

Abstract
Tumor cells exhibit an altered metabolism, characterized by increased glucose uptake and elevated glycolysis, which was first recognized by Otto Warburg 70 years ago. Warburg originally hypothesized that these metabolic changes reflected damage to mitochondrial oxidative phosphorylation. Although hypoxia and hypoxia inducible factor can induce transcriptional changes that stimulate glucose transport and glycolysis, it is clear that these changes can occur in cultured tumor or transformed cells cultured under normoxic conditions, and thus there must be genetic alterations independent of hypoxia that can stimulate aerobic glycolysis. In recent years it has become clear that loss of p53 and activation of Akt can induce all or part of the metabolic changes reflected in the Warburg effect. Likewise, changes in expression of lactate dehydrogenase and other glycolytic control enzymes can contribute to increased or altered glycolysis. It is also clear that changes in lipid biosynthesis occur in tumor cells to support increased membrane biosynthesis and perhaps the altered energy needs of the cells. Changes in fatty acid synthase, Spot 14, Akt, and DecR1 (2,4-dienoylcoenzyme A reductase) may underlie altered lipid metabolism in tumor cells and contribute to the ability of tumor cells to proliferate or metastasize. Although these advances provide new therapeutic targets that merit exploration, there remain critical questions to be explored at the mechanistic level; this work may yield insights into tumor cell biology and identify additional therapeutic targets.
Introduction
For more than 70 years it has been appreciated that cancer cells exhibit an altered metabolism that is characterized by elevated uptake of glucose and an increased glycolytic rate; this observation was first reported by Otto Warburg [1], comparing liver cancer cells with normal liver cells. The observation that cancer cells generated the majority of their ATP by glycolysis, even when grown in the presence of oxygen, caused Warburg to hypothesize that the metabolic shift toward glycolysis observed in cancer cells reflected damage to mitochondrial respiration, which resulted in aerobic glycolysis. In normal cells the presence of oxygen inhibits glycolysis, as first recognized by Pasteur (the Pasteur effect) [2]. Furthermore, Warburg hypothesized that this metabolic change was the origin of cancer, as reflected in the title of his report published in 1956 [3]. It is now clear that the majority of tumor cells in vivo, and transformed cells in vitro, exhibit elevated levels of glucose transport and elevated rates of glycolysis that result in an increase in the production of lactate; this phenomenon is known as the Warburg effect.
Glycolysis is a topic covered in virtually every biochemistry course because of its central role in biology and is summarized in Figure 1. During glycolysis, glucose is metabolized to form two molecules of pyruvate with a net gain of two molecules of ATP from one molecule of glucose. Under normal conditions, pyruvate is converted into acetylcoenzyme A to provide starting material for the citric acid cycle and oxidative phosphorylation, which yields about 34 more molecules of ATP from the molecule of glucose. Despite inefficient use of glucose, tumor cells often convert pyruvate to lactate, which is secreted from the cell, but this change in metabolism is prominent and would appear to result from rather strong selective pressure. Tumor hypoxia (lack of oxygen) will also cause a shift to glycolytic metabolism, because respiration cannot occur without oxygen. Tumor hypoxia and activation of hypoxia inducible factor (HIF) is undoubtedly an important pathway that contributes to tumorigenesis, angiogenesis, increased glycolysis and tumor cell survival. Additionally, HIF can be activated under normoxia by loss of the von Hippel-Lindau tumor suppressor (which normally acts to keep levels of HIF activity low under normoxic conditions) or activation of receptor tyrosine kinase signaling [4]. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in von Hippel-Lindau deficient renal cell carcinoma by repression of c-Myc activity [4]. HIF activation not only stimulates glycolysis but also actively attenuates mitochondrial respiration, making HIF a key regulator of cancer cell metabolism [5,6].
Figure 1.
