

Abstract
Regulators of G protein signaling (RGS) proteins accelerate GTP hydrolysis by Gi but not by Gs class α-subunits. All RGS proteins share a conserved 120-amino acid sequence termed the RGS domain. We have demonstrated that the RGS domains of RGS4, RGS10, and GAIP retain GTPase accelerating activity with the Gi class substrates Giα1, Goα, and Gzα in vitro. No regulatory activity of the RGS domains was detected for Gsα. Short deletions within the RGS domain of RGS4 destroyed GTPase activating protein activity and Giα1 substrate binding. Comparable protein–protein interactions between Giα1–GDP–AlF4− and the RGS domain or full-length RGS4 were detected using surface plasmon resonance.
Keywords: GTP hydrolysis, desensitization
Regulators of G protein signaling (RGS) proteins were first identified in genetic screens in fungi and nematodes as negative regulators of G protein signaling (1–3). A diverse group of more than 20 proteins have been identified in eukaryotic organisms with the single common feature of ≈120 amino acids, referred to as the RGS domain (3). Protein interactions assayed in the yeast two-hybrid system indicated that amino acid sequence including the RGS domain in GAIP contributed to the formation of a complex with Giα3 (4). In addition, in vivo activity of egl-10, an RGS protein expressed in Caenorhabditis elegans, was impaired by mutations that introduced either an amino acid substitution or termination codons within the RGS domain (3). Biochemical characterization of GAIP, RGS4, and RGS10 showed these proteins accelerated GTP hydrolysis by Gi class α-subunits in vitro, thereby providing a molecular mechanism for their role as inhibitors of G protein signaling in vivo (5–7). We show herein that the RGS domains of GAIP, RGS4, and RGS10 retain GTPase activating protein (GAP) activity with Gi class α-subunits in vitro. Short deletions within the RGS domain of RGS4 destroyed GAP activity and its ability to bind Gα substrates.
MATERIALS AND METHODS
Expression Vectors.
cDNAs were PCR amplified (Expand; Boehringer Mannheim) using gene-specific primers to make deletions of RGS4, RGS10, and GAIP. All cDNA clones were sequenced to verify that the correct recombinant RGS proteins were expressed in Escherichia coli JM109. All recombinant proteins were His6 tagged at the amino terminus with the sequence MGH6MG. Recombinant RGS10 was obtained by reverse transcription–PCR: random primed cDNA was made by reverse-transcription (Superscript; BRL) of total RNA from human placenta (2 μg). The RGS domain of RGS10 (amino acids 29–147) (6) was PCR amplified with sense primer TW87 (GATCCATGGGCAAATGGGCGGCATCCCTGGA) and antisense primer TW88 (GATGGATCCTAGTGTTTTAAAAACAAGTCAG). The PCR product was digested and cloned into the NcoI and BamHI sites of H6-pQE60 vector (8). The RGS domain of GAIP (amino acids 86–205) was PCR amplified from plasmid cDNA (5) with sense primer TW104 (GATCCATGGGCAGCTGGGCGCAGTCTTTTGA) and antisense primer TW105 (GCCAAGCTTCTACAGGGCACGGTAGGTGGGAG), digested and cloned into the NcoI and HindIII sites of H6-pQE60 vector. The RGS domain of RGS4 (amino acids 58–177) was PCR amplified from plasmid cDNA (5) with sense primer KY1 (GATCCATGGGCAAATGGGCTGAATCGCTGGAA) and antisense primer KY2 (CGGCTCGAGCTACAGGTCAAGATAGAATCGAGA), digested and cloned into the NcoI and XhoI sites of a modified H6-pQE60 vector. The RGS4 deletion constructs (see Fig. 1) were made with the indicated sense and antisense primers, and cloned into H6-pQE60: RΔ5Δ3, TW59 [GCCGAATTCCATGGGAAG(CT)GAGGAGAACAT(TG)(GC)(AT)C], and TW61 [GCCGGATCCTAGTATGAGTCC(TC)(TG)(TG)T(GC)CAT]; RΔ5, TW59, and KY2 (CGGCTCGAGCTACAGGTCAAGATAGAATCGAGA); RΔ3, KY1 (GATCCATGGGCAAATGGGCTGAATCGCTGGAA), and TW61. The RGS4 internal deletions were made by independent PCR amplification of the 5′ and 3′ segments: iΔ5 5′ segment, KY3 (TGCTTTGTGAGCGGATAACAA) and KY5 (AAGTCAATGTTCTCCTCACTCTTGACTTCTTCTTGGCTCA); iΔ5 3′ segment, KY6 (TGAGCCAAGAAGAAGTCAAGAGTGAGGAGAACATTGACTT) and KY4 (GCGTTCTGAACAAATCCAGAT); iΔ3 5′ segment, KY3 and KY7 (CCGCAGCTGGAAGGATTGGTGTATGAATCCTTTTCCATCA); iΔ3 3′ segment KY8 (TGATGGAAAAGGATTCATACACCAATCCTTCCAGCTGCGG) and KY4. Single bands of 5′ and 3′ segments were excised from low melting point agarose gel and combined in a second PCR to produce a single DNA fragment by overlap extention (9, 10) using outside flanking primers KY3 and KY4. All oligonucleotide sequences are written 5′ to 3′.
