

Abstract
The γc-family cytokine IL-2 activates signaling events that contribute to cell survival and proliferation, the best-studied of which are the STAT-5 and phosphatidylinositol 3-kinase (PI3K) pathways. The starting point of this study was to define genes regulated by the IL-2R-mediated PI3K pathway in T cells. Accordingly, we used an erythropoietin (EPO) receptor chimeric receptor system in which IL-2-dependent HT-2 T cells expressed a mutant EPO-IL-2Rβ construct where Tyr-338 is mutated to Phe. Cells expressing this mutant IL-2Rβ chain fail to induce phosphorylation of PI3K-p85α/β or activate Akt, but mediate normal IL-2-dependent proliferation and activation of JAK1 and STAT-5A/B. Microarray analyses revealed differential regulation of numerous genes compared with cells expressing a wild-type IL-2Rβ, including up-regulation of the IL-17 receptor subunit IL-17RA. Blockade of the PI3K pathway but not p70S6K led to up-regulation of IL-17RA, and constitutive Akt activation was associated with suppressed IL-17RA expression. Moreover, similar to the mutant EPO-IL-2Rβ chimera, IL-15 and IL-21 induced IL-17RA preferentially compared with IL-2, and IL-2 but not IL-15 or IL-21 mediated prolonged activation of the PI3K p85 regulatory subunit. Thus, there are intrinsic signaling differences between IL-2 and IL-15 that can be attributed to differences in activation of the PI3K pathway.
Cytokine receptors exhibit remarkable redundancy in their signaling cascades, in part because of widespread sharing of receptor subunits (1). However, individual cytokines are generally found to have unique, non-redundant activities in vivo, despite considerable overlap in signaling cascades. A striking case in point is interleukin (IL)2-2 and IL-15, members of the “γc-superfamily” of cytokines. IL-2 and IL-15 use identical receptor subunits, IL-2Rβ and the common γ (γc) chain, to mediate downstream signals. Both receptor complexes also contain unique affinity-modulating subunits, IL-2Rα and IL-15Rα, which mediate high affinity ligand binding. However, the “private” α subunits have very short cytoplasmic domains and are generally not thought to contribute directly to signal transduction (reviewed in Refs. 2, 3). Despite sharing signaling subunits, IL-2 and IL-15 mediate distinct events in vivo. Differential expression of their respective private subunits causes NK cells and CD8+ memory cells to be more sensitive to IL-15, whereas newly activated CD4+ T cells and Treg cells express high levels of CD25/IL-2Rα and thus are regulated by IL-2. Moreover, IL-2 and IL-15 exert opposing effects on survival and expansion of the T cell memory population (3).
Because they share IL-2Rβ and γc, IL-2 and IL-15 have long been presumed to trigger the same signaling pathways in target cells, which may then be interpreted differently depending on cell type. IL-2 and IL-15 activate the Janus (JAK) kinases-1 and -3 leading to activation of the STAT-5 transcription factor (3-7), and also the MAPK and phosphatidylinositol 3-kinase (PI3K) pathways (8-10), leading to cell growth and inhibition of apoptosis (reviewed in Refs. 11, 12). STAT-5 can be recruited to any one of three distinct tyrosine residues on the IL-2Rβ chain (9, 13), whereupon it activates a variety of genes, particularly IL-2Rα (14) (Fig. 1A). This event leads to formation of a high affinity IL-2 receptor and preferential expansion of antigen-specific naïve T cells. The PI3K pathway is activated via the Shc adaptor binding to a single tyrosine within IL-12Rβ, leading to recruitment and phosphorylation of the regulatory subunits of PI3K, p85α/β (15). Activation of PI3K leads to phosphorylation of the membrane lipid PIP2 converting it to PIP3, and subsequent recruitment of pleckstrin homology (PH) domain-containing kinases such as PDK1 and Akt/PKB. Akt in turn phosphorylates a number of downstream targets that cumulatively serve to promote cell survival, including GSK3α/β, FOXO proteins, and Bad (reviewed in Ref. 16). The PI3K pathway is also upstream of mTOR and p70 S6 kinase (p70S6K), rapamycin-sensitive signaling molecules that contribute to IL-2-mediated growth signaling (8, 16, 17). Therefore, the JAK-STAT-5 and PI3K pathways mediate distinct signaling events, and are critical components of the IL-2 and IL-15 signaling pathways.
FIGURE 1.

Differential gene expression mediated by the PI3K pathway through IL-2RβY338. A, schematic diagram of the EPOR-IL-2Rβ chimeric receptor construct. The location of JAK-binding site (Box1/Box2) and the pathways activated by individual tyrosine residues are indicated. B, p85 subunit of PI3K is activated through Tyr-338 (1Y). HT-2 cells expressing the indicated EPO-IL-2R chimeras were stimulated for 1 h with EPO or IL-2. Lysates were immunoprecipitated with α-p85α/β Abs, separated by SDS-PAGE, and immunoblotted with α-Ptyr (4G10) Abs (top) or p85α/β Abs (bottom). C, differentially expressed genes in HT-2.EPOβγ cells versus HT-2.EPOβ5Y/γ cells. Gene differences with a p value <0.01 are shown (222 total genes). D, selected genes enhanced in HT-2.EPOβ5Y/γ cells or HT-2.EPOβγ cells. E, IL-17RA surface expression is elevated in HT-2.EPOβ5Y/γ cells. The indicated cell lines were incubated in EPO for 24 h and IL-17RA expression was assessed by flow cytometry. Filled histograms, isotype control.
IL-2 is produced primarily by newly activated CD4+ T cells, although CD8+ T cells and B cells are also sources of IL-2 (11). Recently, a paradigm shift in how T cells produce cytokines occurred with the discovery of a new subset of CD4+ T helper (Th) cells distinct from Th1 and Th2, termed Th17. Differentiation and expansion of these cells is driven by TGFβ, IL-6, and IL-23. As the name implies, these cells produce IL-17 as well as other pro-inflammatory cytokines including IL-17F and IL-22. Interestingly, Th17 cells differentiate in opposition to Treg cells, in part because development of Th17 cells requires TGFβ and IL-6, whereas TGFβ without IL-6 promotes Treg development (reviewed in Refs. 18, 19). It is becoming increasingly clear that the Th17 population plays a major role in driving autoimmunity, and efforts to block IL-17 or its receptor are under serious consideration for anti-cytokine therapy (20, 21). Interestingly, many γc cytokines regulate the IL-17/IL-17 receptor system. For example, IL-4 derived from Th2 cells inhibits Th17 development (22), and IL-2 activation of STAT-5 appears to inhibit Th17 differentiation in favor of Treg expansion, although this is still somewhat controversial (23, 24). Conversely, IL-21 up-regulates the IL-17 receptor (25), and was recently shown to be a mediator of Th17 differentiation (26-28). IL-15 augments IL-17 expression, and both IL-15 and IL-17 are elevated in RA patients (29). However, the connections between γc cytokines and the IL-17 family remain poorly understood.