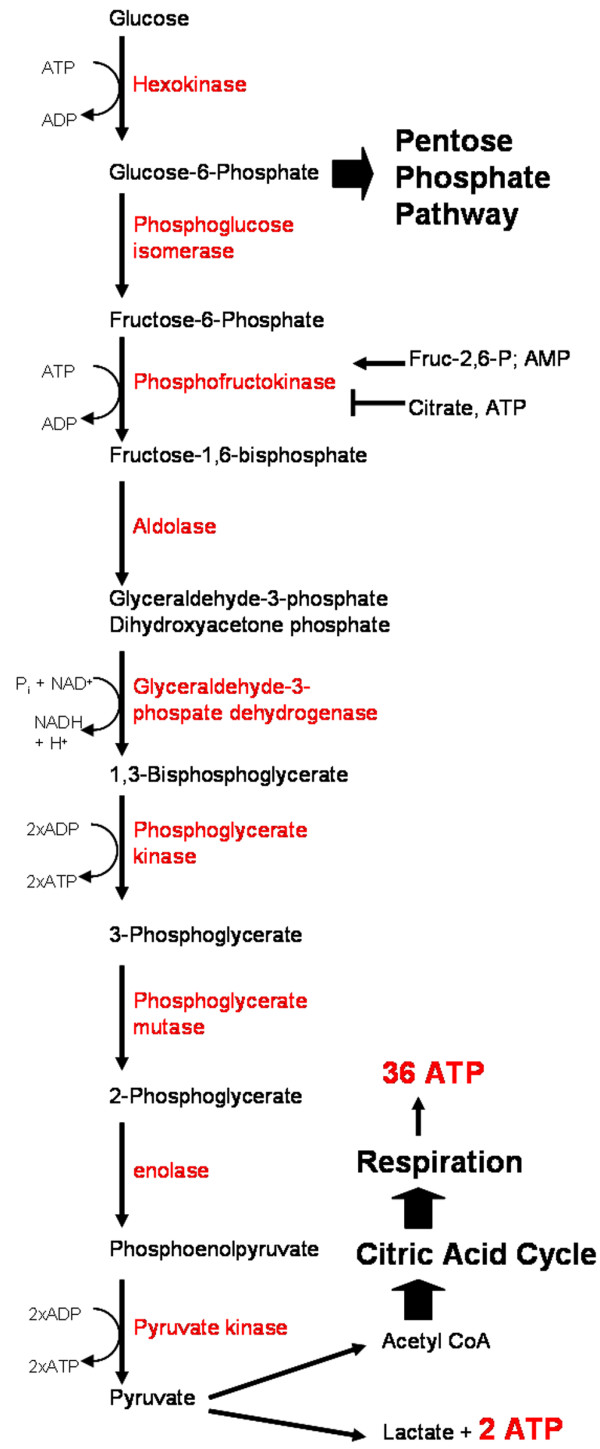
Glycolysis overview. Glycolysis involves the metabolism of glucose to two molecules of pyruvate via nine other intermediates by the actions of nine enzymes (depicted in red). CoA, coenzyme A; NAD, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide, reduced.
Factors that contribute to the Warburg effect, other than tumor hypoxia and HIF, are discussed further in this review; a number of recent comprehensive reviews provide extensive information about HIF activation and its role in cancer [7-12]. Hypoxia cannot be completely responsible for the elevated glucose transport and increased glycolysis observed in tumors cells, because these properties can be induced by oncogene-mediated cell transformation in vitro and are maintained when tumor cells are cultured in vitro under normoxic conditions.
Interest in the Warburg effect has re-emerged in recent years, in part because positron emission tomography imaging has proven to be highly effective in detecting tumors and metastatic lesions in patients and can be used as an indicator of disease progression or remission [13-16]. Although many different tracers can be used in positron emission tomography, the diagnostic detection of tumors primarily utilizes the glucose analog 18fluorodeoxyglucose, which is effective because tumors take up large quantities of glucose. Cuezva and colleagues [17] demonstrated that tumors that transport less glucose (as determined by positron emission tomography) carry better prognosis in terms of patient survival than do tumors that transport more glucose. This group has also defined a 'bioenergetic signature' that is predictive of disease progression, correlates with high rates of glucose transport and decreased mitochondrial metabolism, and may identify therapeutic targets specific to a cancer cells' metabolism [17]. In the present commentary, we review some of the recent advances that have led to new molecular insights regarding the Warburg effect and pose questions that merit attention in the coming years.
The role of Akt in activating glycolysis
Activation of the serine/threonine kinase Akt induces aerobic glycolysis by affecting multiple molecules that are directly involved in glycolysis while maintaining cell survival. Expression of an activated mutant of Akt1, namely myr-Akt1, in hematopoietic cells stimulates accumulation of nicotinamide adenine dinucleotide, reduced (NADH) and lactate (byproducts of glycolysis) and accelerates glucose consumption, and it does so without increasing cellular oxygen consumption, indicating that activated Akt induces aerobic glycolysis without effecting mitochondrial respiration [18]. Similarly, paired glioblastoma cell lines with and without high levels of activated Akt proliferate at a similar rate, but cells with activated Akt consume more glucose and secrete more lactate, which can be inhibited by pharmacologic inhibition of Akt; expression of activated Akt in the cell line lacking Akt activation increases glucose consumption and lactate production [18]. Thus, expression of activated Akt induces aerobic glycolysis in both neoplastic hematopoietic and glial cells.
Analysis of knockout mice lacking specific Akt isoforms has shown that different Akt family members are important for glucose uptake and initiating glycolysis. The insulin-induced uptake of glucose by insulin responsive tissues such as skeletal muscle and adipose tissue requires Akt2 for translocation of glucose transporter (GLUT)4 to the cell surface and subsequent glucose import [19]. Growth factor withdrawal causes interleukin-3 dependent hematopoietic cells to downregulate expression of GLUT1, hexokinase (HK)II, and phosphofructokinase 1, thus decreasing glycolysis by reducing glucose import and diminishing the activity of these two key glycolytic enzymes [20]. The ability of interleukin-3 dependent cells to stimulate plasma membrane localization of GLUT1 and glucose import requires phosphatidylinositol 3-kinase (PI3K) [21,22], but expression of myr-Akt1 can induce GLUT1 plasma membrane localization and HKI activity in the absence of interleukin-3 [23]. In virgin mice, mammary epithelial cells lack basolateral expression of GLUT1; however, conditional expression of myr-Akt1 in the mammary gland in vivo induces basolateral localization of GLUT1 in virgin mammary epithelial cells, whereas mice lacking Akt1 fail to properly induce GLUT1 basolateral localization during pregnancy [24]. Because the Akt pathway is activated in the majority of cancers (via over-expression of receptor tyrosine kinases [25], phosphatase and tensin homolog [PTEN] inactivation [26], mutation of PI3K [27], or mutation of Akt itself [28]), and because most tumors over-express GLUT1 in the plasma membrane and exhibit elevated glucose transport [29], this would suggest a causal role for the PI3K/Akt pathway in stimulating over-expression and membrane localization of GLUT1 in these tumor cells (Figure 2). Further studies are needed to confirm the role played by Akt in regulating glucose transport and glycolysis, especially given that glucose transporters can be over-expressed in breast tumors that do not express activated Akt [29].
