

Abstract
Cytosine DNA methylation is important in regulating gene expression and in silencing transposons and other repetitive sequences 1, 2. Recent genomic studies in Arabidopsis have revealed that many endogenous genes are methylated either within their promoters or within their transcribed regions, and that gene methylation is highly correlated with transcription levels 3-5. However, plants have different types of methylation controlled by different genetic pathways, and detailed information on the methylation status of each cytosine in any given genome is lacking. To this end, we generated a map at single base pair resolution of methylated cytosines for Arabidopsis, by combining bisulfite treatment of genomic DNA with ultra-high-throughput sequencing using the Illumina 1G Genome Analyzer and Solexa sequencing technology 6. This approach, termed BS-Seq, unlike previous microarray-based methods, allows one to sensitively measure cytosine methylation on a genome-wide scale within specific sequence contexts. We describe methylation on previously inaccessible components of the genome along with an analysis of the DNA methylation sequence composition and distribution. We also describe the effect of various DNA methylation mutants on genome-wide methylation patterns, and demonstrate that our newly developed library construction and computational methods can be applied to large genomes such as mouse.
To generate a DNA methylation map at one nucleotide resolution across the genome, we adapted the Illumina 1G Genome Analyzer using Solexa sequencing technology (Illumina GA) for shotgun sequencing of bisulfite-treated Arabidopsis genomic DNA. Sodium bisulfite converts unmethylated cytosines to uracils, but 5-methylcytosines remain unconverted. Hence, after polymerase chain reaction amplification, unmethylated cytosines appear as thymines and methylated cytosines appear as cytosines 7. We created genomic DNA libraries after bisulfite conversion and produced ~3.8 billion nucleotides of high quality sequence which successfully mapped to the genome. We subsequently used several filters to ensure accuracy, including only retaining reads mapping to sequences that are unique in the genome after bisulfite conversion from every possible methylation pattern (see Supplementary Methods and Supplementary Table 1). This resulted in a conservative dataset of ~2.6 billion nucleotides mapping to unique genomic locations with very high confidence, covering ~93% of all cytosines which could theoretically be covered (~92% of the ~43 million cytosines in the ~120 Mbp Arabidopsis genome can be covered uniquely with 31 nucleotide sequences). This represents ~20-fold average coverage, similar to typical coverage in a traditional bisulfite sequencing experiment for a single locus.
Methylation in Arabidopsis exists in three sequence contexts, CG, CHG (where H = A, C, or T), and asymmetric CHH 1. We observed overall genome-wide levels of 24% CG, 6.7% CHG, and 1.7% CHH methylation (Supplementary Fig. 1a). Most CGs were either unmethylated or were highly methylated (80–100%), whereas CHH sites were either unmethylated or methylated at ~10%. CHG sites showed a more uniform distribution between 20–100% (Supplementary Fig. 1b-d). These differences underscore the fact that each type of methylation is under distinct genetic control 1. Our reads also contained 504-fold average coverage of 99.97% of theoretically-coverable cytosines in the unmethylated chloroplast genome 3, 8, giving false positive rates of 0.29% (CG), 0.29% (CHG), and 0.25% (CHH) (Supplementary Fig. 1a, Supplementary Fig. 2). The BS-Seq data were highly consistent with traditional bisulfite sequencing data from individual methylated or unmethylated loci 3 (Supplementary Table 2, Supplementary Fig. 3, and below).
While CG, CHG, and CHH methylation were highly correlated, showing enrichment in repeat-rich pericentromeric regions (Fig. 1a), a striking deviation was found within gene bodies, which contained almost exclusively CG methylation (Fig. 1b). This is consistent with previous studies 3, 4, 9 and with a depletion of short interfering RNAs (siRNAs) in the bodies of genes (Fig. 1b). Conversely, genomic regions corresponding to siRNAs were highly correlated with CG, CHG, and CHH methylation, consistent with the known molecular nature of RNA-directed DNA methylation (Fig. 1c) 1. For methylation of all types there was a strong positive correlation with the length of the methylated sequence (Fig. 1d).
Figure 1. Methylation of different fractions of the Arabidopsis genome.

a, Chromosome-wide distribution of methylation and correlation with repeats in sliding 100 kB windows. b, Methylation levels and siRNA abundance 26 are plotted across different types of repeats and genes. c, High levels of methylation are detected at loci corresponding to siRNAs. d, Relationship between methylation levels and the length of different types of repeats and genes. e, From left to right, methylation levels of the three consecutive cytosines in the (CCCTAAA)n telomeric repeat unit are calculated in wild type and the drm1 drm2 cmt3 mutant, respectively.