IL-17 binds to a specific receptor on target cells composed of IL-17RA and IL-17RC, and IL-17 activates a pro-inflammatory signaling cascade leading to expression of chemokines, cytokines and other inflammatory effector molecules (30, 31). Although the IL-17 receptor is ubiquitously expressed, its function has mainly been evaluated on non-immune cells, particularly epithelial cells and fibroblasts. The role of IL-17 in lymphocytes is unknown, and almost nothing is known about how the IL-17 receptor is regulated.
The initial goal of the present study was to define specific molecular events mediated by the IL-2R-mediated PI3K pathway in T cells. Microarray analyses revealed that activation of a mutant IL-2 receptor that cannot activate PI3K leads to the up-regulation of numerous immune genes, including IL-17RA. Moreover, IL-15 and IL-21 induce IL-17RA preferentially compared with IL-2, and IL-17RA expression is limited by the PI3K pathway. Consistent with this finding, IL-2 but not IL-15 mediates prolonged activation of the PI3K p85 regulatory subunit. Further studies revealed that signaling by PI3K through Akt but not p70S6K leads to up-regulation of IL-17RA. Thus, there are intrinsic signaling differences between IL-2 and IL-15 that can be attributed to differences in molecular activation of the PI3K pathway.
EXPERIMENTAL PROCEDURES
Cell Culture, Cell Lines, and Cytokine Stimulations—HT-2, Jurkat, A20, and primary cells were maintained in RPMI 1640, 10% fetal bovine serum (Gemini Bioproducts, Woodland, CA), 2 mm glutamine, 0.05 mm 2-β-mercaptoethanol, penicillin/streptomycin, and 1 nm recombinant human IL-2 (provided by the Chiron Corporation, Emeryville, CA). HT-2.EPOβγ, HT-2.EPOβ5Y/γ, and HT-2.EPOβ1Y/γ cells were maintained in this medium with human EPO (a kind gift of Amgen, Thousand Oaks, CA) in place of IL-2. ST2 cells were maintained in α-MEM with 10% fetal bovine serum and antibiotics. HT-2 cells were transfected by electroporation (32). For stimulations, HT-2 cells were starved in phosphate-buffered saline, stripped with 10 mm sodium citrate/140 nm NaCl, and incubated in RPMI 1640/1% bovine serum albumin (Sigma) for 2-4 h. Stimulations used IL-2 (5 nm), IL-15 (50 nm), IL-21 (50 nm), IL-17 (100 ng/ml), TNFα (2 ng/ml), or EPO (50 units/ml) unless otherwise indicated. Other cytokines were from R&D Systems (Minneapolis, MN) or Peprotech (Rocky Hill, NJ). Primary CD8+ T cells were obtained from mouse spleen with a CD8+ T Cell Isolation kit (Miltenyi Biotech, Auburn, CA). All animal protocols were approved by the University at Buffalo IACUC.
Microarray Analysis—To generate expression summary values from Affymetrix GeneChips, MAS5.0 software in the “Affy” package of Bioconductor in the R statistical computing environment was used with its default settings, followed by global normalization to bring the median expression values of all four GeneChips to the same scale. This was done by selecting a baseline GeneChip from the dataset and scaling each GeneChip to the median of the baseline GeneChip (m̃) as in Equation 1.
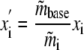 |
(Eq.1) |
For data quality control, MAS5.0 “present calls” was used to filter out probe sets whose expression intensities were close to background noise across the majority of samples before performing differential gene analysis. Filtering of 2 “present calls” was applied to either the EPOβ5Y/γ or EPOβγ group, which led to ∼47% data reduction for the comparisons. Further filtering was performed to reduce variance between replicates of the same condition by first ranking those selected probe sets based on their intensity for each of the 4 GeneChips, followed by filtering out those probe sets with 10% rank order differences for replicated GeneChips in the same group. This led to another ∼1% data reduction. Regularized Student's t test was employed to detect gene expression values significantly different between groups. For multiple test correction, the false discovery rate (FDR) was estimated using SAM, and was 0 for all data sets (33).
Plasmids—EPO-IL-2R chimeras are described in Ref. 7. The Myr-Akt construct was a kind gift of P. Tsichlis (34) and subcloned into the pIRES2-EGFP plasmid (Invitrogen).
Proliferation Assays—[3H[Thymidine incorporation assays were performed as described (32). 5 × 103 cells were washed twice in phosphate-buffered saline and incubated with cytokines for 24-48 h in triplicate. 5 h prior to harvesting, 1 μCi/well of [3H]thymidine (PerkinElmer Life Science Products) was added. Cells were harvested on a Skatron microwell harvester and analyzed on a Wallac Microbeta counter (PerkinElmer).
Real-time RT-PCR, Immunoprecipitations, Western Blotting, and EMSA—Total mRNA was prepared with RNeasy kits (Qiagen). Real time RT-PCR was performed with SYBR-Green as described (35). IL-17RA primers were from SuperArray (Frederick, MD) (35). Immunoprecipitations were performed as described (32) with α-JAK1 (Santa Cruz Biotechnology), α-p85 (Upstate Biotechnologies, Saranac, NY), and protein A- or protein G-agarose (Roche Applied Sciences). Western blots were probed with 4G10, α-p85 (Upstate Biotechnologies), α-SHIP (Santa Cruz Biotechnology), α-PTEN (Cell Signaling, Beverly, MA) or α-JAK1. EMSA were performed as described (7) with 10 μg of nuclear extract and 105 cpm of 32P-labeled probe per lane. STAT-5A/B Abs were a kind gift from L. Hennighausen (9).
