H-15N HSQC spectra between the noncovalent complex of p85 and a native p85 sample. (A) Relative 1H (gray) and 15N (black) chemical shift changes that occur at each residue in the sequence. Residues 84, 107, and 116 are prolines. Residue 50S was not assigned. Additional residues are not included because of uncertainties in assignment resulting from spectral overlap (75R, 102L, 121Q, and 126V). For residues 51 through 56, no readily ideiable complex peaks are observed, indicating a large change in their immediate chemical environments. These residues were assigned a lower limit of 15N chemical shift of 1.0 ppm and 1H chemical shift of 0.1 ppm for the purpose of displaying them on the graph. (B) 15N chemical shift changes mapped on the p85 backbone structure. Residues for which there are no data are depicted in white. Residues that experience no appreciable chemical shift changes are colored gray. Small changes between 0.1 and 0.3 ppm are colored yellow, medium changes with chemical shift changes > 0.3 ppm are colored orange, and residues with changes in crosspeaks that are > the lower limit of 1.0 ppm in 15N and 0.1 ppm in 1H are colored red. Overall, the most significant changes occur at the site of trypsin cleavage (K52) in the BC loop along the interface between the fragments, especially at the ends of strands near the BC loop, and at the N terminus of helix A in the 5-kD fragment. Specific residues of interest at the interface and in phosphopeptide binding are labeled.
Investigation of the sequence specificity of complex formation
The specificity of this interaction between SH2 domain fragments was examined by testing the folding capability of the corresponding N-terminal 5-kD fragment of the human Src SH2 domain with the 9-kD p85 SH2 domain fragment. The Src SH2 domain has high sequence and structure homology (superposition of 5-kD region with 1.5 Å RMSD) to the p85 SH2 domain (Fig. 6A ▶), and the corresponding Src 5- and 9-kD fragments can form a partially active complex (Williams and Shoelson 1993). The sequence identity of the complementary Src 5-kD peptide to the p85 5-kD peptide is 42%, and most of the hydrophobic core residues are conserved in this fragment between the two domains. However, despite the high structure and sequence homology between the two SH2 domains, no complex formation was observed by NMR under a variety of conditions (Fig. 6B,C). Complex formation could not be induced by attempting to refold the complex from either thermal or chaotropic denatured states. Thus, the folding recognition between p85 SH2 domain fragments is not conserved between the homologous domains.
Fig. 6.

(A) Sequence alignment of the human Src and p85 SH2 domain sequences. The two domains have been aligned by the method proposed by Eck et al. (1993), and conserved elements of sequence are shaded in gray. Secondary structural elements and numbering are defined by the crystal and solution structures (Booker et al 1992;Hensmann et al 1994;Xu et al. 1995) and are shown above and below the sequences, respectively. The lysine at which trypsin digestion produces 5- and 9-kD fragments is starred. (B) 1H-15N HSQC spectrum of synthetic 5-kD p85 peptide combined with 15N-labeled 9-kD p85 peptide, both at 180 μM. Native-like crosspeaks are observed. (C) 1H-15N HSQC spectrum of synthetic Src 5-kD peptide combined 1:1 at 40 μM with 15N-labeled 9-kD p85 peptide. The Src 5-kD fragment is unable to complement the p85 9-kD fragment, and a spectrum similar to 9-kD peptide alone is observed.
The inability of the Src 5-kD peptide to form a stable complex when swapped with its complementary p85 fragment could be attributed to destabilization of the complex because of the missing peptide bond between fragments rather than a lack of conserved folding elements between the two domains. To test this possibility, a Src-p85 SH2 domain chimera was designed in which the hybrid would be expressed in Escherichia coli cells as a fusion protein between the 5-kD fragment of Src and the 9-kD fragment of p85. Attempts to induce chimera expression at three different cell densities and temperatures failed to result in any expression of a stable fusion protein, whereas the wild-type p85 and Src SH2 domains express well under all conditions. The only indication of induced protein expression occurred at 16°C; however, the protein was approximately half the target molecular weight and had a low abundance. This suggests that the unfolded Src-p85 chimera protein is rapidly being proteolyzed within the cytoplasm of the cell on expression and cannot be driven into inclusion bodies. Extensive mutation of the Src-like region back to p85 residues such that the interface is 81% identical did allow for the expression of folded full-length protein with significantly reduced stability relative to wild type (Tm = 30°C). Therefore, the inability of the Src 5-kD and p85 9-kD fragments to fold into a stable native-like SH2 domain as either the complex or full-length protein is attributable to missing sequence/structure information or the introduction of a disruptive contact at the interface.