BS-Seq appears to be more sensitive than previously-employed microarray-based methods 3-5. For example, we found a cluster of 5 methylated CG sites in a 34 base pair region and a lone methylated CG site, both within the FWA locus, that were not detected by previous methods (Supplementary Fig. 4). We also found CG methylation within genes previously classified as unmethylated 3, 4 (Supplementary Fig. 5). Finally, in analyzing genes whose expression is de-repressed in DNA methyltransferase mutants, BS-Seq was more accurate in identifying genes with promoter methylation that was otherwise variably detected in previous microarray studies (Supplementary Fig. 6).
BS-Seq can be used to analyze repetitive sequences that are difficult to study with microarrays as they may exceed the dynamic detection range or cross-hybridize. For example, we mapped methylation across the highly repetitive rDNA loci and found high levels of CG, CHG, and CHH methylation, including on the minimal promoter and upstream Sal1 repeats (Supplementary Fig. 7). Further, we detected methylation in telomeric repeat sequences (CCCTAAA)n which have not been previously shown to be methylated (Fig. 1e). Interestingly, the vast majority of methylation occurred at the cytosine in the third position (Fig. 1e).
The single base resolution of BS-Seq allows determination of the precise boundaries between methylated and unmethylated regions. For example, we found that the boundary between tandem repeats and flanking DNA showed a sharp drop in methylation, but DNA methylation extended from inverted repeats into flanking DNA, showing a more gradual reduction (Fig. 1b). This apparent “spreading” of methylation was not correlated with siRNA spreading because siRNA abundance levels drop sharply at the flanks of both tandem and inverted repeats (Fig. 1b).
We analyzed the relationship between sequence context and preference of methylation. We calculated the percent methylation of all possible 7-mer sequences in which the methylated cytosine was either in the fifth position (allowing an analysis of four nucleotides upstream of CG, CHG, and CHH methylation; Fig. 2, Supplementary Table 3) or in the first position (allowing analysis of 6 nucleotides following the methylated cytosine; Supplementary Fig. 8, Supplementary Table 4). To ensure that sequence preferences were not simply 7-mers enriched in particular components of the genome, we analyzed either all of chromosome 1, only sequences previously defined to be methylated by methyl-DNA immunoprecipitation, or a group of 9,507 body-methylated genes containing mostly CG methylation 3 (Fig. 2, Supplementary Fig. 8 and 9). We observed a surprisingly high level of sequence context specificity. The highest and lowest methylated 7-mers showed a 13-fold difference for CG-methylation, an 11-fold difference for CHG methylation, and > 900-fold difference for CHH-methylation (Supplementary Table 3). Sequences with the lowest CG methylation were highly enriched for the sequence ACGT (Fig. 2, Supplementary Fig. 9). Poorly methylated CHG sites were depleted of upstream cytosines but tended to contain cytosine following the methylated C. This trend is consistent with nearest-neighbour analysis of wheat germ DNA that found CAG and CTG sites methylated at a higher level than CCG sites 10. Highly methylated CHH sequences had a very specific configuration, with a tendency for cytosines and CG dinucleotides to be present upstream (Supplementary Table 3) and the sequence TA following the methylated cytosine. In contrast, poorly methylated CHH sequences always contained a cytosine following the methylated cytosine, and frequently contained a cytosine but always lacked an adenine two nucleotides downstream (Fig. 2, Supplementary Fig. 8). These results are consistent with data from individual plant genes showing that cytosines preceding a cytosine are undermethylated while those following a cytosine are more heavily methylated 11-13, and with asymmetric methylation in mammalian genomes that is found at CT and CA sequences more frequently than CC sequences 14. It is also of interest that Arabidopsis telomere sequences (CCCTAAA)n are composed of nearly optimal asymmetric target units, possibly explaining the high methylation of the third cytosines (Fig. 1e). While the molecular basis for these trends is unknown, the results suggest that DNA methyltransferases show strong sequence preferences beyond the CG, CHG, and CHH contexts. Finally, we found that regions with higher concentrations of CG dinucleotides were more heavily methylated at CG sites (Supplementary Fig. 10). Interestingly, this is different from mammalian genomes that show the opposite trend: CGs are depleted in methylated regions and at a higher density in unmethylated CpG islands.
Figure 2. Sequence preferences for methylation in CG, CHG, and CHH contexts.

