

Abstract
The expression of cell-specialization genes is likely to be changing in tumor cells as their differentiation declines. Functional changes in these genes might yield unusual peptide epitopes with anti-tumor potential and could occur without modification in the DNA sequence of the gene. Melanomas undergo a characteristic decline in melanization that may reflect altered contributions of key melanocytic genes such as tyrosinase. Quantitative reverse transcriptase–PCR of the wild-type (C) tyrosinase gene in transgenic (C57BL/6 strain) mouse melanomas has revealed a shift toward alternative splicing of the pre-mRNA that generated increased levels of the Δ1b and Δ1d mRNA splice variants. The spontaneous c2j albino mutation of tyrosinase (in the C57BL/6 strain) changes the pre-mRNA splicing pattern. In c2j/c2j melanomas, alternative splicing was again increased. However, while some mRNAs (notably Δ1b) present in C/C were obligatorily absent, others (Δ3 and Δ1d) were elevated. In c2j/c2j melanomas, the percentage of total tyrosinase transcripts attributable to Δ3 reached approximately 2-fold the incidence in c2j/c2j or C/C skin melanocytes. The percentage attributable to Δ1d rose to approximately 2-fold the incidence in c2j/c2j skin, and to 10-fold that in C/C skin. These differences provide a basis for unique mouse models in which the melanoma arises in skin grafted from a C/C or c2j/c2j transgenic donor to a transgenic host of the same or opposite tyrosinase genotype. Immunotherapy designs then could be based on augmenting those antigenic peptides that are novel or overrepresented in a tumor relative to the syngeneic host.
Keywords: alternative splicing, melanocytic genes, skin grafts, melanoma progression, transgenic mice
Peptides recognized by cytotoxic T lymphocytes from melanoma patients have been found in a number of cases to originate from melanocyte-specific genes required for pigmentation, such as tyrosinase (1–3), tyrosinase-related protein 1/gp75/brown (4), tyrosinase-related protein 2/slaty (5), Pmel17/gp100/silver (6–9), and MART-1/Melan-A (10, 11). In melanoma, these peptides appear to be acting as weak autoantigens. Their effectiveness as immunotherapeutic agents might be enhanced by experimentally augmenting their abundance, novelty, or presentation.
We have recently proposed a “reverse” route toward discovery of melanoma antigens that was prompted by the well known tendency of melanomas to become less pigmented in the course of malignant progression. It seemed possible that the pigmentary decline might be due to a change in RNA processing of some melanocytic genes. If splicing of any of the pre-mRNAs were, in fact, shifted from the constitutive mode toward particular alternative splicing modes, this would decrease the levels of proteins with essential roles in melanogenesis (relative to their incidence in normal melanocytes) and might increase the incidence of specific peptides with antigenic properties. Such functional modifications in RNA processing can occur without mutation in the gene under consideration—an important advantage in treatment designs directed against peptide products of that gene, because all the malignant cells then would be expected to have the same unchanged gene (12).
The possibility of a selective increase in specific alternative transcripts was first investigated for the wild-type (C) tyrosinase gene in murine melanomas as compared with skin melanocytes (12). Tyrosinase is the rate-limiting enzyme in melanin biosynthesis (13). The murine gene has five exons and a variety of splicing patterns (14, 15). Over 19 alternative mRNA splice variants were detected in skin melanocytes of the C57BL/6 inbred strain, and a reliable protocol was established for quantitation of the mRNAs (S.R.K., N.L.F., and B.M., unpublished data). Melanomas in progressive stages of malignancy were then obtained from Tyr-SV40E (C57BL/6 strain) transgenic mice (16–18), in which the disease closely resembles human melanoma (19). Analysis of the tyrosinase transcript profile in the mouse melanomas by quantitative reverse transcriptase (RT)–PCR clearly documented specific increases in alternatively spliced mRNAs, particularly the Δ1b and Δ1d sets, over their very low levels in normal skin melanocytes (12). These transcripts represent deletions within the first exon.