Figure 1.

Schematic diagram of RGS4 full-length and RGS domain deletion constructs. All recombinant proteins were His6-tagged at the amino terminus (see Materials and Methods). The amino acids of RGS4 included in each construct are indicated. The RGS domain is defined by homology to a consensus sequence of 120 amino acids identified in numerous RGS domain proteins (3).
Expression and Purification of Proteins.
For protein expression, 1 liter of T7 medium (8) with ampicillin (100 μg/ml) was inoculated with an overnight culture started from a single colony, isopropyl β-d-thiogalactoside (10 μM) induction was performed at OD600 = 0.6, cultures were shaken overnight, and cells were pelleted, lysed by freezing, and sonicated with TBP buffer (50 mM Tris⋅HCl, pH 8.0/20 mM 2-mercaptoethanol/0.1 mM phenylmethylsulfonyl fluoride). Lysozyme (0.2 mg/ml) and DNase I (5 μg/ml) were added to complete lysis and digest DNA. Soluble proteins [e.g., RGS4, iΔ5, iΔ3, and R10 (see Figs. 1 and 5 for abbreviations) were isolated from total lysate centrifuged at 12,000 × g (30 min at 4°C). The supernatant was applied onto 2 ml Ni-NTA column (Qiagen, Chatsworth, CA) pre-equilibrated with TBP buffer, washed first with 20 ml TBP and 0.2 M NaCl, and finally washed with 10 ml of TBP with 10 mM imidazole (pH 8.0). Protein was eluted with 9 ml elution buffer (TBP containing 150 mM imidazole, pH 8.0) and concentrated with an Ultrafree15 device (Millipore) in buffer A [50 mM Hepes, pH 8.0/1 mM DTT/0.05% C12E10 (Merck)/5 mM EDTA]. SDS/PAGE analysis indicated more than 90% purity by Coomassie blue staining. Insoluble proteins [e.g., RGAIP, RΔ5, RΔ3, and RΔ5Δ3 (R4, prepared in the same manner, retained full GAP activity)] were isolated from cell pellets lysed with 8 M urea buffer (8 M urea/20 mM Tris⋅HCl, pH 8.0/100 mM NaCl). The lysate was sonicated to shear DNA and centrifuged at 22,000 × g (30 min at 4°C). The supernatant was applied onto 2 ml Ni-NTA column. Protein was simultaneously washed and renatured on the column with 100 ml of an 8 M urea to 1 M urea gradient buffer. A final wash with 10 ml TBP buffer removed residual urea. Protein was eluted with 9 ml elution buffer and concentrated with an Ultrafree15 device (Millipore) in buffer A. The purity was 90% as assessed by SDS/PAGE analysis with Coomassie blue staining. Gα protein substrates were purified as described for Giα1, Gsα (short form), Goα (8), and Gzα (11).
Figure 5.
