

SUMMARY
The Gag protein is the major structural determinant of retrovirus assembly. Although a number of cellular factors have been reported to facilitate retrovirus release, little is known about the cellular machinery that directs Gag to the site of virus assembly. Here we report roles for the Golgi-localized γ-ear containing Arf-binding (GGA) and ADP ribosylation factor (Arf) proteins in retrovirus particle assembly and release. Whereas siRNA-mediated depletion of GGA2 and GGA3 led to a significant increase in particle release in a late-domain-dependent manner, GGA overexpression severely reduced retrovirus particle production by impairing Gag trafficking to the membrane. GGA overexpression inhibited retroviral assembly and release by disrupting Arf protein activity. Furthermore, disruption of endogenous Arf activity inhibited particle production by decreasing Gag–membrane binding. These findings identify the GGA proteins as modulators of HIV-1 release, and the Arf proteins as critical cellular cofactors in retroviral Gag trafficking to the plasma membrane.
INTRODUCTION
The Gag polyprotein precursor is the major structural component of retroviral assembly and when expressed alone is sufficient to drive the production of virus-like particles (VLPs). Discrete domains have been identified within Gag that are responsible for particle assembly and release. The matrix (MA) domain directs Gag to the membrane; capsid (CA) and nucleocapsid (NC) domains mediate Gag-Gag interactions during assembly; and late domains promote the release of assembled virus particles from the cell surface [for reviews, see (Freed, 1998; Swanstrom and Wills, 1997; Vogt, 1997)].
It is becoming increasingly clear that retroviruses take advantage of cellular machinery to promote their assembly and release. For example, retrovirus budding requires productive interactions between viral late domains in Gag and the host endosomal sorting machinery. Three retroviral late domains have been characterized thus far: P(T/S)AP, PPPY, and LYPxnLxxL (where x is any amino acid). These late domains all function by interacting with specific host factors in the endosomal sorting pathway (Bieniasz, 2006; Demirov and Freed, 2004; Morita and Sundquist, 2004).
Although components of the endosomal sorting machinery have been clearly demonstrated to promote retroviral budding, the role of host factors in Gag trafficking to the plasma membrane (PM) remains poorly understood. It has been known for many years that the MA domain of Gag contains the viral determinants responsible for PM targeting (Joshi and Freed, 2007), and we reported that the phospholipid phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2] is a key cellular cofactor in directing Gag to the PM (Ono et al., 2004), apparently via a direct Gag–PI(4,5)P2 interaction (Saad et al., 2006; Shkriabai et al., 2006). Several host proteins, including the clathrin adapter protein 3 (AP-3) (Dong et al., 2005), have also been implicated in the trafficking of Gag to the PM. The mechanism by which these proteins regulate the subcellular localization of Gag remains to be defined.
The GGA (for Golgi-localized γ-ear containing Arf-binding) proteins constitute a recently identified family of monomeric clathrin-binding factors that function in the sorting of mannose phosphate receptors (MPRs) between the trans-Golgi network (TGN) and endosomes. In addition to binding clathrin and MPRs, GGAs associate with a variety of factors implicated in protein sorting; these include Tsg101, ubiquitin, and ADP ribosylation factors (Arfs). GGAs are composed of several distinct regions: the N-terminal Vps27, Hrs and STAM homology (VHS) domain interacts with the acidic cluster di-leucine sorting signals present in the cytoplasmic tail of MPRs. The GGA and TOM (GAT) domain is responsible for recruitment of GGAs to membranes by virtue of interaction with Arf-GTP. The hinge region recruits clathrin, and the C-terminal γ-adaptin ear homology (GAE) domain binds proteins with a DFGXØ motif (e.g., rabaptin 5, epsinR, and γ-synergin) (Bonifacino, 2004).
As mentioned above, GGA proteins are recruited to membrane via their direct interaction with GTP-bound Arfs. In mammalian cells there are six members of the Arf family (Arf1–6), which, based on sequence similarity, are organized into three classes. Class I Arf proteins (Arf1–3) control the assembly of coat complexes in the secretory pathway and regulate levels of PI(4,5)P2 in the Golgi. Class II Arfs (Arf4 and Arf5) reportedly regulate protein and membrane transport in the Golgi. Arf6, the sole class III Arf, functions primarily in sorting and signaling at the PM, in part through its ability to regulate levels of PI(4,5)P2 at the cell surface. Like other GTPases, Arfs cycle between a cytosolic GDP-bound inactive state and a membrane-associated, GTP-bound active state. At the membrane, GTP-Arfs encounter effectors such as the GGAs, coatomer (COPI), or adaptor protein complexes (AP-1, AP-3, AP-4), thereby facilitating various cellular processes (D’Souza-Schorey and Chavrier, 2006; Donaldson, 2005; Takatsu et al., 2002).
In the current study we investigated a possible role for the GGA and Arf proteins in retroviral particle assembly and release. Depletion of endogenous GGAs led to a significant increase in retrovirus release in a PT/SAP-dependent manner. In contrast, GGA overexpression severely inhibited retrovirus particle production by disrupting the association of Gag with membrane. Attempts to decipher the precise mechanism by which GGA overexpression inhibited virus particle production revealed a central role for the Arf proteins in retrovirus assembly and release. These findings identify the GGA and Arf proteins as key modulators of retroviral Gag trafficking.
RESULTS
siRNA-mediated depletion of GGA2 and GGA3 increases HIV-1 release in a PTAP-dependent manner
We first determined the effect of siRNA-mediated depletion of the GGA proteins on HIV-1 particle production. Tsg101 depletion, which is known to inhibit virus budding (Garrus et al., 2001) was included as a positive control. Efficient knockdown of GGA and Tsg101 expression was achieved (data not shown) and virus release efficiency was determined by transfection of depleted cells with the full-length HIV-1 molecular clone pNL4-3. Surprisingly, virus production from cells depleted of GGA2 and GGA3 was significantly increased (Figure 1A). In contrast, Tsg101 siRNA led to a ~4-fold inhibition of HIV-1 release (Figure 1A).
Figure 1.