A special feature of the transgenic mouse melanomas is that they can be experimentally elicited in several ways. The transgene alone acts only as an “initiating” stimulus conferring melanoma susceptibility; some additional (“promoting”) stimulus is needed for malignant skin melanomas to result. Conditions associated with wound healing were among those found to supply promoting stimuli. The melanomagenic effect was manifest after a small disc of skin was grafted from one transgenic animal to another (17). When the skin-graft donor is from a transgenic line with high melanoma susceptibility (due to high expression of the transgene) and the host is from a relatively low-susceptibility line (with low expression of the same transgene), the tumors invariably arise in the grafted skin and metastasize into organs of the hosts. This grafting arrangement allows many novel and informative graft-host combinations to be carried out. For example, an allele can first be substituted for a gene of interest in either the donor or the host transgenic line by breeding animals of that line with mice of the relevant syngeneic histocompatible derivative of C57BL/6.
The c2j albino mutation at the mouse tyrosinase locus provides an interesting opportunity of this sort, because the mutation changes the pattern of pre-mRNA splicing in comparison with that of the wild-type tyrosinase gene (20). The c2j mutation arose spontaneously in the C57BL/6 strain. Molecular analysis of the gene in c2j/c2j albino skin disclosed a single change: a base substitution at an alternative splice donor site within exon 1. This abolishes the usage of that site for alternative splicing. Among several consequences shown in Fig. 1 is complete absence of Δ1b mRNAs, which were previously found to be present at very low abundance in skin of the wild-type (C/C) tyrosinase genotype and at relatively higher levels in C/C melanomas (12).
Figure 1.
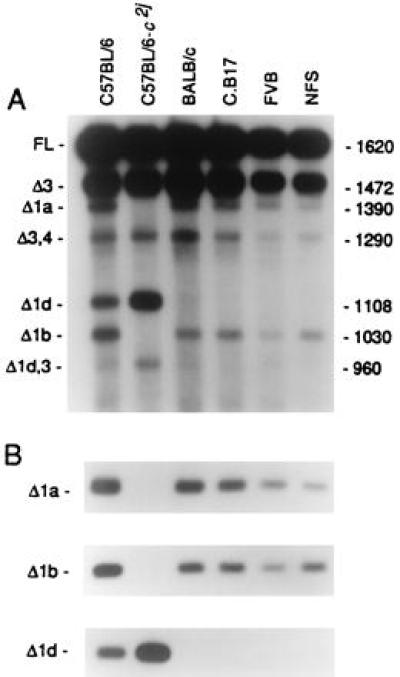
Tyrosinase transcripts in skin of young mice of the wild-type (C/C) C57BL/6 strain, the c2j/c2j albino C57BL/6 strain, and four other inbred strains with the c/c albino genotype. After RT-PCR amplification, transcript levels were analyzed in Southern blots, either (A) with an oligonucleotide probe, prTyr, matching the 3′ end of the cDNAs, which revealed all the transcripts; or (B) with specific oligonucleotide probes for further analysis of each group of alternatively spliced transcripts. Three examples are shown. In A, most of the abundant transcripts (>1%) are identified at the left.
The objectives of the present study were to characterize the tyrosinase transcripts in c2j/c2j albino melanomas relative to c2j/c2j albino skin melanocytes; to compare the transcripts with those of C/C melanomas and skin melanocytes (12); and to describe the graft-host combinations of C/C and c2j/c2j genotypes in which qualitative or quantitative differences in specific alternative mRNAs may be favorable for model immunotherapy experiments.
MATERIALS AND METHODS
Skin Samples.
Dorsal body skin was obtained from inbred-strain mice (Icr sublines) at 2–6 days of age. Mice of the standard C57BL/6 strain are wild type at the tyrosinase locus while BALB/c, FVB, and NFS strains have the classical (c) albino mutation. The albino C.B17 strain was derived in this institution from BALB/c. Mice of the c2j/c2j albino C57BL/6 strain were imported from The Jackson Laboratory, and a breeding colony was established here.
c2j/c2j Melanomas.
