

Abstract
Spinocerebellar ataxia type 1 (SCA1) is a dominantly inherited neurodegenerative disease caused by expansion of a glutamine-encoding repeat in SCA1. In all known polyglutamine diseases, the glutamine expansion confers toxic functions onto the protein. The mechanism by which this occurs remains enigmatic, however, in light of the fact that the mutant protein apparently maintains interactions with its usual partners. Here we show that the expanded polyglutamine tract differentially affects the function of the host protein in the context of different endogenous protein complexes. Polyglutamine expansion in Ataxin1 favors the formation of a particular protein complex containing RBM17, contributing to SCA1 neuropathology via a gain-of-function mechanism. Concomitantly, polyglutamine expansion attenuates the formation and function of another protein complex containing Ataxin1/Capicua, contributing to SCA1 via a partial loss-of-function mechanism. This model provides mechanistic insight into the molecular pathogenesis of SCA1 as well as other polyglutamine diseases.
Expansion of an unstable translated CAG repeat located in different disease genes so far causes nine dominantly inherited neurodegenerative disorders, the so-called polyglutamine diseases: Huntington’s disease (HD), spinobulbar muscular atrophy (SBMA), dentatorubropallidoluysian atrophy (DRPLA), and six autosomal dominant spinocerebellar ataxias (SCAs)1. As would be expected for dominant mutations, polyglutamine expansions confer toxic properties on the host proteins1–3; animal models genetically lacking the polyglutamine-containing proteins do not develop neurodegeneration4–7. However, expansion of the polyglutamine tract is necessary but not sufficient to cause pathology: in the case of SCA1, for example, expanded Ataxin1 (ATXN1) does not produce cerebellar degeneration if it lacks the nuclear localization signal8 or the AXH domain9, or if a serine to alanine substitution prevents phosphorylation at residue 77610. These and other studies in SBMA and HD indicate that protein domains outside of the polyglutamine tract play a significant role in the selective neurotoxicity observed in these diseases11–18. Moreover, they suggest that there is a relationship between the normal functions of the wild-type proteins and the toxic functions of their expanded counterparts. Given that mouse and fly models overexpressing wild-type ATXN1 develop a mild version of SCA119 begs the question of whether the glutamine expansion enhances some interactions to mediate the gain-of-function.
To gain a foothold on this question, we sought to characterize protein partners of ATXN1 that interact with it in a manner dependent on two criteria necessary for toxicity: polyglutamine expansion and phosphorylation at serine 776 (S776). We have identified RBM17 (RNA binding motif protein 17) as a protein that meets these criteria. Here we show that ATXN1 forms at least two distinct, large native complexes, one containing Capicua (CIC) and the other containing RBM17. Polyglutamine expansion alters the proportion of the mutant protein participating in the formation of these complexes in vivo. Furthermore, we show that the ATXN1/RBM17 complex causes disease by a gain-of-function mechanism, whereas the ATXN1/CIC complex causes a loss-of-function. These data not only show that there is an unexpected loss of function that contributes to SCA1 pathology, but provide a molecular mechanism to explain how glutamine expansion causes both gain and loss of function.
RESULTS
RBM17 preferentially interacts with glutamine-expanded ATXN1
To identify proteins that preferentially interact with ATXN1 in a manner dependent on both polyglutamine expansion and S776-phosphorylation, we performed a yeast two-hybrid (Y2H) screen20 using several human ATXN1 constructs with either wild-type [30Q] or expanded [82Q] polyglutamine tracts and varying phosphorylation status at S776 (wild-type [S776], phosphorylation-defective [S776A], or phosphorylation-mimicked [S776D]) (Fig. 1a). This screen yielded a protein called RBM17 that binds specifically to ATXN1-S776D (Fig. 1b). We verified the ATXN1/RBM17 interaction in mammalian cells by co-affinity purification (co-AP) assays using lysates from HEK293T cells transfected with GST-tagged ATXN1 and myc-tagged RBM17 (Fig. 1c, arrow). We also performed co-immunoprecipitation (co-IP) assays on mouse cerebellar extracts using a specific RBM17 antibody (Supplementary Fig. S1). The anti-RBM17 antibody co-immunoprecipitated Atxn1 from cerebellar extracts (Fig. 1d, arrow), suggesting that wild-type ATXN1 and RBM17 do indeed interact in vivo.
Figure 1. ATXN1-S776D but not ATXN1-S776A interacts with RBM17.

(a) Schematic representation of the ATXN1 constructs.
(b) RBM17 specifically interacted with ATXN1-S776D in the Y2H screen. AD (activation domain) and DB (DNA binding domain) of Gal4 were fused to human ATXN1 or RBM17, respectively. Y2H controls are: lane 1, negative control; lane 2, weak positive control; and lanes 3–5, strong positive controls.