GGA depletion leads to an increase in retrovirus release in a late-domain-dependent manner. (A) HeLa cells were transfected with control, GGA or Tsg101 siRNAs. 24 h posttransfection, cells were cotransfected with pNL4-3 and the indicated siRNAs and were metabolically labeled with [35S]Met/Cys. Cells and virus lysates were immunoprecipitated with HIV-Ig and resolved by SDS-PAGE. Data were quantified using a phosphorimager and virus release efficiency calculated as the ratio of virion-associated p24 to total (cell + virion) Gag. Values represent mean ± SD; n=3. The positions of the HIV-1 Env precursor gp160, mature surface glycoprotein gp120, the Gag precursor Pr55Gag (Pr55), Nef, and the CA protein (p24) are shown. (B) HeLa cells were transfected with EIAV proviral DNA bearing a wild-type (YPDL) or PTAP late domain along with the indicated siRNAs. Cell and virus lysates were immunoprecipitated with EIAV serum and subjected to SDS-PAGE. Data represent mean ± SD; n=3. The position of the EIAV Gag precursor (Pr55) is shown. (C) GGAs interact with Tsg101 in coimmunoprecipitation analysis. Lysates derived from cells transfected with HA-tagged, full-length Tsg101 expression vector (TSG-F) along with control plasmid or Myc-tagged GGA expression plasmids were immunoprecipitated with rabbit anti-Myc Ab (IP=anti-Myc) followed by immunoblotting with mouse anti-HA Ab (IB=anti-HA). Molecular mass standards are shown on the left (in kDa). Lower panels of 1C represent GGA or Tsg101 protein expression in the cell lysates (input). The blot shown in the top panel of 1C is a longer exposure than the blots at the bottom of 1C. In lane 1, lysates were obtained from cells transfected with the Tsg-F expression vector alone. (D) Small increases in Tsg101 expression stimulate HIV-1 release. HeLa cells were transfected with a constant amount of pNL4-3 DNA (2 g) along with decreasing amounts of full-length Tsg101 (TSG-F) expression vector. Virus release was determined by phosphorimager analysis as described in the Figure 1A legend. Exogenous and endogenous Tsg101 levels are shown following immunoblotting of cell lysates with anti-Tsg101 Ab. One representative of three independent experiments is presented.
To determine whether the increased virus release observed upon GGA depletion is specific to HIV-1, we examined the production of EIAV particles from cells transfected with GGA siRNAs. We observed that GGA siRNAs did not affect the release of WT EIAV (Figure 1B). Interestingly, however, when the YPDL late domain of EIAV was replaced with that of HIV-1 (EIAV-PTAP) (Li et al., 2002; Shehu-Xhilaga et al., 2004), we again observed an increase in virus production upon GGA depletion (Figure 1B). As expected, substitution of YPDL for the PTAP late domain also rendered EIAV release sensitive to Tsg101 depletion. These data demonstrate that the effect of GGA depletion on retrovirus release is late domain (PTAP)-dependent. We also observed that depletion of GGAs in macrophages led to a significant (p < 0.05) increase in HIV-1 release (Figure S1A). These findings demonstrate a role for the GGA proteins as negative modulators of HIV-1 release both in HeLa cells and in a physiologically relevant cell type.
To explain the PTAP dependence of GGA depletion on virus release, we postulated that in physiological situations GGAs might sequester endogenous Tsg101. Hence, GGA depletion could make additional Tsg101 available to function in promoting HIV-1 release. To verify an interaction between the GGAs and Tsg101, we performed co-immunoprecipitation analyses. These results demonstrated the ability of GGA1, GGA2, and GGA3 to interact with Tsg101 (Figure 1C).
If endogenous GGA expression suppresses virus release by sequestering endogenous Tsg101, one would predict that small increases in Tsg101 expression would stimulate HIV-1 release. To test this hypothesis, HeLa cells were transfected with a constant amount of pNL4-3 along with control plasmid or plasmid expressing full-length Tsg101 over a wide range of input DNA concentrations. As we reported previously (Goila-Gaur et al., 2003; Shehu-Xhilaga et al., 2004), high levels of exogenous Tsg101 expression severely inhibited HIV-1 release (Figure 1D). In contrast, at lower concentrations of Tsg101 a clear increase in virus particle production was observed (Figure 1D). These data demonstrate that although high levels of exogenous Tsg101 severely inhibit HIV-1 release, more modest increases in Tsg101 levels enhance HIV-1 particle production.
GGA overexpression inhibits virus release
We next tested the effect of GGA overexpression on HIV-1 particle production. HeLa cells were transfected with pNL4-3 along with either control plasmid or plasmids expressing full-length GGA1, GGA2 or GGA3, singly or together. Full-length Tsg101 was used as a positive control. Transfected cells were labeled with [35S]Met/Cys; cell and virus lysates were analyzed by radioimmunoprecipitation analysis. As shown in Figure 2A, overexpression of full-length GGA1, GGA2 or GGA3, separately or together, led to a significant inhibition in virus particle production. GGA overexpression also disrupted Gag processing, as indicated by an increase in the ratio of Pr55Gag to p24 levels in cells (Figure 2A). We also noted that GGA1 overexpression led to an increase in the ratio of gp160 to gp120 in cells (Figure 2A) and a decrease in virion-associated gp120 (data not shown). However, the inhibition of particle production mediated by GGA overexpression was not due to its effects on the viral Env, Vpu, or Nef proteins as GGAs also inhibited the release of Env(−), Vpu(−) and Nef(−) HIV-1 mutants (data not shown). As expected, Tsg101 overexpression severely inhibited virus release (Figure 2A). We also observed that GGA overexpression inhibited HIV-1 release in MDMs (Figure S1B). Interestingly, Tsg101 overexpression did not inhibit HIV-1 release in MDMs (Figure S1B) nor did it induce the formation of swollen endosomes in MDMs (data not shown). Together, these results indicate that GGA overexpression severely inhibits HIV-1 particle production both in HeLa cells and in a physiologically relevant cell type.
Figure 2.