Flow Cytometry and ELISA—Apoptosis assays were performed by staining with propidium iodide (PI) and GFP-Annexin V (32). Cells were analyzed on a FACS Calibur using Cellquest software (Becton Dickinson, San Jose, CA). IL-17RA staining was performed with rat anti-mouse IL-17RA M751 mAbs (kindly provided by Amgen, Seattle WA) as described (36). ELISA assays were performed with commercial kits from R&D and eBiosciences (San Diego, CA).
RESULTS
Genes Regulated by the IL-2-induced PI3K Pathway—IL-2 induces multiple signaling events in T cells, the best studied of which are the JAK1/3-STAT-5 and PI3K pathways (37). While many of the downstream targets of these pathways have been defined, these pathways also exert overlapping functions in terms of cellular survival and growth signaling (11). Therefore, we sought to delineate STAT-5-independent functions for the IL-2-induced PI3K pathway in T cells. All known IL-2-dependent signaling events proceed from tyrosine residues located within the IL-2Rβ andγc subunits (11, 32). We have extensively examined the mechanisms by which the IL-2R complex initiates these pathways using a chimeric receptor system, in which the extracellular domains of IL-2Rβ and γc have been replaced with the erythropoietin (EPO) receptor and expressed stably in IL-2-dependent HT-2 T cells. In this system, treating cells with EPO faithfully recapitulates IL-2-dependent signaling (7, 9, 38). Former studies demonstrated that activation of STAT-5 occurs independently through three Tyr residues within IL-2Rβ (Tyr-338, Tyr-392, and Tyr-510, also termed “1Y,” “5Y,” and “6Y,” Fig. 1A) (9, 13). However, activation of PI3K occurs only through Tyr-338 (1Y) via the Shc adaptor protein (Fig. 1B and Ref. 39). Importantly, these chimeric receptors demonstrated that the PI3K pathway is not required for proliferative signaling, as HT-2.EPOβ5Y/γ or HT-2.EPOβ6Y/γ cells can be maintained indefinitely in EPO and show similar growth kinetics to a wild-type receptor (9). To define genes regulated by the PI3K pathway that are not required for proliferation, we compared gene expression in HT-2 cells expressing the EPOβγ (wild-type) chimera to the EPOβ5Y/γ chimera and cultured in EPO, using replicate Affymetrix Mouse Expression Array 430A GeneChips. There was a high replicate correlation between chips (r = 0.98, data not shown). 883 genes were differentially regulated with p values <0.05 (data not shown), and 222 genes were differentially regulated with p values <0.01 (Fig. 1, C and D and supplemental Tables S1 and S2). As expected, many genes were up-regulated in HT-2.EPOβγ cells compared with HT-2.EPOβ5Y/γ cells, indicating that these are direct or indirect targets of the PI3K pathway. The most strongly enhanced of these genes (∼100-fold) was that encoding the E1A enhancer-binding protein (E1AF), an ets oncogene transcription factor involved in Ewings sarcoma (supplemental Table S1) (40). In addition, many genes were up-regulated in HT-2.EPOβ5Y/γ cells, suggesting that signals mediated by IL-2RβY338 inhibit expression of certain genes. The strongest of these (32-fold) was the pregnancy-specific glycoprotein 17 (supplemental Table S2), which is thought to play an immunoregulatory role during pregnancy (41). Interestingly, genes involved in known IL-2-mediated signaling pathways were differentially regulated, including STAT-5A, Bcl-x, and FK506-binding proteins. In addition, T cell receptor (TCR)-associated signaling genes were differentially regulated, including TCRα, Lck-interacting transmembrane protein 1 (LIME-1), PKCθ, Bcl-10, and Cot-2 (Fig. 1D) (42, 43).
Of particular interest to us was the enhancement of the gene encoding IL-17RA, the receptor for IL-17, in HT-2.EPOβ5Y/γ cells (Fig. 1D). Although IL-17RA is expressed ubiquitously (30), its surface expression varies widely (36), and nothing is known about mechanisms of IL-17RA gene regulation. By flow cytometry, HT-2 cells express quite high baseline levels of IL-17RA when cultured in IL-2 (Fig. 1E). Normal CD8+ and CD4+ T cells also express IL-17RA (data not shown, Ref. 25). HT-2.EPOβγ and HT-2.EPOβ1Y/γ cells expressed similar levels of IL-17RA whether grown in EPO or IL-2 (Fig. 1E). However, HT-2.EPOβ5Y/γ cells expressed a higher level of IL-17RA when cultured in EPO compared with IL-2, consistent with the microarray results. Moreover, IL-17RA expression was suppressed when EPOβ5Y/γ cells were co-cultured with EPO and IL-2 together, suggesting that the PI3K pathway (in this case, activated through the endogenous IL-2R) inhibits IL-17RA expression (Fig. 1E).
IL-17RA Is Induced by IL-15 and IL-21 and Is Inhibited by IL-2—In a published microarray screen of CD8+ T cell genes, the γc cytokine IL-21 was reported to up-regulate IL-17RA mRNA, which was the first suggestion that IL-17RA is subject to immune regulation (25). Accordingly, we examined IL-17RA regulation by γc cytokines in HT-2 cells. Indeed, both IL-21 and IL-15 up-regulated IL-17RA compared with IL-2 in a dose-dependent manner (Fig. 2A and supplemental Fig. S1). Moreover, IL-2 dominantly suppressed the effects of either IL-15 or IL-21, at the level of both surface expression and mRNA (Fig. 2).
FIGURE 2.

IL-17RA is induced by IL-15 and IL-21 but not by IL-2. A, IL-2 dominantly suppresses IL-17RA surface expression mediated by IL-15 and IL-21. HT-2 cells were treated for 24 h with the indicated cytokines, and IL-17RA surface expression was assessed by flow cytometry. Filled histograms, isotype control. Average mean fluorescent intensity of IL-17RA expression relative to IL-2-treated cells was determined from five independent experiments (right panel). 2°, secondary Ab alone. B, IL-2 dominantly suppresses IL-17RA mRNA expression mediated by IL-15 and IL-21. HT-2 cells were treated for 24 h with the indicated cytokines, and IL-17RA mRNA levels were determined in triplicate by real-time RT-PCR. *, p < 0.05 compared with IL-2-treated control.
