

Abstract
Phage display is widely used for expression of combinatorial libraries, not least for protein engineering purposes. Precise selection at the single molecule level will provide an improved tool for generating proteins with complex and distinct properties from large molecular libraries. To establish such an improved selection system, we here report the detection of specific interactions between phage with displayed antibody fragments and fluorescently labeled soluble antigen based on Fluorescence Correlation Spectroscopy (FCS). Our novel strategy comprises the use of two separate fluorochromes for detection of the phage–antigen complex, either with labeled antiphage antibody or using a labeled antigen. As a model system, we studied a human monoclonal antibody to the hepatitis-C virus (HCV) envelope protein E2 and its cognate antigen (rE2 or rE1/E2). We could thus assess the specific interactions and determine the fraction of specific versus background phage (26% specific phage). Aggregation of these particular antigens made it difficult to reliably utilize the full potential of cross-correlation studies using the two labels simultaneously. However, with true monomeric proteins, this will certainly be possible, offering a great advantage in a safer and highly specific detection system.
Keywords: Phage-protein interaction, phage display, combinatorial libraries, hepatitis-C virus, monoclonal antibodies, fluorescence correlation spectroscopy
Antibody engineering combined with combinatorial methods has become a new field for producing and selecting specific antibodies. Molecular libraries, both of biological and chemical nature, based on other molecular scaffolds are also widespread. Quick and efficient screening of small quantities of single molecules with improved characteristics, such as increased binding affinity, altered specificity, or catalytic ability is therefore desired. The quantity of either receptor and ligand, such as antigen and antibody, for example, may be limiting in evolutionary strategies using enormous libraries of biological molecules as a source for detecting species with a new character. Large biological libraries of mutants or biologically diverse molecules, such as a repertoire of Ig specificities are available as presentation packages, linking phenotype and genotype in display technologies such as phage or ribosome display (Smith 1985; McCafferty et al. 1990; Hanes and Plückthun 1998).
The extreme sensitivity of Fluorescence Correlation Spectroscopy (FCS) makes it interesting to use for identification of rare clones in molecular libraries. Recent advances in laser techniques and microscopy have allowed this new technique to be utilized with full potential (Rigler and Widengren 1990; Rigler et al. 1993). It is based on the ability to measure single fluorescent molecules excited by monochromatic laser light in extremely small volumes. One records spatiotemporal correlations among intensity fluctuations belonging to the emission of discrete molecules. The sensitivity has been increased so that single molecules can be detected. This opens up the possibility of detecting rare events and allows working concentrations of fmoles or less to be used.
FCS may thus provide substantially improved selection strategies for molecular libraries. To investigate this potential, we have used a model system comprised of a previously isolated Fab fragment displayed on the surface of filamentous M13 phage, and its cognate antigen, the Hepatitis-C Virus (HCV) E2 protein (Allander et al. 2000). Two approaches for detecting phage were utilized (Fig. 1 ▶): Autocorrelated detection of specific phage with fluorescently labeled antigen, or detection of phage with labeled antiphage antibodies.
Fig. 1.

Principle for Fluorescence Correlation Spectroscopy -based detection utilizing two different colors in autocorrelation mode.
Results
Phagestocks
The first phagestocks were produced with colony-forming units (cfu) titers varying between 1010 −1012 cfu/mL. These preparations were polyethylene glycol (PEG) precipitated once and they resulted in unreasonably high backgrounds. Testing all components we determined that the super broth (SB) medium contributed to the large signal (Table 1). Medium components were apparently co-precipitated with the phage during PEG precipitation.
Table 1.
Background values for different components used in phage preparations
| Component | Concentration | I (kHz) |
| SB medium | undiluted | 557.8 |
| Ampicillin | 100 mg/mL | 73.7 |
| Tetracycline | 5 mg/mL | 40.4 |
| IPTG | 800 mM | 5.7 |
| PEG NaCl | 20%/15% | 2.6 |
| PBS buffer | undiluted | 1.1 |
| 1× PEG precipitated phage* | 122 nM | 69.9 |
| 2× PEG precipitated phage* | 1.4 nM | 8.4 |
| CsCl purified phage* | 66 nM | 9.1 |
| RhoGr anti-M13 labeled phage | 170 nM | 81.6 |
I is the mean fluorescence intensity. *The phage were resuspended in PBS buffer after treatment. The background does not depend on the concentration of phages but on phage medium components.