Figure 2.

Regulation of glycolysis and fatty acid synthesis. The regulation of glycolysis and metabolism is far too complicated for one figure, but some of the key players described in the text are pictured. Akt promotes plasma membrane association of the glucose transporter, GLUT1, which transports glucose into the cell; it activates hexokinase (HK) association with the mitochondria; and it phosphorylates ATP citrate lyase (ACL), stimulating its activity of cleaving citrate to form oxaloacetate (OAA) and acetyl-coenzyme A (Ac-CoA), with downstream activation of fatty acid (FA) synthesis, which requires fatty acid synthase (FASN) and nicotinamide adenine dinucleotide phosphate (NADPH). Akt also inhibits fatty acid β-oxidation (β ox) via inhibition of carnitine palmitoyltransferase (CPT)1A, which imports fatty acids for mitochondrial β-oxidation. 2,4-Dienoyl-coenzyme A reductase (DecR1) is a β-oxidation enzyme, and its expression is often decreased in breast cancer. Impaired oxidative phosphorylation (Ox/Phos) leads to Akt activation. HK association with the mitochondria promotes phosphorylation of glucose to form glucose-6-phosphate, which can be metabolized via either glycolysis or the pentose phosphate pathway (PPP). Low levels of nutrients (such as high levels of AMP and low levels of ATP) cause activation of AMP-activated protein kinase (AMPK), which inhibits fatty acid synthesis and promotes fatty acid β-oxidation, and also phosphorylates/activates p53. p53 can inhibit glycolysis by inhibition of phosphoglycerate mutase (PGM) and can also arrest the cell cycle. However, p53 is commonly mutated in cancer cells, leading to a lack of cell cycle arrest and lack of PGM inhibition, as well as decreased expression of two p53 targets: TP53-induced glycolysis and apoptosis regulator (TIGAR) and synthesis of cytochrome oxidase (SCO)2. This drives glycolysis by disfavoring oxidative phosphorylation and PPP. Hypoxia and/or oncogenes can activate hypoxia inducible factor (HIF), which can drive transcription of nearly all glycolysis-related enzymes (most not depicted), including pyruvate kinase (PK) and lactate dehydrogenase (LDH). Some oncogenes can activate PK and LDH independently of hypoxia and HIF.
In addition to stimulating glucose import, Akt stimulates mitochondria-associated HK activity [30], which initiates glycolysis and the pentose phosphate pathway by phosphorylating glucose to form glucose-6-phosphate (Figure 1). Phosphorylation of glucose also allows for further transport of glucose into the cell along its concentration gradient. Growth factors promote the association of HKI and HKII with mitochondria [30], which can also be stimulated by expression of activated myr-Akt1, suggesting that growth factors utilize the PI3K/Akt/mammalian target of rapamycin (mTOR) pathway to regulate localization of HKI and HKII [31,32]. Localization of HKI and HKII to the outer mitochondrial membrane is associated with increased phosphorylation of glucose, suppression of apoptosis and maintenance of mitochondrial membrane potential, and may link glycolytic glucose metabolism to oxidative phosphorylation via use of intramitochondrial ATP to phosphorylate glucose [30]. Both glucose and HK activity are required for Akt-mediated suppression of apoptosis [33], indicating that Akt activation can mediate two hallmarks of cancer (Figure 2): glycolysis and evasion of apoptosis.
Pyruvate can be converted into either cytosolic lactate, which is secreted, or into mitochondrial acetyl-coenzyme A, which is converted into citrate within the mitochondria. Citrate can be processed by the citric acid cycle or exported to the cytoplasm, where it is cleaved by ATP citrate lyase, generating cytosolic acetyl-coenzyme A, which is a building block for cholesterol and fatty acid biosynthesis. Additionally, high concentrations of cytosolic citrate inhibit glycolysis and shunt glucose-6-phosphate into the pentose phosphate pathway; this generates nicotinamide adenine dinucleotide phosphate (NADPH), which is a reducing equivalent necessary for cholesterol and fatty acid biosynthesis. ATP citrate lyase is a substrate for Akt [34], and expression of activated Akt enhances de novo synthesis of fatty acids from either glucose or pyruvate precursors, whereas inhibition of Akt decreases fatty acid synthesis [35]. Inhibition of PI3K/Akt signaling enhances β-oxidation (catabolism) of fatty acids, whereas expression of myr-Akt1 inhibits β-oxidation of fatty acids by reducing expression of carnitine palmitoyltransferase (CPT)1A, a key enzyme that is involved in transporting fatty acids to the mitochondria for β-oxidation [36]. A simplified model that incorporates the above observations would suggest that activation of Akt dynamically reprograms the cancer cell by supplying the cancer cell with energy via enhanced glucose uptake, stimulating hexokinase activity and glycolytic processing of glucose; by suppressing apoptosis; and by suppressing β-oxidation of fatty acids while stimulating the biosynthesis of fatty acids necessary for forming membranes of rapidly dividing cells. These metabolic changes support the energy needs of the cell and shift metabolic precursors to where they are needed to support cell proliferation.