(c) ATXN1 interacted with RBM17 in HEK293T cells by co-AP assays. Top panel shows expression of myc-RBM17 after affinity purification on Glutathione-Sepharose 4B beads, demonstrating the ATXN1/RBM17 interaction (arrow). GST-empty vector was used as a control (−). IB, Immunoblot.
(d) Co-IP of Atxn1 with RBM17 from wild-type mouse cerebellar extracts. The anti-RBM17 antibody co-immunoprecipitated Atxn1 (arrow), but not the long and short isoforms of Capicua, CIC-L and CIC-S, respectively (bracket).
To determine whether the interaction between RBM17 and ATXN1 depends on S776-phosphorylation of ATXN1 in mammalian cells, we transfected myc-RBM17 with GST-ATXN1-S776, -S776A, or -S776D into HEK293T cells. Both wild-type ATXN1 and ATXN1-S776D were able to precipitate RBM17, but ATXN1-S776A showed almost no interaction with RBM17 (Fig. 2a, arrow; supplementary Fig. S2), indicating that the interaction of RBM17 with ATXN1 requires S776-phosphorylation. That RBM17 interacts more robustly with ATXN1-S776D than with ATXN1-S776 suggests that S776-phosphorylation is important for the interaction. We next determined the domains responsible for the interaction between the two proteins using a series of ATXN1 and RBM17 deletion constructs. We found that the C-terminal region of RBM17 interacts with ATXN1’s C-terminal sequence harboring the phosphorylated S776 residue (Supplementary Fig. S3).
Figure 2. Enhanced interaction of RBM17 and ATXN1 depends on S776-phosphorylation and polyglutamine tract expansion.

(a) Wild-type ATXN1 and ATXN1-S776D, but not ATXN1-S776A, interacted with RBM17.
(b) RBM17 bound most strongly to polyglutamine-expanded ATXN1. A bracket showed that there is no obvious difference in S776-phosphorylation depending on polyglutamine expansion in ATXN1 when using a PN1168 antibody (specific to phosphorylated S776 of ATXN1) for IB.
(c,d) RBM17 expression in the nuclei of PCs of 18-week-old mice from either wild-type (FVB)
(c) or SCA1 transgenic (ATXN1[82Q]) homozygotes (d). RBM17 protein (green) was co-immunostained with calbindin (red, PC marker) and Toto3 (blue, nuclear marker).
To test the effect of polyglutamine tract length on ATXN1/RBM17 interaction, we performed co-AP assays by transfecting HEK293T cells with GST-fused ATXN1 containing 2Q, 30Q, or 82Q. The interaction between myc-RBM17 and GST-ATXN1 was strongly enhanced by expansion of the polyglutamine tract (Fig. 2b, arrow; supplementary Fig. S4). We checked the phosphorylation level of S776 in ATXN1 containing a different polyglutamine tract length and found little if any enhancement in S776-phosphorylation of ATXN1 by polyglutamine expansion (Fig. 2b, bracket; supplementary Fig. S5). These data suggest that expansion of the N-terminal polyglutamine tract of ATXN1 alters the conformation of the C-terminal regions of the protein, making it more accessible to RBM17 binding.
We explored RBM17 expression patterns in mouse brain and found RBM17 protein abundant in Purkinje cell (PC) nuclei in wild-type mouse cerebella (Fig. 2c), consistent with the mRNA expression pattern shown in the Allen Brain Atlas21. We also tested RBM17 expression in SCA1 transgenic mice (B05 line)22, which express a polyglutamine-expanded form of human ATXN1 (ATXN1[82Q]) in PCs. RBM17 expression remained high in the PCs of 18-week-old B05 mice, at a time when there is marked PC atrophy (Fig. 2d and supplementary Fig. S6), arguing that RBM17 is not depleted in SCA1.
To ascertain the relevance of ATXN1/RBM17 interaction to ATXN1-induced neuropathology in vivo, we utilized a Drosophila model of SCA1 in which expression of polyglutamine-expanded ATXN1 in the Drosophila eye causes retinal degeneration, ommatidial disorganization and fusion, and interommatidial bristle loss19. We established the physical interaction between ATXN1 and Drosophila RBM17 (dRBM17) (data not shown) and looked for genetic interaction between the two proteins by crossing SCA1 flies with dRBM17 mutant flies (Fig. 3 and supplementary Fig. S7). A heterozygous loss of one dRBM17 allele partially suppressed ommatidial disorganization phenotypes of SCA1 flies at 30°C (Fig. 3b), a temperature at which ATXN1[82Q] causes severe retinal degeneration (Fig. 3a). To verify the specificity of the genetic interaction, we generated two different lines of transgenic flies that expressed either human RBM17 (hRBM17) or dRBM17. Overexpression of either hRBM17 or dRBM17 alone in the Drosophila eye did not cause any obvious external morphological phenotypes at 25°C (Supplementary Fig. S7). Co-expression of hRBM17 (or dRBM17) with ATXN1[82Q] worsened ommatidial disorganization and bristle loss in SCA1 flies (Fig. 3c, d). In contrast, hRBM17 (or dRBM17) had little effect when co-expressed with wild-type ATXN1[30Q] (Fig. 3e, f). These genetic data suggest that RBM17 plays a crucial role in mediating the toxicity of polyglutamine-expanded ATXN1 in vivo.