Cutaneous melanomas of the c2j/c2j albino genotype were experimentally produced by grafting discs of adult full-thickness body skin (17) taken from second-backcross albino transgenic progeny of c2j/c2j mice and transgenic (C/C) mice. Transgenic graft donors were high-susceptibility line 8 hemizygotes or, in one case, a hemizygote of the moderately susceptible line 9. Hosts were low-susceptibility line 12 hemizygotes. Melanomas arose after an average latency of 35 weeks (range: 24–43 weeks) in the line 8 grafts, and after 73 weeks in the less susceptible line 9 graft. The cutaneous tumors averaged 797 mm3 in size when they were collected. All had ulcerated and were invasive. Metastases were obtained in some cases. Pieces of the tumors were frozen in dry ice and stored at −70°C. Fourteen melanoma samples were analyzed; 11 were from separate primary tumors, and three were from metastases (one each from a regional lymph node, a lung, and a liver).
Amplification and Quantitation of Tyrosinase Transcripts.
RNA was isolated from the transgenic tumors, or from dorsal body skin of nontransgenic mice, by the acid guanidinium thiocyanate-phenol-chloroform procedure (21). Tyrosinase cDNAs were generated from 2.5 μg of total RNA, and the products were amplified by RT-PCR as described (12). A hot-start procedure (22) was used to prevent mis-priming. The same internal standard RNA as in the analysis of C/C melanomas (12) was included in the RT reactions to serve as a reference in determining the levels of tyrosinase mRNA isoforms. The standard (corresponding to the full-length tyrosinase transcript with a 98-bp deletion in exon 1) was included at 100, 300, or 600 fg/μg cellular RNA so as to be present at approximately the level of the Δ3 transcript in the Southern blot. Primers for RT and PCR, and probes for hybridizations, have been described (12). Hybridizations were performed according to Church and Gilbert (23). For each tumor sample, it was necessary to determine the number of PCR cycles that would provide sufficient amplification of all transcripts within the limits of the exponential phase. The number of cycles ranged from 21 to 25. Intensities of hybridizing bands were measured with a BAS1000 phosphorimager (Fuji).
RESULTS
Differences in Levels of the Full-Length Tyrosinase Transcript in c2j/c2j Melanomas vs. Skin Melanocytes.
The full-length (constitutively spliced) tyrosinase transcript was found at similar levels in nontransgenic c2j/c2j and C/C skin melanocytes (Fig. 1). The tyrosinase protein was virtually absent in c2j/c2j skin, possibly due to proteolytic degradation (20).
In c2j/c2j primary melanomas and their metastases, absolute levels of the full-length transcript varied but were higher in most cases than in c2j/c2j skin (Figs. 2 and 3 Left). In the c2j/c2j tumors there was an increase in specific alternatively spliced mRNAs (Tables 1 and 2). The combined chief alternative mRNAs were now as much as 29.5% of the total tyrosinase mRNAs in the tumors, as compared with 18.4% in skin (Table 1). A shift from constitutive toward alternative splicing was also noted previously in the C/C primary melanomas and metastases relative to C/C skin (12). Tyrosinase transcripts were not detected in a lung metastasis of c2j/c2j tumor 554P, although present in the skin tumor of origin (Table 2).
Figure 2.
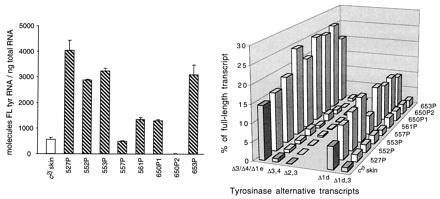
Tyrosinase mRNA expression in cutaneous c2j/c2j melanomas and c2j/c2j mouse skin. Each primary (P) melanoma arose in a separate graft of c2j/c2j transgenic skin, except for the 650P1 and 650P2 tumors, which originated independently in the same skin graft. Transcripts were quantitated after RT-PCR, electrophoresis, and hybridization with the prTyr probe. (Left) Absolute levels of the full-length tyrosinase mRNA relative to total RNA. Results are means and SD for at least two experiments. (Right) Levels of specific alternative mRNAs relative to the full-length mRNA in the same tumors. The most abundant transcripts are represented by the bars, with the Δ3, Δ4, and Δ1e transcripts combined here. Δ3 is the single most abundant transcript, whereas Δ3,4 and Δ2,3 are minor codeletions.