Thus far we have discussed how Akt may stimulate glycolysis, but new evidence points to a pathway by which defects in mitochondrial respiration can lead to activation of Akt [37]. Pelicano and coworkers [37] demonstrated that respiration deficiency induced by mitochondrial mutagenesis, pharmacologic inhibition, or hypoxia causes inactivation of PTEN. Because PTEN is a negative regulator of Akt, inactivation of PTEN by these multiple modes of respiratory deficiency may result in activation of Akt [37]. These cells are highly glycolytic and resistant to a number of apoptotic stimuli, and inhibition of the PI3K/Akt pathway restores sensitivity to drug-induced apoptosis [37]. These findings indicate that impaired respiration can activate Akt, which would be expected to induce glucose import, HK activity, and glycolysis as a means to generate energy as well as induce ATP citrate lyase activity. The discussed findings are somewhat at odds with those of Bauer and coworkers [35], who demonstrated enhanced ATP citrate lyase activity and fatty acid synthesis (which requires NADPH) in cells expressing activated Akt, because Pelicano and coworkers argued that the inactivation of PTEN is a result of excessive NADH, which causes oxidation of PTEN due to a lack of sufficient NADPH (required to maintain PTEN in a reduced state). Further study is needed to clarify how a lack of respiration may affect levels of NADH and NADPH, and how this in turn affects PTEN, cellular metabolism, and other signaling pathways.
Modulation of glycolysis and oxidative phosphorylation by p53
The tumor suppressor p53 is among the genes that are frequently mutated in tumors, and it is regarded as the guardian of the genome because of its role in responding to DNA damage [38]. Less well appreciated is the role of p53 in mediating a metabolic check point for cell cycle progression [39]. Cells that are subjected to nutritional stress exhibit an increase in the ADP/ATP ratio, and under these conditions adenylate kinase is activated and converts two molecules of ADP into one ATP and one AMP. Although this helps to maintain a consistent ADP/ATP ratio, it also results in an increase in the ratio of AMP/ATP, which stimulates activation of AMP-activated protein kinase (AMPK). AMPK acts as a nutrient sensor that is activated under conditions of low nutrients to activate catabolic events such as β-oxidation of fatty acids and autophagy, and inhibit anabolic events such as transcription, translation, and synthesis of nucleotides and fatty acids [40,41]. Recent studies have demonstrated that culturing cells in low glucose causes cell cycle arrest, which is mediated by AMPK in a p53-dependent manner and involves AMPK phosphorylation of p53 at Ser15 (human sequence; Figure 2) [39]. Expression of a constitutively activated mutant of AMPK induces cell cycle arrest even in the presence of high concentrations of glucose and nutrients, whereas loss of p53 prevents cell cycle arrest induced by low glucose [39]. This pathway also requires mTOR inactivation and both tuberous sclerosis 1 and 2 [42], suggesting that disruption of multiple points along this pathway might result in a loss of this metabolic checkpoint.
The finding that p53 was inactivated in the majority of cancers and that the majority of tumors also exhibit the Warburg effect has stimulated two different groups to consider whether p53 may regulate glycolysis [43,44]. Matoba and coworkers [43] demonstrated that synthesis of cytochrome oxidase (SCO)2 is expressed in a p53-dependent manner. SCO2 is required for the assembly of the cytochrome c oxidase complex in the mitochondria, and therefore diminished expression of SCO2 results in impaired mitochondrial respiration. As a target of p53, the level of SCO2 protein and the rate of mitochondrial respiration correlated with p53 gene dosage; p53+/+ cells had higher levels of SCO2 and respiration than did p53+/-, and p53-/- had the lowest of all. Expression of SCO2 in p53-/- cells using a heterologous promoter restored respiration, whereas targeted disruption of SCO2 in p53 wild-type cells impaired mitochondrial respiration. The inhibition of mitochondrial respiration by disrupted expression of SCO2 in p53-deficient cells results in a cell that is dependent upon glycolysis, and the impaired respiration would also be expected to activate Akt, as discussed above, and further stimulate glycolysis (Figure 2).
Following a different approach, Bensaad and coworkers [44] identified TP53-induced glycolysis and apoptosis regulator (TIGAR) as a p53-regulated gene that might regulate the production of reactive oxygen species (ROS). Increased expression of TIGAR decreases the level of fructose-2,6-bisphosphate in a cell, which suppresses glycolysis by diverting the flow of glucose-6-phosphate to the pentose phosphate pathway. Diversion of sugars to the pentose phosphate pathway results in the generation of NADPH, which increases the concentration of reduced glutathione – an antioxidant that can scavenge ROS. Thus, p53-dependent expression of TIGAR decreases glycolytic rates and decreases the level of ROS, whereas loss of p53 function in tumor cells would be expected to decrease expression of TIGAR and thereby stimulate glycolysis and increase the concentration of ROS (Figure 2). This increase in ROS at the expense of NADPH production may cause PTEN oxidation and Akt activation in a manner similar to that discussed by Pelicano and coworkers [37]. Increased Akt activation stimulates glycolytic energy production and fatty acid synthesis, as discussed above.