Figure 3. RBM17 contributes to polyglutamine-expanded ATXN1 toxicity in the Drosophila eye.
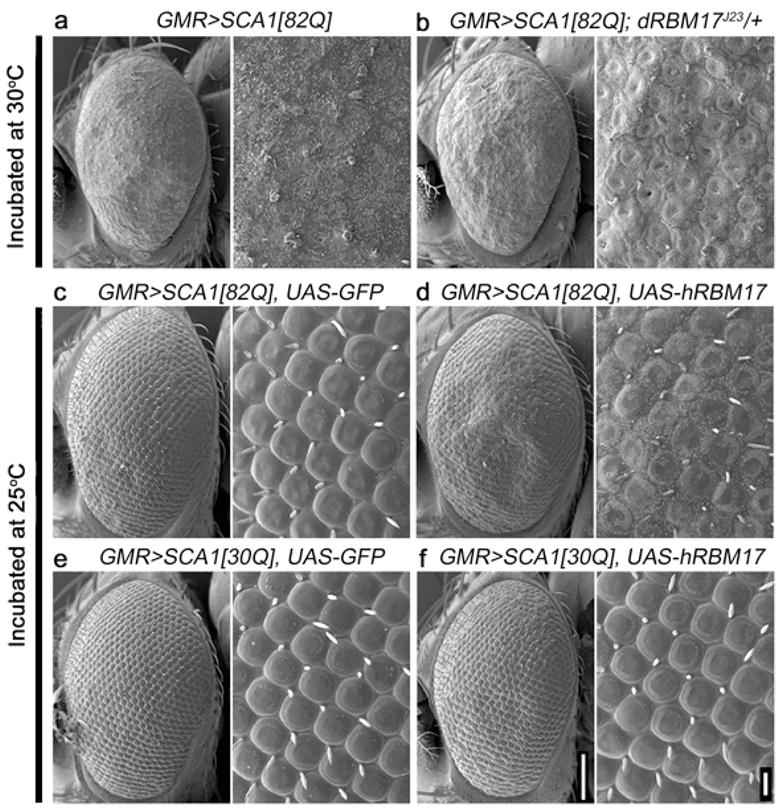
Scanning electron microscopy (SEM) of adult Drosophila eyes. Loss of one dRBM17 allele suppressed ATXN1[82Q]-mediated ommatidial disorganization (a,b), while overexpression of hRBM17 worsened abnormalities induced by ATXN1[82Q] but not by ATXN1[30Q] (c–f). Flies were raised at 30°C (a,b) or 25°C (c–f), and genotypes are: (a) GMR-Gal4>UAS-ATXN1[82Q], (b) GMR-Gal4>UAS-ATXN1[82Q]; dRBM17J23/+, (c) GMR-Gal4>UAS-ATXN1[82Q]; UAS-GFP, (d) GMR-Gal4>UAS-ATXN1[82Q]; UAS-hRBM17, (e) GMR-Gal4>UAS-ATXN1[30Q]; UAS-GFP, and (f) GMR-Gal4>UAS-ATXN1[30Q]; UAS-hRBM17. Magnified images are on the right of each panel. Scale bars are 100μm or 10μm, respectively. Additional data with controls are available in Supplementary Fig. S7.
Native ATXN1 complexes contain RBM17
We next asked whether the ATXN1/RBM17 interaction is transient or whether the two proteins form a stable protein complex in vivo. We first analyzed the elution profile of RBM17 in wild-type mouse cerebellum using gel-filtration chromatography and compared it with the elution profile of ATXN1. RBM17 eluted in a broad range of fractions (8 through 17) with at least two identifiable peaks (Fig. 4a, red and blue boxes): one peak coincided with large protein complexes with an estimated size larger than 4MDa, at fraction 9, and a second peak occurred at fractions 12–13, consistent with small protein complexes at ~700kDa in size. The elution profile of RBM17 in a human cell line, HEK293T cells, also revealed two elution peaks enriched for this protein in fractions similar to those found in wild-type mouse cerebellum, but relatively more RBM17 protein eluted in the large protein complexes detected in fraction 9 (Supplementary Fig. S8a, red and blue boxes). ATXN1 also eluted in a broad range of fractions and was associated with two isoforms of CIC (Fig. 4a and supplementary Fig. S8a, top two rows), consistent with our previous work23.
Figure 4. Polyglutamine expansion enhances RBM17 incorporation into the large ATXN1 native protein complexes.