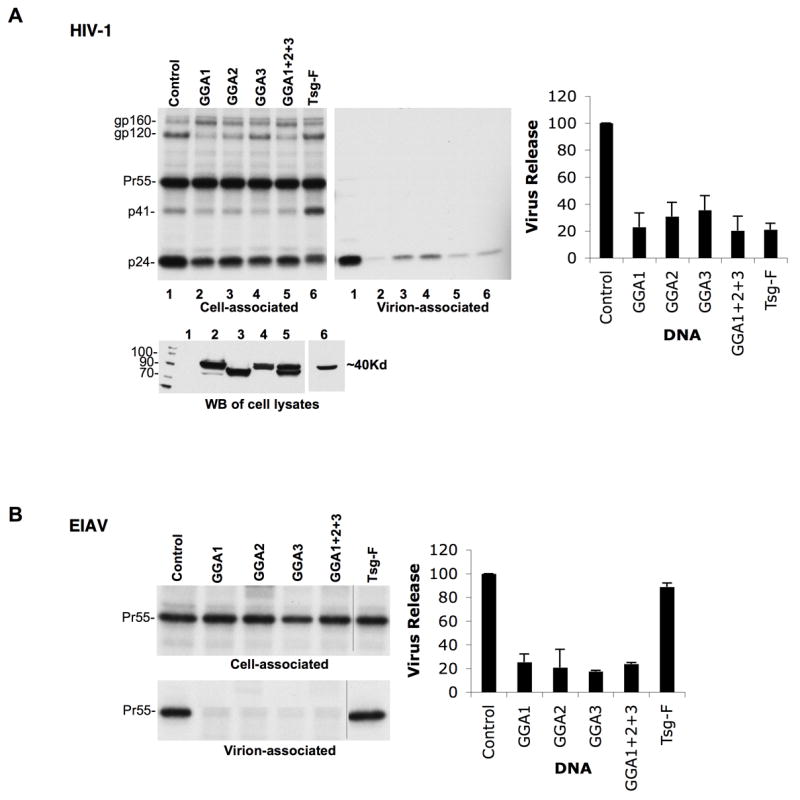
GGA overexpression inhibits retrovirus release. HeLa cells were transfected with (A) HIV-1 (pNL4-3) or (B) EIAV DNA along with expression vectors for GGA1, GGA2, GGA3, all three GGAs, or Tsg101 (TSG-F) at a ratio of 1:1. Cell and virus lysates were radioimmunoprecipitated with HIV-Ig (A) or anti-EIAV antiserum (B). n=3; ± SD. Lower panel in (A) shows GGA (lanes 2-5) and Tsg101 (lane 6) expression detected by anti-Myc or anti-HA Western blot (WB), respectively.
To investigate whether inhibition mediated by GGA overexpression is, like GGA depletion, late domain dependent, we examined EIAV assembly and release (Figure 2B). Interestingly, we observed that GGA overexpression led to a severe inhibition in the production of WT EIAV particles. Consistent with our previous findings (Shehu-Xhilaga et al., 2004), Tsg101 overexpression had no significant effect on EIAV release. MLV particle production was also significantly inhibited by GGA overexpression (data not shown). Thus, unlike the PTAP-specific effects of GGA depletion on virus release, the impact of GGA overexpression was more global and capable of inhibiting retrovirus particle production independent of late domain function.
GGA overexpression disrupts HIV-1 Gag binding to membrane
To delineate the step at which GGA overexpression interferes with HIV-1 particle production, Gag–membrane binding assays were conducted. We observed that GGA overexpression led to a reduction in HIV-1 Gag–membrane binding (Figure 3A and B) without significantly affecting Gag stability (data not shown). These results suggest that GGA overexpression causes defects in HIV-1 particle production by inhibiting the trafficking of newly synthesized Gag to the PM. We note that GGA overexpression caused a 1.5- to 3-fold reduction in Gag expression in this analysis. Because Gag–membrane binding is a cooperative process (Perez-Caballero et al., 2004; Tang et al., 2004), we sought to determine whether reduced Gag expression contributed to the impaired membrane binding induced by GGA overexpression. HeLa cells were transfected with decreasing amounts of pNL4-3 to titrate down levels of Gag expression. We observed that reducing Gag expression by as much as six-fold did not cause any significant difference in the amount of Gag associated with membrane (data not shown). These results indicate that the impaired Gag–membrane binding observed upon GGA overexpression is not due to reduced Gag expression levels.
Figure 3.

GGA overexpression inhibits HIV-1 Gag binding to membrane and an HIV-1 proviral clone bearing a foreign membrane-targeting signal is resistant to the inhibitory effects of GGA overexpression. (A) HeLa cells were transfected with pNL4-3/PR− along with control vector or GGA or Tsg101 (TSG-F) expression constructs. Cells were pulse-labeled with [35S]Met/Cys for 5 min and chased for 15 min in cold medium. Membrane (M) and non-membrane (NM) fractions were separated by membrane flotation centrifugation. The individual fractions were immunoprecipitated with HIV-Ig after denaturation. (B) Quantitation of data; ± SD, n=3. Position of Pr55Gag is indicated (Pr55). (C) Schematic depiction of the Fyn10deltaMA construct, with myristic (Myr) and palmitic (Palm) acid moieties shown. (D) Effect of GGA overexpression on the release of WT or Fyn10deltaMA virus. HeLa cells were transfected with either WT pNL4-3 or Fyn10deltaMA proviral construct along with GGA or Tsg101 (TSG-F) expression plasmids. Cell and virus lysates were radioimmunoprecipitated with HIV-Ig. Means ± SD, n=3.
To determine whether a heterologous membrane-targeting signal would reverse the inhibition of virus production induced by GGA overexpression, we examined the release of VLPs produced by a chimeric Gag molecule in which the MA domain is replaced with the membrane-targeting signal of Fyn (Fyn10deltaMA) (Fig. 3C). Interestingly, the Fyn10deltaMA proviral construct was resistant to the inhibitory effects of GGA overexpression (Figure 3D).
Recently, it was reported that the MA domain of HIV-1 Gag interacts directly with the δ subunit of the adaptor protein complex AP-3, and that this interaction regulates HIV-1 Gag trafficking (Dong et al., 2005). To investigate a possible connection between GGA overexpression and AP-3 function, we examined the effect of GGA overexpression on AP-3 localization. We observed that GGA overexpression induced a partial dissociation of AP-3 from intracellular membranes (Figure S2A), as AP-3 in GGA-overexpressing cells displayed a less punctate and more diffuse staining pattern. We also examined the effect of brefeldin A (BFA) treatment. As expected (Donaldson et al., 1992), we observed that BFA caused a marked loss in AP-3 association with membrane (Figure S2B) but interestingly did not inhibit HIV-1 particle production in HeLa cells (Figure S2C) or in primary monocyte-derived macrophages (data not shown). These data suggest that the effect of GGA overexpression on HIV-1 particle production is not due to dissociation of AP-3 or other Arf-dependent coat proteins from membranes.