Figure 3.

Tyrosinase mRNA expression in three c2j/c2j cutaneous melanomas and a metastasis from each, and in c2j/c2j skin. The numbered primary (P) melanomas and their metastases (M) in lung, lymph node (LN), and liver were analyzed as in Fig. 2. (Left) Absolute levels of the full-length tyrosinase mRNA relative to total RNA. Results are means and SD for at least two experiments. (Right) Levels of specific alternative mRNAs relative to the full-length mRNA in the same tumors. The most abundant transcripts are represented by the bars, with the Δ3, Δ4, and Δ1e transcripts combined as in Fig. 2.
Table 1.
Alternative transcripts as percentages of total tyrosinase transcripts in c2j/c2j primary (P) melanomas
| Sample* | Principal alternative mRNAs† | Δ3/Δ4/Δ1e‡ | Δ1d | Δ1d,3 |
|---|---|---|---|---|
| c2j/c2j skin | 18.4 | 11.8 | 5.0 | 0.9 |
| 527P | 21.9 | 12.4 | 7.3 | 1.5 |
| 552P | 24.4 | 13.8 | 8.1 | 1.7 |
| 553P | 29.5 | 17.3 | 8.9 | 1.7 |
| 557P | 21.8 | 15.7 | 4.2 | 1.3 |
| 561P | 29.4 | 17.6 | 8.2 | 2.1 |
| 650P1 | 28.0 | 17.4 | 7.4 | 2.1 |
| 650P2 | 28.3 | 18.4 | 7.8 | 2.1 |
| 653P | 24.6 | 15.2 | 7.0 | 1.6 |
The 650P1 and 650P2 cases were independent tumors in the same skin graft. All other cases were single tumors in separate skin grafts.
In this group are most of the relatively abundant alternative transcripts detected by the prTyr probe, including the comigrating Δ3/Δ4/Δ1e transcripts, Δ1d, and Δ1d,3, and excluding transcripts of low abundance (<0.1 molecule per ng of RNA).
Δ4 and Δ1e transcripts are only minor components of this comigrating group.
Table 2.
Alternative transcripts as percentages of total tyrosinase transcripts in c2j/c2j primary (P) melanomas and metastases (M)
| Sample | Principal alternative mRNAs* | Δ3/Δ4/Δ1e† | Δ1d | Δ1d,3 |
|---|---|---|---|---|
| c2j/c2j skin | 18.4 | 11.8 | 5.0 | 0.9 |
| 554P | 22.4 | 13.2 | 6.5 | 1.9 |
| M-Lung | — | — | — | — |
| 556P | 27.6 | 20.5 | 3.6 | 1.5 |
| M-LN | 23.9 | 18.6 | 2.7 | 0.8 |
| 559P | 19.5 | 9.7 | 8.0 | 1.4 |
| M-Liver | 30.1 | 22.7 | 4.4 | 1.5 |
LN, lymph node. —, Lack of detectable transcripts.
In this group are most of the relatively abundant alternative transcripts detected by the prTyr probe, including the comigrating Δ3/Δ4/Δ1e transcripts, Δ1d, and Δ1d,3, and excluding transcripts of low abundance (<0.1 molecule per ng of RNA).
Δ4 and Δ1e transcripts are only minor components of this comigrating group.
Specific Increases in the Δ3 and Δ1d Alternative Splice Variants of Tyrosinase in c2j/c2j Primary Melanomas.
