

Abstract
Adherence of Helicobacter pylori to cultured gastric epithelial cells is associated with several cellular events, including the tyrosine phosphorylation of a 145-kDa host protein; the reorganization of the host cell actin and associated cellular proteins, like vasodilator-stimulated phosphoprotein, adjacent to the attached bacterial cell; and the subsequent release of the cytokine, interleukin 8 (IL-8). H. pylori isolated from patients with ulcer disease and gastric cancer contain a DNA insertion, the cag pathogenicity island (PAI), that is not present in bacteria isolated from individuals with asymptomatic infection. Mutations in a number of PAI genes abolish tyrosine phosphorylation and IL-8 synthesis but not the cytoskeletal rearrangements. Kinase inhibition studies suggest there are two distinct pathways operative in stimulating IL-8 release from host cells and one of these H. pylori pathways is independent of the tyrosine phosphorylation step.
Keywords: phosphorylation, interleukin 8, vasodilator-stimulated phosphoprotein
Helicobacter pylori, a human pathogen and class I carcinogen (1), causes acute and chronic inflammation of the stomach (gastritis). Progression of the disease in some, but not all, patients can lead to peptic ulcer disease, gastric cancer, or lymphoma originating in the mucosal-associated lymphoid tissue. Several putative H. pylori virulence factors have been identified, including urease; the vacuolating cytotoxin, VacA; and a cytotoxin-associated antigen, CagA. However, mutations in the genes encoding these factors do not have a measurable effect on the virulence of H. pylori in cell culture or animal infection studies (2, 3).
Most of the clinical isolates of H. pylori isolated from peptic ulcer disease or patients suffering from malignant disease express CagA. Consequently, H. pylori strains have been divided into two broad categories. Type I strains are cag+ and vacA+. Type II strains are cagA− and fail to produce a functional VacA toxin. Yet, DNA sequences homologous to the vacA gene are present in the chromosomes of type II strains. Type I isolates are associated with duodenitis, duodenal ulcers, and gastric cancer (4–6). A pathogenicity island (PAI), called cag1, has been identified exclusively in H. pylori type I strains. The cag1 region contains over 22 open reading frames (ORF) encoded in 40 kb of DNA (7). The majority of the proteins predicted to be encoded by the cag1 ORF appear to be membrane-associated with features similar to bacterial secretion systems, particularly the Type IV system epitomized by Bordetella pertussis toxin secretion (8).
The cytokine interleukin 8 (IL-8) is a neutrophil chemotactic factor that has been shown to be increased in H. pylori-infected patients with active gastritis (9). A correlation between type I strains and the ability to induce IL-8 secretion in gastric epithelial cell lines has been demonstrated by several laboratories (10). Tummuru et al. (11) have identified a gene, picB, with significant amino acid homology to the B. pertussis toxin-secretion protein (PtlC) and have demonstrated that the picB gene product is involved in inducing IL-8 in vitro. The picB gene now is known to be within the H. pylori cag1 PAI. VacA, CagA, and urease are not functionally involved with the induction of IL-8 from cultured gastric cells (12, 13).
We have previously shown (14) that H. pylori attachment to gastric epithelial cells is associated within effacement of microvilli, and rearrangement of the cytoskeleton occurs beneath the attached bacterium (pedestal formation). These findings are similar to that described for the enteropathogenic Escherichia coli (EPEC) which are associated with genes encoded within a PAI (15). Furthermore, like EPEC, H. pylori binding to epithelial cells induces tyrosine phosphorylation of host cell proteins adjacent to the site of bacterial adherence. These effects of H. pylori attachment to cells suggests that alteration of host cell signal transduction might lead to chronic inflammation and perhaps may lead to the oncogenic transformation that are the hallmarks of symptomatic H. pylori infection.
To better understand the mechanism(s) involved in the induction of the host cell response to H. pylori attachment, wild-type type I and type II strains and isogenic type I H. pylori mutants were screened for their ability to attach to gastric epithelial cells, to induce tyrosine phosphorylation of the 145-kDa host protein, and for their capacity to generate an inflammatory reaction in vitro as measured by the induction of IL-8. In addition, we have performed experiments to analyze further the cellular pathways affected by H. pylori infection.
