

Abstract
The process of self-assembly of the triple-helical peptide (Pro-Hyp-Gly)10 into higher order structure resembles the nucleation-growth mechanism of collagen fibril formation in many features, but the irregular morphology of the self-assembled peptide contrasts with the ordered fibers and networks formed by collagen in vivo. The amino acid sequence in the central region of the (Pro-Hyp-Gly)10 peptide was varied and found to affect the kinetics of self-assembly and nature of the higher order structure formed. Single amino acid changes in the central triplet produced irregular higher order structures similar to (Pro-Hyp-Gly)10, but the rate of self-association was markedly delayed by a single change in one Pro to Ala or Leu. The introduction of a Hyp-rich hydrophobic sequence from type IV collagen resulted in a more regular suprastructure of extended fibers that sometimes showed supercoiling and branching features similar to those seen for type IV collagen in the basement membrane network. Several peptides, where central Pro-Hyp sequences were replaced by charged residues or a nine-residue hydrophobic region from type III collagen, lost the ability to self-associate under standard conditions. The inability to self-assemble likely results from loss of imino acids, and lack of an appropriate distribution of hydrophobic/electrostatic residues. The effect of replacement of a single Gly residue was also examined, as a model for collagen diseases such as osteogenesis imperfecta and Alport syndrome. Unexpectedly, the Gly to Ala replacement interfered with self-assembly of (Pro-Hyp-Gly)10, while the peptide with a Gly to Ser substitution self-associated to form a fibrillar structure.
Keywords: triple helix, collagen, self-assembly, fibrils, peptides
Collagen molecules self-assemble into higher order structures that provide mechanical support to the extracellular matrix and mediate cell attachment and other biological processes. The most abundant collagens, e.g., type I collagen, are found in fibrils with a periodicity of D = 670 Å, which arises from the axial staggering of adjacent molecules and is related to the distribution of charged and hydrophobic residues (Hulmes et al. 1973; Hofmann et al. 1980). In addition to fibril forming collagens, there are more than 20 types of nonfibrillar collagens which form distinctive higher order structures required for their specific role in the extracellular matrix (Kielty and Grant 2002; Myllyharju and Kivirikko 2004). For example, type IV collagen forms network-like structures in basement membranes, involving interactions of C-terminal globular domains as well as supercoiling and branching of triple-helical regions (Yurchenco and Ruben 1987, 1988; Yurchenco and Schittny 1990). Supramolecular organizations of other nonfibrillar collagens include hexagonal networks of type VIII collagen in the subendothelial layers, antiparallel arrays of type VII collagen in anchoring fibrils attached to the basal lamina of the skin, and microfibrils of type VI collagen in all connective tissues (Engel et al. 1985; Burgeson 1993; Kielty and Grant 2002; Baldock et al. 2003; Myllyharju and Kivirikko 2004; Stephan et al. 2004). The general principles that allow collagen molecules to form diverse supramolecular structures are not well understood.
Underlying the diversity of supramolecular structures, all collagens are defined by the presence of a triple-helix domain. The molecular conformation of the collagen triple helix consists of three polyproline II-like chains supercoiled around a common axis (Ramachandran and Kartha 1955; Rich and Crick 1961). Each polypeptide chain has a unique repeating (Gly-X1-X2)n sequence, reflecting the requirement for Gly as every third residue to allow close packing and hydrogen bonding between the three chains. A high content of the imino acids Pro and Hyp stabilizes the triple helix by promoting a polyproline II-like conformation in individual chains, while the posttranslationally modified Hyp introduces additional stability through stereo-electronic effects and participation in a hydration network (Bella et al. 1994; Jenkins and Raines 2002). The indentities of the residues in the X1 and X2 positions appear to promote sequence-dependent variations in local stability or conformation along the triple helix (Kramer et al. 2001; Persikov et al. 2005). Features of the basic triple-helix structure as well as sequence-dependent interactions involving side chains are likely to determine the self-assembly process and the molecular organization of higher order structures.
The process of self-assembly of type I collagen into D-periodic fibrils has been extensively studied (Kadler et al. 1987, 1988; Prockop and Fertala 1998). Fibril formation can occur in vitro, and it follows a nucleation–propagation mechanism that increases with temperature. The process is fastest at temperatures just below the Tm value of collagen, leading to the suggestion that micro-unfolding at temperatures close to the melting temperature plays an important role (Kadler et al. 1988; Leikina et al. 2002). Direct measurements of the forces responsible for fibril stability indicate that hydration is a major driving force of collagen self-assembly (Leikin et al. 1995, 1997). In vitro reconstitution of basement membrane-like networks from type IV collagen (Yurchenco and Ruben 1988), hexagonal networks from type VIII collagen (Stephan et al. 2004), and anchoring fibrils from type VII collagen (Chen et al. 2001) have been observed, but these processes, kinetics, and macromolecular organization are much less defined than the formation of D-periodic fibrils.