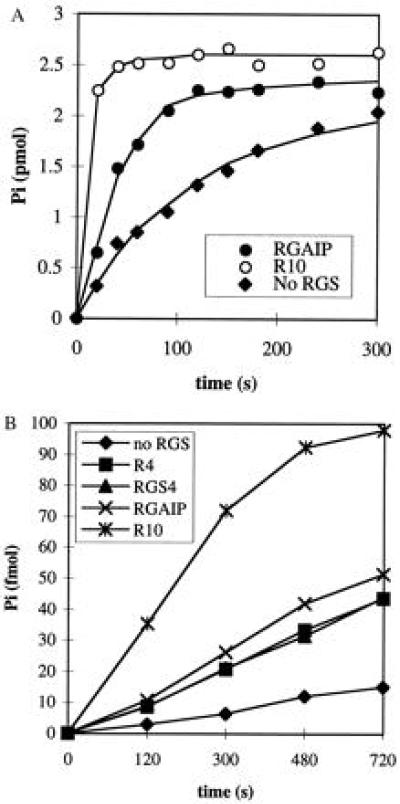
The RGS domains of RGS4, RGS10 and GAIP stimulate hydrolysis of GTP bound to Giα1 and Gzα. (A) Time curves of GTP hydrolysis for Giα1 (500 nM) was preloaded with [γ-32P]GTP at 30°C (estimated concentration of GTP–Giα1 was 215 nM), then incubated at 0°C with R10 (50 nM) or RGAIP (1 μM). (B) Time curves of GTP hydrolysis for Gzα (2.5 nM) at 15°C and 50 nM of RGS4 and each RGS domain from RGS4 (R4), GAIP (RGAIP), and RGS10 (R10).
GTP Binding and Hydrolysis.
All assays were performed in buffer A. Temperatures and protein concentration are described in the figure legends. For measurements of GTP hydrolysis, 20–40% of Giα1 protein was loaded with [γ-32P]GTP (1–2 μM) for 10–20 min at 30°C, and Gsα was loaded for 20 min at room temperature. After GTP loading, the temperature was then lowered to 0°C for 5 min. The GTP hydrolysis reaction was initiated at 0°C by adding 500 mM MgSO4 (10 mM final concentration) and unlabeled GTP (200 μM final concentration) with RGS protein or buffer alone as control. Aliquots of the reaction mix (25 μl) were removed at the indicated times and immediately mixed with 375 μl of 5% (wt/vol) Norit charcoal (Fisher) in 50 mM NaH2PO4. After centrifugation at 1,500 × g for 5 min, 200 μl aliquots of supernatant were mixed with 4 ml scintillation liquid and counted by liquid scintillation spectrometry. Thus, the amount of Pi released at each time point was determined from a 12.5 μl aliquot of the original reaction mix. GTP hydrolysis assays for Gzα were done as described (11).
Kinetic Analysis.
Kinetic parameters of GTP hydrolysis were calculated from Fig. 3 A and B and replicate experiments. The RGS catalyzed GTP hydrolysis was assumed to follow the simplest model of two parallel reactions:
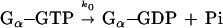 |
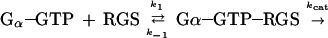 |
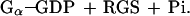 |
The first reaction is a hydrolysis of GTP by the Gα subunit itself, whereas the second reaction is RGS-catalyzed. Under the assumption that [Gα-GTP]o ≪ Km, the observed total kinetic constant of Pi formation is given by the equation kobs = ko + (kcat/Km)[RGS]o. If kcat ≫ k-1, then kcat/Km = k1. Giα1 has a relatively high basal rate of GTP hydrolysis. Therefore, initial rate measurements taken by withdrawing aliquots of reaction mix would be inaccurate. Thus, we calculated kinetic constants from the curves over their entire time course using the biaevaluation software (Pharmacia Biosensor). These programs calculated the best fit value of kobs, the corresponding standard deviation and the extrapolated maximal amount of Pi released in each experiment. Reliable values of kobs were obtained in the interval between 5 and 60 nM RGS4.
Figure 3.