MATERIALS AND METHODS
Bacterial Strains and Cell Lines.
Helicobacter pylori strain 87A300 was obtained from the State of California, Department of Health Services, Berkeley. It is a human clinical isolate producing the vacuolating cytotoxin (vacA), urease, CagA, and a contiguous cag1 PAI. This strain was grown as described (14). Urease mutants of strain 87A300 were described (16). H. pylori strains G27, 314, G50, G198, and their mutants were provided by Antonio Covacci (Immunobiological Research Institute, Siena, Italy) and are described by Censini et al. (7). A summary of all strains used in this study and their respective genotypes is presented in Table 1. AGS cells (ATCC CRL 1739), a human gastric adenocarcinoma epithelial cell line, were grown in DMEM plus 10% fetal calf serum as described (14).
Table 1.
H. pylori strains used in this study
| Strain | Genotype | Phenotype |
|---|---|---|
| 87A300 | Type 1 | cag+, vac+, urease+ |
| Mutants* | ||
| ure1 | urease− | |
| G27 | Type 1 | cag+, vac+, urease+ |
| Mutants† | ||
| 10B4 | cagE::Kan | |
| Mfe:Kan | cagF::Kan | |
| 62A | cagG::Kan | |
| 3F | cagH::Kan | |
| 5-3 | cagI::Kan | |
| 2B | cagL::Kan | |
| 14A-4 | cagN::Kan | |
| H12-10 | cagN::Kan | |
| H12-18A | cagN::Kan | |
| H12-47 | cagM::Kan | |
| H12-4 | cagM::Kan | |
| H12-16 | cagM::Kan | |
| H12-5 | cagM::Kan | |
| 314 | Type 1 | cag+, vac+, urease+ |
| G50 | Type 2 | cag−, vac−, urease+ |
| G104 | Type 2 | cag−, vac−, urease+ |
| G198 | Type 2 | cag−, vac−, urease+ |
Isolates generated by ethyl methanesulfonate mutagenesis.
Isolates generated by kanamycin insertion into the gene.
Antibodies.
Rabbit polyclonal anti-H. pylori antibodies were produced against whole heat-killed H. pylori strain 87A300. Working dilution of the polyclonal anti-H. pylori was 1:100. Monoclonal anti-phosphotyrosine PY20 and anti-vasodilator-stimulated phosphoprotein (VASP) were purchased from Transduction Laboratories (Lexington, KY) and used at a working dilution of 1:1000. Anti-mouse IgG crystalline tetramethylrhodamine isothiocyanate conjugate and anti-rabbit IgG fluorescein isothiocyanate conjugate was obtained from Sigma. Anti-mouse IgG 7-amino-4-methylcoumarin-3-acetic acid conjugate was obtained from Vector Laboratories.
Immunofluorescence (IF) Microscopy.
For IF studies, 1 × 106 AGS cells were plated per well into a four-chamber tissue culture slide (Falcon). The next morning the monolayers were washed twice with PBS, and 3 × 106 bacteria added per well in a final volume of 400 μl DMEM (multiplicity of infection, 10:1). The slides were incubated, and at appropriate time points the wells aspirated, washed five times with PBS (pH 7.4) to remove nonadherent bacteria, and processed for IF. IF microscopy was done on an Applied Precision (Issaquah, WA) DeltaVision Deconvolution System with an Olympus IX-70 inverted microscope.
Infection Assays and Determination of Tyrosine Phosphorylation of Host Cell Proteins.
AGS cells were cultured for 24 hr in 60-mm tissue culture dishes containing DMEM as described (14). The cells were washed once with PBS (pH 7.4), and 2 ml of fresh media was added to each dish. Approximately 1 × 107 H. pylori from a liquid overnight culture was added to the AGS cells at a multiplicity of infection of 10:1. After incubation in a 5% CO2 incubator for 2 hr, cell lysates were made as described (14) and stored at −80°C until needed. Whole cell lysates of H. pylori were made by pelleting the bacteria and suspending the pellet in an equal amount of PBS and 2× SDS lysis buffer (250 mM Tris, pH 6.8/4% SDS/20% glycerol/0.002% bromophenol blue/10% 2-mercaptoethanol). SDS, PAGE, and electrotransfer was performed as described (14). Anti-phosphotyrosine binding and detection was done using the ECL system (Amersham) according to the manufacturer’s instructions.