The up-regulation of alternatively spliced transcripts in the c2j/c2j tumors was due mainly to specific increases in Δ3, Δ1d, and Δ1d,3 alternative transcripts over their levels in skin; the sole exception was a slight decrease in Δ1d of tumor 557P (Table 1). [Δ3 represents deletion of exon 3; Δ1d is a deletion within exon 1 (15); and Δ1d,3 is a relatively minor codeletion of both Δ1d and Δ3. Δ1d and Δ1d,3 transcripts would have the same ORF and would therefore generate the same target peptide.] The Δ3 mRNA was the most abundant splice variant in skin. It was further overexpressed in all the c2j/c2j tumors. Δ3 largely accounted for the comigrating Δ3/Δ4/Δ1e transcript group, which rose as high as 20.5% of total tyrosinase transcripts in the skin melanomas, as compared with 11.8% in skin melanocytes; Δ1d increased to a maximum of 8.9%, as compared with 5.0% in skin; and Δ1d,3 reached 2.1%, as compared with 0.9% in skin (Tables 1 and 2). In the cityscapes (Figs. 2 and 3 Right), alternative transcript levels are depicted relative to those of the full-length mRNAs in the same tumors; for these determinations, an equal amount of the full-length mRNA was loaded in each lane of the Southern blots.
The c2j albino mutation, occurring at an alternative 5′ splice donor site within exon 1 of the tyrosinase gene, precludes the usage of that site for pre-mRNA splicing. This eliminates the possibility of producing any of the Δ1a or Δ1b groups of transcripts, both of which are obtained in skin with the wild-type tyrosinase gene. At the same time, the usage of the next downstream alternative splice donor site within exon 1 is enhanced approximately 7-fold in c2j over that in wild-type skin, resulting in increased levels of Δ1c and Δ1d mRNAs in c2j/c2j skin. However, the absolute levels of Δ1c transcripts are low in any case (20). The results for skin melanocytes that are relevant for the melanoma analyses are included in Fig. 1. In the c2j/c2j melanomas, Δ1a and Δ1b transcripts are obligatorily absent and the Δ1d transcript set is notably increased (Tables 1 and 2).
Relative Decline in Δ3 and Δ1d Alternative Splice Variants of Tyrosinase in c2j/c2j Melanoma Metastases.
Comparison of three metastases with their skin melanomas of origin reveals a relative decrease in Δ1d transcripts in two metastases and complete loss of this and other tyrosinase transcripts in the lung metastasis of tumor 554 (Fig. 3 Right and Table 2). The transcript group due mainly to Δ3 was also decreased in two of the metastases but was increased, surprisingly, in the liver metastasis of tumor 559 (Table 2).
DISCUSSION
The tyrosinase transcripts found here in malignant mouse melanomas of the c2j/c2j albino genotype were all present in c2j/c2j skin melanocytes. A similar conclusion was reached for transcripts of the wild-type tyrosinase gene in C/C melanomas as compared with C/C melanocytes (12). In both melanoma genotypes, there was a shift toward alternative splicing of the pre-mRNA, thereby increasing particular alternative splice variants in the tumors relative to skin melanocytes. Thus, in melanomas, the tyrosinase locus yields characteristic quantitative changes in mRNA isoforms that are allele-specific and are not due to de novo mutation in either of the investigated alleles. The high degree of consistency of these changes in the melanomas lends preliminary support to the general hypothesis that modified mRNA expression of lineage-specific normal genes may be common in many kinds of tumors; this, in turn, may enable candidate peptide targets for immunotherapy to be predicted (12). Moreover, reliance upon overexpressed products of intact genes circumvents the problem that would be posed if a treatment strategy depended on genes subject in tumors to ongoing mutation.
Some melanomas farthest advanced in malignant progression may fail to express tyrosinase transcripts. This is evident from analyses of c2j/c2j tumors (Fig. 3 and Table 2) as well as C/C tumors (12). It therefore will be necessary to identify other melanocyte-lineage genes with overexpressed mRNA splice variants in these late stages to devise a vaccine cocktail of peptide antigens encompassing tumor cells in all stages. Judging from the RT-PCR data for C/C melanomas, it appears that tyrosinase-based antigens may, in fact, be maximally effective only in the early stages of malignancy (12). Northern and immunoblot analyses of C/C melanomas from Tyr-SV40E transgenic mice have led to the same conclusion for tyrosinase mRNA and protein, respectively (24). Those analyses also have identified at least one melanocytic gene, tyrosinase-related protein 2, which was expressed in all 10 tumor samples examined.