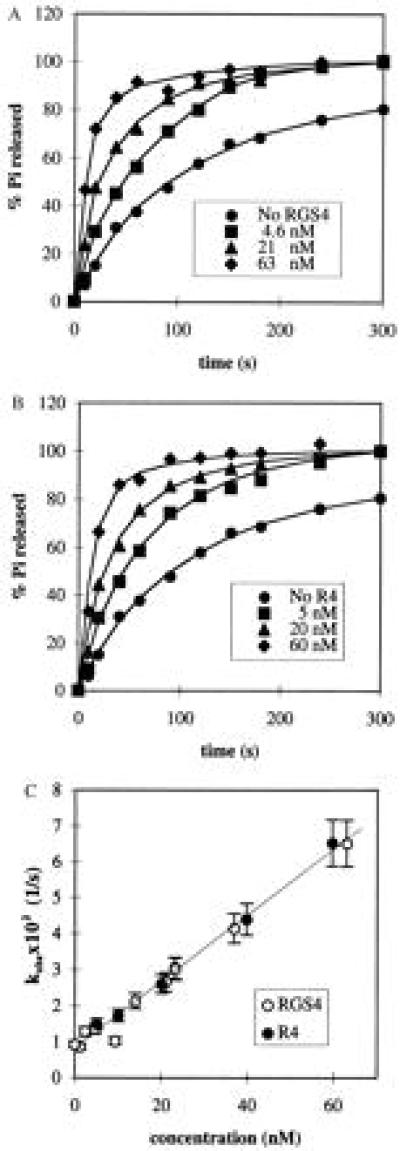
The RGS domain and full-length RGS4 have similar catalytic activities. To obtain kinetic curves of GTP hydrolysis, Giα1 (500 nM) was preloaded with [γ-32P]GTP at 30°C (estimated concentration of GTP–Giα1 was 215 nM), then incubated at 0°C with different concentrations of (A) full-length RGS4 and (B) the RGS domain of RGS4 (R4). (C) A plot of the observed first order kinetic constant vs. initial concentration of RGS4 and R4 derived from A and B and additional experiments. Each data point corresponds to one time course curve, error bars indicate one standard deviation in each kobs calculation.
Biosensor Measurements.
Surface plasmon resonance measurements were carried out using the BIAcore 1000 instrument at 25°C (Pharmacia Biosensor). RGS proteins were coupled to the sensor chip surface using the amine chemistry (N-hydroxyl succinimide activation of the carboxyl groups of the chip) in accordance with the manufacturer’s instructions to a density of 2,300–2,800 response units (RU). Giα1 was injected with a flow rate 5 μl/min at different concentrations (Fig. 4) in buffer containing 10 mM Hepes (pH 8.0), 150 mM NaCl, 1 mM AlCl3, 10 mM NaF, 5 mM MgSO4, 1 mM GDP, 1 mM 2-mercaptoethanol, and 0.05% C12E10. Regeneration of the RGS protein on the biosensor chip after each binding cycle was carried out using 10 μl injections of 0.05% SDS in 20 mM Hepes (pH 8.0) with 150 mM NaCl. Biphasic association curves, detected in all cases (Fig. 4), may reflect heterogeneity of chemically immobilized proteins on the biosensor chip surface (12). Calculations of ka reported in Table 1 were made using the portion of the sensorgrams corresponding to the faster interaction, during which time the majority of Gα substrate bound to RGS protein on the biosensor chip. The data fitted the monophasic pseudo-first-order equation well and consistently for different concentrations of Giα1 in solution. kd was calculated across the interval beginning several seconds after the end of sample injection (12) and ending 400 sec later. Kinetic parameters were extracted from the sensorgrams following subtraction of blank control values using the biaevaluation 2.1 software (Pharmacia Biosensor).
Figure 4.