The disparity in signaling between IL-2 and IL-15 was unexpected, because these cytokines use identical signaling subunits (2). However, a recent study suggested that IL-2 and IL-15 differ in the ability to activate the PI3K pathway, at least with respect to p70S6K activation (44). To determine the basis for signaling differences between IL-2 and IL-15 in HT-2 cells, we examined signaling events known to be activated by these cytokines. IL-2, IL-15, and IL-21 all activated JAK1 phosphorylation and STAT-5 nuclear import essentially equivalently (Fig. 3, A and B). Therefore, differences between IL-2 and IL-15 are likely not due to JAK or STAT activation.
FIGURE 3.

IL-2 and IL-15 differentially activate the PI3K pathway but not the JAK-STAT-5 pathway or proliferation in HT-2 cells. A, IL-2, IL-15, and IL-21 activate JAK1 equivalently in HT-2 cells. HT-2 cells were stimulated for 15 min with the indicated cytokines. Lysates were immunoprecipitated with α-JAK1 Abs, separated by SDS-PAGE, and immunoblotted with α-PTyr (4G10) Abs (top) or α-JAK1 Abs (bottom). B, IL-2, IL-15 and IL-21 activate STAT-5 equivalently in HT-2 cells. HT-2 cells were stimulated for 15 min with the indicated cytokines. Nuclear extracts were subjected to EMSA with a 32P-labeled STAT oligonucleotide (7) (which binds to STAT-1, 5, 6 and to a lesser extent STAT-3). FP, free probe. Supershifting with α-STAT-5 Abs was performed in lanes 4, 6, and 8. Arrows indicate shifted and supershifted complexes. C, IL-2, IL-15, but not IL-21 drive proliferation and inhibit apoptosis in HT-2 cells. HT-2 cells were cultured for 24, 48, or 72 h in the indicated cytokines and concentrations. Proliferation was assessed by [3H]thymidine incorporation in triplicate ± S.D., *, p < 0.05. Apoptosis was determined by FACS staining with propidium iodide and GFP-Annexin V, and apoptotic cells were defined as PI-negative/GFP-Annexin V-positive, as described in Ref. 32, 60. Data are representative of three experiments. D, signaling from PI3K via IL-2 and Y338 promotes increased cell size. HT-2 cells (top panel) or HT-2 cells expressing the indicated EPO-IL-2Rβ chimeras (bottom 3 panels) were cultured in the indicated cytokines for 24 h, and cell size was assessed by forward scatter analysis by flow cytometry.
The γc-family cytokines have many redundant functions, including proliferation and protection from apoptosis. However, prior studies using the EPO chimeric system indicate that signaling through IL-4, IL-7, and IL-9 do not support proliferation in HT-2 cells, although all receptors effectively activate their respective STAT complexes. In addition, IL-7 and IL-9 can delay apoptosis induced by IL-2 withdrawal (45-47). Here, we found that IL-21 failed to promote proliferation or protect cells from apoptosis (Fig. 3C), nor did IL-21 delay a G2 cell cycle arrest (not shown). IL-15 has never been tested directly in these cells, as the EPO chimeric system has long been assumed to reflect both IL-2 and IL-15 signaling (7). Because we had observed signaling differences between IL-2 and IL-15, we assessed the ability of IL-15 to inhibit apoptosis and drive proliferation. Indeed, both IL-2 and IL-15 protected HT-2 cells from apoptosis caused by IL-2 starvation and triggered nearly identical proliferative responses in short- and long-term culture (Fig. 3C). Therefore, IL-2 and IL-15 are indistinguishable with respect to proliferative and survival responses in HT-2 cells, whereas IL-21 does not support proliferation.
In performing these studies, we noted that HT-2 cells cultured in IL-15 or IL-21 appeared smaller in size than when cultured in IL-2. This observation was confirmed by forward scatter FACS profiles (Fig. 3D). These data confirm that IL-2 and IL-15 have indistinguishable mitogenic activities, whereas IL-2 is a more potent growth factor (44). Notably, we made a similar observation with the chimeric receptors; namely, HT-2.EPOβγ cells and HT-2.EPOβ1Y/γ cells are the same size when grown in EPO or IL-2, whereas HT-2.EPOβ5Y/γ cells are smaller in EPO (Fig. 3D). Therefore, despite an equivalent ability to promote proliferation, IL-2 induces cellular growth and metabolism in a manner distinct from IL-15 and IL-21. Moreover, signaling by IL-15 appears to be similar to HT-2.EPOβ5Y/γ, suggesting that the PI3K pathway may be differentially controlled by IL-2 and IL-15 in the same cell background.
IL-2 and IL-15 Differentially Induce the PI3K Pathway—Accordingly, to assess activation of the PI3K pathway by IL-2 and IL-15, the p85 regulatory subunit of PI3K was examined (Fig. 4A). To reduce activation of PI3K to baseline, cells were starved without IL-2 for 2 h, and then restimulated with IL-2, IL-15, or IL-21 for various time points, and phosphorylation of p85 was monitored. The kinetics and magnitude of p85 phosphorylation were similar at a 1-h time point (Fig. 4A). However, starting at 4 h and continuing to 24 h, phosphorylation of p85 was substantially reduced in IL-15-treated or IL-21-treated cells compared with IL-2-treated cells (Fig. 4A). Therefore, IL-15 and IL-21 are unable to sustain PI3K activation over a time frame that correlates with IL-17RA gene regulation. Although many T cell lines have defects in the PI3K pathway (48), we verified that HT-2 cells express normal levels of both PTEN and SHIP and do not exhibit constitutive activation of Akt (Fig. 4B and data not shown) (32).
FIGURE 4.

The PI3K pathway inhibits IL-17RA expression. A, sustained phosphorylation of p85 induced by IL-2 but not IL-15 or IL-21. HT-2 cells were cultured in IL-2, IL-15, or IL-21 for the indicated time points, and phosphorylation of the p85α/β was assessed as in Fig. 1. B, HT-2 cells express PTEN and SHIP. Lysates from HT-2, Jurkat T cells or the A20 B cell line were immunoblotted with Abs to PTEN or IKKγ (top panels) or immunoprecipitated with α-SHIP Abs and probed for SHIP (bottom panel). C, PI3K inhibitors up-regulate IL-17RA. HT-2 cells were incubated in IL-2 ± LY294002 or Wortmannin for 20 h. IL-17RA expression was assessed by flow cytometry. D, rapamycin inhibits p70S6K but not IL-15-induced IL-17RA expression. HT-2 cells were incubated with IL-2 or IL-15 ± Rapamycin for 20 h. IL-17RA (top) or p70S6K (bottom) were assessed by flow cytometry.