GGA1- but not GGA2- or GGA3-induced compartments accumulate predominantly unprocessed Gag and VLPs
To evaluate the localization pattern of exogenously expressed GGAs, and to determine the impact of GGA overexpression on subcellular Gag localization, we performed immunofluorescence microscopy analysis with cells transfected with GGA1, GGA2, or GGA3 expression vectors. In contrast to the Golgi localization pattern observed for endogenous GGAs (Dell’Angelica, 2000) (data not shown), high levels of exogenous GGA expression caused the accumulation of enlarged abnormal vacuolar or aggresome-like compartments (Figure 4A). To examine Gag localization, we transfected HeLa cells with pNL4-3 along with either control or GGA expression plasmids and stained with an anti-p24 Ab that recognizes both processed and unprocessed Gag. As shown in Figure 4A, there was a high degree of colocalization (R > 0.8) between GGA1-induced compartments and HIV-1 Gag. This Gag/GGA1 colocalization was not observed with an anti-p17 (MA) monoclonal Ab that is specific for processed MA (data not shown), suggesting that the Gag present in the GGA1-induced structures was predominantly unprocessed. In contrast to results obtained with GGA1, little or no significant colocalization was evident between Gag and GGA2- or GGA3-induced structures (Figure 4A). Analysis of cells expressing GGAs and HIV-1 by electron microscopy (EM) revealed large numbers of VLPs in vacuolar compartments in cells overexpressing GGA1 (Figure 4B). Notably, GGA overexpression did not induce tethering of virus particles to the cell surface, as observed for overexpression of full-length Tsg101 (TSG-F) or the Tsg101 C-terminal fragment (TSG-3′) (Figure 4B).
Figure 4.

GGA1-induced compartments sequester unprocessed HIV-1 Gag and VLPs. (A) HeLa cells were transfected with pNL4-3 DNA along with control vector or Myc-tagged GGA expression plasmids. Cells were fixed and immunostained with anti-Myc (red) and anti-HIV-1 p24 (green) Abs. R = Pearson coefficient of correlation. (B) HeLa cells were transfected with pNL4-3 along with vectors expressing the indicated proteins, fixed, and analyzed by EM. Bar = 100 nm.
The GAT domain is responsible for the defect in HIV-1 particle production induced by GGA overexpression
To determine which GGA domain(s) are responsible for the inhibition of HIV-1 particle production, we cotransfected HeLa cells with pNL4-3 and vectors expressing full-length GGA3 or the GGA3 VHS, GAT, hinge, or GAE domains (Figure 5A). As shown in Figure 5B and C, the different GGA3 fragments varied widely in their ability to disrupt HIV-1 particle production when normalized for fragment expression levels. The GAT domain exhibited the most potent inhibition, with an effect comparable to that observed upon overexpression of full-length GGA3. To confirm these results, and exclude effects mediated by disruption of Gag processing, we examined the impact of GGA fragment overexpression on the release of unprocessed Gag VLPs produced by the PR-defective molecular clone pNL4-3/PR−. As we observed with WT pNL4-3, overexpression of the GAT domain alone severely inhibited release of Gag VLPs produced by pNL4-3/PR− (Fig. S3). We also analyzed the localization of the different GGA3 fragments in HeLa cells by fluorescence microscopy. The VHS, hinge, and GAE domains displayed diffuse staining throughout the cytoplasm. In contrast, the GAT domain exhibited a staining pattern similar to that observed for full-length GGA3 (Figure 5D). These data indicate that the GAT domain is primarily responsible for the defect in HIV-1 particle production induced by GGA overexpression.
Figure 5.
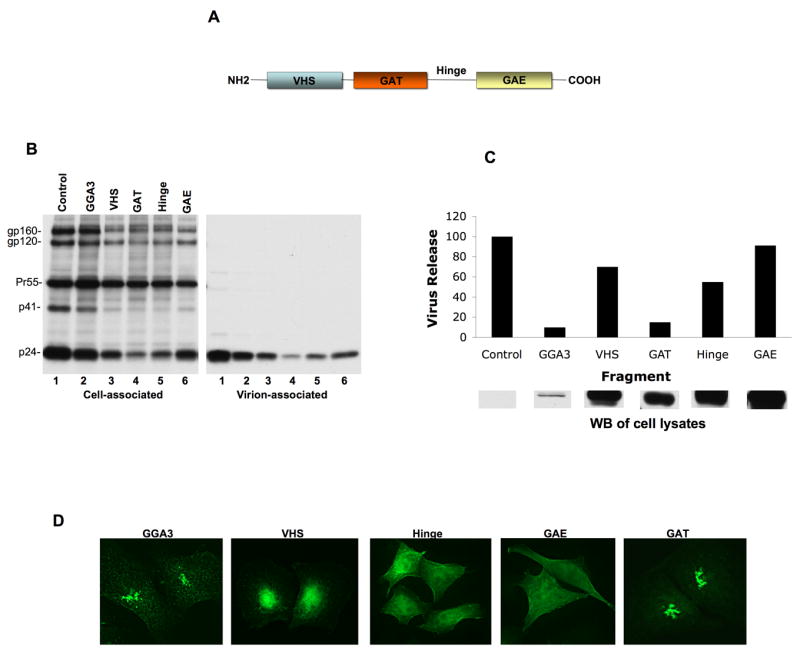
Overexpression of the GAT domain alone inhibits HIV-1 particle production. (A) Domain organization of GGAs. VHS, Vps27, Hrs and STAM homology; GAT, GGA and Tom; GAE, γ-adaptin ear homology. (B) HeLa cells were transfected with pNL4-3 along with vectors expressing full-length GGA3 or the indicated GGA domains at a 10:1 DNA ratio. Virus release was determined by radioimmunoprecipitation analysis. (C) Quantitation of virus release efficiency (top panel) normalized to GGA fragment expression levels, determined by quantitative Western blotting (WB) with anti-Myc Ab (lower panel). Data from one representative experiment are shown. (D) HeLa cells transfected with vectors expressing full-length GGA3 or individual GGA domains were fixed and immunostained with anti-Myc Ab (green).
Mutation of the Arf-binding site reverses the defect in particle production caused by GGA2 and GGA3 overexpression
As demonstrated above, the inhibition of HIV-1 particle production induced by GGA overexpression mapped largely to the GAT domain, a region of the protein that encompasses binding sites for several cellular proteins including Tsg101, ubiquitin, rabaptin5, and the Arf proteins (Bonifacino, 2004). To investigate a role for the Arf proteins in the observed inhibition, we introduced into GGA1, GGA2, and GGA3 a mutation (N194A, here abbreviated “NA”) previously reported to abolish GGA–Arf binding (Puertollano, 2001). We then tested the effect of overexpressing the mutant GGA proteins on HIV-1 particle production. Interestingly, GGA2 and GGA3 NA mutants were significantly less potent than the WT proteins in inhibiting HIV-1 particle production (Figure 6A and B). Loss of inhibition with the NA mutants was not due to differences in WT versus mutant protein expression (Figure 6A). Intriguingly, introduction of the NA mutation into GGA1 had no significant effect on its ability to inhibit particle production (Figure 6A and B). However, the defect in Env processing induced by GGA1 overexpression was reversed by the GGA1 NA mutation (Figure 6A). Together, these findings suggest that the ability of GGA overexpression to disrupt HIV-1 particle production is linked to Arf activity. We observed that overexpression of the NA GGA1 mutant still led to the trapping of Gag in intracellular structures (data not shown), as did overexpression of WT GGA1 (Figure 4A), providing an explanation for the ability of this mutant to inhibit HIV-1 particle production. We also tested the ability of overexpressed GGA-NA (Arf-binding) mutants to inhibit the production of EIAV virions. We observed that the Arf-binding (NA) mutation in GGA3 completely reversed the ability of GGA3 overexpression to inhibit EIAV particle production (Fig. S4). In contrast, WT GGA1 and GGA1-NA overexpression inhibited EIAV production to a similar extent. The NA mutation in the context of GGA2 partially reversed the inhibition of EIAV particle production (Fig. S4).