Representative westerns of gel-filtration fractions of (a) wild-type (Atxn1 +/+) and (b) SCA1 knock-in (Atxn1 154Q/+) mouse cerebellar extracts analyzed for CIC, Atxn1, and RBM17. Right top panels show the elution profiles for RBM17 plotted as the average percent protein (± standard error) in each fraction. Right bottom panel shows the relative ratio of RBM17 incorporation into large (red box, fractions 8–10) to small (blue box, fractions 12–13) protein complexes and the dramatic increase in RBM17 incorporation into large protein complexes in Atxn1 154Q/+ mice (*p<0.005, n=4 for Atxn1 +/+ and n=3 for Atxn1 154Q/+). Ex, extract.
Given that the proteins showed broadly overlapping elution profiles, we sought to establish the fractions in which ATXN1 and RBM17 interact. We transfected HEK293T cells with Flag-ATXN1[82Q], fractionated cell extracts, and immunoprecipitated Flag-ATXN1[82Q] from each of the fractions using anti-Flag antibody (Supplementary Fig. S8b). We found that ATXN1[82Q] immunoprecipitated endogenous RBM17 proteins from the large protein complexes (peak fraction 9, red box at the bottom panel), but not from the small protein complexes (peak fractions 12–13, blue box at the bottom panel). This suggests that ATXN1 and RBM17 interact in the large protein complexes. Incorporation of RBM17 and ATXN1 into large protein complexes was not caused by differential phosphorylation of ATXN1 at S776 (Supplementary Fig. S9). Of note, Atxn1 was not required for the presence of RBM17 in the large protein complexes, since RBM17 was still present in fractions 8–10 in Atxn1 knock-out (Atxn1 −/−) mouse cerebellum (Supplementary Fig. S10). This suggests that RBM17 associates with other proteins in the large protein complex in addition to forming complexes with Atxn1.
Glutamine expansion enhances RBM17 incorporation in native complexes
The findings that polyglutamine expansion enhanced the interaction of ATXN1 with RBM17 (Fig. 2b and supplementary Fig. S4) and that ATXN1/RBM17 associated in vivo into large protein complexes (Supplementary Fig. S8b) suggested to us that more RBM17 molecules might incorporate into the large protein complexes in the presence of polyglutamine-expanded ATXN1. To test this hypothesis, we examined the elution profile of RBM17 in cerebellar protein extracts from SCA1 knock-in mice that carry the polyglutamine-expanded (Atxn1[154Q]) allele24 (Fig. 4b).
RBM17 from SCA1 knock-in mouse cerebellar extracts fractionated into both large (Fig. 4b, red box) and small (Fig. 4b, blue box) protein complexes, but the relative ratio of RBM17 incorporation into the large protein complexes was much greater in the SCA1 knock-in mice than in wild-type mice (Fig. 4). These data strongly support the hypothesis that more RBM17 is incorporated into large protein complexes because of enhanced interaction with polyglutamine-expanded ATXN1.
Two distinct large ATXN1 protein complexes
A major portion of wild-type ATXN1 co-elutes and forms large native protein complexes with two isoforms of CIC in mouse cerebellum23 (Fig. 4a, top two rows). We therefore asked whether in vivo native protein complexes containing ATXN1/CIC contain RBM17 as well. Interestingly, despite the fact that both RBM17 and CIC interact with ATXN1, the two proteins were not detectable in the same ATXN1 complexes in either mouse cerebellum (Figs. 1d and 5a) or in HEK293T cells (Supplementary Fig. S11). This suggests that there are at least two different ATXN1-associated large protein complexes in vivo: one containing ATXN1 and RBM17, the other containing ATXN1 and CIC. Interestingly, these two proteins, and possibly the protein complexes associated with them, have contrary effects on SCA1 pathology: extra RBM17 augments expanded ATXN1 toxicity in Drosophila (Fig. 3), but extra CIC represses it23. Consistent with these results as well as the accumulating evidence that nuclear inclusions serve a protective role in SCA1-affected cells25, we observed that CIC enhanced, while RBM17 slightly suppressed, ATXN1 nuclear inclusion formation (Supplementary Fig. S12). Interestingly, overexpression of Ataxin1-like (a paralog of ATXN1), which can interact with ATXN1 and CIC, also increases sequestration of mutant ATXN1 into inclusions in PC nuclei of SCA1 knock-in mice25.
Figure 5. RBM17 and CIC form two distinct protein complexes that compete with each other.

(a) The CIC anti-serum co-immunoprecipitated Atxn1 (arrow), but not RBM17 (bracket) from mouse cerebellar extracts.
(b) ATXN1 co-affinity purified more CIC protein when RBM17 expression was reduced. The interaction of GST-ATXN1[82Q] with endogenous CIC was strongly increased (red arrows, compare lane 2 with lane 3) when endogenous RBM17 expression was decreased (blue arrow) in Neuro-2a cells. Right panel shows the normalized levels of co-affinity purified CIC-L and CIC-S. Mean relative levels (siRNA control [Lamin A/C siRNA]=100%) and standard error are shown (n=4, *P=0.056, **P<0.006).