The main mRNA splice variants consistently overrepresented in the c2j/c2j melanomas were attributable to Δ3 and the Δ1d set (Figs. 2 and 3 Right; Tables 1 and 2). The alternative mRNAs generally overexpressed in C/C melanomas were the Δ1b and Δ1d sets, whereas the Δ3 variant was overexpressed in two-thirds of the tumors (12). While Δ1d mRNAs were elevated in both genotypic groups of melanomas, the comparative levels were unexpected. The Δ1d level in c2j/c2j primary melanomas increased an average of 1.4-fold over the low basal level in c2j/c2j skin. This was not as high as the 2.4-fold average increase of Δ1d in C/C primary melanomas over the level in C/C skin. One might have expected a greater increase in c2j/c2j melanomas in view of the fact that the Δ1d level was 7-fold higher in c2j/c2j skin than in C/C skin—the result of increased activation of a splice donor site in exon 1 located downstream of the site inactivated by the c2j mutation (20).
The c2j albino mutation in the C57BL/6 strain makes it possible to carry out model immunotherapy experiments based on quantitative as well as qualitative tyrosinase mRNA differences between a skin graft—hence, a melanoma actually developing in the graft—and its host. Other albino mutations, e.g., c (Fig. 1), are known that alter pre-mRNA splicing due to a nucleotide change in the tyrosinase coding sequence, but the well characterized mutations are not in strains histocompatible with C57BL/6. The need for graft–host histocompatibility might be circumvented by using a melanoma transplant line or cell line and inoculating it into an immunoincompetent host (e.g., nude or scid); however, this would bypass the role of the immune system. In addition, results would pertain to a long-established tumor whose evolution was quite different from that of a primary tumor in its natural place. Radiation-induced albino mutations in mice also might be considered for experiments intended to exploit differences between melanomas and their hosts. But they are present in strains other than C57BL/6, and also would be problematical because of chromosomal deletions extending upstream and/or downstream of the tyrosinase gene (25). Thus, a region at ≈15 kb upstream of the transcription initiation site that controls tyrosinase expression (26) could be compromised.
In Table 3 are shown experimental arrangements in which a host of either the C/C or c2j/c2j tyrosinase genotype would have an opportunity to interact with a developing or progressing melanoma of the same or opposite syngeneic genotype. In all the genotypic combinations, specific tyrosinase alternative transcripts would be overrepresented in the melanoma cells relative to the host’s melanocytes. In the last of the listed combinations, there also would be transcripts in the melanoma cells that are absent in the host; these are designated as “foreign.” The hosts then might be actively immunized against unusual peptides predicted from proteolytically processed products of the overexpressed or “foreign” splice variants that may bind well to class I major histocompatibility proteins of the C57BL/6 strain (27).
Table 3.
Experimental mouse models with tyrosinase alternative transcripts in melanoma cells that are overrepresented or “foreign” relative to transcripts in melanocytes of the host
| Tyrosinase genotype
|
Alternative transcripts in melanoma cells
|
||
|---|---|---|---|
| Melanocytes in host | Melanoma cells in skin graft | Overrepresented* | “Foreign” |
| C/C | C/C | Δ1b, Δ1b,3, Δ1d, Δ1d,3, (Δ3) | |
| C/C | c2j/c2j | Δ1d, Δ1d,3, Δ3 | |
| c2j/c2j | c2j/c2j | Δ1d, Δ1d,3, Δ3 | |
| c2j/c2j | C/C | Δ1d, Δ1d,3, (Δ3) | Δ1a, Δ1a,3, Δ1b, Δ1b,3 |
If the hosts bearing these skin grafts were to develop a strong spontaneous autoimmune response against antigenic peptides on melanocytes within the grafts, it is evident that all melanocytes in the grafted skin would be destroyed and no melanomas would develop. We have by now carried out three of the four graft-host combinations in Table 3 (excluding grafts from c2j/c2j donors to c2j/c2j hosts) and have in fact obtained melanomas in a large percentage of the grafts (W.K.S. and B.M., unpublished data). This need not signify that the host’s immune system fails to recognize melanocytic antigens that may be present, nor that the animal is tolerant of them. The occurrence of partial or incomplete tolerance has long been recognized. For example, immunological tolerance of histocompatibility antigens in murine skin allografts was clearly shown not to be an all-or-none phenomenon. All degrees of tolerance were found, ranging from a total and persistent inability to reject allografts to a minor delay in graft destruction (28).