Discussion
Fragment reconstitution of proteins has been shown to provide valuable information in protein-folding and recognition studies. However, few high-resolution structural studies have been performed on these fragment reconstitution systems, including the complete structures of RNaseS (Kim et al. 1992) and the chymotrypsin inhibitor-2 noncovalent complex (Neira et al. 1996). The p85 SH2 domain complementation system is potentially an excellent model in which to study these folding and recognition events. When the p85 SH2 domain is cleaved into two peptide fragments with trypsin, the isolated fragments do not retain their native phosphopeptide-binding activity. However, on reconstitution of the peptide fragments, not only is native activity partially restored (Williams and Shoelson 1993), but we have shown by high-resolution NMR that the native structure is restored as well. Chemical shift changes of the backbone amide resonances in the complex relative to the native protein reveal structural differences between the noncovalent complex and the native domain that are quite modest. As expected, the most dramatic changes are observed at the cleavage site. Chemical shifts in residues flanking the cleavage site at position 52 differ so dramatically in the complex that comparison with the native domain does not allow for assignment. Resonances for residues found along the interface near the site of cleavage and the terminus of one helix shift slightly, yet can be assigned readily by comparison with the native spectrum.
Comparison of the observed chemical shift changes between the noncovalent complex and the native p85 SH2 domain reveals small structural differences which explain the reduced phosphopeptide-binding activity of the complex (Williams and Shoelson 1993). Cleavage of the BC loop of the protein has been proposed to be responsible for reduced binding affinity for its cognate phosphopeptide. This loop creates the pocket that specifically binds the phosphate of the tyrosine, and significant chemical shift changes were observed in the loop on phosphopeptide binding in the solution structure of the bovine p85 SH2 domain (Hensmann et al. 1994a). The greatest chemical shift changes in the noncovalent complex localize to this region (Fig. 5A,B). A significant chemical shift change is also observed for R29 located in the N terminus of the 5-kD fragment α-helix. This arginine is highly conserved among SH2 domains and has been observed to form a hydrogen bond to the phosphate and a stabilizing cation-π interaction with the aromatic ring of the tyrosine in the phosphopeptide in the Src/peptide structure (Waksman et al. 1993). This residue has also been shown to undergo a chemical shift change in p85 when bound to a phosphopeptide (Hensmann et al. 1994a). A change in the chemical environment for this arginine may indicate a slight structural change that leads to loss of affinity for the phosphopeptide. Significant chemical shift changes in the noncovalent complex, however, were not observed for K71, which forms the second cation-π interaction with the opposite face of the tyrosine ring. A slight perturbation in chemical shift of the noncovalent complex is also observed at L69, which lines the hydrophobic pocket of the peptide-binding site. The reduction in binding affinity for the phosphopeptide to the noncovalent p85 SH2 domain does not appear to be because of a loss of specificity for the cognate peptide. No significant chemical shift changes were observed for F81, which has been proposed to provide specificity of binding by interaction with the +3 methionine of the phosphopeptide (Waksman et al. 1993; Hensmann et al. 1994a). Thus, loss of phosphopeptide-binding activity in the noncovalent p85 complex is mostly attributed to slight structural perturbations at residues involved in binding the phosphate and tyrosine ring rather than those involved in binding the remaining residues of the peptide ligand.
Studies of the individual peptides have revealed that the p85 complex forms from unfolded fragment states; thus, the peptides fold on binding to create the noncovalent complex. The interactions that stabilize the complex are likely to be the same as those that stabilize the protein. Residual structures in protein fragments have been observed previously in proteins that are capable of fragment reconstitution (Sancho et al. 1992; de Prat-Gay and Fersht 1994; Kanaya and Kanaya 1995; Wang and Shortle 1997; Llinas and Marqusee 1998; Honda et al. 1999; Kobayashi et al. 1999; Neira and Fersht 1999a Neira and Fersht 1999b; Tasayco et al. 2000) and have been proposed to be nucleation sites for protein folding in which the structural m serves as the basis for molecular recognition (Wright et al. 1988; Dyson et al. 1992a, 1992b). No evidence for residual structure in the SH2 domain fragments has been found by NMR using chemical shift analysis, denaturation studies, and heteronuclear NOE data. The fact that the p85 peptides are predominantly unstructured makes this an interesting protein–protein recognition problem and an unusual fragment complementation system. Recognition of a disordered state suggests that the fragments are induced to cooperatively fold as opposed to recognition of a stable structural m that is preformed in one of the fragments.
The important interactions for formation of a fragment complex with native structure likely occur along the buried surface area at the interface between the p85 peptide fragments in the noncovalent complex. This buried surface area between the fragments, as measured on the native structure, is quite large, i.e., 2.36 × 103 Å2 of buried surface area measured using MOLMOL (Koradi et al. 1996), and traverses the hydrophobic core of the domain. In addition to the interactions at the interface between the β2 and β3 strands, extensive side-chain contacts between the 5- and 9-kD fragments also occur between the β1 strand (W22, Y23) and the α2 helix (V90, V91, I94), between the α1 helix (V32, N33, L36) and the β2 strand. Only small chemical shift perturbations were observed for these additional interfacial residues with the exception of N33, which lies close to the surface of the protein near N67 and R62. These important interactions in the native domain appear to be intact in the noncovalent complex. Because of the high degree of sequence and structural conservation between the p85 and Src SH2 domains, in particular at the interface (50%), we tested whether a p85/Src chimera is capable of folding. The 5-kD peptide of the Src SH2 domain is not able to fold into a native-like complex with the 9-kD fragment of the p85 SH2 domain. Even a chimeric Src 5-kD-p85 9-kD fusion protein was incapable of folding into a stable domain. Thus, the results of the fragment complementation experiments mimic the behavior of the full-length protein, establishing that fragment complementation is a valuable tool for probing protein stability. The lack of stability in both the chimeric complex and chimeric protein suggests that either specific interactions at the interface between the 5- and 9-kD fragments through the hydrophobic core of the protein are required to form a stable fold, or that an unfavorable interaction has been introduced by the swap. These interactions can be probed easily along this large interface for their impact on complex formation and protein stability, and further work in this area is currently in progress.