IL-8 Assay.
Experiments were performed with the AGS cell line grown and infected with H. pylori as described above. After incubation in a 5% CO2 incubator for 2 hr, the medium was removed from the dish, spun to remove any remaining bacteria (10,000 rpm for 10 min at 4°C), and the aliquots were frozen at −80°C. IL-8 levels of these extracts was determined by ELISA (Amersham).
Kinase Inhibition Assays.
The concentration of kinase inhibitors used was as follows: 1 μM staurosporine, 250 μM genistein, 2 μM bisindolymaleimide I, 9.8 μM H-89 (dihydrochloride), and 47 μM KT5823. All inhibitors were purchased from Calbiochem. For the assay, AGS cells were seeded as described above. The medium was removed before the assay and replaced with fresh DMEM with the appropriate inhibitor and incubated for 30 min. H. pylori was added as above and incubated for an additional 3 hr. Lysates were prepared and analyzed for tyrosine phosphorylation as described earlier.
RESULTS
H. pylori Attachment to Cultured Gastric Epithelial Cells and Host Cell Phosphorylation.
Qualitative attachment of H. pylori strains and of their isogenic mutants to AGS cells was estimated visually using light and transmission electron microscopy. Quantitative assessment of H. pylori attachment to AGS cells was determined by performing viable counts of bacteria that had attached to an AGS monolayer. No differences were observed in the ability of wild-type strains or their mutants to attach to AGS cells (data not shown).
We also examined host cell cytoskeletal changes associated with H. pylori attachment using confocal microscopy. Type I strains induced the expected reorganization of host cell actin (Fig. 1A) and the adjacent cellular proteins at the site of microbial attachment. Type II strains induced a minimal focal pattern of actin rearrangement at the site of attachment (Fig. 1B). The cag1 mutants we examined induced cytoskeletal changes similar to those observed for type I H. pylori (Fig. 1C). Thus, the attachment of H. pylori cag1 mutants was not dramatically changed as compared with their parental strain.
Figure 1.
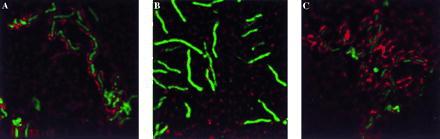
Deconvolution IF of spiral H. pylori strains attached to AGS cells (0.2-μm image). Each field is stained for H. pylori (green) and actin (red). Colocalization of H. pylori with actin appears yellow. (A) H. pylori strain G27. (B) H. pylori strain G50. (C) H. pylori mutant 10B4. Imaged on an Applied Precision DeltaVision Deconvolution System.
Tyrosine phosphorylation of host cell proteins occurs subsequent to the attachment of type I H. pylori to gastric cells (14). Therefore, we examined the ability of type II strains and mutants in the cag1 PAI to mediate the tyrosine phosphorylation of host cell proteins. Cell lysates were prepared at various times after bacterial attachment and subjected to Western Blot analysis using a mAb against phosphotyrosine. Typical results are shown in Fig. 2 and summarized in Table 2. Type I strains induced tyrosine phosphorylation of the 145 kDa protein but type II strains did not. Transposon insertion mutants in eight PAI genes described by Censini et al. (7) were examined for their capacity to mediate tyrosine phosphorylation of host cell proteins. Seven of the mutants, including those in genes cagE, cagF, cagG, cagH, cagI, cagL, and cagM, could no longer induce the phosphorylation of the 145-kDa host cell protein (Table 2). However, none of the three independent insertions into cagN affected the microbes ability to phosphorylate the 145-kDa host cell protein.
Figure 2.