To test the hypothesis that the PI3K pathway is responsible for IL-2-mediated suppression of IL-17RA, HT-2 cells were cultured in IL-2 in the presence of PI3K inhibitors, Wortmannin or LY294002. IL-17RA was up-regulated in the presence of both these inhibitors, further demonstrating the ability of the PI3K pathway to limit IL-17RA expression (Fig. 4C). In contrast, the inhibitor rapamycin, which blocks mTOR, p70S6K activation, and IL-2-induced proliferation (16), did not relieve IL-2-induced suppression of IL-17RA, although rapamycin did effectively inhibit phosphorylation of p70S6K. Activation of p70S6K was much stronger in IL-2-induced cells than in IL-15-induced HT-2 cells (Fig. 4D). However, because rapamycin had no effect on IL-15-mediated induction of IL-17RA, it is unlikely that this arm of the PI3K pathway is involved in the differential regulation of IL-17RA by these cytokines. Together, these data indicate that the downstream target of PI3K with respect to IL-17RA regulation is apparently not p70S6K.
To determine if Akt, also a PI3K target, is involved in IL-17RA regulation, HT-2 cells were stably transfected with a myristoylated form Akt that is constitutively active (34). Clonal lines expressing Myr-Akt showed constitutive Akt phosphorylation and prolonged survival following IL-2 withdrawal (Fig. 5, A and B), confirming the functionality of Myr-Akt. As shown in Fig. 4A, depriving HT-2 cells of IL-2 caused a reduction in PI3K activation. This was accompanied by a concomitant up-regulation in IL-17RA expression after 22 h of IL-2 starvation (Fig. 5C and data not shown). However, in Myr-Akt cells, there was almost no enhancement of IL-17RA following IL-2 withdrawal, consistent with a dominant suppressive effect of the PI3K/Akt signaling pathway on IL-17RA expression (Fig. 5C).
FIGURE 5.

A constitutively active form of Akt promotes IL-17RA expression. A, HT-2 cells stably express Myr-Akt. Lysates from 3 HT-2 subcloned stably transfected with Myr-Akt were immunoblotted with α-pAkt (Ser-473). B, Myr-Akt delays apoptosis induced by cytokine withdrawal. HT-2 cells expressing Myr-Akt (clones A26 and A39) were incubated with or without IL-2 for the indicated time points, and cell survival (PI-negative) or apoptosis (GFP-Annexin V-positive) cells were assessed by flow cytometry. C, expression of a constitutively active form of Akt prevents up-regulation of IL-17RA induced by cytokine withdrawal. HT-2 or HT-2.MyrAktA39 cells were deprived of IL-2 for the indicated times, and IL-17RA expression was assessed by flow cytometry.
Differential signaling between IL-2 and IL-15 has been reported in primary CD8+ T cells previously with respect to cell size, growth, and gene expression (44). To determine whether γc cytokines differentially regulate IL-17RA in vivo, we stimulated CD8+ T cells purified by magnetic beads with IL-2, IL-15, and IL-21 and stained for IL-17RA (Fig. 6). As shown, IL-21 induced IL-17RA to high levels, which is consistent with a previous report (25). However, when cultured with the PI3K inhibitor LY294002, IL-15 up-regulated IL-17RA significantly more strongly than IL-2. Although these data do not precisely mirror findings in HT-2 cells, they nonetheless indicate differential use of the PI3K pathway by these cytokines in terms of IL-17RA expression. It should be noted that the lineage of HT-2 cells is unclear, as they express both CD4 and CD8.3 Because purified CD8+ T cells probably contain multiple distinct subsets of cells with potentially different capacities for responsiveness to cytokines, the finding that IL-2 and IL-15 can regulate distinct responses in a single cell line provides compelling evidence that these cytokines are capable of mediating distinct biochemical signals.
FIGURE 6.

Regulation of IL-17RA in primary CD8+ T cells. Splenic CD8+ T cells were purified by magnetic beads and stimulated for 6 h with the indicated cytokines with DMSO (top) or LY294002 (bottom). IL-17RA was assessed by flow cytometry.
IL-17 Mediates Down-regulation of IL-17RA—IL-17RA is expressed ubiquitously, but its functions have been determined primarily in nonimmune cell types such as mesenchymal and epithelial cells (31). T and B cell lines express high levels of IL-17RA (Ref. 30),3 but the functional significance of this expression is unknown. Because HT-2 cells express a high baseline level of IL-17RA, we treated cells with IL-17 and evaluated the consequences to gene expression. Surprisingly, there was no expression of known IL-17 target genes (35) in either HT-2 cells or primary CD8+ T lymphocytes, including IL-6, LIX/CXCL5, Groα/KC/CXCL1, or 24p3 (Fig. 7, A and B and data not shown) following IL-17 stimulation. Therefore, T cells appear refractory to IL-17 signaling, at least in terms of known gene expression. This is at least partly due to a lack of detectable IL-17RC mRNA expression in this cell line by RT-PCR (data not shown).
FIGURE 7.

IL-17 does not induce detectable signaling in HT-2 cells or primary T lymphocytes. A and B, IL-17 does not induce canonical target genes such as IL-6 or LIX/CXCL5. HT-2 cells, splenocytes, or purified CD8+ T cells were incubated with the indicated cytokines for 2-24 h, and conditioned supernatants were assayed in triplicate for IL-6 and LIX/CXCL5 by ELISA (36).