Conclusion
Reconstitution of protein fragments is an excellent tool for probing protein–protein and protein-folding interactions. The human p85α N-terminal SH2 domain is well-suited for these studies for several reasons. First, it is a small monomeric domain that contains no disulfide bonds and does not require additional ligands to form a stable noncovalent complex. Second, a wealth of sequence and structure information is available on SH2 domains, because nearly 200 of these domains have been ideied by sequence homology, and several crystal and solution structures are available. High-resolution structural characterization of this noncovalent complex system has shown that the structure of the fragment complex is nearly identical to the native p85 SH2 domain. In contrast, the peptide fragments are predominantly unstructured in solution, establishing complex formation as a folding-on-binding event. Complex formation is sequence specific because fragment peptides from two highly conserved domains were unable to structurally complement either as a noncovalent complex or in the intact chimeric protein. Examination of the specific interactions that enable the noncovalent complex to form will provide insight as to the nature of recognition elements necessary for formation of a stable protein core.
Materials and methods
Materials
All chemicals used for protein expression, purification, and sample preparation were purchased from Fisher Scieic and Sigma Chemical Company. Uniformly 15N-labeled (NH4)2SO4 and 13C-labeled glucose were obtained from Cambridge Isotope Laboratories. Preloaded amino acid resins for peptide synthesis were purchased through Calbiochem-Novabiochem. Additional peptide synthesis reagents and N-terminal Fmoc-protected amino acids were purchased from PE Applied Biosystems. Expression vectors were obtained from Novagen. Oligonucleotides were purchased from New England Biolabs or GibcoBRL Life Technologies. Restriction enzymes were purchased from Promega, GibcoBRL, or NEB.
Expression and purification of p85 and His-tagged p85
The p85α N-terminal SH2 domain, i.e., residues 323–438 of the p85α subunit gene product, which correspond to residues 12–129 as defined by Hensmann et al. (1994a), was expressed in BL21(DE3). E. coli cells transformed with a pET21a expression vector containing the recombinant gene, which had been subcloned into the vector using NdeI and EcoRI restriction enzyme sites (p85 ANNMSLQNAEWYWGDISREEVNEKLRDTADGTFLVRDA STKMHGDYTLTLRKGGNNKLIKIFHRDGKYGFSDPLTFSSV VELINHYRNESLAQYNPKLDVKLLYPVSKYQQDQVVKED). Cultures were grown 12–16 hr after induction with 1 mM IPTG at 25°C. Cells were lysed by French press and protein was precipitated with 90% (w/v) ammonium sulfate following a 60% (w/v) cut. Protein was further purified by cation exchange chromatography on a 22 mL Pharmacia SP-Sepharose column in 20 mM potassium phosphate at pH 6.0. Eluted fractions of protein were > 99% pure as verified by SDS-PAGE. Eluted fractions were combined, dialyzed into deionized distilled water, and lyophilized. Samples were reconstituted in an appropriate buffer.
A His-tagged construct of the p85 SH2 domain was also expressed in E. coli cells via a pET21a vector (ANNMSLQNAEWY WGDISREEVNEKLRDTADGTFLVRDASTKMHGDYTLTLR KGGNNKLIKIFHRDGKYGFSDPLTFSSVVELINHYRNESLA QYNPKLDVKLLYPVSKYQQDQVVKEDNQNSSSVSQLAAA LEHHHHHH). Cell cultures were induced with 1 mM IPTG at 25°C at mid-log phase and grown 12–16 hr. The cell lysate was made to 10 mM Tris at pH 8.0 and passed over a Pharmacia metal chelating column bound with Ni2+. Nonspecific protein interactions were disrupted with 50 mM imidazole and the > 99% pure p85 His-tagged protein was eluted in 250 mM imidazole. Samples were dialyzed into deionized distilled water, lyophilized, and reconstituted in an appropriate buffer.