New phagestocks prepared with one or two PEG precipitations as a means of purification were tested in FCS. The background was reduced for 2× PEG precipitated material but the degree of aggregation had increased compared to 1× PEG precipitation. Aggregates of varying sizes are a problem in FCS measurements because identification of the various species in the reactions relies on diffusion times.
An aliquot of the phagestock was used for buffer exchange using an Amicon 100 concentrator. The background was reduced by 75% compared with the same volume of unconcentrated phage, but the phage titer was unfortunately also affected.
Finally we tested CsCl gradient centrifugation after PEG precipitation, with good results, even though the degree of aggregation was still a problem for some batches. In addition to reducing aggregates, CsCl gradient purification also allowed concentration of the phage, as is desired for certain applications (e.g., affinity determinations). Some measurements were performed with phagestocks concentrated up to 10,000× compared to original culture volumes. New batches of phage were tested by ELISA for specificity and were also titrated on indicator bacteria for estimating the phage concentration before use in FCS experiments. ELISA revealed a lack of stability of the Fab fragments in thawed phagestocks, necessitating freshly thawed material for each day's measurements.
FCS-based approach for the detection of specific interactions with phage displayed antibodies
Figure 1 ▶ illustrates the novel approach for the phage-specific detection of the HCV rE2 protein. We used a combination of two colors. The Cy5-labeled antigen allowed the direct detection of specific phage via cognate interaction between the displayed antibody fragment on the minor coat protein p3 and the fluorophore-labeled antigen. The rhodamine-green-labeled antiphage antibody (RhoGr anti-M13) allowed detection of all phages. Because we introduced two colors, quantification could be performed by comparing the autocorrelated fluorescence signals of the samples. Absolute values were obtained in terms of the number of interacting phages using labeled anti-phage antibodies, or by using fluorescently labeled antigen binding, the displayed antibody fragments. Thus the fraction of specific phages in our stocks was calculated to be approximately 26% (Tables 2,3).
Table 2.
Results from FCS measurements using rhodamine-green-labeled M13 antibody (RhoGr anti-M13) and specific phage
| Sample | Conc (nM) | τ 1 (ms) | % | τ 2 (ms) | % | τ 3 (ms) | % | N |
| RhoGr | 2.4 | 0.0648 | 100 | — | — | — | — | 0.939 |
| RhoGr anti-M13 | 0.55 | 0.0648 | 41.6 | 0.389 | 58.4 | — | — | 0.214 |
| RhoGr anti-M13 + phase | 11.2 | 0.0648 | 69.7 | 0.389 | 5.57 | 1.34 | 24.7 | 4.45 |
| displacement | 4.2 | — | — | 0.389 | 34.33 | — | — | 1.56 |
τ represents the different diffusion times (measured in ms), % (percentage) is the relative fraction of each component in the reaction and N gives the average number of molecules in the volume element. The concentration in this table is calculated from the N values and gives total number of labeled molecules.
Table 3.
Binding and displacement of the Cy5-labelled antigen rE2 to E2 specific phage
| Sample | Conc (nM) | τ 1 (ms) | % | τ 2 (ms) | % | τ 3 (ms) | % | N |
| Free Cy5 | 0.82 | 0.0652 | 100 | — | — | — | — | 0.252 |
| Cy5-streptavidin | 3.95 | 0.0652 | 51 | 0.319 | 49 | — | — | 0.997 |
| E2—Cy5 | 2.14 | 0.0652 | 34.4 | 0.319 | 0 | 0.548 | 65 | 0.539 |
| phage + E2Cy5 | 2.89 | 0.0652 | 47.7 | 0.548 | 37.5 | 1.8 | 14.8 | 0.730 |
| displacement | 3.52 | 0.0652 | 58 | 0.548 | 36.5 | 1.8 | 5.5 | 0.889 |
τ represents the different diffusion times (measured in ms), % (percentage) is the relative fraction of each component in the reaction and N gives the average number of molecules in the volume element. The concentration in this table is calculated from the N values and gives total number of labeled molecules.