DISCUSSION
A major advance in understanding T cell differentiation occurred with the recognition of a distinct population of IL-17-secreting T cells, “Th17.” Th17 cells are now recognized to be the major drivers of autoimmunity (49). However, the functions of IL-17 and other Th17-derived cytokines are still being defined. The IL-17 receptor subunit IL-17RA is highly expressed on non-immune cells such as epithelial cells and fibroblasts, where it acts similarly to innate immune cytokines and Toll-like receptors to amplify inflammation via expression of IL-6, chemokines, and other pro-inflammatory effectors (35). Studies of IL-17RA-deficient mice have identified defects in neutrophil development and recruitment, leading to increased susceptibility to infectious disease but resistance to chronic, autoimmune conditions (50, 51). However, IL-17RA is expressed on lymphocytes as well, where its function and regulation are poorly defined. A prior microarray study identified IL-21 as an inducer of IL-17RA in CD8+ T cells (25). Here, we confirm and extend that finding using chimeric IL-2 receptor mutants defective in PI3K activation. We further show that IL-2, IL-15, and IL-21 differentially regulate IL-17RA through the PI3K-Akt pathway.
Detailed studies of Th17 differentiation have implicated the γc cytokine family. Th17 differentiation is driven by TGFβ in combination with IL-6, and the STAT-3 transcription factor is essential for this event. IL-23 serves as a proliferation signal to specifically expand Th17 cells, which exclusively express the IL-23R (49). Recently, it was shown that IL-21, which also activates both STAT-3, drives Th17 differentiation downstream of IL-6 and is necessary for optimal Th17 function in vivo (26-28). Conversely, Th1 and Th2 cytokines (IFNγ and IL-4, respectively) inhibit Th17 development (52, 53). IL-2 has also been shown to inhibit Th17 differentiation, favoring expansion of the T regulatory cell (Treg) lineage. This is mediated by STAT-5 (23). Although IL-15 has not been implicated in Th17 differentiation, there is evidence that IL-15 can enhance IL-17 expression (29). Therefore, our finding that IL-2 limits IL-17RA expression whereas IL-15 and IL-21 promote it (Fig. 2A) is consistent with the overall model of Th17 differentiation.
The function of IL-17 in T cells is unknown. Lymphocytes, both primary cells and lymphocyte cell lines, express IL-17RA (Fig. 1 and data not shown). However, canonical IL-17 target genes such as IL-6 and chemokines are not induced, either in HT-2 cells or primary T cells (Fig. 7, A and B). In HT-2 cells this appears to be due to a lack of IL-17RC expression,3 an IL-17R superfamily member that was recently shown to be an essential component of the IL-17R complex (54). However, IL-17RC mRNA is present in primary CD8+ populations,3 though we do not know whether this subunit is expressed at the surface. Nor do we find that IL-17 can promote proliferation, survival, or apoptosis in T cells.3 It is possible that IL-17 target genes are completely non-overlapping in T cells compared with fibroblasts and epithelial cells. Alternatively, IL-17RA may bind to another, unknown ligand. Further experimentation will be necessary to determine the function of IL-17RA in T cells.
There is increasing evidence that IL-2 and IL-15 exhibit differential effects on target cells, despite using identical signaling subunits in their respective receptor complexes (44, 55, 56). However, most studies to date have been performed in primary lymphocytes, where subpopulations may express different levels of IL-15Rα and IL-2Rα and hence respond differentially to the same cytokines. One microarray study showed highly similar gene expression induced by IL-2 and IL-15, suggesting few or no signaling differences between these cytokines (57).
Here, we examined γc cytokine signaling in a controlled and uniform system with the IL-2-dependent T cell line, HT-2. We show for the first time that an IL-2Rβ mutant that cannot activate the PI3K pathway inhibits expression of many immunomodulatory genes, including IL-17RA (Fig. 1D, supplemental Tables S1 and S2). Surprisingly, despite the use of identical signaling subunits, IL-15 promotes IL-17RA expression and fails to maintain long-term activation of the PI3K-p85 pathway (Fig. 3A). Moreover, HT-2 cells grown in IL-15 or HT-2.EPOβ5Y/γ cells grown in EPO were reproducibly smaller than cells cultured in IL-2 or HT-2.EPOβγ and HT-2EPOβ1Y/γ cells grown in EPO (Fig. 3D); thus, cell size correlates with activation of the PI3K pathway.
The reason that IL-15 does not sustain PI3K is not certain. However, unlike IL-2, IL-15 can be presented in a “transsignaling” mode, wherein IL-15Rα on a neighboring cell binds IL-15 and induces signaling on a target cell lacking IL-15Rα (58). It is believed that this mode of presentation permits IL-15 to have a longer stimulatory half-life compared with soluble IL-15 (59), which may influence its signaling properties (e.g. the duration in which PI3K is activated). We also observed slightly more enhanced JAK1 activation by IL-2, which could possibly contribute to a more potent downstream signal (Fig. 3A).
Although our studies in primary T cells do not precisely mirror effects in HT-2 cells, we nonetheless observe differences in signaling between IL-2 and IL-15, which are revealed most strongly when the PI3K pathway is blocked (Fig. 6). Clearly IL-21 is a more potent inducer of IL-17RA in primary CD8+ T cells, which may be biologically relevant in light of the fact that IL-21 is an autocrine inducer of the Th17 pathway (26-28).
The present studies suggest that IL-2-induced PI3K activation limits IL-17RA gene expression, but HT-2 cells still maintain a fairly high baseline level of IL-17RA even in the presence of IL-2. Thus, there are likely to be separate regulatory elements that govern baseline versus inducible levels of the IL-17RA gene. The promoter for IL-17RA is uncharacterized, and few clues can be gleaned from phylogenetic alignments of its proximal promoter region.3 However, it is clear that IL-17RA is subject to modulation by inflammatory factors such as γc cytokines including IL-21, IL-15, and IL-4 (25, 52, 53). Therefore, future studies will be directed at understanding this process in molecular detail.
Supplementary Material
Acknowledgments
We thank J. Ernst for Annexin V-GFP, L. Hennighausen for α-STAT-5 Abs, and P. Tshichlis for the Myr-Akt plasmid. Anti-IL-17RA Abs and EPO were generously provided by Amgen. IL-2 was a kind gift from Chiron Corp.
This work was supported, in whole or in part, by National Institutes of Health Grants AI05439 and AR050458 (to S. L. G.). This work was also supported by Training Grant DE007034 from the Department of Oral Biology at SUNY Buffalo (to M. J. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1 and Tables S1 and S2.