Generation and purification of peptide fragments
Fragments were generated by limited proteolytic digest with trypsin as described by Williams and Shoelson (1993). Lyophilized p85 or p85 His-tagged protein was dissolved in 0.1 M sodium bicarbonate at pH 8.0 to a concentration of 120 μM. Trypsin was added to a final concentration of 0.2% (w/w). Digests were performed at 20°C for 1 hr. Immediately following the digest, fragments were purified by reverse-phase HPLC chromatography on a Vydac C18 semipreparative column with an acetonitrile gradient in 0.1% TFA. Fractions were combined, lyophilized, and reconstituted in the appropriate buffer. The mass of the 9-kD, His-tagged 9-kD, and 5-kD fragments was confirmed by MALDI TOF mass spectrometry (5-kD p85 fragment ANNMSLQNAEWYW GDISREEVNEKLRDTADGTFLVRDASTK, 9-kD p85 fragment MHGDYTLTLRKGGNNKLIKIFHRDGKYGFSDPLTFSSVVE LINHYRNESLAQYNPKLDVKLLYPVSKYQQDQVVKED, Histagged 9-kD p85 fragment MHGDYTLTLRKGGNNKLIKIFHR DGKYGFSDPLTFSSVVELINHYRNESLAQYNPKLDVKLLY PVSKYQQDQVVKEDNQNSSSVSQLAAALEHHHHHH).
NMR experiments
Uniformly 15N-isotopically labeled protein was produced by expression in minimal media containing 6.7 g/L Na2HPO4, 3 g/L KH2PO4, 1.5 g/L NaCl, 2 g/L glucose, 10 mL/L Basal Medium Eagle Vitamin solution from GibcoBRL, 162.2 μg/L FeCl3, 286 μg/L H3BO4, 1.5 mg/L CaCl2.2H2O, 4 μg/L CoCl2.6H2O, 20 μg/L CuSO4.5H2O, 20.8 mg/L MgCl2.6H2O, 0.2 μg/L MoO3, 20.8 μg/L ZnCl2, and 1.5 g/L (15NH4)2SO4. Uniformly 15N, 13C-labeled protein was produced under the same conditions using D-glucose (6-13C, 99%).
All NMR samples were prepared in 50 mM potassium phosphate buffer at pH 5.8, 50 mM sodium chloride, 0.02% sodium azide, and 10% deuterium oxide unless otherwise specified. Experiments were run on a Varian Unity Inova 500 MHz spectrometer and spectra were processed with NMRPipe software using a cosine apodization function and one round of zero filling (Delaglio et al. 1995). All experiments were run at 25°C unless otherwise specified. Typical 1H-15N HSQC spectra were obtained with 2048 points and 128 t1 increments using a gradient sensitivity-enhanced pulse sequence (Kay et al. 1992). The 1H-15N heteronuclear NOE experiment used 2048 points, 128 t1 increments, and 2.5 sec for the NOE to build on proton saturation (Farrow et al. 1994).
Reassignment of the 1H-15N HSQC assignments published by Hensmann et al. (1994a) was necessary because of consistent differences reported for 15N and 1H chemical shifts (Hensmann et al. 1994aHensmann et al. 1994b). Backbone amides and Cα carbons of the p85 SH2 domain were assigned using standard multidimensional NMR techniques. The experiments performed included a gradient sensitivity-enhanced HNCA (Ikura et al. 1990; Grzesiek and Bax 1992; Kay et al. 1994; Muhandiram and Kay 1994) using 1024 points, 64 t1 increments, and 32 t2 increments, a gradient sensitivity-enhanced HN(CO)CA (Shaka 1985; Patt 1992; Yamazaki et al. 1994) using 1024 points, 64 t1 increments, and 32 t2 increments, and an HSQC-TOCSY and HSQC-NOESY spectra using 1024 points, 64 t1 increments, 128 t2 increments, and 10 DIPSI-3 cycles with 69.1 msec TOCSY mixing time and 125 msec NOE mixing time, respectively (Marion et al. 1989; Talluri and Wagner 1996). Assignments are available as supplemental material.
Chemical shift analysis of the noncovalent p85 α N-terminal SH2 domain complex
Chemical shift values in 1H-15N HSQC spectra were measured using the Pipp peak-picking software package (Garrett et al. 1991). The noncovalent p85 SH2 domain complex chemical shift assignments were made by superposition of the complex 1H-15N HSQC spectrum with the native domain spectrum. The great similarity between these two spectra allowed a quick assignment of nearly all the crosspeaks by visual inspection. Peaks shifted > 1.0 ppm in 15N and 0.1 ppm in 1H, when compared to the native spectrum, were not assigned but are indicated in Figure 4 ▶ with a lower limit of 1.0 and 0.1 ppm, respectively.
Dynamic light scattering
12 mg/mL samples of native p85 and 9-kD fragment were prepared in 50 mM potassium phosphate buffer at pH 5.8 and 50 mM NaCl and filtered through a 0.2 micron filter. Samples were examined by dynamic light scattering using a Brookhaven Instruments Corp. (BIC) system with a BI-200SM goniometer and BI-9000AT digital correlator and with a 532 nm vertically polarized solid-state laser (Uniphase, μGreen laser) with 125-mW power output. Light scattering was detected at a 90° angle over the course of 10 min using delay times from 175–500 msec. Three data sets of each sample were collected as an autocorrelation function, which was analyzed by cumulants analysis to give an average effective solution diameter (Koppel 1972).