Collagen-like peptides with Gly as every third residue and a high content of imino acids form stable triple helices, and have proved amenable to systematic stability studies, X-ray crystallography, and NMR characterization (Baum and Brodsky 1999; Li et al. 2007). A recent report from this laboratory showed that the triple-helical peptide (Pro-Hyp-Gly)10 (abbreviated as [POG]10) assembles into higher order structures in a process that resembles that of collagen fibril formation (Kar et al. 2006). Self-assembly of (POG)10 follows nucleation-growth kinetics, with lag, growth, and plateau phases, and the rate of association increases with temperature, being fastest at temperatures close to the Tm of the peptide. The similarities between the self-association of collagen and this simple peptide were striking, including the effect of pH, sugars, temperature, and concentration. However, the morphology of the higher order structure generated from (POG)10 shows an irregular aggregated structure, indicating a lack of specificity in self-association compared with the highly ordered D-periodic fibrils of collagen.
Here, studies on the self-association of peptides are extended to better clarify sequence dependence and specificity. Some residue changes in (POG)10 resulted in no self-association. Several single amino acid substitutions introduced into the (POG)10 peptide are found to affect the kinetics but not the final morphology of the associated structure. One sequence change, involving the introduction of a hydrophobic Hyp-rich sequence from type IV collagen, dramatically affected the morphology but not the kinetics of higher order structure formation. The peptide with the type IV sequence formed a fibrous structure of regular diameter, with supercoiling of the fibers leading to branching in some cases. These results suggest that small changes in amino acid sequence can direct the kinetics and affect the morphology of the self-assembled structure.
Results
Ability of triple helices to self-associate
The inherent ability of triple-helical peptides to self-associate was investigated. Previous results showed that incubation of the triple-helical peptide (POG)10 at 7 mg/mL resulted in self-association and precipitation within 20 min, with the rate of association increasing with higher temperature (Kar et al. 2006). It was also shown that (Pro-Pro-Gly)10 did not undergo any aggregation even after incubation for 1 wk at high concentrations (up to 14 mg/mL) at 1°C–4°C below its Tm. Here, the potential of triple-helical peptides to undergo self-assembly is extended to four peptides homologous to (POG)10 that were previously found to form stable triple helices (Table 1). Each peptide chain contains 10 tripeptide units, with 3–4 (Pro-Hyp-Gly) triplets on the C terminus and 3–4 (Pro-Hyp-Gly) tripeptides on the N terminus. The central amino acid sequences of these peptides contain charged residues (GPKGEK, GQKGEK, GPKGEKGEO) or a hydrophobic, imino acid poor sequence near the collagen cleavage site of type III collagen (GITGARGLA; peptide T3–785); these peptides had a C-terminal Tyr to allow concentration determination. Each peptide was incubated at a concentration of 7 mg/mL at temperatures 2°C–5°C below their Tm and monitored by the absorbance at 313 nm to detect turbidity indicating aggregation. None of these four triple-helical peptides showed any indication of thermally induced self-assembly under these conditions (Table 1).
Table 1.
List of collagen peptides that do not undergo self-assembly at 7 mg/mL in PBS at pH 7 and temperatures just below their Tm value, with their sequence and thermal stability (Tm)

In light of the inability of the above four peptides to self-associate, a more systematic stepwise approach was undertaken to vary sequences in (POG)10 to design other self-associating triple-helical peptides. A peptide was synthesized, where one Hyp residue in (POG)10 was replaced by Ala (peptide O14A), and peptides were designed to replace one Pro residue in (POG)10 by an Ala residue (peptide P13A) or by a Leu residue, peptide P13L (Table 2).
Table 2.
List of collagen peptides that show self-assembly at 7 mg/mL in PBS buffer at pH 7 with their sequences, melting temperatures (Tm), and self-association time at its optimal incubation temperature

Circular dichroism spectroscopy studies show peptide O14A forms a stable triple helix with Tm = 52°C, a value somewhat lower than (POG)10 (Tm = 59°C) as expected for the replacement of a Hyp by an Ala residue. An increase in turbidity is observed as a function of incubation time at 52°C, with a lag, growth, and plateau phase, indicating a nucleation-growth self-assembly process. As seen for (POG)10, the rate of self-assembly for peptide O14A accelerates with increasing temperature and the maximum rate is observed near its Tm = 52°C (see Fig. 2A). At the optimum temperature, the kinetics are similar to that observed for (POG)10, with a slightly longer lag phase (∼10 min longer) (Fig. 1B). The rates of self-assembly for peptide O14A obtained at different temperatures fit a linear Arrhenius plot (see Supplemental material) with an activation energy Ea = 158 ± 8 kJ/mol, a value higher than the Ea observed for (POG)10 (137 ± 10 kJ.mol−1). Differential scanning calorimetry at a heating rate of 1°C/min shows two distinct transitions: a large one at 59°C, and a small one at 75°C (Fig. 1C). The first transition at 59°C represents the melting of triple helices, and appears at a higher temperature than the CD Tm value because of the higher DSC heating rate under nonequilibrium conditions (Persikov et al. 2004b). Consistent with the previous analysis of (POG)10 (Kar et al. 2006), the second DSC transition at 75°C coincides with a loss of turbidity, and is therefore likely to represent the melting of aggregated structures of O14A formed during heating. The ∼25°C difference between the molecular unfolding and the aggregate melting is similar for peptides O14A and (POG)10. The critical concentration of polymerization was somewhat higher for peptide O14A than seen for (POG)10 (Table 2).