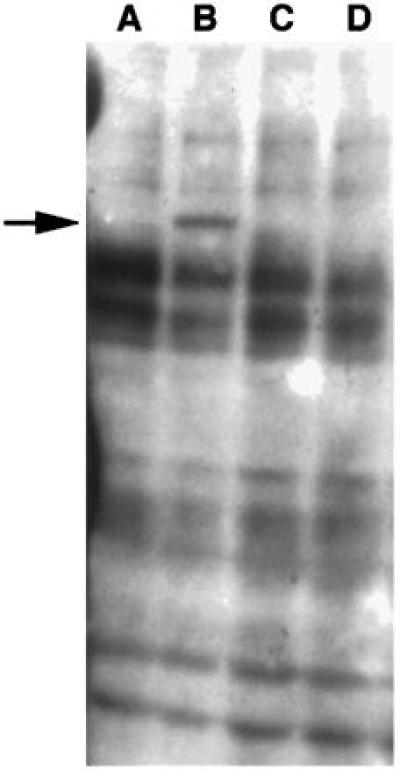
Immunoblot analysis of AGS cells infected with H. pylori and probed with anti-phosphotyrosine antibody. Lanes: A, extract of AGS cells; B, extract of AGS cells to which H. pylori strain 87A300 had attached for 4 hr; C, extract of AGS cells to which H. pylori strain G104 had attached for 4 hr; D, extract of AGS cells to which H. pylori strain G198 had attached for 4 hr.
Table 2.
Characterization of the ability to induce phosphorylation of host proteins by H. pylori
| H. pylori strain | Phosphotyrosine |
|---|---|
| 87A300 | + |
| ure1 | + |
| G27 | + |
| cagE | − |
| cagF | − |
| cagG | − |
| cagH | − |
| cagI | − |
| cagL | − |
| cagN | + |
| cagM | − |
| 314 | + |
| G50 | − |
| G104 | − |
| G198 | − |
Ability to Induce IL-8 Secretion.
It has been shown in vivo and in vitro (17, 18) that H. pylori infection induces IL-1a, IL-1b, and IL-8, and that this process is independent of the expression of CagA, VacA, urease, or lipopolysaccharide (13, 19–21). The ability of H. pylori strains and mutants to induce IL-8 secretion from AGS cells is shown in Table 3. All type I isolates induced IL-8 secretion while type II isolates did not. Of the cag1 PAI mutants tested, only isolates with insertions into cagN retained their ability to induce IL-8; all other isolates failed to maintain this trait. Hence, all of the mutants in cag1 PAI showed an absolute correlation between their ability to phosphorylate host cell proteins and their ability to induce IL-8 synthesis.
Table 3.
Comparison of the ability to induce IL-8 secretion and tyrosine phosphorylation of host cell proteins by H. pylori
| Strain | Phosphotyrosine* | IL-8, ng/ml |
|---|---|---|
| Negative control | 0.5 ± 0.866 | |
| 87A300 | + | 44.6 ± 2.97 |
| G50 | − | 0.0 |
| G198 | − | 1.5 |
| 314 | + | 17.0 |
| G27 | + | 35.25 ± 0.354 |
| 10B4 | − | 2.5 |
| Mfe/Kan | − | 3.5 |
| 14A4 | + | 22.5 |
| H12-18A | + | 34.0 |
| ure1 | + | 46.8 |
Tyrosine phosphorylation of 145-kDa host cell protein.
Kinase Inhibition Studies.
We employed kinase inhibitors of varying substrate specificity to gain further insight into the possible host cell pathways that might participate in the phosphorylation of the 145-kDa host cell proteins and their effects on IL-8 induction by AGS cells. Two broad-range inhibitors with overlapping specificities were used: staurosporine [inhibits Ca2+/calmodulin kinase, myosin light chain kinase, protein kinase A (PKA), protein kinase C (PKC), and protein kinase G (PKG)] (22) and genistein (inhibits protein tyrosine kinases including epidermal growth factor receptor, PKA and PKC) (23). In addition, three inhibitors with distinct substrate profiles were used: bisindolymaleimide I (inhibits PKC) (24), H-89 (inhibits PKA) (25), and KT5823 (inhibits PKG) (26). The effects of the various kinase inhibitors on IL-8 induction by H. pylori and tyrosine phosphorylation of the 145-kDa host protein after H. pylori infection are summarized in Table 4. Exposure of AGS cells to staurosporine, genistein, and KT5823 inhibited IL-8 induction by H. pylori to a greater extent than the presence of bisindolymaleimide I or H-89. Genistein and KT5823 were also able to inhibit tyrosine phosphorylation of the 145-kDa protein, while staurosporine, bisindolymaleimide I, and H-89 had no measurable effect.