SH2 domain fragment swapping experiment
The Src SH2 domain 5-kD peptide (residues 143–183 of the human Src gene product) was synthesized on an ABI 433A peptide synthesizer using standard Fmoc chemistry for batchwise solid-phase peptide synthesis on an Fmoc-Lys(Boc)-Wang resin (for review, see Fields and Noble 1990). N-terminal Fmoc-protecting groups were removed in 50% piperidine/DMF. Activation of amino acids before coupling was achieved by HBTU/HOBt and DIEA methodologies. Final deprotection of sidechains and cleavage from the resin were performed by treatment of the resin with trifluoracetic acid for 3 hr. The peptide was precipitated with diethyl ether and resolubilized in 10% ACN, 10% MeOH, and 0.1% TFA. The peptide was then purified by reverse-phase HPLC on a Vydac C18 semipreparative column using an acetonitrile gradient in 10% MeOH and 0.1% TFA. Fractions were lyophilized and the mass of the peptide confirmed by MALDI TOF mass spectrometry (Src 5-kD peptide DSIQAEEWYFGKITRRESERLLLNAEN PRGTFLVRESETTK). An equal molar amount of the 5-kD Src SH2 peptide was combined with 15N-labeled 9-kD C-terminal fragment of the p85 SH2 domain in the same NMR solution conditions described above. 1H-15N HSQC spectra were collected for each sample and examined for evidence of complex formation. Additional samples were taken up in 5 M GndHCl and slowly dialyzed back into NMR buffer to help induce complex formation.
Design and expression of the p85/Src chimera
Primers were designed containing NdeI and NsiI restriction sites to amplify the gene encoding the 5-kD fragment of the Src SH2 domain (Src 5-kD fragment AWYFGKITRRESERLLLNAEN PRGTFLVRESETTK). Standard recombinant techniques were used to create a pET21a plasmid coding for the N-terminal 5-kD fragment of the Src SH2 domain fused to the C-terminal 9-kD fragment of the p85 SH2 domain using the existing pET21a vector containing the p85 gene. BL21(DE3) cells were transformed with the plasmid and single colonies were grown to an O.D.600 of ∼0.4, 0.7, and 1.0 on which time they were induced with 1 mM IPTG. Timepoints were collected over the course of 16 hr at 16°, 25°, and 37°C and analyzed for induction of protein expression by SDS PAGE.
Electronic supplemental material
The complete 1H, 15N, and 13Cα backbone chemical shift assignments for the p85 SH2 domain.
Acknowledgments
We thank Drs. Tony Pawson and Gerry Gish for donating the vectors harboring the genes for the p85α N-terminal SH2 domain and the Src SH2 domain and Drs. Steven Cabe and Theodore Randolph for instruction and use of the DLS instrumentation. We acknowledge Jonathan Bleyhl, Sarah Lehto, and Daniel Strauss for contributions in subcloning work. We thank Drs. Arthur Pardi and Fiona Jucker for thoughtful comments on the manuscript. We gratefully acknowledge funding for this work, which was given by the Research Corporation (RI0155), the Petroleum Research Fund (32218-G), a Beckman Young Investigator Award, and a National Science Foundation Career Award (MCB 9875663).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
SH2, Src homology 2
NMR, nuclear magnetic resonance
HSQC, heteronuclear single quantum coherence
p85, N-terminal SH2 domain of the p85α
subunit of phosphatidylinositol 3` kinase
CD, circular dichroism
MALDI TOF, matrix assisted laser desorption ionization time of flight
DLS, dynamic light scattering
ACN, acetonitrile
GndHCl, guanidine hydrochloride
IPTG, isopropyl thiogalactoside
SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis
Fmoc, 9-fluorenylmethoxycarbonyl
HBTU, 2–(1-H-benzotriazole-1-yl)-1, 1, 3, 3-tetramethyluronium hexafluorophosphate
HOBt, N-hydroxybenzotriazole
DMF, N, N-dimethylformamide
DIEA, N, N-diisopropylethylamine
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.18701.
Supplemental material: See www.proteinscience.org.