Figure 2.
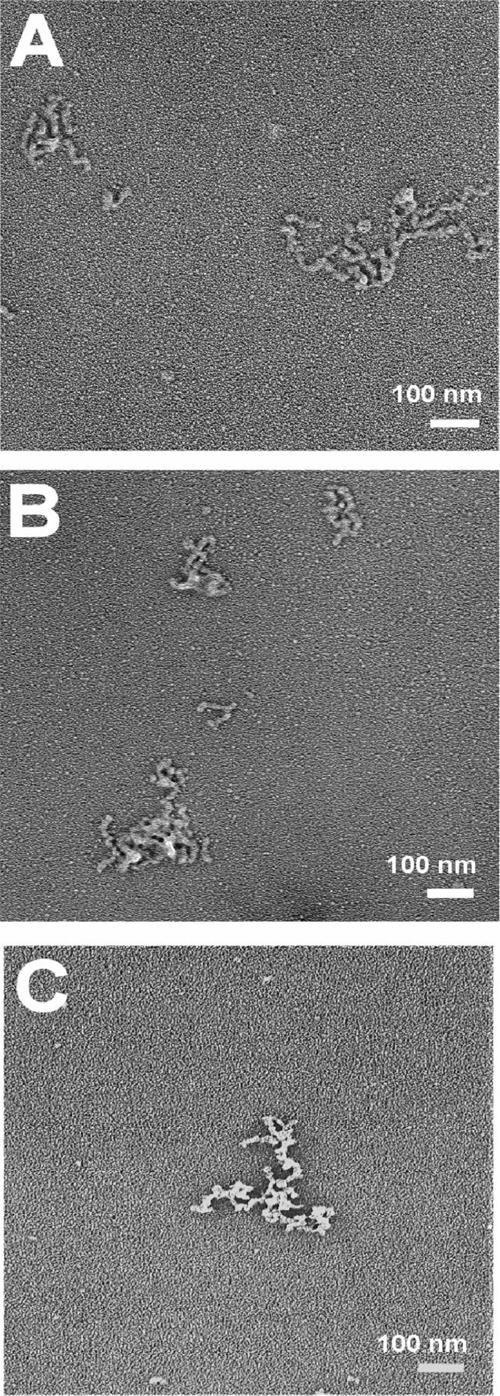
Electron micrographs of the self-assembled structures formed by the collagen peptides: (A) (Pro-Hyp-Gly)10, (B) O14A, and (C) P13L. Each peptide (7 mg/mL, PBS, pH 7) was incubated at its optimum temperature for aggregation. The sample was collected after the onset of the plateau phase for microscopy.
Figure 1.

(A) The temperature dependence of the self-assembly process for peptide O14A (7 mg/mL, PBS, pH = 7) monitored by turbidity measurements (absorbance at 313 nm). (B) Turbidity curves showing temperature induced self-assembly of collagen peptides (7 mg/mL, PBS, pH = 7): (1) (POG)10 at 58°C, (2) O14A at 52°C, (3) P13L at 51°C (dashed line), and (4) P13A at 51°C. The inset shows turbidity (absorbance at 313 nm) measured as a function of time at 51°C for the peptide P13L at a higher concentration of ∼16 mg/mL. (C) DSC profile of the thermal unfolding of (POG)10 (○), O14A (■), and P13L (▲).