Logos of sequence contexts that are preferentially methylated at the highest or lowest levels for 7-mer sequences in which the methylated cytosine is in the fifth position. In a, all genomic 7-mers in chromosome 1 were analyzed, while in b sequences were restricted to previously-defined methylated sequences 3. The logo graphically displays the sequence enrichment at a particular position in the alignment of 7-mers in each class, measured in bits. The maximum sequence conservation per site is 2 bits (i.e., 1 base) when a site is perfectly conserved, and 0 if there is no preference for a nucleotide.
We used autocorrelation analysis to examine the correlation between methylation in different sequence contexts and methylation at adjacent residues. We observed significant correlation between methylated cytosines for distances up to 5,000 nucleotides or more, a likely reflection of regional foci of methylation throughout the genome and of large blocks of pericentromeric heterochromatin (Supplementary Fig. 11, Supplementary Table 5). We also found a high correlation of CHG and CHH methylation within several nucleotides downstream of methylated CG sites, and a tendency for CHH methylation four nucleotides downstream of methylation at CHG sites (Fig. 2, Supplementary Fig. 12, Supplementary Table 5). These data suggest complex interactions between the different types of methylation.
We analyzed the propensity for full methylation of the strand-symmetrical CG and partially symmetrical CHG sequences. As expected, CG methylation on one strand was highly correlated with CG methylation on the opposing strand. We also saw a high correlation for CHG methylation of the two strands, showing that, like CG methylation, CHG sites show a strong tendency for symmetrical methylation (Supplementary Fig. 12). Unexpectedly, we observed a correlation between CHH methylation on one strand, and methylation at the cytosine three nucleotides downstream and on the opposite strand (Supplementary Fig. 12, Supplementary Table 5). Since the sequence of such sites is CHHG, this shows that “asymmetric” methylation shows a propensity for symmetrical methylation at these sites, even though methylation on CHHG sites is not particularly prominent in the genome (Supplementary Fig. 8, Supplementary Table 4).