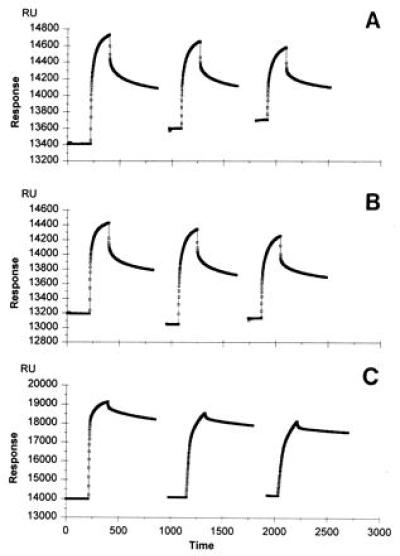
RGS4 and RGS10 binding to Giα1–GDP–AlF4− on the BIAcore sensor chip. Sensorgrams of (A) RGS4, (B) RGS domain of RGS4 (R4), and (C) RGS domain of RGS10 (R10). In each experiment, RGS proteins were immobilized on the sensor chip and subjected to three injections of Giα1-GDP-AlF4− at concentrations of 500 nM, 250 nM, and 125 nM, respectively. Following each cycle of Giα1 binding, the flow cell was regenerated by injecting 5–10 μl of 0.05% SDS. Time (sec) and response units (RU) are indicated at multiple points on each sensogram.
Table 1.
Kinetic and thermodynamic parameters of RGS protein binding to Giα1–GDP–A1F4−
| RGS protein | ka, (M−1⋅s−1) | kd, s−1 | Kd, nM |
|---|---|---|---|
| RGS4 | 7.3 ± 3.6 × 105 | 4.9 ± 1.0 × 10−4 | 0.67 |
| R4 | 7.2 ± 2.7 × 105 | 4.2 ± 0.4 × 10−4 | 0.58 |
| R10 | 2.4 ± 1.1 × 105 | 1.4 ± 0.4 × 10−4 | 0.59 |
ka, observed kinetic association constant; kd, observed kinetic dissociation constant; and Kd = kd/ka, observed thermodynamic dissociation constant. The ka and kd values are shown with standard errors calculated from sensorgrams according to BIAcore manual.
RESULTS AND DISCUSSION
The RGS Domain Is the Minimal Sequence Required for GAP Activity of RGS4.
RGS4 exhibits striking sequence homology with several recently identified proteins that each accelerate hydrolysis of GTP by heterotrimeric G protein α-subunits of the Gi class (5–7). Sequence similarity between these proteins is restricted to ≈120 amino acids referred to as the RGS domain (3). To test the in vitro GAP activity of this conserved domain, we constructed a series of deletions that either retained or removed portions of the RGS domain from RGS4 (Fig. 1) and expressed the truncated proteins in E. coli. The RGS domain of RGS4 retained GAP activity with the substrates Giα1 (Fig. 2) and Goα (data not shown). Thus, the amino acid sequences flanking either side of the RGS domain are not essential for GAP activity in vitro.
Figure 2.
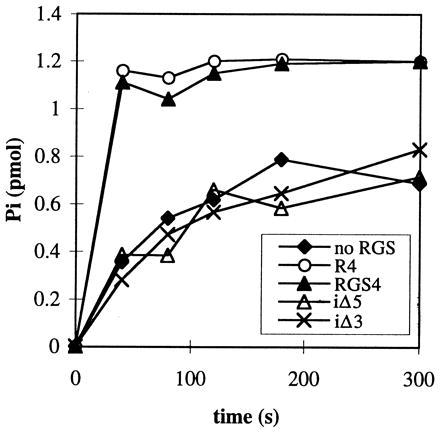
RGS domain of RGS4 retains GAP activity. Giα1 (375 nM) was preloaded with [γ-32P]GTP at 30°C (estimated concentration of GTP-Giα1 was 110 nM), then incubated at 0°C with or without 1 μM of the indicated recombinant RGS4 protein (see Fig. 1 for a description of RGS4, R4, iΔ5, and iΔ3).
Integrity of the RGS Domain Is Necessary for its GAP Function.
The most highly conserved amino acid residues within RGS domains are found at their amino and carboxyl ends (3) and provide important contacts in the RGS4–Giα1–GDP–AlF4− crystal structure (13). We tested whether RGS4 retained GAP activity in the absence of these conserved residues. In these constructs, either the amino or carboxyl terminus of the RGS domain was removed as an internal deletion with the remainder of the sequence intact (Fig. 1; residues 58–84 and 166–177, deletion iΔ5 and iΔ3, respectively). These internal deletions of RGS4 were expressed as soluble proteins of the expected size (assayed by SDS/PAGE) but did not stimulate GTP hydrolysis by Giα1 (Fig. 2). We assayed truncated RGS4 proteins that were missing portions of the RGS domain and flanking sequences (Fig. 1, deletions RΔ5, RΔ3, and RΔ5Δ3) but they were also inactive in the GAP assay (data not shown). Mixing the amino and carboxyl-terminal truncated RGS domain proteins (RΔ5 and RΔ3) also failed to stimulate GTP hydrolysis in vitro. Gsα catalyzed GTP hydrolysis was not stimulated by full-length RGS4 (5) nor any of the truncated RGS4 proteins described herein. These observations provide independent confirmation that the integrity of the RGS domain is essential for catalytic activity of RGS4.