CD studies indicate peptides P13A and P13L form stable triple-helical structures with a somewhat lower stability than (POG)10 (Tm = 59°C for [POG]10; Tm = 52.5°C for P13A; Tm = 50.3°C for P13L), as predicted for the replacement of a Pro by and Ala or Leu residue. After incubation of peptides P13A and P13L at 50°C and 51°C, respectively, no increase in turbidity was observed initially (Fig. 1B), but a gradual increase was seen after about 12 h. Self-association of both peptides included a very long lag phase (∼12 h) followed by a growth and plateau phase (Supplemental material). Increasing the concentration of P13L from 7 mg/mL to ∼16 mg/mL resulted in faster kinetics of the process of self-assembly, decreasing the lag time from 12 h to 1.5 h (Fig. 1B, inset). Thus, substitution of one Pro residue by either Ala or Leu significantly decreases the rate of self-assembly compared with (POG)10 and O14A, with the main effect appearing to be a delay of the nucleation process in the lag phase. A DSC scan of the peptide P13L (7 mg/mL) at a heating rate of 1°C/min shows only one transition representing the melting of triple helix at 58°C. The very slow kinetics for P13L self-association means that aggregation would not occur at the DSC heating rate, so a second higher transition is not observed. The critical concentration of polymerization for peptide P13A was twice as high as that seen for (POG)10, suggesting the replacement of one Pro led to a decreased tendency for aggregation.
Electron microscopy studies were carried out on rotary shadowed specimens to visualize the higher order structures formed. Both peptides O14A and P13L show a branched irregular morphology very similar to the aggregated structures of (POG)10 (Fig. 2). Thus, modification of (POG)10 by changing single residues in the X1 or X2 position did not affect the nature of the higher order structures formed.
Self-association of the triple-helical peptides with a type IV collagen sequence
To extend these studies, self-association was investigated for a triple-helical peptide containing a hydrophobic, Hyp-rich sequence from type IV collagen. A typical Hyp-rich sequence in the α5 chain of human type IV collagen (residue 491–499) (GPOGQOGLOGLOGPO) was embedded in Gly-Pro-Hyp flanking sequences to form peptide T4α5–491 (Table 2). CD studies indicate that peptide T4α5–491 forms a stable triple helix (Tm = 41.3°C), and this stability is close to the one predicted from the effect of replacing three Pro residues by Gln and two Leu residues (Persikov et al. 2005). Monitoring the turbidity of this peptide at 40°C indicates little initial change, but shows a growth and plateau phase after ∼24 h, suggesting a substantial delay in the lag phase (Fig. 3A, inset).
Figure 3.

(A) Turbidity curves showing self-assembly (7 mg/mL, PBS, pH = 7) of a set of triple-helical peptides with a type IV sequence, differing only in their terminal residues: (1) T4α5–491Y at T = 40°C, (2) T4α5–491Y′ at T = 44°C, (3) T4α5–491 at T = 40°C. Inset is the turbidity curve for the peptide T4α5–491, showing the slower kinetics of self-assembly with a timescale of hours. (B) Turbidity curves showing the temperature dependence of self-assembly for peptide T4α5–491Y (7 mg/mL, PBS, pH = 7). (C) DSC profiles of thermal unfolding of peptide T4α5–491Y′ showing two distinct transitions.
In order to more accurately measure concentration, a Tyr was added to the C terminus of peptide T4α5–491 to generate the sequence POGPOGPOGQOGLOGLOGPOGPOGPOGPOGY, which is denoted as peptide T4α5–491Y (see Table 2). A peptide was also constructed in a modified format with Gly at the N terminus and a Tyr at the C terminus: GPOGPOGPOGQOGLOGLOGPOGPOGPOGPOGY, denoted as peptide T4α5–491Y′. CD studies indicate both peptides with Tyr form stable triple-helix structures (Tm = 44.5°C for T4α5–491Y′, Tm = 41°C for T4α5–491Y). Turbidity studies show that both Tyr-containing peptides exhibit strong temperature-dependent self-assembly with much faster kinetics than seen for T4α5–491 without the Tyr residue (Fig. 3A). Precipitation occurs within an hour of incubation, generating a typical turbidity curve with a lag phase, growth phase, and plateau phase similar to that of (POG)10. The rate of self-assembly is greatly enhanced with increasing temperature, with an optimum near its Tm. Association of peptide T4α5–491Y′ occurs at any temperature above a critical temperature of 30°C at 7 mg/mL in PBS, and the maximal rate of assembly is at 44°C (Tm = 44.5°C). The rates obtained from temperature-dependent turbidity curves were used to calculate activation energy from the Arrhenius plot, Ea = 148 (±10) kJ.mol−1 (see Supplemental material). The values of critical concentration for these T4 peptides were similar to those seen for (POG)10 (Table 2).
Thermal unfolding of T4α5–491Y′ by DSC (heating at 1°C/min) under aggregating conditions shows two distinct peaks (Fig. 3C). The large first transition at 49°C corresponds to triple-helix melting. The second small transition at 87°C corresponds to a loss of turbidity and is likely to reflect dissociation of aggregated structures formed during the DSC scan. The 38°C difference between the triple helix unfolding and the aggregate dissociation is greater than that seen for the other peptides, suggesting there may be additional interactions stabilizing the higher order structure.