It has also been observed that p53 downregulates expression of phosphoglycerate mutase (PGM; Figure 1) and that loss of p53 results in an increase in PGM expression and enhanced glycolysis [45]. Over-expression of PGM resulted in immortalization of primary mouse embryo fibroblasts and protected them from Ras-induced senescence, although expression of PGM did not result in cellular transformation. Consistent with this model, pharmacologic inhibition of PGM inhibits glycolytic flux and results in cell cycle arrest [46]. Loss of p53 is a common occurrence in tumor cells and has profound affects on DNA damage repair, cell cycle arrest, and apoptosis [38]. However, loss of p53 also results in increased glycolysis via loss of SCO2 expression, which impairs the respiration machinery, leaving glycolysis as the major source of ATP production; via loss of TIGAR expression, which results in reduced shuttling of glucose to the pentose phosphate pathway and more glycolysis; and via increased PGM expression, which also activates glycolysis (Figure 2).
Lactate dehydrogenase as a regulator of glycolysis and mitochondrial physiology
The conversion of pyruvate to lactate, mediated by lactate dehydrogenase (LDH), normally only occurs under conditions in which oxygen is limited, because this prevents extraction of the maximum amount of energy from glucose. The notable exception is tumor cells where lactate accumulates, reflecting the altered balance between glycolysis and oxidative phosphorylation. Fantin and colleagues [47] explored the effect of reducing expression of LDH-A in Neu-transformed mammary tumor cells derived from MMTV-Neu transgenic mice [48]. The proliferation of tumor cells lacking expression of LDH-A and control tumor cells were identical under normoxic conditions, but tumor cells lacking LDH-A proliferated at a greatly reduced rate under hypoxic conditions, which correlated with reduced cellular concentration of ATP and increased mitochondrial respiration. Using a tumor cell mammary gland transplant model, a very significant decrease in tumor growth rate and an increase in animal survival were observed using LDH-A knockdown tumor cells in comparison with tumorigenesis induced by the unmodified control tumor cells [47] (Figure 2). Increased expression of LDH in tumor cells may be a critical part of the metabolic reprogramming that occurs during tumorigenesis, and it has been shown that expression of LDH can be directly induced by oncogenes such as c-Myc [48], or indirectly via activation of HIF-1α [49]. Targeting expression of LDH or inhibiting LDH could help to restore metabolic balance, but more importantly it may diminish the ability of tumor cells to survive under conditions of hypoxia.
Another key enzyme involved in glycolysis that may accelerate glucose metabolism in cancer cells is pyruvate kinase type M2 (M2-PK). M2-PK is characteristically expressed in cells of the lung, cells with high rates of nucleic acid synthesis, and tumor cells [50]. Expression of M2-PK can be stimulated by hypoxia and oncogenes such as Ras, Src, and human papilloma virus gene E7 [50]. This as a theme reminiscent of LDH described above, in which oncogene-induced expression of LDH or M2-PK could be further enhanced by hypoxia resulting from increased tumor volume driven by pro-growth oncogenes.
Tumor cells have altered lipid metabolism
Although the Warburg effect has been recognized since the 1920s, less well appreciated are alterations in lipid metabolism and the high rates of de novo fatty acid biosynthesis exhibited by many tumors [51]. Labeling studies indicate that the majority of fatty acids in cancer cells are derived from de novo synthesis regardless of the concentration of extra-cellular lipids, which primarily reflects dietary fats [52,53]. The enzymatic activity of ATP citrate lyase [35,54] and fatty acid synthase (FASN) are increased to support the synthesis of fatty acids. FASN was identified as the breast tumor associated protein OA-519, and numerous studies have shown that it is over-expressed in tumors, including breast carcinomas [55-58]. Although it is thought that increased fatty acid biosynthesis is required to support the synthesis of new membranes, experimental evidence to support this contention is lacking [56,59-61]. Lipogenic breast tumors, defined by high level expression of FASN, carry a poor clinical prognosis [62], but it is not clear whether expression of FASN plays a causal role in disease or whether it reflects other underlying metabolic changes in such tumors. Discussed above is the role of PI3K/Akt in stimulating fatty acid synthesis via activation of ATP citrate lyase and inhibition of fatty acid β-oxidation via reduced expression of CPT1A. Along a similar line as CPT1A, mouse mammary tumor models and human primary breast cancer often exhibit diminished expression of DecR1 (2,4-dienoyl-coenzyme A reductase), another enzyme involved in fatty acid β-oxidation [63]. Ectopic expression of DecR1 in a mouse mammary carcinoma model reduces tumor growth and decreases de novo fatty acid synthesis, although it does not reduce glucose uptake by tumor cells [63], reinforcing the therapeutic potential of targeting tumor cell fatty acid synthesis.