Binding and displacement of rhodamine green-labeled anti-M13 antibody
RhoGr anti-M13 antibodies bound to the surface of phage particles. This binding was displaced using an excess of free, unlabeled anti-M13 antibodies, proving the specificity of the binding (Table 2; Fig. 2 ▶). The addition of unlabeled antibody/antigen was conducted in small volumes compared to the total reaction volumes in order not to affect the equilibrium by dilution effects.
Fig. 2.

Fluorescence Correlation Spectroscopy curves depicting recorded values for free rhodamine green, rhodamine green anti-M13, binding between the phage and the labeled antibody and displacement using free unlabeled antibody in excess. The phage concentration was 66 nM, using 11.5 nM total anti-M13 antibody and the displacement was performed using 670 nM unlabeled antibody.
In a first example, the phage had been purified by only one PEG precipitation (data not included). We used 86 nM phage and 170 nM of total anti-M13 antibodies incubated for 1 hr before analysis. The displacement was measured after 5 min using 670 nM free antibody. In this first example, there was a clear difference in the amount of free RhoGr anti-M13 antibody, and we could thus prove binding and displacement despite not being able to identify displacement looking directly at the labeled phage population. The RhoGr anti-M13 constituted 59.4% of the sample with a diffusion time of 0.367 ms, which corresponds well with the diffusion time of a full antibody. After binding, the species with 0.367 ms diffusion time had decreased to 30% with a subsequent increase to 40% after displacement with free antibody. In addition, we detected a species with huge molecular weight corresponding to 160 ms, which constituted approximately 23% of the sample. After displacement, the diffusion time was further increased. Wild-type phage have a molecular weight of about 10 MD–13 MD and their size corresponds to a τ (diffusion time) of ∼1.3 ms (Table 2). The huge species apparently (i.e.160ms) was clearly either an aggregate of phage or an immunocomplex.
In experiment 2, both binding and displacement were demonstrated using FCS analysis (Table 2; Fig. 2 ▶). In this case we had used the additional step of CsCl centrifugation. The amount of labeled antibody before binding was 58.4%, that after binding 5.6%, with a subsequent increase to 34.4% after displacement. In this example, the species corresponding to 1.3 ms (i.e., labeled single phage) appears after binding to an extent of 24.7%. After competing unlabeled antibody was added, labeled single phages were again reduced to 3.5%.
Binding and displacement of rE2-Cy5– and rE1/E2-Cy5–labeled antigen
FCS analysis using the second model system was performed using two slightly different antigens, rE2 and rE1/E2, both of which contain the E2 protein for which the antibody was specific. We produced several different batches of rE2–biotin–streptavidin–Cy5 and observed that the FCS results that allowed successful analyses were obtained when using preparations of rE2–biotin–streptavidin–Cy5 devoid of aggregates. Under such conditions both binding and displacement of the labeled rE2 protein was observed (Table 3). The Cy5-streptavidin used for labeling the biotinylated recombinant proteins had a diffusion time of 0.319 ms, which corresponds well with a size of ∼70 kD for streptavidin. This species disappeared completely after conjugation, and a larger species of 0.548 ms comprising the rE2–Cy5 complex was evident. This species was present to an extent of 65%. The size of this conjugate corresponds well with eight streptavidin–Cy5 molecules per rE2 molecule, which was the ratio used in the conjugation step. After binding had taken place, 14.8% of labeled phages were detected (diffusion time of 1.8 ms); after displacement, only 5.5% remained labeled.