Electron microscopy of rotary shadowed specimens of T4α5–491 peptides after self-association shows morphology very different from the higher order structures obtained for the peptides (POG)10, P13L, and O14A (Fig. 4A). Two of the T4α5–491 peptides showed regular, linear fibers with a diameter ∼20 nm. The third peptide T4α5–491Y′ showed fibers of similar diameter, that were sometimes supercoiled around each other, producing a branching appearance (Fig. 4C). Compared to the irregular worm-like associated structures of (POG)10, P13L, and O14A, the self-assembled structures of T4α5–491Y′ appear to be regular, with a longer linear coherence length.
Figure 4.

Electron micrograph of the self-assembled structures formed by the set of collagen peptides with a type IV sequence. (A) T4α5–491, (B) T4α5–491Y, and (C) T4α5–491Y′. A magnified view of a portion of C is shown in D, with small arrows indicating the branching and supercoiled nature of the structure.
Effect of replacement of one Gly residue on peptide self-association
The replacement of one Gly by Ala in (POG)10, changes this peptide from a self-associating triple helix to a nonassociating triple helix (c = 7 mg/mL, at 30°C). However, the substitution of one Gly in (POG)10 by Ser did allow self-association. The self-association took more than 12 h, placing it in the category of slower associating peptides. The Gly-Ser peptide (7 mg/mL) incubated at 25°C in PBS at pH 7 resulted in a less turbid solution than seen for other aggregating peptides, with absorbance increasing from 0.001 to 0.23 after ∼16 h. Fiber-like structures were formed, with uniform diameter. These appear more rigid than the (POG)10 structure, but have some indication of flexibility (Fig. 5).
Figure 5.

Electron micrograph of the self-assembled structure formed by the peptide (POG)10 with one Gly replaced by Ser. The peptide (7 mg/mL, PBS, pH = 7) undergoes aggregation resulting in a slightly turbid solution when incubated at 25°C for 1 d.
Discussion
It was previously reported that the triple-helix peptide (POG)10 undergoes a thermally induced self-assembly process following a nucleation–propagation mechanism similar to collagen (Kar et al. 2006), but the higher order structure formed lacks the extended linear nature and order seen in type I collagen fibers or type IV collagen networks. This simple repeating peptide appears to contain intrinsic information essential for triple-helical molecules to self-associate, but to lack critical interactions needed for the specific intermolecular packing found in in vivo supramolecular structures. We hypothesize that self-association of the (POG)10 peptide involves the nonspecific interactions that promote lateral self-assembly of triple-helical collagen molecules. Experimental data suggest this nonspecific driving force for collagen fibrillogenesis is dominated by hydrogen bonded hydration networks bridging adjacent molecules (Leikin et al. 1995, 1997; Kuznetsova et al. 1997).
The packing of collagen model peptides within crystals provides some insight into the interactions between collagen triple helices, since the crystal packing often shows intermolecular distance and quasi-hexagonal packing reminiscent of the organization of collagen molecules in native fibrils in rat tail tendon, skin, and other tissues (Bella et al. 1994). Most intermolecular interactions between adjacent triple-helix molecules in peptide crystal structures are water mediated, while direct contacts are seen between Hyp residues and between charge side chains and backbone C=O groups (Kramer et al. 2000; Vitagliano et al. 2001). In contrast to simple repeating peptides, it is likely that hydrophobic, electrostatic, and hydrogen bonding interactions specify the axial relationships between collagen molecules which lead to well-defined supramolecular assemblies such as periodic fibrils or networks (Hulmes et al. 1973; Hofmann et al. 1980; Leikin et al. 1995).
Amino acid sequences of the triple-helix peptides were varied to clarify the nonspecific driving force for self-assembly and to investigate the consequences of introducing hydrophobic and charged residues. Monitoring the ability of the peptides reported here to self-associate by turbidity indicate three types of triple-helical peptides: those which self-assemble rapidly at 7 mg/mL (lag phase of <60 min), those which self-associate slowly (lag phase ∼12–24 h), and those which do not self-associate even after 1 wk of incubation. Rapid self-association may depend on having Pro residues in the X1 positions, since replacement of GPO triplets by GAO, GLO, or GQOGLOGLO led to slow association, while the replacement of one Hyp in the X2 position by Ala (GPA) still led to rapid kinetics. Unlike collagen, the activation energy for aggregation (Ea ∼ 137 kJ/mol) of the peptide (POG)10 is of a similar magnitude to its unfolding activation energy (Ea ∼ 135 kJ/mol), raising the possibility of a major conformational change upon self-association of the peptide. The values for the critical concentration of polymerization were much higher for peptides than seen for collagen (Kadler et al. 1988), and some sequence-dependent variations were observed.