The identification of two biochemically and functionally distinct large ATXN1 protein complexes led us to test whether RBM17 and CIC compete for interaction with ATXN1. We found that RBM17 and CIC do indeed compete for ATXN1 interaction (Fig. 5b and supplementary Fig. S11). These data, along with results described above, strongly suggest that there are at least two distinct endogenous protein complexes associated with ATXN1.
SCA1 involves gain and partial loss of ATXN1 function
Next, we wished to address the question of how the RBM17- and CIC-containing endogenous protein complexes affect each other’s formation and function, and how their interactions influence SCA1 neuropathology. Given that ATXN1/RBM17 interactions are polyglutamine-length dependent and that RBM17 and CIC can compete with each other to form distinct ATXN1-containing protein complexes, we infer that polyglutamine expansion in ATXN1 favors the formation of the RBM17-containing protein complex, leaving mostly the wild-type ATXN1 to form a CIC-containing protein complex. Bearing in mind that the expression level of wild-type ATXN1 in SCA1 knock-in mice and in human patients is reduced to half the levels in healthy controls and that CIC expression is also significantly reduced in SCA1 knock-in (Atxn1 154Q/+) mice (Supplementary Fig. S13), formation of expanded ATXN1/RBM17-containing complexes in soluble fractions is increased but the formation of wild-type ATXN1/CIC-containing protein complexes is decreased (Fig. 4). These considerations raise the interesting possibility that SCA1 is caused not only by a toxic gain-of-function mechanism but also by a partial loss of wild-type ATXN1 function.
To ascertain whether SCA1 neuropathology is affected by the presence or absence of the wild-type ATXN1 protein, we crossed Atxn1 heterozygote (Atxn1 +/−) animals with the SCA1 knock-in (Atxn1 154Q/+) mice. Atxn1 154Q/− mice showed a worsened performance on the rotarod than Atxn1 154Q/+ animals (Fig. 6a). Furthermore, Atxn1 154Q/− mice had a shortened life span compared with Atxn1 154Q/+ animals (Fig. 6b). Given that Atxn1 heterozygotes (Atxn1 +/−) have no discernable phenotype on their own, these data strongly suggest that wild-type ATXN1 and its interacting proteins are protective and that loss of the wild-type ATXN1 worsens SCA1 neuropathology.
Figure 6. Loss of wild-type Atxn1 function worsens SCA1 neuropathology in mice.

(a,b) Atxn1 154Q/− animals showed a worsened rotarod performance (*P<0.05, **P<0.005) and earlier lethality (P<1×10−6) than Atxn1 154Q/+ mice.
(c) Model for SCA1 neuropathology. In wild-type individuals, there are at least two distinct and mutually exclusive ATXN1-associated endogenous protein complexes in vivo. The formation of one of these complexes (ATXN1/RBM17) appears to be regulated in that it requires phosphorylation of ATXN1. Polyglutamine expansion in ATXN1 favors formation of the RBM17-containing complex, thereby enhancing one endogenous function and contributing to neuropathology via a gain-of-function mechanism. Polyglutamine expansion concomitantly decreases the formation of CIC-containing complex, resulting in a partial loss of function.
DISCUSSION
The results presented in this study provide a mechanistic explanation of how polyglutamine expansion can cause both gain and loss of normal protein function: the expansion differentially affects the interactions of the host protein in the context of distinct endogenous protein complexes in vivo. Previous work showed that polyglutamine-expanded ATXN1, like its wild-type counterpart, interacts with several nuclear proteins and incorporates into native complexes23. Some complexes, like those containing CIC, are relatively stable, whereas ATXN1’s interactions with Gfi-1 and Tip60/RORa are more transient9,23,26. RBM17 is unique among these factors, however, in that it is the only protein discovered so far whose interaction with ATXN1 is regulated by the length of the latter’s polyglutamine tract and phosphorylation status at S776, two features that are absolute requirements for SCA1 pathogenesis.
Previously we have shown that wild-type and polyglutamine-expanded ATXN1 associate into large and small protein complexes (Fig. 4b, second row) and that incorporation of polyglutamine-expanded ATXN1 into the large complexes causes SCA1 neuropathology23,25. In this study, we found that there are at least two distinct large native protein complexes associated with ATXN1: one containing CIC and the other RBM17. Polyglutamine expansion in ATXN1 strongly enhanced the formation of an ATXN1/RBM17-containing protein complex in both cell culture and in SCA1 knock-in mouse cerebellum, providing a mechanistic explanation for the toxic gain-of-function. RBM17 is expressed in the nuclei of a broad range of neuronal and non-neuronal tissues21,27. It is interesting that both RBM1728,29 and ATXN130 appear to be involved in regulation of RNA metabolism. One potential molecular function of this protein complex might thus be RNA splicing. This hypothesis receives some support from the observation that ATXN1 and RBM17 incorporation into large protein complexes was strongly decreased after RNase treatment (Supplementary Fig. S14).