Using the rE1/E2–biotin–streptavidin–Cy5 protein we observed binding of antigen to phage and displacement of the labeled antigen using an excess of unlabelled antigen (Fig. 3 ▶). The Cy5-labeled monomeric rE1/E2 protein had a diffusion time of 0.385 ms and constituted 18.7% of the sample, an additional 36.2% constituting a species of labeled protein with a diffusion time of 1.32 ms. After binding had occurred, a species of 2.78 ms corresponding to labeled phage appeared to 43.3%. This was again decreased to 14.9% after competition with free protein. The labeled species that remained was most likely huge aggregates of the rE1/E2 protein itself, but these aggregates were decreased by the presence of specific antigen (only 14.9% after competition), which presumably affects the equilibrium of the nonspecific complex. Native PAGE supported the notion that unbiotinylated rE1/E2 protein was not monomeric (data not included). The FCS analyses proved specific binding was accomplished using rE1/E2 or rE2 protein. The rE2 measurements were one in a series of measurements using decreasing concentrations of phage (ranging from 66 nM to 0.4 nM), but with constant amounts of antigen (0.33 nM). We obtained clearly identifiable binding and displacement using the highest concentration of phage alone and concluded that it was necessary to perform binding experiments using higher phage concentrations.
Fig. 3.

Fluorescence Correlation Spectroscopy curves for interactions between phage and rE1/E2 protein with free Cy5, binding and displacement. The biotinylated rE1/E2 was labeled with Cy5–streptavidin. The concentrations used were 237 nM phage and 28 nM rE1/E2–Cy5 antigen. The displacement was performed using 222 nM free antigen and analyzed after 2 hr.
Discussion
We report the novel detection of specific interactions between phage with displayed antibody fragments and soluble antigen using fluorescence correlation spectroscopy. High background values and amounts of aggregates were reduced in the phagestocks using PEG precipitation combined with CsCl gradients. This purification method also allowed concentration of the phage.
Unequivocal data were obtained proving the detection of interactions between phage-displaying antibodies and soluble antigen. We could also evaluate the fraction of specific phages in our stocks.
Admittedly, certain details need to be improved to exploit the full potential of FCS-based selection. Thus the use of two colors simultaneously in cross-correlation experiments would offer a great advantage in a safer and more specific detection system, differentiating target organism from indigenous or background populations. This is not possible as long as there are aggregates in the conjugates, which give rise to high background values. These aggregates seemed to originate from certain batches of the actual antigen preparation per se. Other target molecules may prove much easier to use in this respect.
There are ∼2800 units of major coat protein per M13 phage, and consequently there could be several labeled or unlabeled antibodies bound per phage. The monoclonal anti–major coat protein antibodies used here were IgG antibodies (i.e., with more than one binding site), and could therefore create large complexes of phage and antibody molecules with uncontrollable size and diffusion times. This was evident in our experiments in which the size of the complexes increased with increasing concentrations of available antibody. This presents a problem when analyzing the FCS curves, as the labeled population cannot be unambiguously identified. The problem could be circumvented by using lower concentrations of the labeled antibody for detection (as in Fig.2 ▶) or using antibody fragments instead of complete Ig molecules.
Affinity measurements using FCS have been reported elsewhere (Briggs et al. 1981; Thompson and Axelrod, 1983; Rauer et al., 1996; Rigler et al. 1999) and for those studies phage antibodies and their antigens were limited by the requirement for monomeric protein preparations.
Using the described FCS approach, detection and affinity determinations are made in solution. The values obtained should therefore be reliable with respect to no impact of factors, such as rebinding at high antigen concentrations or distortion of the antigen, which can occur in solid-phase systems such as ELISA, RIA, or BIAcore analyses. Accordingly, FCS analysis may offer an improvement in this respect.
Our next goal is to combine our FCS-based detection approach with sorting devices to allow the enrichment of specific phage clones by using the antigen–phage interaction at a single molecule/phage level. We suggest that this could be one approach for improving enrichment factors beyond those obtained using solid-phase selection methods.