For the GQOGLOGLO peptides, the presence of Tyr at the C-terminal of these type IV model peptides greatly accelerated the nucleation process, changing them from a slow to a fast self-association category. However, if a peptide does not have the intrinsic potential to self-associate, the presence of Tyr did not confer this ability (Table 1). A recent publication reports fibril formation by the peptide (POG)10 with a pentafluorophenylalanine on the N terminus and a Phe on the C terminus, with the suggestion that the terminal aromatic residues play a role in formation of the supramolecular structure (Cejas et al. 2007). The effect of terminal aromatic residues on triple-helix self-assembly could be related to the catalytic effect of non-helical terminal telopeptides of type I collagen on the kinetics of collagen fibril formation (Prockop and Fertala 1998; Kuznetsova and Leikin 1999).
The morphology of the aggregated structures formed by (POG)10, O14A, and P13L had an irregular appearance, giving the appearance of some underlying elongated structure which coils back on itself. A number of recent reports on the self-association of collagen model peptides with repeating Gly-Pro-Hyp sequences (Kishimoto et al. 2005; Kotch and Raines 2006) showed similar morphology. A vertical polymer-like disorder is seen in the crystal structure of (POG)10, due to the repetition of Pro-Hyp-Gly units in the sequence (Nagarajan et al. 1999; Berisio et al. 2000), and it may be that the same lack of sequence discrimination leads to the irregular morphology in these non-crystalline aggregated structures.
In contrast to (POG)10, O14A, and P13L, the T4α5–491 peptides with the sequence GPOGQOGLOGLOGPO generate more regular fibril structures of uniform diameter, which appear extended and less flexible. In some cases, two of these fibers supercoil around each other, in a manner reminiscent of that seen for type IV collagen basement membrane networks (Yurchenco and Ruben 1988), generating branching in the peptide supramolecular structures (Fig. 4C,D). Type IV collagen contains long stretches of sequences with Hyp in every third position, where the most frequent tripeptide sequences are Gly-Pro-Hyp (10%) and Gly-Leu-Hyp (9%). The single crystal structure for a triple-helical peptide containing a Gly-Leu-Hyp-Gly-Leu-Hyp sequence was reported recently (Okuyama et al. 2007). Hydrophobic interactions are seen between adjacent triple helices near the Leu sites and no polymer-type disorder is present. Similar hydrophobic interactions generated by the Gly-Leu-Hyp-Gly-Leu-Hyp sequences in the T4α5–491 peptides could specify axial relationships between collagen molecules and lead to well-defined supramolecular assemblies. A staggered arrangement of parallel or antiparallel triple-helical molecules must be proposed in order to promote elongation of the fibrillar structures formed from the peptides, and some interaction must limit the fibril diameter. The peptide results suggest the frequent Gly-Leu-Hyp sequences in type IV collagen could play a role in its network structure and could relate to the observed supercoiling and branching. This hypothesis is consistent with the computational analysis of Knupp and Squire (2001) who suggested the distribution of hydrophobic residues on the surface of the type VI collagen triple helix is responsible for the supercoiling of these molecules.
A recent publication by Rele et al. (2007) shows the ability of a triple-helix peptide with positively charged residues at the N-terminal end, and negatively charged residues on the C-terminal end, flanking central Gly-Pro-Hyp sequences, to self-associate to form periodic fibers, resembling banded collagen fibrils. It appears that hydrophobic and electrostatic interactions may independently or together have the ability to promote specific supramolecular structures in peptides. Although the results reported here and in the study by Rele et al. (2007) show that hydrophobic and electrostatic interactions can generate more specific self-association of triple-helical molecules to higher order structures, it was observed that several peptides with a hydrophobic sequence from type III collagen or short stretches of charged residues did not undergo self-assembly (Table 1). These results indicate the importance of the distribution of the charged residues and/or hydrophobic residues and suggest the need for multiple reinforcing interactions to attain higher order assembly.
The effect of replacing one Gly by Ser and Ala in the repeating (Gly-X1-X2)n sequence on self-association was also studied here, to model the Gly mutations seen in collagen diseases, such as osteogenesis imperfecta (OI) (Byers and Cole 2002; Marini et al. 2007). Gly-Ser mutations are the most frequent missense mutations observed in OI, in contrast with Gly-Ala mutations, which are underrepresented (Persikov et al. 2004a; Marini et al. 2007). Despite the similar effects that Ala and Ser substitutions have on triple-helix stability and hydrogen bonding within triple-helical peptides (Beck et al. 2000; Bhate et al. 2002), the replacement of one Gly residue in (POG)10 by Ala prevents self-assembly, while the replacement of one Gly by Ser allows self-association and the formation of fibrillar structures. The ability of Ser to form additional hydrogen bonds through its side chain may play a role in the self-association of the Gly-Ser peptide, and could have implications for the participation of OI collagens with Gly-Ser mutations in fibril formation.