Footnotes
The abbreviations used are: IL, interleukin; PI3K, phosphatidylinositol 3-kinase; JAK, Janus kinase; STAT, signal transducer and activator of transcription; EPO, erythropoietin; PH, pleckstrin homology; Th, T helper; GFP, green fluorescent protein; Abs, antibodies; EMSA, electrophoretic mobility shift assay; FACS, fluorescent-activated cell sorting; ELISA, enzyme-linked immunosorbent assay; PI, propidium iodide.
M. J. Lindemann, unpublished observations.
References
- 1.Ozaki, K., and Leonard, W. J. (2002) J. Biol. Chem. 277 29355-29358 [DOI] [PubMed] [Google Scholar]
- 2.Ma, A. (2000) Mod. Asp. Immunobiol. 1 102-104 [Google Scholar]
- 3.Waldmann, T. (2006) Nat. Rev. 6 595-601 [DOI] [PubMed] [Google Scholar]
- 4.Johnston, J. A., Kawamura, M., Kirken, R. A., Chen, Y. Q., Blake, T. B., Shibuya, K., Ortaldo, J. R., McVicar, D. W., and O'Shea, J. J. (1994) Nature 370 151-153 [DOI] [PubMed] [Google Scholar]
- 5.Russell, S. M., Johnston, J. A., Noguchi, M., Kawamura, M., Bacon, C. M., Friedmann, M., Berg, M., McVicar, D. W., Whitthuhn, B. A., Silvennoinen, O., Goldman, A. S., Schmalsteig, F. C., Ihle, J. N., O'Shea, J. J., and Leonard, W. J. (1994) Science 266 1042-1045 [DOI] [PubMed] [Google Scholar]
- 6.Hou, J., Schindler, U., Henzel, W. J., Wong, S. C., and McKnight, S. L. (1995) Immunity 2 325-330 [DOI] [PubMed] [Google Scholar]
- 7.Gaffen, S. L., Lai, S. Y., Xu, W., Gouilleux, F., Groner, B., Goldsmith, M. A., and Greene, W. C. (1995) Proc. Natl. Acad. Sci., U. S. A. 92 7192-7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karnitz, L. M., Burns, L. A., Sutor, S. L., Blenis, J., and Abraham, R. T. (1995) Mol. Cell. Biol. 15 3049-3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaffen, S. L., Lai, S. Y., Ha, M., Liu, X., Hennighausen, L., Greene, W. C., and Goldsmith, M. A. (1996) J. Biol. Chem. 271 21381-21390 [DOI] [PubMed] [Google Scholar]
- 10.Hatakeyama, M., Kawahara, A., Mori, H., Shibuya, H., and Taniguchi, T. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 2022-2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaffen, S. L. (2001) Cytokine 14 63-77 [DOI] [PubMed] [Google Scholar]
- 12.Gaffen, S. L., Goldsmith, M. A., and Greene, W. C. (1998) in The Cytokine Handbook, Third Edition (Thomson, A., ed) pp. 73-103, Academic Press, Ltd., London, England
- 13.Friedmann, M. C., Migone, T.-S., Russell, S. M., and Leonard, W. J. (1996) Proc. Natl. Acad. Sci., U. S. A. 93 2077-2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakajima, H., Liu, X.-W., Wynshaw-Boris, A., Rosenthal, L. A., Imada, K., Finbloom, D. S., Henninghausen, L., and Leonard, W. J. (1997) Immunity 7 691-701 [DOI] [PubMed] [Google Scholar]
- 15.Lord, J. D., McIntosh, B. C., Greenberg, P. D., and Nelson, B. H. (1998) J. Immunol. 161 4627-4633 [PubMed] [Google Scholar]
- 16.Manning, B. D., and Cantley, L. C. (2007) Cell 129 1261-1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nourse, J., Firpo, E., Flanagan, W. M., Coats, S., Polyak, K., Lee, M.-H., Massague, J., Crabtree, G. R., and Roberts, J. M. (1994) Nature 372 570-573 [DOI] [PubMed] [Google Scholar]
- 18.Weaver, C. T., Harrington, L. E., Mangan, P. R., Gavrieli, M., and Murphy, K. M. (2006) Immunity 24 677-688 [DOI] [PubMed] [Google Scholar]
- 19.Cua, D. J., and Kastelein, R. A. (2006) Nat. Immunol. 7 557-559 [DOI] [PubMed] [Google Scholar]
- 20.Kikly, K., Liu, L., Na, S., and Sedgwick, J. D. (2006) Curr. Opin. Immunol. 18 670-675 [DOI] [PubMed] [Google Scholar]
- 21.Afzali, B., Lombardi, G., Lechler, R., and Lord, G. (2007) Clin. Exp. Immunol. 148 32-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong, C. (2006) Nat. Rev. 6 329-333 [DOI] [PubMed] [Google Scholar]
- 23.Laurence, A., Tato, C. M., Davidson, T. S., Kanno, Y., Chen, Z., Yao, Z., Blank, R. B., Meylan, F., Siegel, R., Hennighausen, L., Shevach, E. M., and O'Shea, J. J. (2007) Immunity 26 371-381 [DOI] [PubMed] [Google Scholar]
- 24.Annunziato, F., Cosmi, L., Santarlasci, V., Maggi, L., Liotta, F., Mazzinghi, B., Parente, E., Fili, L., Ferri, S., Frosali, F., Giudici, F., Romagnani, P., Parronchi, P., Tonelli, F., Maggi, E., and Romagnani, S. (2007) J. Exp. Med. 204 1849-1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng, R., Spolski, R., Finkelstein, S. E., Oh, S., Kovanen, P. E., Hinrichs, C. S., Pise-Masison, C. A., Radonovich, M. F., Brady, J. N., Restifo, N. P., Berzofsky, J. A., and Leonard, W. J. (2005) J. Exp. Med. 201 139-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korn, T., Bettelli, E., Gao, W., Awasthi, A., Jager, A., Strom, T. B., Oukka, M., and Kuchroo, V. K. (2007) Nature 448 484-487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nurieva, R., Yang, X. O., Martinez, G., Zhang, Y., Panopoulos, A. D., Ma, L., Schluns, K., Tian, Q., Watowich, S. S., Jetten, A. M., and Dong, C. (2007) Nature 448 480-483 [DOI] [PubMed] [Google Scholar]