Table 4.
Effects of kinase inhibitors on IL-8 and tyrosine phosphorylation induced by H. pylori
| Inhibitor | IL-8 inhibition, % | Phosphotyrosine inhibition |
|---|---|---|
| Staurosporine | 100.0 | − |
| Genestein | 75.0 | + |
| Bisindolymaleimide I | 48.5 | − |
| H-89 | 50.0 | − |
| KT5823 | 85.0 | + |
Colocalization of VASP with Attached H. pylori.
VASP is a known substrate for both cAMP- and cGMP-dependent protein kinases (27), placing VASP phosphorylation at the junction of two signal transduction pathways. VASP and profilin, which bind in vivo (28, 29), appear to act conjointly to relay signal transduction to the actin cytoskeleton. In consideration of the involvement of VASP with PKG and its role in cellular signal transduction pathways, we investigated the effect of H. pylori attachment to gastric cells on VASP using IF microscopy. In Fig. 3 it is shown that VASP colocalizes focally at the site of H. pylori attachment. Identical results were observed for both type I and type II isolates, and for cagI PAI mutants (data not shown).
Figure 3.
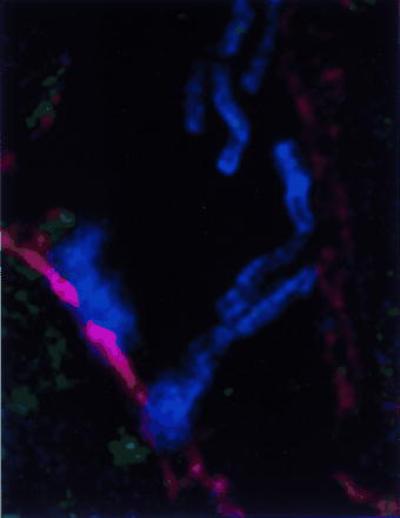
Deconvolution IF of spiral H. pylori 87A300 attached to AGS cells (0.2-μm image). Field is stained for H. pylori (blue), actin (red), and VASP (green). Where colocalization of H. pylori and actin occurs, magenta is produced. Where colocalization of actin and VASP occurs, yellow is produced. Imaged on an Applied Precision DeltaVision Deconvolution System.
DISCUSSION
PAIs identified in a number of enteric bacteria are associated with the contact-dependent modification of host cell regulatory pathways (15, 30). The 40-kb cag1 PAI found by Censini et al. (7) exclusively in type I H. pylori strains led us to investigate whether the cag1 genotype correlated with the ability of type I strains to induce tyrosine phosphorylation of a host cell 145-kDa protein and induce IL-8 secretion. Type 1 H. pylori associated with peptic ulcer disease and gastric cancer induce phosphorylation and IL-8, while type II strains more often associated with asymptomatic gastritis do not (Table 2).
Our analysis of isogenic mutants generated within the cag1 region showed that insertions within several genes of the cag1 PAI resulted in the loss of both IL-8 induction and host cell phosphorylation of the 145-kDa and 40-kDa protein. Although we report here the IL-8 phenotype of only three gene disruptions, these results agree with those reported in Censini et al. (7) who showed that insertion mutants in cagE, cagG, cagH, cagI, cagL, and cagM all lost their ability to induce an IL-8 response in Kato-3 cells, while cagN mutants were not affected. Similarly, these mutants no longer induce the phosphorylation of host cell proteins except for mutants in cagN. The similarity of the cag-encoded genes to known components of bacterial secretory pathways suggests that the expression of H. pylori virulence is triggered by contact with the host cell. As seen for other pathogenic bacteria, the effector molecules of virulence are actively secreted on the host cell surface or even translocated across the mammalian host cell membrane (30, 31).