RGS Domain of RGS4 Retains Full GAP Activity In Vitro.
Full-length RGS4 protein was shown to catalytically accelerate the rate of GTP hydrolysis by members of the Gi class at least 40-fold (5). To compare the catalytic activity of the RGS4 domain and full-length RGS4 protein, dilutions of each protein were incubated with 215 nM GTP–Giα1 (Fig. 3). Comparison of the kinetic constants derived from these time course curves (Fig. 3C) was used to estimate k1 to be 9 × 105 M−1⋅s−1 at 0°C (see Materials and Methods) for either the RGS domain or full-length RGS4 proteins. Based on this value, we calculated that acceleration of GTP hydrolysis was about 90-fold above the basal rate with either the RGS domain or full-length RGS4 (extrapolated to 1 μM), consistent with previous observations (11). Thus, GAP activity of the RGS domain is similar to that of full-length RGS4 protein in vitro.
RGS Domain and Full-Length RGS4 Have Similar Affinities for Giα1.
RGS4 and Giα1 substrate interactions were further characterized by using the surface plasmon resonance technique (Pharmacia Biosensor). Full-length RGS4 was previously shown to form a high affinity complex specifically with Giα1–GDP–AlF4− (11). The GDP–AlF4−–Mg2+ bound form of Giα1 is thought to mimic a transition state complex between Giα1 and GTP–Mg2+ in the hydrolysis reaction (14, 15). We found that both the RGS domain and full-length RGS4 proteins immobilized onto the biosensor chip surface bound Giα1–GDP–AlF4− with similar affinity, about 0.6 nM (Fig. 4). The RGS domain of RGS10, treated in the same manner, showed a similar Kd value but both slower association and dissociation rates compared with the RGS4 proteins (Fig. 4 and Table 1). RGS10 also bound Gzα–GDP–AlF4− on the biosensor chip (Kd = 0.6 nM). Protein–protein interactions were observed with these RGS proteins in the reciprocal experiment with Giα1 coupled to the sensor chip. In contrast, the truncated RGS4 proteins iΔ5, iΔ3, RΔ5, and RΔ3 that lack portions of the RGS domain did not bind Giα1–GDP–AlF4− (data not shown). The protein–protein interactions detected by surface plasmon resonance are consistent with interactions detected by the yeast two-hybrid assay (4), co-immunoprecipitation (6) column chromatography (11), and the RGS4–Giα1–GDP–AlF4− crystal structure (13). These physical interactions are also consistent with our observations that the complete RGS domain of RGS4 retains full GAP activity, whereas further truncations of conserved residues within the RGS domain are inactive (Fig. 2). No interactions were detected with RGS4 when Giα1 was prebound with GDP, GTP, or guanosine 5′-[γ-thio]triphosphate (GTPγS) (data not shown).
RGS Domains of RGS10 and GAIP Are GAPs for Gi Class α-Subunits.