Figure 6.
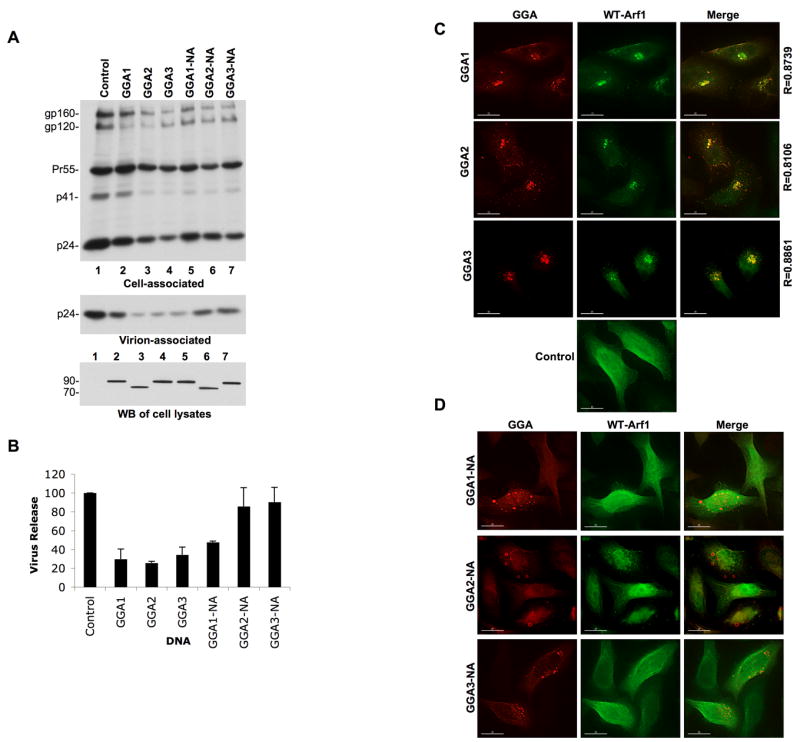
GGA2 and GGA3 mutants defective in Arf binding do not inhibit HIV-1 particle production and GGA overexpression sequesters WT Arf1. (A) HeLa cells were cotransfected with pNL4-3 and either WT GGA expression plasmids or their Arf-binding-defective (NA) counterparts. Virus release was determined by radioimmunoprecipitation analysis. Lower panel shows GGA expression levels detected by Western blotting (WB) with an anti-Myc Ab; molecular mass standards (in kDa) are shown on the left. (B) Quantitation was performed by phosphorimager analysis, mean ± SD, n=3. (C and D) HeLa cells were cotransfected with vectors expressing Myc-tagged WT GGAs (C) or NA mutants defective in Arf binding (D) and low amounts of a vector expressing HA-tagged Arf1 and immunostained with anti-Myc (red) and anti-HA (green) Abs. Degree of colocalization was determined; R = Pearson coefficient of correlation. Lower panel in C; Arf1 localization in cells singly transfected with the HA-tagged Arf1 expression vector.
GGA overexpression leads to Arf sequestration
Based on the data presented above, we focused on the possibility that GGA overexpression might disrupt endogenous Arf function. Thus, we examined the localization of Arf1 in GGA-overexpressing cells by immunofluorescence microscopy. As shown in Figure 6C, Arf1 in control HeLa cells displayed a predominantly diffuse cytosolic distribution. In contrast, GGA overexpression led to Arf1 sequestration in the GGA-induced structures, with a high degree of colocalization (R > 0.8) between the GGAs and Arf1 (Figure 6C). Interestingly, the GGA-NA mutants defective in Arf binding did not lose their ability to form aberrant intracellular compartments, though these mutants displayed an increase in hazy cytosolic staining, presumably representing non-membrane-bound protein (Figure 6D). Consistent with the loss of Arf binding induced by the NA mutation, the mutant GGAs failed to alter the localization of Arf1 (Figure 6D). The sequestration of Arf1 by GGA1, GGA2, and GGA3 (Figure 6C) required GGA overexpression, since endogenous GGAs did not significantly colocalize with Arf1 (Figure S5). These results support the hypothesis that GGA overexpression impairs HIV-1 particle production by disrupting or sequestering Arf activity.
Disruption of endogenous Arf function inhibits retrovirus release
To examine directly whether Arf activity is required for retrovirus particle production, we sought to determine the effect of disrupting the activity of endogenous Arf proteins. We measured the effect of dominant-active Arf isoforms, known to disrupt Arf activity (Dascher and Balch, 1994; Takatsu et al., 2002), on retroviral particle production. Significantly, disruption of endogenous Arf function markedly inhibited HIV-1, EIAV and MLV particle production (Figure 7A). To characterize further the defect in retroviral production imposed by expression of dominant-active Arfs, Gag membrane binding assays were performed. As observed for GGA overexpression, the dominant-active Arfs caused a significant decrease in the fraction of HIV-1 Gag bound to membrane (Figure 7B). Finally, and also consistent with the defect induced by GGA overexpression (Figure 3D), VLP production driven by Fyn10deltaMA was resistant to inhibition by the dominant-active Arf mutants (Figure 7C). The defects in retrovirus particle production described herein did not appear to be due to global depletion or relocalization of cellular PI(4,5)P2 observed upon disruption of class III Arf (Arf6) (Brown et al., 2001; Ono et al., 2004) since the dominant-active class I and II Arfs did not cause a discernible redistribution of cellular PI(4,5)P2 (Figure S6).
Figure 7.