The observations reported here suggest there may be wide variations in the potential for different sequences along a collagen chain to promote higher order structure and/or specific supramolecular structures. An increased understanding of the relation between collagen sequence and its ability to self-associate is key for understanding normal and pathological self-association of collagens and will be important for the design of collagen based biomaterials.
Material and Methods
Peptides
The peptides (Pro-Hyp-Gly)10, and (Pro-Pro-Gly)10 were obtained from Peptides International. The collagen model peptides (see Tables 1 and 2) denoted as PKGEKG, QKGEKG, KGEKGE, Gly → Ala, T3–785, O14A, P13L, P13A, T4α5–491, T4α5–491Y, and T4α5–491Y′ were synthesized by Tufts University Core facility (Boston, Massachusetts). The peptide Gly → Ser was obtained from Alta Biosciences. The peptides were purified on a C-18 column using a reverse-phase HPLC system (Shimadzu) and the purity was ensured by mass spectrometry using MALDI-TOF (DE-PRO mass spectrometer). Concentrations of the peptide solutions were measured by monitoring the absorbance at 214 nm using the ε214 = 2200 cm−1·M−1 per peptide bond. For peptides having Tyr residues, peptide concentration was determined using an extinction coefficient of ε280 nm = 13,980 M −1cm1 (Gill and von Hippel 1989). Buffers used included 20 mM PBS buffer (10 mM NaH2PO4, 10 mM Na2HPO4, and 150 mM NaCl) for pH 7; acetate buffer (20 mM with 150 mM NaCl) for pH 3.0.
Turbidity measurement
Turbidity curves monitoring the process of self-assembly were obtained using the optical density at 313 nm as a function of time on a Beckman DU 640 spectrophotometer with a Peltier temperature controller. A peptide solution of 600 μL was kept in a 2-mm cell sealed to avoid evaporation and then subjected to the desired constant temperature. The activation energy for the self-assembly process was calculated following the previously reported method using the Arrhenius equation, k = A · e −Ea/RT (Kar et al. 2006). The turbidity curves obtained at different temperatures were used to obtain the value of the rate constant, k, from the slope, dA 313 /dt, of the linear growth phase of the turbidity curves. The plot of ln(dA 313/dt) versus 103 · 1/T (K −1) resulted in a linear Arrhenius plot for the studied peptides, and its slope was used in the calculation of activation energy.
Circular dichroism spectroscopy
Circular dichroism (CD) measurements were carried out using an Aviv Model 62DS spectrophotometer (Aviv Biomedical, Inc.). Prior to CD measurements each sample was incubated at 4°C for 2–3 d to allow the formation of triple helix. The characteristic triple-helix CD maximum at 225 nm was used to monitor thermal transitions, refolding curves, reversibility of the process of aggregation, and measurements of critical concentration. For determining the critical concentration, peptide samples at the end of the plateau phase of aggregation were centrifuged and the supernatant analyzed by the mean residue ellipticity at 225 nm in the CD spectrum to determine the percentage of the peptide remaining in solution. Following centrifugation, each supernatant was incubated at 4°C for 2 d to allow complete formation of the native triple helix of all peptides in solution prior to the CD measurement.
Rotary shadowing and electron microscopy
Electron microscopy was carried out on rotary shadowed samples of the peptides to visualize the morphology of the self-assembled higher order structures formed. A small aliquot of the sample was placed on a 400-mesh copper grid following the method as described earlier (Kar et al. 2006). Samples were rotary shadow cast with tungsten using a JEOL (JEE-400) vacuum evaporator and examined by transmission electron microscopy (Phillips 420 TEM). Micrographs were taken at a magnification ranging from 40,000× to 80,000×.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) measurements were performed on a Nano-DSC II, Model 6100 scanning calorimeter from Calorimetry Sciences Corp. All DSC profiles were obtained at a scan rate of 1°C/min and each curve was baseline subtracted before the data analysis. Prior to all measurements, peptide solutions were dialyzed. ΔH cal was obtained by integrating the excess heat capacity curve.
Electronic supplemental material
Arrhenius plots for peptides and the turbidity curve for the slower self-association process for peptide P13L are available.
Acknowledgments
This work was funded by NIH Grants GM60048 (B.B.), and CA085826 and CA113863 (Y-H.W.). We thank Peter Yurchenco for helpful discussions.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Barbara Brodsky, Department of Biochemistry, Robert Wood Johnson Medical School, University of Medicine and Dentistry of New Jersey, 675 Hoes Lane, Piscataway, NJ 08854, USA; e-mail: brodsky@umdnj.edu; fax: (732) 235-4783.
Abbreviations: CD, circular dichroism; DSC, differential scanning calorimetry; OI, osteogenesis imperfecta; PBS, phosphate buffer saline; (POG)10, (Pro-Hyp-Gly)10, and O is used to represent hydroxyproline in the one-letter amino acid code.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.083441308.