Our observations of pigmented (C/C) transgenic skin grafts (in C/C or c2j/c2j transgenic mouse hosts) suggest that a weak and delayed autoimmune response may be taking place. This appears to be indicated partly by the occasional regression of a tumor and partly by the gradual blanching of hairs in some grafts (W.K.S. and B.M., unpublished data). The latter effect is reminiscent of the patchy coats in mice immunized with a tyrosinase-related protein 1/gp75 antigen (29). Moreover, pale skin patches in human vitiligo patients with melanoma have been thought to be related to the presence of anti-tyrosinase antibodies (30).
Many practical issues remain to be resolved before an effective peptide-antigen tumor vaccine is obtained. That is the case whether the peptides are identified through T cell recognition (31) or by prediction from overexpressed mRNA isoforms in tumors. The recent identification of a human melanoma rejection antigen recognized by T cells as the product of an alternative transcript of the tyrosinase-related protein 1/gp75 gene (32) implies that products of alternative transcripts may indeed play a role in tumor–host relations. The new murine models described here to evaluate tyrosinase-based antigens in melanoma provide a natural biological context in which to examine effects of vaccines on tumor development, progression, or metastasis. Comparable syngeneic mouse models with allelic differences between skin graft and host can be constructed to evaluate candidate melanoma antigens based on other loci, or to investigate antigens in other kinds of skin malignancies.
Acknowledgments
W.K.S. is a Visiting Scientist from the University of Pennsylvania. S.R.K. is the recipient of a fellowship from the Fox Chase Cancer Center Board of Associates. This work was supported by an American Cancer Society Special Research Grant for Metastatic Melanoma (NP-917) to B.M. and by U.S. Public Health Services Grants CA-06927 and RR-05539 to the Fox Chase Cancer Center.
ABBREVIATION
- RT
reverse transcriptase
References
- 1.Brichard V, Van Pel A, Wölfel T, Wölfel C, De Plaen E, Lethé B, Coulie P, Boon T. J Exp Med. 1993;178:489–495. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wölfel T, Van Pel A, Brichard V, Schneider J, Seliger B, Meyer zum Büschenfelde K-H, Boon T. Eur J Immunol. 1994;24:759–764. doi: 10.1002/eji.1830240340. [DOI] [PubMed] [Google Scholar]
- 3.Brichard V G, Herman J, Van Pel A, Wildmann C, Gaugler B, Wölfel T, Boon T, Lethé B. Eur J Immunol. 1996;26:224–230. doi: 10.1002/eji.1830260135. [DOI] [PubMed] [Google Scholar]
- 4.Wang R-F, Robbins P F, Kawakami Y, Kang X-Q, Rosenberg S A. J Exp Med. 1995;181:799–804. doi: 10.1084/jem.181.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang R-F, Appella E, Kawakami Y, Kang X, Rosenberg S A. J Exp Med. 1996;184:2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox A L, Skipper J, Chen Y, Henderson R A, Darrow T L, Shabanowitz J, Engelhard V H, Hunt D F, Slingluff C L., Jr Science. 1994;264:716–719. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- 7.Bakker A B H, Schreurs M W J, de Boer A J, Kawakami Y, Rosenberg S A, Adema G J, Figdor C G. J Exp Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakker A B H, Schreurs M W J, Tafazzul G, de Boer A J, Kawakami Y, Adema G J, Figdor C G. Int J Cancer. 1995;62:97–102. doi: 10.1002/ijc.2910620118. [DOI] [PubMed] [Google Scholar]