Autocorrelation analysis also revealed a striking periodicity of 10 nucleotides (the length of one helical DNA turn) for CHH methylation (Fig. 3a, b). We confirmed this period in data from the whole genome and from regions previously defined to be methylated, and confirmed that the periodicity was not due to our computational filtering of the data (Supplementary Fig. 13). We observed this period both when looking at average methylation of cytosines in the genome (Fig. 3a, b, Supplementary Fig. 13) and when individual reads are directly examined (Supplementary Fig. 14). Mammalian Dnmt3a was recently shown to act as a tetramer with Dnmt3L, and two active sites methylate two CG sequences spaced ~8–10 nucleotides apart 15. Since DRM2 is the main enzyme controlling asymmetric methylation and is the ortholog of Dnmt3a 16, these data suggest that the mechanism of action of these enzymes may be conserved between plants and mammals.
Figure 3. Methylation shows periodic patterns.
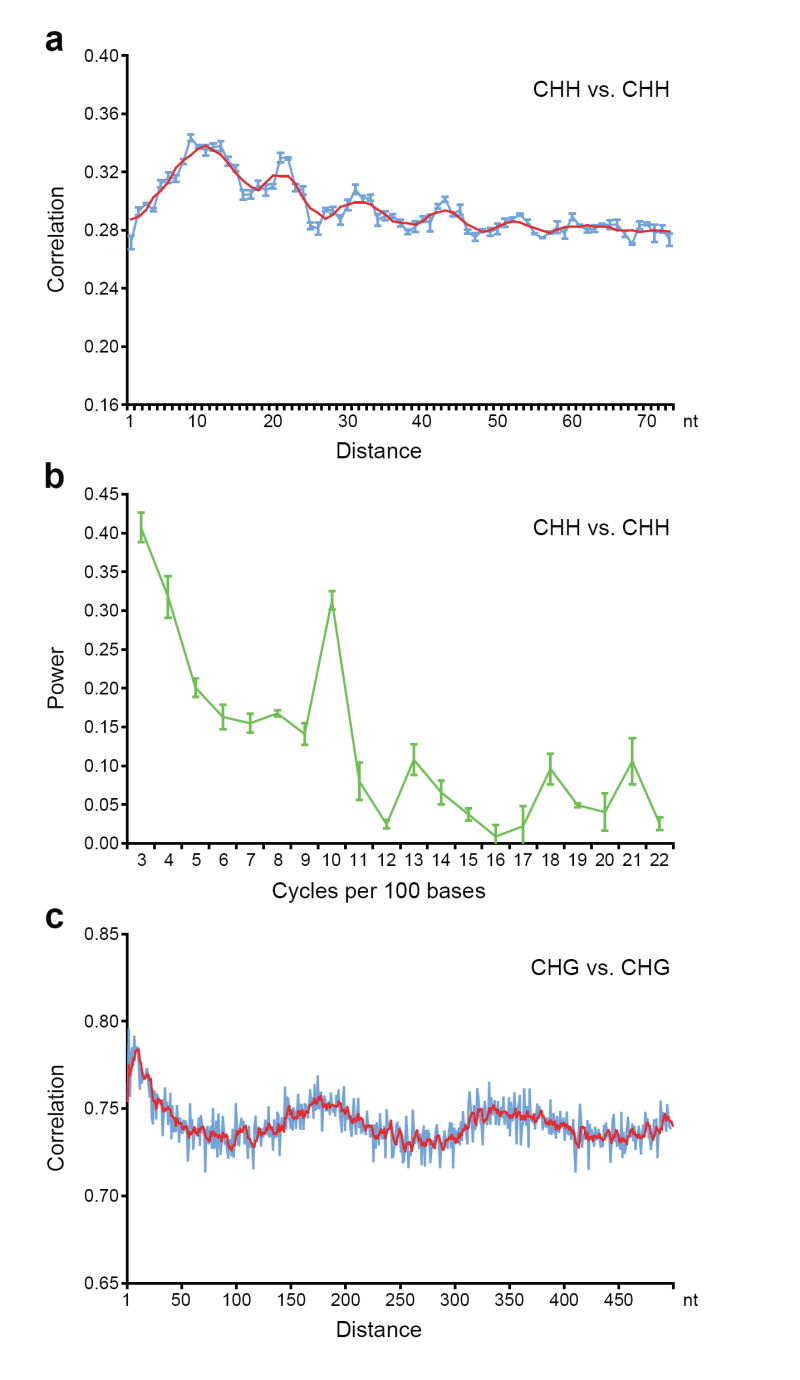
a, c, Correlation of the methylation status of cytosines in a CHH (a) and CHG (c) context. The x-axis indicates the distance between the two cytosines. The y-axis indicates the level of autocorrelation in methylation. The red line is a running average of windows that are ±2 bases around a single base. b, Fourier transform analysis of CHH methylation correlation. The x-axis indicates the number of cycles per 100 bases. The y-axis is the amplitude of the corresponding frequency. The peak at position 10 represents a periodicity of ten nucleotides, with a p-value smaller than 10−108 for observing this periodicity value by chance in random permutations of the genome. In a-c, Monte Carlo sampling of three datasets each consisting of half the data was used to compute the mean and standard deviations of the autocorrelations and Fourier transforms. Mean values are shown and error bars (a and b) represent standard deviations. In a and b, methylation from the whole genome was analyzed, while in c the analysis was restricted to previously-defined methylated sequences 3 (see Supplementary Fig. 15 for details).
Autocorrelation also showed a period of 167 nucleotides (Fig. 3c, Supplementary Fig. 15), which is similar to, but slightly shorter than, estimates of the average spacing of nucleosomes in plant chromatin 17-19. One explanation for this period is that nucleosomes or particular histone modifications might dictate access to the DNA by methyltransferase proteins. Furthermore, the slightly shorter length of 167 nucleotides relative to most estimates of plant nucleosome repeat length (175-185 nt) 17-19 suggests that DNA methylated chromatin may be more compact because of shorter linker regions or depletion in linker histones 20.