Several full-length RGS proteins, including recombinant RGS4, GAIP, and RGS10, have been shown to accelerate GTP hydrolysis by Giα1 in vitro (5–7). To investigate whether the RGS domains from these RGS proteins also retain their GAP activity, we expressed and purified the complete RGS domain of RGS10 and GAIP, in addition to RGS4. As with RGS4, the GAP activity of the RGS domain of RGS10 was at least as high as that of the full-length RGS10 (data not shown). GAP activity of the RGS domain of GAIP, RGS10, and RGS4 was observed with Giα1 and Gzα (Fig. 5), but not for Gsα (data not shown). Specificity of these RGS domains for Gi class α-subunits is similar to the reported activity of full-length RGS proteins (5–7). The specific activity of the RGS domain of RGS10 is higher than other RGS domain proteins for both Giα1 and Gzα (Fig. 5). As calculated from the data of Fig. 5, the RGS domain of RGS10 (at 1 μM) would accelerate GTP hydrolysis by Gzα about 325-fold, 5–7 times faster than observed with other RGS proteins. In summary, we have demonstrated that the RGS domain of RGS4, RGS10, and GAIP is required and sufficient for GAP activity in vitro. Several lines of evidence indicates that the RGS domain of RGS4 is fully active, including similarities between the RGS domain and full-length protein in regards to affinities toward Giα1–GDP–AlF4−, catalytic activities and acceleration values. Consistent with our in vitro results are the genetic observations that deletion of the RGS domain results in a loss of function in sst2, flbA, and egl-10 (1–3), all genes that normally suppress G protein signaling in vivo.
Acknowledgments
We thank Al Gilman and David Berman for helpful suggestions, Stephen Anderson and the Green Center at the University of Texas Southwestern Medical Center for providing human RNA samples, Karen Chapman for preparing [γ-32P]GTP, and Elliott Ross, Meg Phillips, and Stephan Sprang for comments on the manuscript. This work was supported by grants to T.M.W. from the Leukemia Association of North Central Texas and the National Institutes of Health (DK47890).
ABBREVIATIONS
- RGS
regulators of G protein signaling
- GAP
GTPase activating protein
Note Added in Proof
A truncation of RET–RGS1 that includes the RGS domain also retains GAP activity in vitro (16).
References
- 1.Dohlman H G, Apaniesk D, Chen Y, Song J P, Nusskern D. Mol Cell Biol. 1995;15:3635–3643. doi: 10.1128/mcb.15.7.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee B N, Adams T H. Mol Microbiol. 1994;14:323–334. doi: 10.1111/j.1365-2958.1994.tb01293.x. [DOI] [PubMed] [Google Scholar]
- 3.Koelle M R, Horvitz H R. Cell. 1996;84:115–125. doi: 10.1016/s0092-8674(00)80998-8. [DOI] [PubMed] [Google Scholar]
- 4.De Vries L, Mousli M, Wurmser A, Farquhar M G. Proc Natl Acad Sci USA. 1995;92:11916–11920. doi: 10.1073/pnas.92.25.11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman D, Wilkie T M, Gilman A G. Cell. 1996;86:445–452. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 6.Hunt T W, Fields T A, Casey P J, Peralta E G. Nature (London) 1996;383:175–177. doi: 10.1038/383175a0. [DOI] [PubMed] [Google Scholar]
- 7.Watson N, Linder M E, Druey K M, Kehrl J H, Blumer K J. Nature (London) 1996;383:172–175. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- 8.Lee E, Linder M E, Gilman A G. Methods Enzymol. 1994;237:147–165. doi: 10.1016/s0076-6879(94)37059-1. [DOI] [PubMed] [Google Scholar]
- 9.Horton R M, Hunt H D, Ho S N, Pullen J K, Pease L R. Gene. 1989;77:61–66. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Puder M, Soberman R J. BioTechniques. 1996;20:954–955. doi: 10.2144/96206bm01. [DOI] [PubMed] [Google Scholar]
- 11.Berman D, Kozasa T, Gilman A G. J Biol Chem. 1996;271:27209–27212. doi: 10.1074/jbc.271.44.27209. [DOI] [PubMed] [Google Scholar]
- 12.O’Shannessy D J O, Brigham-Burke M, Soneson K K, Hensley P, Brooks I. Anal Biochem. 1993;212:457–468. doi: 10.1006/abio.1993.1355. [DOI] [PubMed] [Google Scholar]
- 13.Tesmer J J G, Berman D M, Gilman A G, Sprang S R. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 14.Sondek J, Lambright D G, Noel J P, Hamm H E, Sigler P B. Nature (London) 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- 15.Coleman D E, Berghuis A M, Lee E, Linder M E, Gilman A G, Sprang S R. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 16.Favrobert E, Hurley J B. Proc Natl Acad Sci USA. 1997;94:2945–2950. doi: 10.1073/pnas.94.7.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]