Materials and methods
FCS measurements
The fluorescence auto and cross-correlation spectrometer is a prototype, which was built by Carl Zeiss, Inc. The experimental setup of the completely computer-controlled device was described in detail in Rigler et al. 1998. The excitation laser light at the two wavelengths 488 nm (argon–ion laser) for the rhodamine green dye and 633 nm (helium–neon laser) for the Cy5 dye were transmitted by one optical fiber to the inverse microscope (Zeiss Axiovert 135 TV™) by means of a telescope. The beams were of nearly perfect Gaussian intensity profiles at the exit of the polarization preserving single-mode fiber. The telescope adapted the laser light from the fiber to the diameter of the microscope objective's entrance pupil. The light was imaged to two spatially superimposed, nearly diffraction limited foci within the sample (illumination foci), that is, 100 μm above the surface of the coverglass (glass base). All FCS measurements were conducted in Lab-Tek 8 chambered coverslips (Nalge-Nunc, IL, USA, # 136439) at ∼18°C using a 20 μL total volume. Dilutions were performed in phosphate buffered saline (PBS) or water. Fluorescence light coming from the sample molecules was detected by confocal optical geometry. Essential components of the optical setup were the excitation and the detection branches. The excitation branch consisted of fiber, beam collimation objective, dichroic mirror 2, microscope objective (C-Apochromat 40× /1,2W Korr, numerical aperture 1.2, Carl Zeiss), and coverglass. The dichroic mirror 2 reflected the excitation laser light (488 nm and 633 nm) in two wavelength regions toward the sample and also transmitted the emitted fluorescence light of the sample molecules in two wavelength regions (see Rigler et al. 1998 for the key element of the optical setup). The detection branch consisted of coverslip, microscope objective, dichroic mirror 2, pinhole, additional lenses, dichroic beamsplitter, focusing lenses, and silicon avalanche photodiodes operating in photon counting mode (SPCM 131 AQ, EG&G). The signal output transistor-transistor-logic (TTL pulses) of the detectors were directly used for the hardware correlator (ALV5000™ E, ALV) on a PC board.
The unit of pinhole and detectors were aligned in three dimensions relative to the illumination volumina. The illumination foci were imaged by the microscope objective and an additional lens on to the pinhole, which was located in the optical conjugate point of the foci. A pinhole size of 30 μm was used in the experiments. Although light near the focal spot transmitted through the pinhole, light coming from other sample areas was strongly attenuated by the pinhole. Both illumination foci and both detection profiles were spatially superimposed by chromatic correction of the excitation (chromatic correction for the two excitation wavelengths 488 nm and 633 nm) and the detection branches (chromatic correction from 400 nm to 700 nm) of the optical system. Before each series of measurements, the excitation power levels were chosen as a compromise between fluorescence photon count rate and background signal from the sample preparations. The excitation powers before the microscope objective were between 30 μW and 60 μW (blue laser light) and between 150 μW and 300 μW (red laser light). It was optimized by maximizing the number of detected absolute fluorescence photon counts per dye molecule and second. The photon counts per molecule and second at blue excitation (488 nm) and green emission (peak maximum at 540 nm) were typically about 40 × 103 for rhodamine green and at red excitation (633 nm) and red emission (peak maximum at 685 nm) ∼ 75 × 103 for Cy5. In background measurements of the sample preparations, average count rates of fluorescence quanta had to be detected below 10 KHz (see Table 3), whereas the range of the average output count rates of the dye-labeled molecules was >100 KHz. All FCS measurements were performed in a total sample volume of 20 μL, which was placed in the Lab-Tek 8 chambered coverslips (Nalge-Nunc, IL, USA, # 136439) at ∼18°C. The measurement time was 60 s. The emitted fluorescence intensity fluctuations from fluorescent molecules moving by three-dimensional diffusion in a volume element of about 1fl (10−15l) within the sample were autocorrelated. The theoretical expression for the normalized intensity auto-correlation function G(τ) is
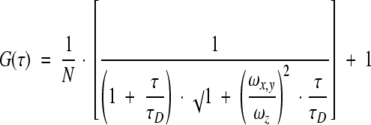 |
1 |
where N is the average number and τD is the characteristic diffusion time of the fluorescent molecules. The biochemical reactions did not equilibrate within the time scale of the diffusion process. The Equation 1 can be easily expanded to three components (molecule species) occupying the volume element with the fractions y and z of fluorescent component 2 and 3. Sections through the intensity profiles allowed fits to ideal Gaussian beam profiles, which were assumed for data evaluation. The Gaussian volume elements are rotation symmetric around the z axis and have a radius ωx,y and a half-length ωz. From measurements with the free dyes rhodamine-green and Cy5, the axis ratio ωz/ωx,y and the effective dimensions (ωx,y;green and ωx,y;red) of the green and red auto-correlated volume elements were determined for each series of experiments. Data analysis was performed with software based on the Marquardt nonlinear least-squares parameterization for calculating the normalized mean square deviation between experimental data and model.