We utilized BS-Seq to study the genome-wide effects of a variety of methyltransferase mutants on DNA methylation (Fig. 4). The MET1, CMT3, and DRM1/DRM2 DNA methyltransferase enzymes are mostly responsible for CG, CHG, and CHH methylation, respectively, though at many loci CHG and CHH methylation is redundantly controlled by CMT3 and DRM1/DRM2 1, 12. We sequenced and mapped ~90 million nucleotides of BS-Seq data from each of several combinations of DNA methyltransferase mutants (Supplementary Table 1) including met1 single mutants, cmt3 single mutants, drm1 drm2 double mutants, met1 cmt3 double mutants, met1 drm1 drm2 triple mutants, and drm1 drm2 cmt3 triple mutants 21. We then analyzed the effect of these mutants on global methylation, the methylation on genes and chromosomes, and the methylation on rDNA and telomeres (Supplementary Table 6, Fig. 1e, Fig. 4, Supplementary Fig. 7, 16). The met1 single mutant, or any mutant combination containing met1, essentially eliminated CG methylation throughout the genome. For instance, gene body methylation, which is almost exclusively CG, was eliminated in all met1-containing strains (Fig. 4a). Surprisingly, in the met1 drm1 drm2 triple mutant, we observed a marked hypermethylation of CHG sites in the bodies of genes (Fig. 4a). This methylation was skewed toward the 3’ end and in this way assumed a pattern of methylation similar to the missing CG methylation. Although previous studies have suggested that the drm1 drm2 cmt3 triple mutant eliminates CHG and CHH methylation 12, BS-Seq data shows residual methylation (Supplementary Table 6), particularly in pericentromeric heterochromatin (Fig. 4b), suggesting that another enzyme is involved 22. Furthermore, the met1 cmt3 double mutant was equally effective in reducing CHH methylation as was drm1 drm2 cmt3 (Supplementary Table 6), suggesting that CHH methylation depends in part on the presence of CG and CHG methylation. These compensating behaviours suggest that the different DNA methyltransferases act redundantly, and help explain the viability of these mutant combinations whereas the met1 cmt3 drm1 drm2 quadruple mutant causes embryonic lethality 21.
Figure 4. BS-Seq profiling of methylation mutants in Arabidopsis and mouse.

a, BS-Seq data mapping to protein-coding genes was plotted in 500 nucleotide sliding windows. Two vertical blue lines mark the boundaries between upstream regions and gene bodies (left) and between gene bodies and downstream regions (right). b, Distribution of methylation along chromosome 4 in 25 nucleotide sliding windows. In a and b, a horizontal blue line indicates zero percent methylation. c, Comparison of the amount of CG methylation in wild type and mUhrf1−/− embryonic stem cells, represented as the average number of CGs appearing per million sequenced nucleotides.
The BS-Seq procedure described here should be generally useful in other organisms. For example we applied BS-Seq to quantify the overall genomic methylation difference between wild type mouse embryonic stem cells and cells carrying a mutation in the UHRF1 gene recently shown to control maintenance of CG methylation 23, 24. By analyzing ~60 million nucleotides of shotgun sequencing data from each, we found that Uhrf1−/− cells contained only 25% of the CpG methylation level of wild type (Fig. 4c). Furthermore, to demonstrate that the complete analysis pipeline used for Arabidopsis is applicable to larger genomes, we produced a library from mouse germ cell tissue and generated ~46 million nucleotides of high quality mapped BS-seq data. Approximately 66% of the reads mapped uniquely, a level only slightly lower than that of Arabidopsis (Supplementary Table 1), suggesting that it is practical to apply BS-Seq to entire mammalian genomes.
In summary, BS-Seq analysis of wild type and methyltransferase mutants has allowed a more detailed characterization of the Arabidopsis methylome. In addition, the computational approaches developed in this study should be generally useful for other short read sequencing genomics approaches. An installation of the UCSC browser allowing community access to detailed methylation patterns of individual genes and a source code distribution of the computational methods are available at http://epigenomics.mcdb.ucla.edu/BS-Seq/.
METHODS SUMMARY
Construction and sequencing of DNA libraries
Bisulfite treatment of DNA was performed as previously described 25, except that adaptor sequences and PCR conditions were modified and optimized for this study. Library generation and ultra-high-throughput sequencing were carried out according to manufacturer instructions (Illumina).
Processing of sequence data and mapping of reads
Raw data from Illumina GA was processed using the initial stages of the Solexa software pipeline (Illumina) into short reads, except that per-lane per-cycle multidimensional Gaussian mixture models (GMMs) were developed to optimize base call A–vs.–C–vs.–G–vs.–T probability distribution accuracies at each sequenced base compared to the Solexa software pipeline’s _ prb files. Sequenced reads were mapped to reference genomes fully using per-base probabilities from the GMMs using highly-optimized novel C++ tools. Sequences that mapped to more than one position with similar scores (within 1% of the maximum likelihood mapping) were removed in order to retain only reads that map uniquely. To eliminate unconverted bisulfite reads, a filter discarded reads with three or more consecutive methylated cytosines when each of these was in a CHH context, resulting in a loss of ~0.23% of reads. This filter was effective and with only minimal loss of true CHH methylation (Supplementary Table 1, Supplementary Fig. 13, 17, and 18).
Validation of BS-Seq results
Traditional bisulfite sequencing was employed to validate BS-Seq results at select loci (Supplementary Table 2, Supplementary Fig. 4, 6, 17). The PCR primers used in validation are listed in Supplementary Table 7.