References
- Booker, G.W., Breeze, A.L., Downing, A.K., Panayotou, G., Gout, I., Waterfield, M.D., and Campbell, I.D. 1992. Structure of an SH2 domain of the p85-alpha subunit of phosphatidylinositol-3-OH kinase. Nature 358 684–687. [DOI] [PubMed] [Google Scholar]
- Braun, D., Wider, G., and Wüthrich, K. 1994. Sequence-corrected N-15 random coil chemical shifts. J. Am. Chem. Soc. 116 8466–8469. [Google Scholar]
- Bundi, A. and Wüthrich, K. 1979. H-1-NMR parameters of the common amino-acid residues measured in aqueous-solutions of the linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers 18 285–297. [Google Scholar]
- Chaffotte, A.F., Li, J.-H., Georgescu, R.E., Goldberg, M.E., and Tasayco, M.L. 1997. Recognition between disordered states: Kinetics of the self-assembly of thioredoxin fragments. Biochemistry 36 16040–16048. [DOI] [PubMed] [Google Scholar]
- Chakshusmathi, G., Ratnaparkhi, G.S., Madhu, P.K., and Varadarajan, R. 1999. Native-state hydrogen-exchange studies of a fragment complex can provide structural information about the isolated fragments. Proc. Natl. Acad. Sci. 96 7899–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Prat-Gay, G. 1996. Association of complementary fragments and the elucidation of protein folding pathways. Protein Eng. 9 843–847. [DOI] [PubMed] [Google Scholar]
- de Prat-Gay, G. and Fersht, A.R. 1994. Generation of a family of protein fragments for structure-folding studies. 1. Folding complementation of two fragments of chymotrypsin inhibitor-2 formed by cleavage at its unique methionine residue. Biochemistry 33 7957–7963. [DOI] [PubMed] [Google Scholar]
- de Prat-Gay, G., Ruiz-Sanz, J., Davis, B., and Fersht, A.R. 1994. The structure of the transition state for the association of two fragments of the barley chymotrypsin inhibitor 2 to generate native-like protein: Implications for mechanisms of protein folding. Proc. Natl. Acad. Sci. 91 10943–10946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe—A multidimensional spectral processing system based on UNIX Pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., Merutka, G., Waltho, J.P., Lerner, R.A., and Wright, P.E. 1992a. Folding of peptide fragments comprising the complete sequence of proteins. Models for initiation of protein folding. I. Myohemerythrin. J. Mol. Biol. 226 795–817. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., Sayre, J.R., Merutka, G., Shin, H.C., Lerner, R.A., and Wright, P.E. 1992b. Folding of peptide fragments comprising the complete sequence of proteins. Models for initiation of protein folding. II. Plastocyanin. J. Mol. Biol. 226 819–835. [DOI] [PubMed] [Google Scholar]
- Eck, M.J., Shoelson, S.E., and Harrison, S.C. 1993. Recognition of a high-affinity phosphotyrosyl peptide by the Src homology-2 domain of p56(Lck). Nature 362 87–91. [DOI] [PubMed] [Google Scholar]
- Eustance, R.J. and Schleif, R.F. 1996. In vivo association of protein fragments giving active AraC. Proteins 25 501–505. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33 5984–6003. [DOI] [PubMed] [Google Scholar]
- Fields, G.B. and Noble, R.L. 1990. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 35 161–214. [DOI] [PubMed] [Google Scholar]
- Garrett, D.S., Powers, R., Gronenborn, A.M., and Clore, G.M. 1991. A common-sense approach to peak picking in 2-dimensional, 3-dimensional, and 4-dimensional spectra using automatic computer-analysis of contour diagrams. J. Magn. Reson. 95 214–220. [DOI] [PubMed] [Google Scholar]
- Gegg, C.V., Bowers, K.E., and Matthews, C.R. 1997. Probing minimal independent folding units in dihydrofolate reductase by molecular dissection. Protein Sci. 6 1885–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu, R.E., Braswell, E.H., Zhu, D., and Tasayco, M.L. 1999. Energetics of assembling an aicial heterodimer with an α/β m: Cleaved versus uncleaved Escherichia coli thioredoxin. Biochemistry 38 13355–13366. [DOI] [PubMed] [Google Scholar]
- Goldberg, J.M. and Baldwin, R.L. 1999. A specific transition state for S-peptide combining with folded S-protein and then refolding. Proc. Natl. Acad. Sci. 96 2019–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1992. Improved 3D triple-resonance NMR techniques applied to a 31-kDA protein. J. Magn. Reson. 96 432–440. [Google Scholar]
- Hensmann, M., Booker, G.W., Panayotou, G., Boyd, J., Linacre, J., Waterfield, M., and Campbell, I.D. 1994a. Phosphopeptide binding to the N-terminal SH2 domain of the p85α subunit of PI 3`-kinase: A heteronuclear NMR study. Protein Sci. 3 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1994b. Erratum: Phosphopeptide binding to the N-terminal SH2 domain of the p85a subunit of PI 3`-kinase: A heteronuclear NMR study. Protein Sci. 3 1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homandberg, G.A. and Laskowski, Jr., M. 1979. Enzymatic resynthesis of the hydrolyzed peptide bond(s) in ribonuclease S. Biochemistry 18 586–592. [DOI] [PubMed] [Google Scholar]