We used kinase inhibition studies to identify possible signal transduction pathways involved in gastric cell response to H. pylori. Two broad range kinase inhibitors, staurosporine and genistein, demonstrated differing effects on the cellular response. Staurosporine did not inhibit phosphorylation of the 145-kDa protein, yet totally inhibited IL-8 induction. Genistein inhibited both tyrosine phosphorylation of the host 145-kDa protein and induction of IL-8. While staurosporine and genistein are often regarded as general kinase inhibitors, their specificities are not identical (22, 23). Genistein inhibits tyrosine kinases to a greater extent than staurosporine, which, in turn, exhibits expanded inhibition of serine/threonine (Ser/Thr) kinases as compared with tyrosine kinases. In as much that genistein was able to inhibit both IL-8 induction and tyrosine phosphorylation, we believe that the site of genistein action occurs after the target(s) of staurosporine inhibition. Additional investigation using kinase inhibitors specific for PKA, PKC, or PKG suggests that PKG, a Ser/Thr kinase, is part of the signal transduction pathways leading to IL-8 induction and tyrosine phosphorylation of the 145-kDa protein. The specificity of KT5823 for Ser/Thr kinases could indicate that there is activation of a tyrosine kinase by a Ser/Thr kinase. A precedent for this has been described in the activation of the tyrosine/threonine kinase MEK (mitogen-activating protein/epidermal growth factor receptor kinase), following activation by the MEK Ser/Thr kinase (c-raf) (32).
Our studies suggest that there are at least three distinct cellular responses to H. pylori attachment. First, there is a cytoskeletal response to bacterial adherence that is common to all type I strains tested. Although type I and type II H. pylori appear to attach to similar extents, type II induces a minimal actin rearrangement upon binding to host cells as compared with type I. This difference might be indicative of the involvement of different adhesins or might result from an alternative secretory pathway used by type II strains, which are lacking the secretory genes present in the cagI PAI. We have shown that VASP accumulation is induced by type I strains in an apparently identical manor as in type II. Second, type I strains, but not type II strains, mediate the tyrosine phosphorylation of a 145-kDa host cell protein as well as other minor host cell proteins. Finally, there is a pathway leading to IL-8 synthesis. While cag PAI mutants show a concomitant loss in both IL-8 synthesis and tyrosine phosphorylation, it is clearly possible to distinguish between these activities using kinase inhibitors. Whether this reflects that there are two distinct pathways or distinct steps in the same pathway is not clear. However, we have recently discovered that certain H. pylori mutants deficient in hemolysin expression can no longer bring about host cell phosphorylation but can still induce IL-8 following attachment to cultured AGS gastric cells (E.D.S., unpublished data). We believe that the further investigation of H. pylori virulence factors and their precise effect on host cell signal transduction pathways will provide a better understanding of the spectrum of clinical responses seen to H. pylori infection.
Acknowledgments
This research was supported by National Institutes of Health Grant AI23796 (to L.S.T. and S.F.) and Digestive Disease Center Grant of the National Institute of Diabetes and Digestive and Kidney Diseases (DK 38707) (to E.D.S., L.S.T., and S.F.).
ABBREVIATIONS
- PAI
pathogenicity island
- IL-8
interleukin 8
- VASP
vasodilator-stimulated phosphoprotein
- IF
immunofluorescence
- PKA
protein kinase A
- PKC
protein kinase C
- PKG
protein kinase G
References
- 1.Logan R P. Lancet. 1994;344:1078–1079. doi: 10.1016/s0140-6736(94)91729-9. [DOI] [PubMed] [Google Scholar]
- 2.Ghiara P, Marchetti M, Blaser M J, Tummuru M K, Cover T L, Segal E D, Tompkins L S, Rappuoli R. Infect Immun. 1995;63:4154–4160. doi: 10.1128/iai.63.10.4154-4160.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McColm, A. A., Bagshaw, J., O’Malley, C. & McLaren, A. (1991) Microb. Ecol. Health Dis. 4, Suppl., S145 (abstr.).