Supplementary Material
Full methods (including extensive algorithmic and computational details) are available in the Supplementary Methods section at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Acknowledgments
We thank Yana Bernatavichute for nuclear DNA isolation protocols, Amander Clarke for providing ES cell DNA, Angelique Girard and Greg Hannon for providing mouse germ cell DNA, Jonathan Hetzel for technical assistance, and Carey Fey Li for assistance with rDNA annotation. This work was supported in part by a grant from the NSF Plant Genome Research Program (award number 0701745) and some aspects of the work were performed in the UCLA DNA Microarray Facility. S.F. is a Howard Hughes Medical Institute Fellow of the Life Science Research Foundation. X.Z. was supported by a fellowship from the Jonsson Cancer Center Foundation. S.E.J. is an investigator of the Howard Hughes Medical Institute.
References
- 1.Henderson IR, Jacobsen SE. Epigenetic Inheritance in Plants. Nature. 2007;447:418–424. doi: 10.1038/nature05917. [DOI] [PubMed] [Google Scholar]
- 2.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, et al. Genome-wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell. 2006;126:1189–201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–9. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 5.Vaughn MW, et al. Epigenetic Natural Variation in Arabidopsis thaliana. PLoS Biol. 2007;5:e174. doi: 10.1371/journal.pbio.0050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–52. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Frommer M, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–31. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ngernprasirtsiri J, Kobayashi H, Akazawa T. DNA methylation as a mechanism of transcriptional regulation in nonphotosynthetic plastids in plant cells. Proc Natl Acad Sci U S A. 1988;85:4750–4. doi: 10.1073/pnas.85.13.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran RK, et al. DNA methylation profiling identifies CG methylation clusters in Arabidopsis genes. Curr Biol. 2005;15:154–9. doi: 10.1016/j.cub.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Gruenbaum Y, Naveh-Many T, Cedar H, Razin A. Sequence specificity of methylation in higher plant DNA. Nature. 1981;292:860–2. doi: 10.1038/292860a0. [DOI] [PubMed] [Google Scholar]
- 11.Meyer P, Niedenhof I, ten Lohuis M. Evidence for cytosine methylation of non-symmetrical sequences in transgenic Petunia hybrida. Embo J. 1994;13:2084–8. doi: 10.1002/j.1460-2075.1994.tb06483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao X, Jacobsen SE. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16491–8. doi: 10.1073/pnas.162371599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dieguez MJ, Vaucheret H, Paszkowski J, Mittelsten Scheid O. Cytosine methylation at CG and CNG sites is not a prerequisite for the initiation of transcriptional gene silencing in plants, but it is required for its maintenance. Mol Gen Genet. 1998;259:207–15. doi: 10.1007/s004380050806. [DOI] [PubMed] [Google Scholar]
- 14.Ramsahoye BH, et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–42. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007 doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao X, et al. Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc Natl Acad Sci U S A. 2000;97:4979–84. doi: 10.1073/pnas.97.9.4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ideker T, et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–34. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- 18.Vershinin AV, Heslop-Harrison JS. Comparative analysis of the nucleosomal structure of rye, wheat and their relatives. Plant Mol Biol. 1998;36:149–61. doi: 10.1023/a:1005912822671. [DOI] [PubMed] [Google Scholar]
- 19.Fulnecek J, Matyasek R, Kovarik A, Bezdek M. Mapping of 5-methylcytosine residues in Nicotiana tabacum 5S rRNA genes by genomic sequencing. Mol Gen Genet. 1998;259:133–41. doi: 10.1007/s004380050798. [DOI] [PubMed] [Google Scholar]
- 20.Fan Y, et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123:1199–212. doi: 10.1016/j.cell.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Jacobsen SE. Genetic analyses of DNA methyltransferases in Arabidopsis thaliana. Cold Spring Harb Symp Quant Biol. 2006;71:439–47. doi: 10.1101/sqb.2006.71.047. [DOI] [PubMed] [Google Scholar]
- 22.Henderson IR, et al. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nature Genetics. 2006 doi: 10.1038/ng1804. in press. [DOI] [PubMed] [Google Scholar]
- 23.Bostick M, et al. UHRF1 Plays a Role in Maintaining DNA Methylation in Mammalian Cells. Science. 2007 doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 24.Sharif J, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007 doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 25.Meissner A, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–77. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006;20:3407–25. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full methods (including extensive algorithmic and computational details) are available in the Supplementary Methods section at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.