- 9.Zarour H, De Smet C, Lehmann F, Marchand M, Lethé B, Romero P, Boon T, Renauld J-C. J Invest Dermatol. 1996;107:63–67. doi: 10.1111/1523-1747.ep12298177. [DOI] [PubMed] [Google Scholar]
- 10.Kawakami Y, Eliyahu S, Delgado C H, Robbins P F, Rivoltini L, Topalian S L, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castelli C, Storkus W J, Maeurer M J, Martin D M, Huang E C, Pramanik B N, Nagabhushan T L, Parmiani G, Lotze M T. J Exp Med. 1995;181:363–368. doi: 10.1084/jem.181.1.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Fur N, Kelsall S R, Silvers W K, Mintz B. Proc Natl Acad Sci USA. 1997;94:5332–5337. doi: 10.1073/pnas.94.10.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hearing V J, Jimenez M. Int J Biochem. 1987;19:1141–1147. doi: 10.1016/0020-711x(87)90095-4. [DOI] [PubMed] [Google Scholar]
- 14.Ruppert S, Müller G, Kwon B, Schütz G. EMBO J. 1988;7:2715–2722. doi: 10.1002/j.1460-2075.1988.tb03125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter S, Mintz B. Gene. 1991;97:277–282. doi: 10.1016/0378-1119(91)90063-h. [DOI] [PubMed] [Google Scholar]
- 16.Bradl M, Klein-Szanto A, Porter S, Mintz B. Proc Natl Acad Sci USA. 1991;88:164–168. doi: 10.1073/pnas.88.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mintz B, Silvers W K. Proc Natl Acad Sci USA. 1993;90:8817–8821. doi: 10.1073/pnas.90.19.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mintz B, Silvers W K, Klein-Szanto A J P. Proc Natl Acad Sci USA. 1993;90:8822–8826. doi: 10.1073/pnas.90.19.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark W H, Jr, Elder D E, Guerry D, IV, Epstein M N, Greene M H, Van Horn M. Hum Pathol. 1984;15:1147–1165. doi: 10.1016/s0046-8177(84)80310-x. [DOI] [PubMed] [Google Scholar]
- 20.Le Fur N, Kelsall S R, Mintz B. Genomics. 1996;37:245–248. doi: 10.1006/geno.1996.0551. [DOI] [PubMed] [Google Scholar]
- 21.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 22.Chou Q, Russell M, Birch D E, Raymond J, Bloch W. Nucleic Acids Res. 1992;20:1717–1723. doi: 10.1093/nar/20.7.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Church G M, Gilbert W. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orlow S J, Hearing V J, Sakai C, Urabe K, Zhou B-K, Silvers W K, Mintz B. Proc Natl Acad Sci USA. 1995;92:10152–10156. doi: 10.1073/pnas.92.22.10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharan S K, Holdener-Kenny B, Ruppert S, Schedl A, Kelsey G, Rinchik E M, Magnuson T. Genetics. 1991;129:825–832. doi: 10.1093/genetics/129.3.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter S, Larue L, Mintz B. Dev Genet. 1991;12:393–402. doi: 10.1002/dvg.1020120604. [DOI] [PubMed] [Google Scholar]
- 27.Falk K, Rötzschke O, Stevanovic S, Jung G, Rammensee H G. Nature (London) 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 28.Billingham, R. E. & Silvers, W. K. (1962) J. Cell. Comp. Physiol. 60, Suppl. 1, 183–200.
- 29.Naftzger C, Takechi Y, Kohda H, Hara I, Vijayasaradhi S, Houghton A N. Proc Natl Acad Sci USA. 1996;93:14809–14814. doi: 10.1073/pnas.93.25.14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song Y-H, Connor E, Li Y, Zorovich B, Balducci P, Maclaren N. Lancet. 1994;344:1049–1052. doi: 10.1016/s0140-6736(94)91709-4. [DOI] [PubMed] [Google Scholar]
- 31.Robbins P F, Kawakami Y. Curr Opin Immunol. 1996;8:628–636. doi: 10.1016/s0952-7915(96)80078-1. [DOI] [PubMed] [Google Scholar]
- 32.Wang R-F, Parkhurst M R, Kawakami Y, Robbins P F, Rosenberg S A. J Exp Med. 1996;183:1131–1140. doi: 10.1084/jem.183.3.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]