Mice lacking wild-type Atxn1 but carrying polyglutamine-expanded Atxn1 suffered more severe disease and greater lethality, suggesting that either wild-type ATXN1 protects against the mutant protein or is essential for neuronal integrity. It is noteworthy that Atxn1 null mice do not develop SCA1 features4 and that polyglutamine-expanded ATXN1 does not differentially interact with wild-type ATXN1 (Supplementary Fig. S15) arguing against loss-of-function or dominant-negative mechanisms. Our data suggest that wild-type ATXN1 competes with polyglutamine-expanded ATXN1 to decrease formation of a toxic protein complex containing RBM17 (Supplementary Fig. S16). Nevertheless, we cannot rule out the possibility that ATXN1/CIC-containing complex is also neuroprotective. We found that glutamine-expanded ATXN1 preferentially forms a complex with RBM17, whereas wild-type ATXN1 is in the CIC-containing complex. Therefore, when one allele is glutamine-expanded, the levels of wild-type ATXN1 decrease and less wild-type ATXN1/CIC complexes can be formed. Our previous findings23 that the repressive activity of CIC is more robust in the presence of wild-type ATXN1 than with glutamine-expanded ATXN1, argue that a net effect of decreased ATXN1/CIC activity and the decreased co-repressive activity due to mutant ATXN1 might also contribute to SCA1 pathogenesis. Although these data indicate that ATXN1/CIC complexes cause a partial loss of wild-type ATXN1 function, we cannot exclude the possibility that a mutant ATXN1/CIC complex might actively contribute to SCA1 pathogenesis. In sum, we propose a two-pronged model of SCA1 neurodegeneration in which augmented function of a particular stable endogenous protein complex is combined with a simultaneous loss of function of other, stable endogenous protein complexes (Fig. 6c).
This dual model may apply equally well to several other dominant neurodegenerative diseases caused by “gain-of-function” mutational mechanism including other polyglutamine diseases31–38, prion disease39,40, and Alzheimer disease (AD)41,42. Relatively recent work indicates that some of the polyglutamine diseases might also involve a partial loss-of-function of the wild-type host proteins31–38. However, the clear mechanism by which polyglutamine expansion causes both toxic gain-of-function and simultaneously loss-of-function of the disease-causing protein has not been clarified yet. In HD, loss of Huntingtin (Htt) function intensifies neurodegeneration in transgenic models bearing the expanded protein32–34. Htt regulates REST/NRSF activity differentially depending on whether it is wild-type or polyglutamine-expanded37. While the mechanism has not been fully described, the mutant Htt may exert a dominant deleterious effect upon REST/NRSF activity by losing its ability to sequester REST/NRSF in the cytoplasm. In the dominantly inherited prion diseases, mutant prion proteins cause toxicity mainly via a toxic gain-of-function mechanism by interfering with essential cellular processes and activating cell death pathways40. In addition, some pathogenic mutations might also impair certain physiological functions of the normal host protein, contributing to disease by loss of a neuroprotective function of wild-type prion protein39,40. In the case of Alzheimer disease (AD), increased production and aggregation of the amyloid beta peptide by AD-related presenilin mutations is widely accepted to play a key role in AD through a dominant gain-of-function mechanism42. However, recent work demonstrates that inactivation of presenilin in the adult cerebral cortex causes progressive memory loss and neurodegeneration that strikingly resemble AD41. Whether presenilin mutations contribute to AD through a gain- and/or a loss-of-function mechanism is not yet understood, but our finding that the interactions of the mutant protein with its usual partners are differentially affected by the polyglutamine expansion offers a mechanistic explanation for how mutant proteins can gain and lose function simultaneously.
METHODS SUMMARY
Y2H mating type screens43 of human open reading frame (ORF) clones were performed using the hORFeome v1.1 as described20. The co-AP and co-IP experiments were performed using cell lysates or mouse cerebellar extracts as described20,23. Gel-filtration chromatography was performed using soluble extracts from about 20-week-old mouse cerebellum or cells as described23. Drosophila SCA1 model and dRBM17 mutant flies were used to test a genetic interaction between the two proteins. The dRBM17J23 allele is a strong hypomorphic or amorphic allele of dRBM17 whereas Df(2Lh)D1 is a deficiency that spans the dRBM17 region28. Processing and image acquisition of Drosophila eye for scanning electron microscopy were performed as described23. Immunofluorescence staining and rotarod analysis were performed as described24,25.