Spot 14 is a key regulator of de novo lipid synthesis
Spot 14 was originally identified as a protein induced by thyroid hormone [64], which is highly expressed in tissues that actively synthesize fatty acids such as the adipose, liver, and the lactating mammary gland [65-67]. Spot 14 knockout mice exhibit lower levels of medium chain fatty acids in the milk, which is thought to be due to suppression of de novo synthesis of fatty acids in mammary epithelial cells [67]. Spot 14 expression is elevated in the mammary glands of MMTV-myr-Akt1 transgenic mice, which exhibit precocious lipid biosynthesis during pregnancy (Rudolph MC, Anderson SM, unpublished data) and which have a milk fat content that is two to three times that of wild-type mice [68]. The mechanism by which Akt activates lipid synthesis is unclear but may require Spot 14 and/or sterol response element binding protein, which is a transcription factor that induces cholesterol and fatty acid biosynthesis downstream of Akt activation [69]. Analysis of human breast tumors has revealed that expression of Spot 14 is correlated with higher metastatic potential and poorer clinical outcome [70,71]. As with the expression of FASN in tumors, there is no mechanistic explanation for why expression of Spot 14 would correlate with increased metastatic potential and poor clinical outcome, although their influence on fatty acid biosynthesis may be part of the reason. It remains to be determined whether Spot 14 functions to modulate FASN activity, or whether it can regulate transcription of fatty acids.
Conclusion
It is clear that cancer cells have altered metabolism, and although progress has been made in recent years toward identifying molecules that may underlie the changes in metabolic regulation, it is also becoming clear that each of these molecules may interact in overlapping and reinforcing manners to ensure that these metabolic changes continue in tumor cells. Tumorigenesis is an increasingly complex dance of molecules in which pathways regulating metabolism intersect with many other known pathways, such as oncogene signaling, evasion of apoptosis, and angiogenesis. Clearly, more details remain to be elucidated and the critical therapeutic targets remain to be identified. Otto Warburg was the first to appreciate the aerobic glycolytic phenotype of cancer cells, and although people have pointed out that his hypothesis was wrong, one can see that there are mechanisms by which such mitochondrial damage does promote the program of metabolic changes that may favor tumorigenesis or metastasis. In the absence of knowledge about the importance of oncogenes and tumor suppressor genes as genetic causes of tumorigenesis, his insight is remarkable and new data demonstrate that Warburg may not have been as far off base as some have suggested in recent years. The fact that loss of p53 causes a loss of mitochondrial oxidative phosphorylation through decreased expression of SCO2 demonstrates a mitochondrial basis for the Warburg effect [43].
In spite of the advances discussed above, there are numerous questions that merit investigation. What other oncogenes or tumor suppressor genes reduce expression of mitochondrial enzymes that could result in suppression of mitochondrial respiration? How frequently are mitochondria nonfunctional in tumor cells? Are the enzymes of citric acid cycle or respiratory chain enzymes targeted frequently? Does hypoxia supersede the effects of the PI3K/Akt/mTOR pathway on metabolism? Is GLUT1 the critical glucose transporter in tumor cells or do other transporter make major contributions to the Warburg effect? What factors activate other glucose transporters? Does loss of functional BRCA1 or BRCA2 stimulate the Warburg effect? Is the metabolism of 'cancer stem cells' altered, or does that occur later during tumor progression? Does secretion of lactate by tumor cells, and the resulting extracellular acidosis, play a role in tumor cell invasion and metastasis? Would blocking lactate transport result in intracellular acidosis and kill the tumor cell? Is inhibition of β-oxidation of fatty acids required to redirect lipid metabolism? Does DecR1 play a universal role in modifying lipid biosynthesis? Is the lipogenic phenotype of tumor cells driven by transcriptional events? Is there a role for sterol response element binding protein or SPOT 14 in tumorigenesis or metastasis? Do steroid hormones alter the metabolism of breast cancer cells? Finally, what metabolic changes are universal in tumorigenesis and what changes are organ-specific?
The answers to these questions will provide new insights into tumor cell biology and may identify critical therapeutic targets that inhibit tumor cell proliferation as well as tumor invasion and metastasis. Warburg would have loved to have known the answers to these questions.
Abbreviations
AMPK = AMP-activated protein kinase; CPT = carnitine palmitoyltransferase; DecR1 = 2,4-dienoyl-coenzyme A reductase; FASN = fatty acid synthase; GLUT, glucose transporter; HIF = hypoxia inducible factor; HK = hexokinase; LDH = lactate dehydrogenase; M2-PK = pyruvate kinase type M2; NADH = nicotinamide adenine dinucleotide, reduced; NADPH = nicotinamide adenine dinucleotide phosphate; PGM = phosphoglycerate mutase; PI3K = phosphatidylinositol 3-kinase; PTEN = phosphatase and tensin homolog; ROS = reactive oxygen species; SCO = synthesis of cytochrome oxidase; TIGAR = TP53-induced glycolysis and apoptosis regulator.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
The authors should like to thank Lisa Litzenberger for her artistic talent and contribution to generating figures, as well as the members of the Anderson laboratory and the members of the Mammary Gland Program Project Grant at the University of Colorado Health Sciences Center for discussions and encouragement of our research. This work was supported by the grants BCTR0600727 from the Susan G Komen Foundation, and P01HD38129 from the Public Health Service to SMA. CDY is supported by a Predoctoral Fellowship from the US Army Breast Cancer Research Program (BC051149).