- Honda, S., Kobayashi, N., Munekata, E., and Uedaira, H. 1999. Fragment reconstitution of a small protein: Folding energetics of the reconstituted immunoglobulin binding domain B1 of streptococcal protein G. Biochemistry 38 1203–1213. [DOI] [PubMed] [Google Scholar]
- Ikura, M., Kay, L.E., and Bax A. 1990. A novel-approach for sequential assignment of H-1, C-13, and N-15 spectra of larger proteins - Heteronuclear triple-resonance 3-dimensional NMR-spectroscopy - Application to calmodulin. Biochemistry 29 4659–4667. [DOI] [PubMed] [Google Scholar]
- Johnsson, N. and Varshavsky, A. 1994. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. 91 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan, M. and Searle, M.S. 2000. Cooperative assembly of a nativelike ubiquitin structure through peptide fragment complexation: Energetics of peptide association and folding. Biochemistry 39 12355–12364. [DOI] [PubMed] [Google Scholar]
- Kanaya, E. and Kanaya, S. 1995. Reconstitution of Escherichia coli RNase HI from the N-fragment with high helicity and the C-fragment with a disordered structure. J. Biol. Chem. 270 19853–19860. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Keifer, P., and Saarinen, T. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114 10663–10665. [Google Scholar]
- Kay, L.E., Xu, G.Y., and Yamazaki, T. 1994. Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J. Magn. Reson. Ser. A 109 129–133. [Google Scholar]
- Kim, E.E., Varadarajan, R., Wyckoff, H.W., and Richards, F.M. 1992. Refinement of the crystal structure of ribonuclease S. Comparison with and between the various ribonuclease A structures. Biochemistry 31 12304–12314. [DOI] [PubMed] [Google Scholar]
- Kobayashi, N., Honda, S., Yoshii, H., Uedaira, H., and Munekata, E. 1995. Complement assembly of 2 fragments of the streptococcal protein G B1 domain in aqueous solution. FEBS Lett. 366 99–103. [DOI] [PubMed] [Google Scholar]
- Kobayashi, N., Honda, S., and Munekata, E. 1999. Fragment reconstitution of a small protein: Disulfide mutant of a short C-terminal fragment derived from streptococcal protein G. Biochemistry 38 3228–3234. [DOI] [PubMed] [Google Scholar]
- Koppel, D.E. 1972. Analysis of macromolecular polydispersity in intensity correlation spectroscopy: The method of cumulants. J. Chem. Phys. 57 4814–4820. [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Ladurner, A.G., Itzhaki, L.S., de Prat-Gay, G., and Fersht, A.R. 1997. Complementation of peptide fragments of the single domain protein chymotrypsin inhibitor 2. J. Mol. Biol. 273 317–329. [DOI] [PubMed] [Google Scholar]
- Llinas, M. and Marqusee, S. 1998. Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci. 7 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion, D., Driscoll, P.C., Kay, L.E., Wingfield, P.T., Bax, A., Gronenborn, A.M., and Clore, G.M. 1989. Overcoming the overlap problem in the assignment of H-1-NMR spectra of larger proteins by use of 3-dimensional heteronuclear H-1-N-15 Hartmann-Hahn multiple quantum coherence and nuclear Overhauser multiple quantum coherence spectroscopy - Application to interleukin-1-beta. Biochemistry 28 6150–6156. [DOI] [PubMed] [Google Scholar]
- Muhandiram, D.R. and Kay, L.E. 1994. Gradient-enhanced triple-resonance 3-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. Ser. B 103 203–216. [Google Scholar]
- Neira, J.L., Davis, B., Ladurner, A.G., Buckle, A.M., de Prat-Gay, G., and Fersht, A.R. 1996. Towards the complete structural characterization of a protein folding pathway: The structures of the denatured, transition and native states for the association/folding of two complementary fragments of cleaved chymotrypsin inhibitor.2. Direct evidence for a nucleation-condensation mechanism. Folding Des. 1 189–208. [DOI] [PubMed] [Google Scholar]
- Neira, J.L. and Fersht, A.R. 1999a. Acquisition of native-like interactions in C-terminal fragments of barnase. J. Mol. Biol. 287 421–432. [DOI] [PubMed] [Google Scholar]
- ———. 1999b. Exploring the folding funnel of a polypeptide chain by biophysical studies on protein fragments. J. Mol. Biol. 285 1309–1333. [DOI] [PubMed] [Google Scholar]
- Oas, T.G. and Kim, P.S. 1988. A peptide model of a proten folding intermediate. Nature 336 42–48. [DOI] [PubMed] [Google Scholar]
- Patt, S.L. 1992. Single-frequency-shifted and multiple-frequency-shifted laminar pulses. J. Magn. Reson. 96 94–102. [Google Scholar]
- Pecorari, F., Guilbert, C., Minard, P., Desmadril, M., and Yon, J.M. 1996. Folding and functional complementation of engineered fragments from yeast phosphoglycerate kinase. Biochemistry 35 3465–3476. [DOI] [PubMed] [Google Scholar]
- Pelletier, J.N., Campbell-Valois, F.-X., and Michnick, S.W. 1998. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc. Natl. Acad. Sci. 95 12141–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietveld, A.W.M. and Ferreira, S.T. 1998. Kinetics and energetics of subunits dissociation/unfolding of TIM: The importance of oligomerization for conformational persistence and chemical stability of proteins. Biochemistry 37 933–937. [DOI] [PubMed] [Google Scholar]