FULL METHODS
Yeast two-hybrid screens
Yeast two-hybrid (Y2H) mating type screens43 of human open reading frame (ORF) clones were performed using the hORFeome v1.1 as described20. For re-testing ATXN1/RBM17 interaction in yeast, MaV103 (MATa) yeast strain transformed with various AD-ATXN1 constructs was individually mated against MaV203 (MATα) containing DB-RBM17. Resulting interactions were tested on 3-AT plates (synthetic complete media plates lacking leucine, tryptophan, and histidine and containing 20mM 3-amino-,2,4-triazole (3-AT)) as well as by β-Gal filter lift assay. Y2H controls are the following: Lane 1 expresses AD and DB without any fusion, acting as a negative control. Lane 2 expresses AD-E2F1 and DB-pRB and shows a weak two-hybrid phenotype. Lanes 3, 4, and 5 express AD-Jun and DB-Fos, AD and Gal4p, and AD-E2F1 and DB-DP, respectively, and exhibit relatively strong two-hybrid phenotypes.
Co-affinity purification and Co-immunoprecipitation assays
The co-affinity purification (co-AP) experiments were performed as described20. Briefly, HEK293T cells were plated the day before transfection at 1 × 105 cells per well in 6-well plates. The following day, cDNA constructs were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturers instructions and cells were cultured in DMEM medium with 10% fetal bovine serum. Two days later, cells were harvested and lysed with lysis buffer (0.5% NP-40, 20mM Tris-HCl [pH 8.0], 150~180mM NaCl, 1mM EDTA, and complete protease inhibitor cocktail [Roche]) for 15min on ice. After centrifugation, soluble protein complexes were purified using Glutathione Sepharose 4B beads (Amersham), washed with lysis buffer, and analyzed by SDS-PAGE and western blot. GFP expression was used as a transfection and loading control. The co-immunoprecipitation (co-IP) assays were performed using mouse cerebellar extracts as described23.
Column fractionation and analysis
Gel-filtration (sizing) chromatography was performed using soluble extracts from about 20-week-old mouse cerebellum or HEK293T or Neuro-2a cells using the Amersham Pharmacia LCC-500 FPLC system as described23. Protein signal in each fraction (measured by densitometry) is divided by the total signal in all the fractions to determine the percentages. Thyroglobulin (669kDa) and ADH (150kDa) were used for gel-filtration standards. Monoclonal anti-HA agarose conjugate clone HA-7 (Sigma, A2095) and anti-FLAG M2 affinity gel freezer-safe (Sigma, A2220) were used for IP after column fractionation.
Immunofluorescence and confocal microscopy
Immunofluorescence staining was performed as described25. Sections from frozen fixed cerebella of Atxn1 +/+, Atxn1 +/−, Atxn1 154Q/+, Atxn1 154Q/−, and SCA1 transgenic (ATXN1[82Q]) mice were co-stained with the following antibodies: rabbit anti-RBM17 (1:400), rabbit anit-Atxn1 (11NQ, 1:1000), and mouse anti-calbindin (1:1000; Sigma) antibodies. Fluorescent images were scanned using Zeiss LSM510 confocal microscope and processed with ImageJ software and Adobe Photoshop.
Rotarod and statistical analyses
We performed t-tests (two-tailed, not assuming equal variances) and calculated standard error of the mean (s.e.m.) and 95% confidence intervals using Microsoft Excel. Rotarod analysis was performed as described previously24 using 7-week old animals. We analyzed rotarod performance by repeated-measures analysis of variance (ANOVA) using SPSS 11 software for Mac OSX.
Drosophila strains and scanning electron microscopy
Full-length hRBM17 and dRBM17 cDNAs were cloned into the pUAST-dest vector using the Gateway system to generate UAS-hRBM17 or UAS-dRBM17 transgenic fly lines, respectively. The mutant and transgenic flies were used in this study are dRBM17J23, Df(2Lh)D1 (ref. 28), UAS-ATXN1[82Q]F7, and UAS-ATXN1[30Q]F1 (ref. 19). Other strains were obtained from the Bloomington Stock Center (Flybase; www.flybase.org). For the genetic interaction, more than 50 adult flies per each genotype were examined at day 2 after eclosion. Processing and image acquisition of Drosophila eye for scanning electron microscopy were performed as described23.
Supplementary Material
Acknowledgments
We are grateful to Dr. Marc Vidal for the Human ORFeome and Y2H screening technology; Drs. Juan Valcárcel and William Perry for anti-RBM17 antibody; Dr. Helen Salz for dRBM17 mutant flies; Yuchun He for generating the RBM17 transgenic flies; Drs. Hugo Bellen and Hamed Jafar-Nejad, and members of the Zoghbi laboratory for comments on the manuscript, and Vicky Brandt for editorial input. This research was supported by the NIH grants (H.Y.Z., H.T.O., M.V.), cores of the BCM-MRDDRC, and the Ellison Foundation and the W.M. Keck Foundation awarded to M.V. and D.E.H. H.Y.Z. is an investigator with the Howard Hughes Medical Institute.