References
- Warburg O. The Metabolism of Tumors. London: Constable; 1930. [Google Scholar]
- Pasteur L. Experiences et vues nouvelles sur la nature des fermentations. Comp Rend Acad Sci. 1861;52:1260–1264. [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of c-myc activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metabolism. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Kim Jw, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metabolism. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993–998. doi: 10.1016/S0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P, Zhong H. 'The metabolism of tumours': 70 years later. Novartis Found Symp. 2001;240:251–260. [PubMed] [Google Scholar]
- Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71–77. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUM JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, Thompson CB. The transcription factor HIF-1α plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran A, Pio BS, Khatibi B, Czernin J, Phelps ME, Silverman DHS. 18F-FDG PET for staging breast cancer in patients with inner-quadrant versus outer-quadrant tumors: comparison with long-term clinical outcome. J Nucl Med. 2005;46:1455–1459. [PubMed] [Google Scholar]
- Fueger B, Weber W, Quon A, Crawford T, Allen-Auerbach M, Halpern B, Ratib O, Phelps M, Czernin J. Performance of 2-deoxy-2-[F-18]fluoro-d-glucose positron emission tomography and integrated PET/CT in restaged breast cancer patients. Mol Imaging Biol. 2005;7:369–376. doi: 10.1007/s11307-005-0013-4. [DOI] [PubMed] [Google Scholar]
- Czernin J, Phelps ME. Positron emission tomography scanning: current and Future applications. Ann Rev Med. 2002;53:89–112. doi: 10.1146/annurev.med.53.082901.104028. [DOI] [PubMed] [Google Scholar]
- Yap CS, Seltzer MA, Schiepers C, Gambhir SS, Rao J, Phelps ME, Valk PE, Czernin J. Impact of whole-body 18F-FDG PET on staging and managing patients with breast cancer: the referring physician's perspective. J Nucl Med. 2001;42:1334–1337. [PubMed] [Google Scholar]
- Lopez-Rios F, Sanchez-Arago M, Garcia-Garcia E, Ortega AD, Berrendero JR, Pozo-Rodriguez F, Lopez-Encuentra A, Ballestin C, Cuezva JM. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007;67:9013–9017. doi: 10.1158/0008-5472.CAN-07-1678. [DOI] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Hill MM, Clark SF, Tucker DF, Birnbaum MJ, James DE, Macaulay SL. A role for protein kinase Bbeta/Akt2 in insulin-stimulated GLUT4 translocation in adipocytes. Mol Cell Biol. 1999;11:7771–7781. doi: 10.1128/mcb.19.11.7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger A, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002;13:2276–2288. doi: 10.1091/mbc.01-12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley J, Itchayanan D, Barnes K, McIntosh E, Tang X, Downes CP, Holman GD, Whetton AD, Owen-Lynch PJ, Baldwin SA. Interleukin-3-mediated cell survival signals include phosphatidylinositol 3-kinase-dependent translocation of the glucose transporter GLUT1 to the cell surface. J Biol Chem. 2003;278:39337–39348. doi: 10.1074/jbc.M305689200. [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer RB, Stairs DB, Dugan KD, Notarfrancesco KL, Portocarrero CP, Keister BA, Belka GK, Cho H, Rathmell JC, Thompson CB, Birnbaum MJ, Chodosh LA. Isoform-specific requirement for Akt1 in the developmental regulation of cellular metabolism during lactation. Cell Metab. 2006;4:475–490. doi: 10.1016/j.cmet.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7:389–397. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor supression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–929. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Godoy A, Ulloa V, Rodriguez F, Reincke K, Yanez AJ, Garcia MDLA, Medina RA, Carrasco M, Barberis S, Castro T, martinez F, Koch X, Vera JC, Poblete MT, Figueroa CD, Peruzzo B, Perez F, Nualart F. Differential subcellular distribution of glucose transporters GLUT1-6 and GLUT9 in human cancer: Ultrastructural localization of GLUT1 and GLUT5 in breast tumor tisues. J Cell Physiol. 2006;207:614–627. doi: 10.1002/jcp.20606. [DOI] [PubMed] [Google Scholar]
- Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–4696. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Robey RB, Hay N. Akt Inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004;24:730–740. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by Akt Is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277:33895–33900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–6322. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281:37372–37380. doi: 10.1074/jbc.M608372200. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Xu Rh, Du M, Feng L, Sasaki R, Carew JS, Hu Y, Ramdas L, Hu L, Keating MJ, Zhang W, Plunkett W, Huang P. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol. 2006;175:913–923. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Carling D. AMPK. Curr Biol. 2004;14:R220. doi: 10.1016/j.cub.2004.02.048. [DOI] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade: a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 Regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- Engel M, Mazurek S, Eigenbrodt E, Welter C. Phosphoglycerate mutase-derived polypeptide inhibits glycolytic flux and induces cell growth arrest in tumor cell lines. J Biol Chem. 2004;279:35803–35812. doi: 10.1074/jbc.M402768200. [DOI] [PubMed] [Google Scholar]
- Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, laFavera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci USA. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72. doi: 10.1016/S0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue IV: a study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13:27–29. [PubMed] [Google Scholar]
- Ookhtens M, Kannan R, Lyon I, Baker N. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Physiol Regul Integr Comp Physiol. 1984;247:R146–R153. doi: 10.1152/ajpregu.1984.247.1.R146. [DOI] [PubMed] [Google Scholar]
- Sabine JR, Abraham S, Chaikoff IL. Control of lipid metabolism in hepatomas: insensitivity of rate of fatty acid and cholesterol synthesis by mouse hepatome BW7756 to fasting and to feedback control. Cancer Res. 1967;27:793–799. [PubMed] [Google Scholar]
- Szutowicz A, Kwiatkowski J, Angielski S. Lipogenic and glycolytic anzyme activites in carcinoma and nonmalignant diseases of the human breast. Br J Cancer. 1979;39:681–687. doi: 10.1038/bjc.1979.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 2000;16:202–208. doi: 10.1016/S0899-9007(99)00266-X. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR. Fatty acid synthesis: a potential selctive taret for antineoplastic therapy. Proc Natl Acad Sci USA. 1994;91:6379–6383. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen JV, Vanderhoydonc F, Elgamal AA, Eelen M, Vercaeren I, Joniau S, Van Poppel H, Baert L, Goossens K, Heyns W, Verhovoeven G. Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int J Cancer. 2000;88:176–179. doi: 10.1002/1097-0215(20001015)88:2<176::AID-IJC5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, Bubley G, Balk S, Loda M. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol Cancer Res. 2003;1:707–715. [PubMed] [Google Scholar]
- Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, Verhoeven G. Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem Biophys Res Commun. 2003;302:898–903. doi: 10.1016/S0006-291X(03)00265-1. [DOI] [PubMed] [Google Scholar]
- De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res. 2003;63:3799–3804. [PubMed] [Google Scholar]
- Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-α gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res. 2005;65:6719–6725. doi: 10.1158/0008-5472.CAN-05-0571. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP, Piantadosi S, Pasternack GR. Haptoglobin-related protein (Hpr) epitopes in breast cacner as a predictor of recurrance of the disease. N Engl J Med. 1989;321:636–641. doi: 10.1056/NEJM198909073211003. [DOI] [PubMed] [Google Scholar]
- Ursini-Siegel J, Rajput AB, Lu H, Sanguin-Gendreau V, Zuo D, Papavasiliou V, Lavoie C, Turpin J, Cianflone K, Huntsman DG, Muller WJ. Elevated expression of DecR1 impairs ErbB2/Neu-induced mammary tumor development. Mol Cell Biol. 2007;27:6361–6371. doi: 10.1128/MCB.00686-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig S, Liaw C, Towle H, Oppenheimer J. Thyroid hormone attenuates and augments hepatic gene expression at a pre-translactional level. Proc Natl Acad Sci USA. 1981;78:4733–4737. doi: 10.1073/pnas.78.8.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinlaw WB, Tron P, Friedmann AS. Nuclear localization and hepatic zonation of rat 'spot 14' protein: immunohistochemical investigation employing anti-fusion protein antibodies. Endocrinology. 1992;131:3120–3122. doi: 10.1210/en.131.6.3120. [DOI] [PubMed] [Google Scholar]
- Kinlaw WB, Church JL, Harmon J, Mariash CN. Direct evidence for a role of the 'Spot 14' protein in the regulation of lipid synthesis. J Biol Chem. 1995;270:16615–16618. doi: 10.1074/jbc.270.28.16615. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Anderson GW, Mucha GT, Parks EJ, Metkowski JK, Mariash CN. The Spot 14 protein is required for de novo lipid synthesis in the lactating mammary gland. Endocrinology. 2005;146:3343–3350. doi: 10.1210/en.2005-0204. [DOI] [PubMed] [Google Scholar]
- Schwertfeger KL, McManaman JL, Palmer CA, Neville MC, Anderson SM. Expression of constitutively activated Akt in the mammary gland leads to excess lipid synthesis during pregnancy and lactation. J Lipid Res. 2003;44:1100–1112. doi: 10.1194/jlr.M300045-JLR200. [DOI] [PubMed] [Google Scholar]
- Sundqvist A, goechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCFFbw7. Cell Metab. 2005;1:379–391. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Wendy AW, Gary NS, Peter MM, Bernard FC, Jennifer JG, William BK. Expression of Spot 14 (THRSP) predicts disease free survival in invasive breast cancer: immunohistochemical analysis of a new molecular marker. Breast Cancer Res Treat. 2006;98:231–240. doi: 10.1007/s10549-005-9154-z. [DOI] [PubMed] [Google Scholar]
- Kinlaw WB, Quinn JL, Wells WA, Roser-Jones C, Moncur JT. Spot 14: a marker of aggressive breast cancer and a potential therapeutic target. Endocrinology. 2006;147:4048–4055. doi: 10.1210/en.2006-0463. [DOI] [PubMed] [Google Scholar]