- 4.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N, Rappuoli R. Proc Natl Acad Sci USA. 1993;90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weel J F, van der Hulst R W, Gerrits Y, Roorda P, Feller M, Dankert J, Tytgat G N, van der Ende A. J Infect Dis. 1996;173:1171–1175. doi: 10.1093/infdis/173.5.1171. [DOI] [PubMed] [Google Scholar]
- 6.Xiang Z, Censini S, Bayeli P F, Telford J L, Figura N, Rappuoli R, Covacci A. Infect Immun. 1995;63:94–98. doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Censini S, Lange C, Xiang Z, Crabtree J E, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. Proc Natl Acad Sci USA. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winans S C, Burns D L, Christie P J. Trends Microbiol. 1996;4:64–68. doi: 10.1016/0966-842X(96)81513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crabtree J E, Peichl P, Wyatt J I, Stachl U, Lindley I J. Scand J Immunol. 1993;37:65–70. doi: 10.1111/j.1365-3083.1993.tb01666.x. [DOI] [PubMed] [Google Scholar]
- 10.Crabtree J E, Covacci A, Farmery S M, Xiang Z, Tompkins D S, Perry S, Lindley I J, Rappuoli R. J Clin Pathol. 1995;48:41–45. doi: 10.1136/jcp.48.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tummuru M K, Sharma S A, Blaser M J. Mol Microbiol. 1995;18:867–876. doi: 10.1111/j.1365-2958.1995.18050867.x. [DOI] [PubMed] [Google Scholar]
- 12.Crabtree J E, Xiang Z, Lindley I J, Tompkins D S, Rappuoli R, Covacci A. J Clin Pathol. 1995;48:967–969. doi: 10.1136/jcp.48.10.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma S A, Tummuru M K, Miller G G, Blaser M J. Infect Immun. 1995;63:1681–1687. doi: 10.1128/iai.63.5.1681-1687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segal E D, Falkow S, Tompkins L S. Proc Natl Acad Sci USA. 1996;93:1259–1264. doi: 10.1073/pnas.93.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee C A. Infect Agents Dis. 1996;5:1–7. [PubMed] [Google Scholar]
- 16.Segal E D, Shon J, Tompkins L S. Infect Immun. 1992;60:1883–1889. doi: 10.1128/iai.60.5.1883-1889.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crabtree J, Plusa S, Farmery S, Peichl P, Lindley I L D. Acta Gastro-Enterol Belg. 1993;56:52. (abstr.). [Google Scholar]
- 18.Peek R J, Miller G G, Tham K T, Perez P G, Zhao X, Atherton J C, Blaser M J. Lab Invest. 1995;73:760–770. [PubMed] [Google Scholar]
- 19.Tummuru M K, Cover T L, Blaser M J. Infect Immun. 1994;62:2609–2613. doi: 10.1128/iai.62.6.2609-2613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang J, O’Toole P W, Doig P, Trust T J. Infect Immun. 1995;63:1732–1738. doi: 10.1128/iai.63.5.1732-1738.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crowe S E, Alvarez L, Dytoc M, Hunt R H, Muller M, Sherman P, Patel J, Jin Y, Ernst P B. Gastroenterology. 1995;108:65–74. doi: 10.1016/0016-5085(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 22.Omura S, Sasaki Y, Iwai Y, Takeshima H. J Antibiot. 1995;48:535–548. doi: 10.7164/antibiotics.48.535. [DOI] [PubMed] [Google Scholar]
- 23.Peterson G. J Nutr. 1995;125:784S–789S. doi: 10.1093/jn/125.suppl_3.784S. [DOI] [PubMed] [Google Scholar]
- 24.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charon D, Kirilovsky J. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 25.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 26.Gadbois D M, Crissman H A, Tobey R A, Bradbury E M. Proc Natl Acad Sci USA. 1992;89:8626–30. doi: 10.1073/pnas.89.18.8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haffner C, Jarchau T, Reinhard M, Hoppe J, Lohmann S M, Walter U. EMBO J. 1995;14:19–27. doi: 10.1002/j.1460-2075.1995.tb06971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch B M, Walter U. EMBO J. 1995;14:1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reinhard M, Jouvenal K, Tripier D, Walter U. Proc Natl Acad Sci USA. 1995;92:7956–7960. doi: 10.1073/pnas.92.17.7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mecsas J J, Strauss E J. Emerg Infect Dis. 1996;2:270–288. doi: 10.3201/eid0204.960403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galan J E, Bliska J B. Annu Rev Cell Dev Biol. 1996;12:221–255. doi: 10.1146/annurev.cellbio.12.1.221. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava A K. Mol Cell Biochem. 1995;149:87–94. doi: 10.1007/BF01076567. [DOI] [PubMed] [Google Scholar]