Experimental conditions for measurement of RhoGr anti-M13 labeled phage by FCS
The first experiment in which we illustrate binding was performed with 86 nM phage and 170 nM total anti-M13. The components had been incubated for 1 hr at 18°C before FCS analysis, and displacement was measured after 5 min with 670 nM free antibody. In the second reported experiment we had 66 nM phage and 11.5 nM total anti-M13. Measurements were performed after 75 min. Displacement was performed with 670 nM unlabeled antibody and subject to immediate FCS analysis.
Experimental conditions for rE2-Cy5 and rE1/E2-Cy5 analysis
We present results from two different experimental setups. The conditions used for the experiment performed with recombinant E2–Cy5 (rE2–Cy5) protein were 66 nM phage incubated together with 0.33 nM total rE2–Cy5 protein. FCS curves were recorded after incubation for 60 min and displacement was obtained with 94 nM rE2 protein after 30 min.
In the second example we used a rE1/E2 complex, to which the 1:7 antibody binds better than to rE2 alone (Allander et al. 2000). The E1/E2 complex is considered to resemble the native state of the envelope protein. Here we incubated 237nM phage with 28nM rE1/E2–Cy5 complex and the binding was recorded after ∼48 hr. 222 nM free rE1/E2 complex was used to prove the specificity and FCS analysis was performed after 2 hr.
Conjugation of anti-M13 antibodies with rhodamine-green
The mouse monoclonal anti-M13 antibody (PharmaciaBiotech #27–9420, 1 mg/mL) was dissolved in an equal volume of 0.5 mL 0.5 M sodium carbonate buffer (pH 9.3). Rhodamine-green (Molecular Probes Inc. Germany #R-6113) was dissolved at 1 mg/mL in water-free DMSO (Aldrich # 27.685–5). For a 1:1 molar ratio of dye to antibody; 4.14 mg of this dye was used and 0.5 mg of antibody, for the 10:1 conjugate 41.4 mg was used. The mix was incubated with slow mixing for 15 min at room temperature with light protection.
The conjugate was purified on a PA6 size-exclusion column (BioRad #732–2010) preequilibrated with 2 × 13 mL of 25 mM Tris, 25 mM imidazol, 100 mM NaCl, pH 7.4. After the 1 mL of conjugate had entered the column, a further 2 mL of buffer was loaded and allowed to enter. Additional elution buffer was added to the top of the column. The labeled protein was then collected in the main fraction, which was identified measuring absorbance at A280 and A503. The labeled protein was stored at −20°C and tested using FCS to evaluate the remaining free dye. After a single round of size exclusion chromatography on a PA-6 size-exclusion column, the remaining free dye was on average 30% in the main fraction. By repurification of the fractions, we obtained ∼20% remaining free dye.
Conjugation of rE2–Cy5 or rE1/E2–Cy5 with biotin followed by labeling with streptavidin–Cy5
The rE1/E2 and rE2 proteins were kind gifts from Michael Houghton, Chiron Corporation, CA, USA (Spaete et al. 1992). The proteins were biotinylated with NHS LC-biotin (#21335, Pierce) using standard procedures with an approximate biotin excess of 10 per lysine residue. The biotinylated protein was added to streptavidin–Cy5 (#PA45001, Amersham Life Sciences, Inc.) at a molar ratio of 1 : 8 to 1 : 10. No further purification was performed before testing for free dye and labeled aggregates. We tested several different batches of biotinylated rE2 or rE1/E2 complex by ELISA and native polyacrylamide gel electrophoresis (PAGE). Some could be used for further conjugation with streptavidin–Cy5; however, most batches turned out to be suboptimal for FCS measurements as they contained a substantial amount of aggregates of varying sizes, making it difficult to evaluate the obtained FCS curves.