Disruption of endogenous Arf function and Arf depletion inhibit retrovirus release. (A) HeLa cells were cotransfected with pNL4-3 or vectors expressing MLV Gag-Pol or EIAV Gag along with the indicated dominant-active Arf expression plasmids (1:1 DNA ratio). Virus release efficiency was determined by radioimmunoprecipitation with HIV-Ig, anti-MLV Gag or anti-EIAV antisera, followed by phosphorimager analysis. Data represent means ± SD; n=3. (B) HeLa cells were cotransfected with pNL4-3/PR−DNA along with control plasmid or vectors expressing dominant-active Arf (Q71L) mutants. The percentage of membrane-bound Gag was determined. Data represent means ± SD; n=3. (C) HeLa cells were transfected with either WT pNL4-3 or the Fyn10deltaMA proviral construct in the presence or absence of dominant-active Arf expression vector. Virus release efficiency was determined by radioimmunoprecipitation and phosphorimager analysis. Mean ± SD; n=3. (D) HeLa cells were cotransfected with pNL4-3 and the indicated Arf-specific siRNAs. Virus release efficiency; mean ± SD; n=4. (E) Efficiency of siRNA-mediated Arf depletion in HeLa cells as determined by Western blot analysis. Exogenous Arf expression was quantitated with an Alpha Innnotech digital imager, Mean ± SD.
We further confirmed the importance of Arf function in retroviral particle production by depleting HeLa cells of endogenous Arfs by using specific siRNAs (Fig. 7D). Transfection of Arf 1, 3, 5 and 6 siRNAs led to > 80% depletion of the respective Arfs, which significantly inhibited HIV-1 particle production (Figure 7D and E). Some reductions (up to 3.5- to 4-fold) in total HIV-1 Gag expression were observed upon overexpression of dominant-active Arfs (Fig. 7A) or following Arf depletion (Fig. 7D). To confirm that the impaired virus production observed under these conditions was the result of Arf disruption and not simply a consequence of reduced Gag expression, we performed a titration experiment in which HeLa cells were transfected with decreasing concentrations of pNL4-3 DNA in parallel with pNL4-3 cotransfections with dominant-active Arfs or Arf siRNAs. Analysis of virus release efficiency demonstrated that reductions in total Gag expression of six-fold had no effect on virus release efficiency. Indeed, in a Gag titration that spanned an even larger range of pNL4-3 DNA input, a 17-fold reduction in Gag expression did not affect the efficiency of virus particle production (data not shown). In contrast, the dominant-active Arfs and Arf siRNAs reduced total Gag expression by only two-fold and significant reductions in particle production were observed (Fig. S7A). These results demonstrate that effects of Arf disruption on Gag expression levels do not explain the impaired virus production efficiency. Finally, we also tested the impact of dominant-active Arf overexpression or Arf depletion in the context of the PR-defective molecular clone pNL4-3/PR−. We again observed a marked reduction in VLP production, with no appreciable reduction in Gag expression (Fig. S7B). Altogether, these data demonstrate a role for Arf function in retrovirus particle production and strongly suggest that the defect imposed by GGA overexpression is mediated through disruption of endogenous Arf activity.
DISCUSSION
In the current study, we investigated the role of the GGA and Arf proteins in retrovirus assembly and release. Our results indicate that depletion of endogenous GGAs, specifically GGA2 and GGA3, leads to a significant increase in retrovirus release in a PTAP-dependent manner. These results demonstrate that endogenous GGA2 and GGA3 act as negative regulators of retrovirus budding. In contrast, GGA overexpression severely inhibited the assembly and release of a wide variety of retroviruses, independent of viral late domain. The defect in HIV-1 particle production induced by GGA overexpression was due to disrupted Gag–membrane association and was dependent on the MA domain of Gag. Our finding that overexpression of the GAT domain of GGA3 impaired HIV-1 production and that GGA mutants defective in Arf binding lost their inhibitory effect revealed a connection between the Arf proteins and the inhibition mediated by GGA overexpression. This connection was strengthened by the observations that GGA overexpression sequestered WT Arfs, dominant-active Arf mutants disrupted retroviral particle production and Gag–membrane association, and Arf depletion inhibited HIV-1 assembly and release. These data identify the GGA and Arf families of proteins as cellular cofactors in the late stages of retroviral replication.
The role of the ESCRT-1 component Tsg101 in HIV-1 particle release is well established (Bieniasz, 2006; Demirov and Freed, 2004; Morita and Sundquist, 2004) and it is clear that either reducing (Garrus et al., 2001) or significantly increasing (Goila-Gaur et al., 2003; Shehu-Xhilaga et al., 2004) intracellular Tsg101 levels is disruptive to PTAP-mediated particle release. These results indicate that a defined range of Tsg101 expression levels is optimal for virus release. Indeed, in this study, we observed that low levels of exogenous Tsg101 expression increased HIV-1 particle production; the magnitude of this effect was similar to that observed upon GGA depletion (Figure 1A). We speculate that the ability of GGA2 and GGA3 depletion to stimulate particle release in both HeLa cells and in primary macrophages could be due to increased Tsg101 availability for virus budding. The observation of an optimal range of Tsg101 levels for virus budding, and the modulation of PTAP-dependent release by the GGA proteins, could have implications for HIV-1 replication in vivo. The possibility that in vivo (as in HeLa cells) levels of endogenous Tsg101 expression may be suboptimal for promoting virus release could provide explanations for the evolution of PTAP duplications detected in virus isolates from HIV-1-infected patients (Peters et al., 2001; Whitehurst et al., 2003) and the presence of a secondary late domain in Gag that allows HIV-1 to bud via an interaction with Alix when Gag–Tsg101 binding is disrupted (Fisher et al., 2007; Usami et al., 2007).
Despite the high degree of homology between GGA1, GGA2, and GGA3 (Boman et al., 2000; Bonifacino, 2004; Dell’Angelica et al., 2000), there are intriguing differences in the spectrum of effects induced by disrupting these three members of the mammalian GGA family: 1) Overexpression of GGA1 inhibited gp160 processing (Figure 2A); in contrast, overexpression of GGA3 had no effect on Env processing. An intermediate phenotype with respect to gp160 processing was observed upon GGA2 overexpression. 2) Introduction of the NA mutation, which blocks GGA–Arf binding, reversed the inhibition of HIV-1 particle production induced by GGA2 and GGA3 overexpression but did not prevent GGA1 overexpression from disrupting particle production (Figure 6). 3) The intracellular compartment induced by GGA1 overexpression sequestered HIV-1 Gag and VLPs, whereas the compartments induced by GGA2 or GGA3 did not (Figure 4A and B). Interestingly, the Arf-binding-defective GGA1 mutant still trapped HIV-1 Gag in intracellular structures (unpublished results), accounting for the ability of this mutant to disrupt particle production. In addition, depletion of GGA2 or GGA3 significantly enhanced particle release, whereas GGA1 depletion did not, despite comparable depletion efficiencies. Further characterization of these differences is likely to provide additional insights into the host cell machinery involved in regulating retroviral particle production.