References
- 1.Orr HT, Zoghbi HY. Trinucleotide Repeat Disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 2.Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–33. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- 3.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–54. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 4.Matilla A, et al. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J Neurosci. 1998;18:5508–16. doi: 10.1523/JNEUROSCI.18-14-05508.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeh S, et al. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci U S A. 2002;99:13498–503. doi: 10.1073/pnas.212474399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11:155–63. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 7.Kiehl TR, et al. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem Biophys Res Commun. 2006;339:17–24. doi: 10.1016/j.bbrc.2005.10.186. [DOI] [PubMed] [Google Scholar]
- 8.Klement IA, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 9.Tsuda H, et al. The AXH Domain of Ataxin-1 Mediates Neurodegeneration through Its Interaction with Gfi-1/Senseless Proteins. Cell. 2005;122:633–44. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Emamian ES, et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–87. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 11.McManamny P, et al. A mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11:2103–11. doi: 10.1093/hmg/11.18.2103. [DOI] [PubMed] [Google Scholar]
- 12.Katsuno M, et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–54. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- 13.Chevalier-Larsen ES, et al. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24:4778–86. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sopher BL, et al. Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron. 2004;41:687–99. doi: 10.1016/s0896-6273(04)00082-0. [DOI] [PubMed] [Google Scholar]
- 15.Graham RK, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–91. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 16.Warby SC, et al. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14:1569–77. doi: 10.1093/hmg/ddi165. [DOI] [PubMed] [Google Scholar]
- 17.Luo S, Vacher C, Davies JE, Rubinsztein DC. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol. 2005;169:647–56. doi: 10.1083/jcb.200412071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steffan JS, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–4. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 19.Fernandez-Funez P, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–6. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 20.Lim J, et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–14. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 21.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–76. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 22.Burright EN, et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–48. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- 23.Lam YC, et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–47. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 24.Watase K, et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34:905–19. doi: 10.1016/s0896-6273(02)00733-x. [DOI] [PubMed] [Google Scholar]
- 25.Bowman AB, et al. Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes. Nat Genet. 2007;39:373–379. doi: 10.1038/ng1977. [DOI] [PubMed] [Google Scholar]
- 26.Serra HG, et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127:697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- 27.Sampath J, et al. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am J Pathol. 2003;163:1781–90. doi: 10.1016/S0002-9440(10)63538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaouki AS, Salz HK. Drosophila SPF45: A Bifunctional Protein with Roles in Both Splicing and DNA Repair. PLoS Genet. 2006;2:e178. doi: 10.1371/journal.pgen.0020178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lallena MJ, Chalmers KJ, Llamazares S, Lamond AI, Valcarcel J. Splicing regulation at the second catalytic step by Sex-lethal involves 3′ splice site recognition by SPF45. Cell. 2002;109:285–96. doi: 10.1016/s0092-8674(02)00730-4. [DOI] [PubMed] [Google Scholar]
- 30.Yue S, Serra HG, Zoghbi HY, Orr HT. The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract. Hum Mol Genet. 2001;10:25–30. doi: 10.1093/hmg/10.1.25. [DOI] [PubMed] [Google Scholar]
- 31.Zuccato C, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J Neurosci. 2007;27:6972–83. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Auerbach W, et al. The HD mutation causes progressive lethal neurological disease in mice expressing reduced levels of huntingtin. Hum Mol Genet. 2001;10:2515–23. doi: 10.1093/hmg/10.22.2515. [DOI] [PubMed] [Google Scholar]
- 33.Van Raamsdonk JM, et al. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:1379–92. doi: 10.1093/hmg/ddi147. [DOI] [PubMed] [Google Scholar]
- 34.Leavitt BR, et al. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. 2001;68:313–24. doi: 10.1086/318207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6:919–30. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 36.Thomas PS, Jr, et al. Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Hum Mol Genet. 2006;15:2225–38. doi: 10.1093/hmg/ddl148. [DOI] [PubMed] [Google Scholar]
- 37.Zuccato C, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 38.Friedman MJ, et al. Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci. 2007;10:1519–28. doi: 10.1038/nn2011. [DOI] [PubMed] [Google Scholar]
- 39.Li A, Piccardo P, Barmada SJ, Ghetti B, Harris DA. Prion protein with an octapeptide insertion has impaired neuroprotective activity in transgenic mice. Embo J. 2007;26:2777–85. doi: 10.1038/sj.emboj.7601726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–7. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 41.Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–9. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Broeck B, Van Broeckhoven C, Kumar-Singh S. Current Insights into Molecular Mechanisms of Alzheimer Disease and Their Implications for Therapeutic Approaches. Neurodegener Dis. 2007;4:349–365. doi: 10.1159/000105156. [DOI] [PubMed] [Google Scholar]
- 43.Rual JF, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–8. doi: 10.1038/nature04209. NATURE: 2007-08-08406B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.