Conjugation of rE1/E2 with Cy5
The rE1/E2 molecule was dissolved in an equal volume of 0.5 M sodium carbonate buffer (pH 9.3). Fluorolink Cy5 monofunctional dye (#PA 25001, Amersham Pharmacia Biotech) was dissolved at 1 mg/mL in water-free DMSO (Aldrich #27.685–5). The protein to be labeled and the dye were added at a 1 : 1 molar ratio and the mix was incubated with slow mixing for 15 min at room temperature with light protection. The free dye was removed by extensive dialysis against PBS. FCS determined all the remaining free dye. From previous studies it is known that the exact conditions used in the conjugation procedure of coupling NHS ester-derivatized molecules to proteins could be of importance for avoiding aggregates (Bark et al. 1999).
Production and purification of phagestocks
Plasmids containing HCV E2-specific Fab antibody genes in the pComb3H vector were electroporated into XL1 Blue using standard procedures, and single colonies were used to inoculate a 1l culture of SB containing 30 mL 36% glucose, 500 μL ampicillin (100 mg/mL) and 2000 μL tetracycline (5 mg/mL; Barbas and Wagner 1995). The cultures were grown at 37°C until an OD600 of 0.35–0.5 was reached. The cultured cells were centrifuged for 15 min at 4000 rpm using a Beckmann centrifuge and JA-10.5 rotor. The bacteria were resuspended in 1l SB medium containing 50 mg ampicillin, 10 mg tetracycline, and 1240μL IPTG 800 mM, 20 mL of VCS M13 helperphage were added (∼1011 pfu/mL; Stratagene) and incubated at 30°C, shaking for 5 hr or overnight.
The bacteria were pelleted by centrifugation for 15 min at 4000 rpm and the phage precipitated from the supernatant by addition of 0.25 volumes of a 15% NaCl/20% PEG solution. In some experiments we treated the cell culture with DNase I before pelleting the cells by incubating on ice for 30 min and then recentrifuging for 20 min at 9000 rpm with slow deceleration. The supernatant was poured off and the last remains of the growth medium allowed to drain by keeping the bottles in an inverted position. The pellet was dissolved in PBS. Concentration and further purification were performed by gradient centrifugation in 31%w/w CsCl for 48 hr at 37,000 rpm using a SW40I rotor. The collected phage were washed in PBS and recentrifuged for 4 hr and the pellet finally dissolved in PBS containing 1% BSA. The phages were aliquoted and stored at 20°C.
The colony-forming units were calculated after titration on ampicillin-containing agar plates and the concentration of phage calculated by estimating pfu/mL as 100× cfu/mL.
Concentration of phagestocks using microconcentrators
To 50 μL phagestock 450 μL of PBS-1% BSA were added and centrifuged for 12 min at 3000 × g in a benchtop centrifuge using Amicon microconcentrators. The concentrate was recovered by spinning the inverted membrane at 1000 × for 3 min.
ELISA for specificity testing
ELISA wells were coated with E1/E2 protein at a concentration of 0.1 μg/mL in PBS (0.95 nM). Phagestocks were diluted in PBS-0.1% Tween 20 and binding was detected using a mouse-anti-M13 antibody (1 : 1000; PharmaciaBiotech #27–9420, 1 mg/mL, Sweden) and an AP-labeled goat–antimouse antibody (1 : 1000; Dakopatts, Denmark).
Competition experiments were performed using 0.5 μg/mL –0.6 μg/mL free E1/E2–Cy5 conjugate (4.8 nM–5.5 nM).