- 28.Zhou, L., Ivanov, II, Spolski, R., Min, R., Shenderov, K., Egawa, T., Levy, D. E., Leonard, W. J., and Littman, D. R. (2007) Nat. Immunol. 8 967-974 [DOI] [PubMed] [Google Scholar]
- 29.Ziolkowska, M., Koc, A., Luszczukiewicz, G., Ksiezopolksa-Pietrzak, K., Klimczak, E., Chwalinska-Sadowska, H., and Maslinski, W. (2000) J. Immunol. 164 2832-2838 [DOI] [PubMed] [Google Scholar]
- 30.Yao, Z., Fanslow, W. C., Seldin, M. F., Rousseau, A.-M., Painter, S. L., Comeau, M. R., Cohen, J. I., and Spriggs, M. K. (1995) Immunity 3 811-821 [DOI] [PubMed] [Google Scholar]
- 31.Gaffen, S. L., Kramer, J. M., Yu, J. J., and Shen, F. (2006) Vitam. Horm. 74 255-282 [DOI] [PubMed] [Google Scholar]
- 32.Lindemann, M. J., Benczik, M., and Gaffen, S. L. (2003) J. Biol. Chem. 278 10239-10249 [DOI] [PubMed] [Google Scholar]
- 33.Tusher, V. G., Tibshirani, R., and Chu, G. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 5116-5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmed, N. N., Grimes, H. L., Bellacosa, A., Chan, T. O., and Tsichlis, P. N. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 3627-3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen, F., Ruddy, M. J., Plamondon, P., and Gaffen, S. L. (2005) J. Leukoc. Biol. 77 388-399 [DOI] [PubMed] [Google Scholar]
- 36.Shen, F., Hu, Z., Goswami, J., and Gaffen, S. L. (2006) J. Biol. Chem. 281 24138-24148 [DOI] [PubMed] [Google Scholar]
- 37.Liu, K. D., Gaffen, S. L., and Goldsmith, M. A. (1998) Cur. Opin. Immunol. 10 271-278 [DOI] [PubMed] [Google Scholar]
- 38.Goldsmith, M. A., Lai, S. Y., Xu, W., Amaral, M. C., Kuczek, E. S., Parent, L. J., Mills, G. B., Tarr, K. L., Longmore, G. D., and Greene, W. C. (1995) J. Biol. Chem. 270 21729-21737 [DOI] [PubMed] [Google Scholar]
- 39.Nelson, B. H., Lord, J. D., and Greenberg, P. D. (1996) Mol. Cell Biol. 16 309-317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shindoh, M., Higashino, F., and Kohgo, T. (2004) Cancer Lett. 216 1-8 [DOI] [PubMed] [Google Scholar]
- 41.Ha, C., Waterhouse, R., Wessells, J., Wu, J., and Dveksler, G. (2005) J. Leukoc. Biol. 77 948-957 [DOI] [PubMed] [Google Scholar]
- 42.Wang, D., Matsumoto, R., You, Y., Che, T., Lin, X., Gaffen, S., and Lin, X. (2004) Mol. Cell Biol. 24 164-171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hur, E. M., Son, M., Lee, O. H., Choi, Y. B., Park, C., Lee, H., and Yun, Y. (2003) J. Exp. Med. 198 1463-1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cornish, G. H., Sinclair, L. V., and Cantrell, D. A. (2006) Blood 108 600-608 [DOI] [PubMed] [Google Scholar]
- 45.Bauer, J., Liu, K. D., Lai, S. Y., You, Y., and Goldsmith, M. A. (1998) J. Biol. Chem. 273 9255-9260 [DOI] [PubMed] [Google Scholar]
- 46.Lai, S. Y., Molden, J., Liu, K. D., Puck, J. M., White, M. D., and Goldsmith, M. A. (1996) EMBO J. 15 4506-4514 [PMC free article] [PubMed] [Google Scholar]
- 47.Lai, S. Y., Molden, J., and Goldsmith, M. A. (1997) J. Clin. Investig. 99 169-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Astoul, E., Edmunds, C., Cantrell, D. A., and Ward, S. G. (2001) Trends Immunol. 22 490-496 [DOI] [PubMed] [Google Scholar]
- 49.Weaver, C. T., Hatton, R. D., Mangan, P. R., and Harrington, L. E. (2007) Annu. Rev. Immunol. 25 821-852 [DOI] [PubMed] [Google Scholar]
- 50.Yu, J., and Gaffen, S. L. (2008) Front. Biosci. 13 170-177 [DOI] [PubMed] [Google Scholar]
- 51.Kolls, J. K., and Linden, A. (2004) Immunity 21 467-476 [DOI] [PubMed] [Google Scholar]
- 52.Harrington, L. E., Hatton, R. D., Mangan, P. R., Turner, H., Murphy, T. L., Murphy, K. M., and Weaver, C. T. (2005) Nat. Immunol. 6 1123-1132 [DOI] [PubMed] [Google Scholar]
- 53.Park, H., Li, Z., Yang, X. O., Chang, S. H., Nurieva, R., Wang, Y. H., Wang, Y., Hood, L., Zhu, Z., Tian, Q., and Dong, C. (2005) Nat. Immunol. 6 1133-1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toy, D., Kugler, D., Wolfson, M., Vanden Bos, T., Gurgel, J., Derry, J., Tocker, J., and Peschon, J. J. (2006) J. Immunol. 177 36-39 [DOI] [PubMed] [Google Scholar]
- 55.Chu, C. L., Chen, S. S., Wu, T., Kuo, S. C., and Liao, N.-S. (1999) J. Immunol. 162 1896-1903 [PubMed] [Google Scholar]
- 56.Tagaya, Y. (2006) Blood 108 409-410 [Google Scholar]
- 57.Kovanen, P. E., Rosenwald, A., Fu, J., Hurt, E. M., Lam, L. T., Giltnane, J. M., Wright, G., Staudt, L. M., and Leonard, W. J. (2003) J. Biol. Chem. 278 5205-5213 [DOI] [PubMed] [Google Scholar]
- 58.Dubois, S., Mariner, J., Waldmann, T. A., and Tagaya, Y. (2002) Immunity 17 537-547 [DOI] [PubMed] [Google Scholar]
- 59.Sato, N., Patel, H. J., Waldmann, T. A., and Tagaya, Y. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 588-593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaffen, S. L., Lai, S. Y., Longmore, G. D., Liu, K. D., and Goldsmith, M. A. (1999) Blood 94 74-86 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.