- Remy, I. and Michnick, S.W. 1999. Clonal selection and in vivo quantitation of protein interactions with protein-fragment complementation assays. Proc. Natl. Acad. Sci. 96 5394–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, F., Charlton, C.A., and Blau, H.M. 1997. Monitoring protein–protein interactions in intact eukaryotic cells by β-glactosidase complementation. Proc. Natl. Acad. Sci. 94 8405–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho, J. and Fersht, A.R. 1992. Dissection of an enzyme by protein engineering: The N-terminal and C-terminal fragments of barnase form a native-like complex with restored enzymatic activity. J. Mol. Biol. 224 741–747. [DOI] [PubMed] [Google Scholar]
- Sancho, J., Neira, J.L, and Fersht, A.R. 1992. An N-terminal fragment of barnase has residual helical structure similar to that in a refolding intermediate. J. Mol. Biol. 224 749–758. [DOI] [PubMed] [Google Scholar]
- Shaka, A.J. 1985. Composite pulses for ultra-broadband spin inversion. Chem. Phys. Lett. 120 201–205. [Google Scholar]
- Talluri, S. and Wagner, G. 1996. An optimized 3D NOESY-HSQC. J. Magn. Reson. Ser. B 112 200–205. [DOI] [PubMed] [Google Scholar]
- Tasayco, M.L. and Carey, J. 1992. Ordered self-assembly of polypeptide fragments to form native like dimeric Trp repressor. Science 255 594–597. [DOI] [PubMed] [Google Scholar]
- Tasayco, M.L., Fuchs, J., Yang, X.-M., Dyalram, D., and Georgescu, R.E. 2000. Interaction between two discontiguous chain segments from the β-sheet of Escherichia coli thioredoxin suggests an initiation site for folding. Biochemistry 39 10613–10618. [DOI] [PubMed] [Google Scholar]
- Tsuji, T., Yoshida, K., Satoh, A., Kohno, T., Kobayashi, K., and Yanagawa, H. 1999. Foldability of barnase mutants obtained by permutation of modules or secondary structure units. J. Mol. Biol. 286 1581–1596. [DOI] [PubMed] [Google Scholar]
- Vainshtein, I., Atrazhev, A., Eom, S.Y., Elliott, J.F., Wishart, D.S., and Malcolm, B.A. 1996. Peptide rescue of an N-terminal truncation of the Stoffel fragment of Taq DNA polymerase. Protein Sci. 5 1785–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, K. and Chmielewski, J. 1994. Rapid and efficient resynthesis of proteolyzed triose phosphate isomerase. J. Am. Chem. Soc. 116 11163–11164. [Google Scholar]
- Vogel, K., Cook, J., and Chmielewski, J. 1996. Subtilisin-catalyzed religation of proteolyzed hen egg-white lysozyme: Investigation of the role of disulfides. Chem. Biol. 3 295–299. [DOI] [PubMed] [Google Scholar]
- Waksman, G., Shoelson, S.E., Pant, N., Cowburn, D., and Kuriyan, J. 1993. Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain—Crystal structures of the complexed and peptide-free forms. Cell 72 779–790. [DOI] [PubMed] [Google Scholar]
- Wallace, C.J. 1995. Peptide ligation and semisynthesis. Curr. Opin. Biotechnol. 6 403–410. [DOI] [PubMed] [Google Scholar]
- Wang, Y. and Shortle, D. 1997. Residual helical and turn structure in the denatured state of staphylococcal nuclease: Analysis of peptide fragments. Folding Des. 2 93–100. [DOI] [PubMed] [Google Scholar]
- Williams, K.P. and Shoelson, S.E. 1993. Cooperative self-assembly of SH2 domain fragments restores phosphopeptide binding. Biochemistry 32 11279–11284. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Bigam, C.G., Holm, A., Hodges, R.S., and Sykes, B.D. 1995. H-1, C-13 and N-15 random coil NMR chemical-shifts of the common amino-acids. 1. Investigations of nearest neighbor effects. J. Biomol. NMR 5 67–81. [DOI] [PubMed] [Google Scholar]
- Wright, P.E., Dyson, H.J., and Lerner, R.A. 1988. Conformation of peptide fragments of proteins in aqueous solution: Implications for initiation of protein folding. Biochemistry 27 7167–7175. [DOI] [PubMed] [Google Scholar]
- Wu, L.C., Laub, P.B., Elove, G.A., Carey, J., and Roder, H. 1993. A noncovalent peptide complex as a model for an early folding intermediate of cytochrome c. Biochemistry 32 10271–10276. [DOI] [PubMed] [Google Scholar]
- Xu, R.X., Word, J.M., Davis, D.G., Rink, M.J., Willard, D.H., and Gampe, R.T. 1995. Solution structure of the human pp60(c-Src) SH2 domain complexed with a phosphorylated tyrosine pentapeptide. Biochemistry 34 2107–2121. [DOI] [PubMed] [Google Scholar]
- Yamazaki, T., Lee, W., Arrowsmith, C.H., Muhandiram, D.R., and Kay, L.E. 1994. A suite of triple resonance NMR experiments for the backbone assignment of 15-N, 13-C, 2-H labeled proteins with high sensitivity. J. Am. Chem. Soc. 116 11655–11666. [Google Scholar]
- Yu, W.-F., Tung, C.-S., Wang, H., and Tasayco, M.L. 2000. NMR analysis of cleaved Escherichia coli thioredoxin (1–73/74–108) and its P76A variant: Cis/trans peptide isomerization. Protein Sci. 9: 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]