Native PAGE
14% Tris-Glycine polyacrylamide gels (Novex, USA) were used for determining the sizes of the recombinant E1/E2 and protein preparations. Samples were separated using the Tris-glycine native running and sample buffer system from the same supplier.
Acknowledgments
We thank Drs. Michael Houghton and Steven Coates for providing the rE2 and rE1/E2 proteins. The Swedish Research Council for Engineering Sciences, The Swedish Cancer Society, the Swedish MRC, The Swedish National Board for Laboratory Animals and the Torsten and Ragnar Söderberg Foundations provided financial support. Dr Robert A. Harris assisted with linguistic advice.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.5701
References
- Allander, T., Drakenberg, K., Beyene, A., Rosa, D., Abrignani, S., Houghton, M., Widell, A., Grillner, L., Persson, M.A.A. 2000. Recombinant human monoclonal antibodies against different conformational epitopes of the E2 envelope glycoprotein of hepatitis C virus that inhibit its interaction with CD81. J. Gen. Virol. 81 2451–2459. [DOI] [PubMed] [Google Scholar]
- Barbas, III, C.F. and Wagner, J. 1995. Synthetic human antibodies: Selecting and evolving functional proteins. Methods 8 94–103. [Google Scholar]
- Bark, N., Földes-Papp, Z., and Rigler, R. 1999. The incipient stage in thrombin induced fibrin polymerization detected by FCS at the single molecule level. Biochem. Biophys. Res. Comm. 260 35–41. [DOI] [PubMed] [Google Scholar]
- Briggs, J., Elings, V.B., and Nicoli, D.F. 1981. Homogeneous fluorescent immunoassay. Science 212 1266–1267. [DOI] [PubMed] [Google Scholar]
- Hanes, J. and Plückthun, A. 1998. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. 94 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty, J., Griffiths, A.D., Winter, G.W., and Chiswell, D.J. 1990. Phage-antibodies: filamentous phage displaying antibody variable domains. Nature 348 552–553. [DOI] [PubMed] [Google Scholar]
- Rauer, B., Neumann, E., Widengren, J., and Rigler, R. 1996. Fluorescence correlation spectroscopy of interaction kinetics of tetramethylrhodamine-bungarotoxin with Torpedo californica acetylcholine receptor. Biophys. Chem. 58 3–12. [DOI] [PubMed] [Google Scholar]
- Rigler, R., Földes-Papp, Z., Meyer-Almes, F.J., Sammet, C., Volcker, M., and Schnetz, A. 1998. Fluorescence cross-correlation: a new concept for polymerase chain reaction. J. Biotechnol. 63 97–109. [DOI] [PubMed] [Google Scholar]
- Rigler, R., Mets, U., Widengren, S., and Kask, P. 1993. Fluorescence correlation spectroscopy with high countrate and low background: Analysis of translational diffusion. Eur. Biophys. J. 22 169–175 [Google Scholar]
- Rigler, R., Pramanik, A., Jonasson, P., Kratz, G., Jansson, O.T., Nygren, P-Å., Ståhl, S., Ekberg K., Johansson B-L, Uhlén, M., Jörnvall, H., and Wahren, J. 1999. Specific binding of proinsulin C-peptide to human cell membranes. Proc. Natl. Acad. Sci. 96 13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigler, R. and Widengren, J. 1990. Ultrasensitive detection of single molecules by fluorescence correlation spectroscopy. Bioscience 3 180–183. [Google Scholar]
- Smith, G.P. 1985. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 228 1325–1327. [DOI] [PubMed] [Google Scholar]
- Spaete, R. R., Alexander, D., Rugroden, M. E., Choo, Q. L., Berger, K., Crawford, K., Kuo, C., Leng, S., Lee, C., Ralston, R., et al. 1992. Characterization of the hepatitis C virus E2/NS1 gene product expressed in mammalian cells. Virology 188 819–30. [DOI] [PubMed] [Google Scholar]
- Thompson, N.L. and Axelrod, D. 1983. Immunglobulin surface binding kinetics studied by total internal reflection with fluorescence correlation spectroscopy. Biophys. J. 43 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]