Membrane-binding assays suggested that the defect in particle production induced by GGA overexpression and Arf disruption is imposed primarily at the level of Gag trafficking to the PM. Overexpression of either the GGAs or dominant-active Arfs impaired HIV-1 Gag–membrane association. This inhibition was independent of cell type, as we observed that GGA overexpression disrupted HIV-1 particle production both in HeLa cells and in primary macrophages. An effect on Gag–membrane binding was further supported by the finding that the defect in virus particle production induced by GGA or dominant-active Arf overexpression could be reversed by replacing the native membrane-targeting signal in the MA domain of Gag with a heterologous membrane-binding signal from Fyn.
We observed that in some experiments disruption of GGA or Arf proteins reduced overall levels of Gag expression. This raised the possibility that reduced Gag expression could contribute to impaired membrane binding and reduced particle production imposed by GGA or Arf disruption. This does not appear to be the case for the following reasons: 1) Reductions in Gag expression of 6- to 17-fold (levels of Gag that are several-fold lower than observed upon GGA overexpression or Arf disruption) caused no significant reduction in the amount of HIV-1 Gag associated with membrane or virus release efficiency (unpublished results and Fig. S7A), and 2) GGA overexpression (Fig. 2B) or dominant-active Arf overexpression (Fig. 7A) both markedly impaired particle production without reducing EIAV Gag expression. We also stress that our method for calculating virus release efficiency is based on normalization for total (cell plus virus) Gag expression. We therefore conclude that the impact of GGA overexpression or Arf disruption on retroviral particle production reflects the activity of these host factors in virus assembly/release rather than being a secondary consequence of effects on Gag expression levels. The defects in retroviral particle production observed in this study are distinct from those induced by disruption of ESCRT and associated endosomal sorting machinery which blocks the pinching-off of virus particles from the PM.
Although it appears that GGA overexpression and Arf disruption impair the trafficking of Gag to the PM, the precise mechanism by which the inhibition in Gag–membrane binding occurs remains to be elucidated. Arf proteins function in part by recruiting coatomer (e.g., COPI) and adapter protein (e.g., AP-1, AP-3, and AP-4) complexes to various membranes within the cell (D’Souza-Schorey and Chavrier, 2006). We observed that GGA overexpression, dominant-active Arf1, 3, and 5 expression, and depletion of Arf1, Arf3, Arf4, and Arf5 (unpublished results) caused a small increase in the cytosolic localization of AP-3 (Figure S2A) and other adapter protein complexes (e.g., AP-1; unpublished results). However, BFA treatment causes a dramatic shift in the localization of adapter protein complexes (e.g., AP-3) from highly punctate to cytosolic (Figure S2B), yet had no effect on HIV-1 particle production in either HeLa cells (Figure S2C) or primary MDMs (unpublished results). These results imply that loss of AP-3 association, or the association of other adapter protein complexes, with intracellular membranes mediated by Arf disruption is not responsible for the defects in Gag–membrane association observed here. Arf proteins also regulate intracellular trafficking pathways by activating phosphatidylinositol-4-phosphate 5-kinase, which in turn leads to the generation of PI(4,5)P2. Previously, our lab demonstrated a role for PM PI(4,5)P2 in HIV-1 Gag trafficking, and showed that expression of a dominant-active class III (Arf6) mutant induced a redistribution of both PI(4,5)P2 and virus assembly to intracellular endosomal-like structures (Ono et al., 2004). Although in this study we confirmed the effect of dominant-active Arf6 expression on PI(4,5)P2 localization, we observed no clear shift in PI(4,5)P2 localization upon expression of dominant-active class I or class II Arfs (Figure S6). It thus appears that disruption of class I or class II Arfs does not inhibit Gag trafficking by inducing a major relocalization of PI(4,5)P2. With respect to the mechanism by which Arfs function in retroviral particle production, it is interesting to note that we have identified Arf1 as an EIAV Gag-interacting protein in a yeast two-hybrid screen (unpublished results). Moreover, siRNA-mediated Arf depletion inhibited HIV-1 (Figure 7D) and EIAV (unpublished results) particle production. It is therefore possible that Arfs play a direct role in recruiting retroviral Gag proteins to membranes.
The data presented here identify the GGA proteins as modulators of retrovirus release, and the Arf proteins as cellular cofactors in retroviral Gag trafficking (Figure S8). These results provide insights into the role of host cell machinery in retroviral assembly and release.
EXPERIMENTAL PROCEDURES
Cell lines and transfections
HeLa cells were maintained in Dulbecco-modified Eagle medium (DMEM) supplemented with 5% heat-inactivated fetal bovine serum (FBS, HyClone) and 2mM glutamine (Gibco) as described previously (Joshi et al., 2006). HeLa transfections were performed with the Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA). MDMs were obtained by culturing elutriated monocytes (Freed et al., 1995) in RPMI 1640 medium supplemented with 10% FBS and 2mM glutamine for 6 days. On day 7, cells were transfected using the Amaxa Electroporator (Program Y-10).
Radioimmunoprecipitation, co-immunoprecipitation, Western blotting, membrane-binding assays immunofluorescence, and EM
Metabolic labeling of cells and virions with [35S]Met/Cys was performed as described (Freed and Martin, 1994; Willey et al., 1988). In brief, 24 h posttransfection, cells were starved for 30 min in media lacking Met and Cys. Cells were then cultured for 2–3 h in labeling media supplemented with 5% FBS and 250 μCi/well of [35S]Met/Cys. Radiolabeled cell and virus lysates were then immunoprecipitated using HIV-Ig. The precipitated complexes were resolved by SDS-PAGE and viral protein expression was quantitated by phosphorimager analysis. Virus release efficiency was calculated as the amount of virion p24 divided by total Gag (cell Pr55Gag + cell p24 + virion p24). For Western blot analysis, protein lysates were resolved by SDS-PAGE and transferred onto polyvinylidine difluoride membranes (Millipore). After incubation with appropriate primary and secondary antibodies, protein bands were visualized on X-ray film using the enhanced chemiluminescence detection reagent (PerkinElmer, Boston, MA). Quantitation of western blot signal was performed by using an Alpha Innnotech digital imager. Co-immunoprecipitation assays were conducted using the ProFound™ Mammalian IP/Co-IP Kit (Pierce). Gag membrane binding assays were conducted as described before (Joshi et al., 2006; Ono and Freed, 1999). Additional details are provided in the supplemental text.
Supplementary Material
Acknowledgments
We would like to thank Dr. R. Kahn (Emory University, Atlanta) for kindly providing the GGA1 antibody, Dr. K. Nakayama (Kyoto University, Kyoto, Japan) and J. Donaldson (NIH, Bethesda, MD) for generously providing the HA-tagged WT and dominant-active Arf expression constructs, Dr. A. Ono (University of Michigan, Ann Arbor, Michigan) for providing the Fyn10deltaMA Gag chimera, and Dr. E. S. Sztul (University of Alabama at Birmingham, Birmingham) for generously providing the GFP-250 construct. The anti-EIAV serum was kindly provided by Dr. R. Montelaro (University of Pittsburgh) and HIV-Ig was generously provided by the NIH AIDS Research and Reference Reagent Program. We thank F. Soheilian for EM support, S. Ablan for technical assistance, and Drs. A. Ono, V. Pathak, W.-S. Hu, V. KewalRamani, and members of the Freed lab for critical review of manuscript. This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH, and by the Intramural AIDS Targeted Antiviral Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344:55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Boman AL, Zhang C, Zhu X, Kahn RA. A family of ADP-ribosylation factor effectors that can alter membrane transport through the trans-Golgi. Mol Biol Cell. 2000;11:1241–1255. doi: 10.1091/mbc.11.4.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol. 2004;5:23–32. doi: 10.1038/nrm1279. [DOI] [PubMed] [Google Scholar]
- Brown FD, Rozelle AL, Yin HL, Balla T, Donaldson JG. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J Cell Biol. 2001;154:1007–1017. doi: 10.1083/jcb.200103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- Dascher C, Balch WE. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J Biol Chem. 1994;269:1437–1448. [PubMed] [Google Scholar]
- Dell’Angelica EC, Puertollano R, Mullins C, Aguilar RC, Vargas JD, Hartnell LM, Bonifacino JS. GGAs: a family of ADP ribosylation factor-binding proteins related to adaptors and associated with the Golgi complex. J Cell Biol. 2000;149:81–94. doi: 10.1083/jcb.149.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106:87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Donaldson JG. Arfs, phosphoinositides and membrane traffic. Biochem Soc Trans. 2005;33:1276–1278. doi: 10.1042/BST0331276. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature. 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, Peters TR, Dermody TS, Woodruff E, Wang JJ, Spearman P. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell. 2005;120:663–674. doi: 10.1016/j.cell.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Fisher RD, Chung HY, Zhai Q, Robinson H, Sundquist WI, Hill CP. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell. 2007;128:841–852. doi: 10.1016/j.cell.2007.01.035. [DOI] [PubMed] [Google Scholar]
- Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- Freed EO, Englund G, Martin MA. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J Virol. 1995;69:3949–3954. doi: 10.1128/jvi.69.6.3949-3954.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Martin MA. HIV-1 infection of non-dividing cells. Nature. 1994;369:107–108. doi: 10.1038/369107b0. [DOI] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Goila-Gaur R, Demirov DG, Orenstein JM, Ono A, Freed EO. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J Virol. 2003;77:6507–6519. doi: 10.1128/JVI.77.11.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Freed EO. HIV-1 Gag trafficking. Future HIV Ther. 2007;1:427–438. [Google Scholar]
- Joshi A, Nagashima K, Freed EO. Mutation of dileucine-like motifs in the human immunodeficiency virus type 1 capsid disrupts virus assembly, gag-gag interactions, gag-membrane binding, and virion maturation. J Virol. 2006;80:7939–7951. doi: 10.1128/JVI.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Chen C, Puffer BA, Montelaro RC. Functional replacement and positional dependence of homologous and heterologous L domains in equine infectious anemia virus replication. J Virol. 2002;76:1569–1577. doi: 10.1128/JVI.76.4.1569-1577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A. 2004;101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol. 1999;73:4136–4144. doi: 10.1128/jvi.73.5.4136-4144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Caballero D, Hatziioannou T, Martin-Serrano J, Bieniasz PD. Human immunodeficiency virus type 1 matrix inhibits and confers cooperativity on gag precursor-membrane interactions. J Virol. 2004;78:9560–9563. doi: 10.1128/JVI.78.17.9560-9563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters S, Munoz M, Yerly S, Sanchez-Merino V, Lopez-Galindez C, Perrin L, Larder B, Cmarko D, Fakan S, Meylan P, Telenti A. Resistance to nucleoside analog reverse transcriptase inhibitors mediated by human immunodeficiency virus type 1 p6 protein. J Virol. 2001;75:9644–9653. doi: 10.1128/JVI.75.20.9644-9653.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Randazzo PA, Presley JF, Hartnell LM, Bonifacino JS. The GGAs promote ARF-dependent recruitment of clathrin to the TGN. Cell. 2001;105:93–102. doi: 10.1016/s0092-8674(01)00299-9. [DOI] [PubMed] [Google Scholar]
- Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A. 2006;103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu-Xhilaga M, Ablan S, Demirov DG, Chen C, Montelaro RC, Freed EO. Late domain-dependent inhibition of equine infectious anemia virus budding. J Virol. 2004;78:724–732. doi: 10.1128/JVI.78.2.724-732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry. 2006;45:4077–4083. doi: 10.1021/bi052308e. [DOI] [PubMed] [Google Scholar]
- Swanstrom R, Wills JW. Synthesis, Assembly, and Processing of Viral Proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. New York: Cold Spring Harbor Laboratory Press; 1997. pp. 263–334. [PubMed] [Google Scholar]
- Takatsu H, Yoshino K, Toda K, Nakayama K. GGA proteins associate with Golgi membranes through interaction between their GGAH domains and ADP-ribosylation factors. Biochem J. 2002;365:369–378. doi: 10.1042/BJ20020428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A. 2004;101:517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usami Y, Popov S, Gottlinger HG. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J Virol. 2007;81:6614–6622. doi: 10.1128/JVI.00314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt VM. Retroviral virions and genomes in "Retroviruses". Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. [PubMed] [Google Scholar]
- Whitehurst N, Chappey C, Petropoulos C, Parkin N, Gamarnik A. Polymorphisms in p1-p6/p6* of HIV type 1 can delay protease autoprocessing and increase drug susceptibility. AIDS Res Hum Retroviruses. 2003;19:779–784. doi: 10.1089/088922203769232575. [DOI] [PubMed] [Google Scholar]
- Willey RL, Smith DH, Lasky LA, Theodore TS, Earl PL, Moss B, Capon DJ, Martin MA. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J Virol. 1988;62:139–147. doi: 10.1128/jvi.62.1.139-147.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.