References
- Baldock, C., Sherratt, M.J., Shuttleworth, C.A., Kielty, C.M. The supramolecular organization of collagen VI microfibrils. J. Mol. Biol. 2003;330:297–307. doi: 10.1016/s0022-2836(03)00585-0. [DOI] [PubMed] [Google Scholar]
- Baum, J., Brodsky, B. Folding of peptide models of collagen and misfolding in disease. Curr. Opin. Struct. Biol. 1999;9:122–128. doi: 10.1016/s0959-440x(99)80016-5. [DOI] [PubMed] [Google Scholar]
- Beck, K., Chan, V.C., Shenoy, N., Kirkpatrick, A., Ramshaw, J.A., Brodsky, B. Destabilization of osteogenesis imperfecta collagen-like model peptides correlates with the identity of the residue replacing glycine. Proc. Natl. Acad. Sci. 2000;97:4273–4278. doi: 10.1073/pnas.070050097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bella, J., Eaton, M., Brodsky, B., Berman, H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 Å resolution. Science. 1994;266:75–81. doi: 10.1126/science.7695699. [DOI] [PubMed] [Google Scholar]
- Berisio, R., Vitagliano, L., Mazzarella, L., Zagari, A. Crystal structure of a collagen-like polypeptide with repeating sequence Pro-Hyp-Gly at 1.4 Å resolution: Implications for collagen hydration. Biopolymers. 2000;56:8–13. doi: 10.1002/1097-0282(2000)56:1<8::AID-BIP1037>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Bhate, M., Wang, X., Baum, J., Brodsky, B. Folding and conformational consequences of glycine to alanine replacements at different positions in a collagen model peptide. Biochemistry. 2002;41:6539–6547. doi: 10.1021/bi020070d. [DOI] [PubMed] [Google Scholar]
- Burgeson, R.E. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J. Invest. Dermatol. 1993;101:252–255. doi: 10.1111/1523-1747.ep12365129. [DOI] [PubMed] [Google Scholar]
- Byers, P.H., Cole, W.G. Osteogenesis imperfecta. In: Royce P.M., Steinmann B., editors. Connective tissue and its heritable disorders. Wiley-Liss; New York: 2002. pp. 385–430. [Google Scholar]
- Cejas, M.A., Kinney, W.A., Chen, C., Leo, G.C., Tounge, B.A., Vinter, J.G., Joshi, P.P., Maryanoff, B.E. Collagen-related peptides: Self-assembly of short, single strands into a functional biomaterial of micrometer scale. J. Am. Chem. Soc. 2007;129:2202–2203. doi: 10.1021/ja066986f. [DOI] [PubMed] [Google Scholar]
- Chen, M., Keene, D.R., Costa, F.K., Tahk, S.H., Woodley, D.T. The carboxyl terminus of type VII collagen mediates antiparallel dimer formation and constitutes a new antigenic epitope for epidermolysis Bullosa acquisita autoantibodies. J. Biol. Chem. 2001;276:21649–21655. doi: 10.1074/jbc.M100180200. [DOI] [PubMed] [Google Scholar]
- Engel, J., Furthmayr, H., Odermatt, E., von der Mark, H., Aumailley, M., Fleischmajer, R., Timpl, R. Structure and macromolecular organization of type VI collagen. Ann. N. Y. Acad. Sci. 1985;460:25–37. doi: 10.1111/j.1749-6632.1985.tb51154.x. [DOI] [PubMed] [Google Scholar]
- Gill, S.C., von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Hofmann, H., Fietzek, P.P., Kühn, K. Comparative analysis of the sequences of the three collagen chains α1(I), α2 and α1(III). Functional and genetic aspects. J. Mol. Biol. 1980;141:293–314. doi: 10.1016/0022-2836(80)90182-5. [DOI] [PubMed] [Google Scholar]
- Hulmes, D.J., Miller, A., Parry, D.A., Piez, K.A., Woodhead-Galloway, J. Analysis of the primary structure of collagen for the origins of molecular packing. J. Mol. Biol. 1973;79:137–148. doi: 10.1016/0022-2836(73)90275-1. [DOI] [PubMed] [Google Scholar]
- Jenkins, C.L., Raines, R.T. Insights on the conformational stability of collagen. Nat. Prod. Rep. 2002;19:49–59. doi: 10.1039/a903001h. [DOI] [PubMed] [Google Scholar]
- Kadler, K.E., Hojima, Y., Prockop, D.J. Assembly of collagen fibrils de novo by cleavage of the type I pC-collagen with procollagen C-proteinase. Assay of critical concentration demonstrates that collagen self-assembly is a classical example of an entropy-driven process. J. Biol. Chem. 1987;262:15696–15701. [PubMed] [Google Scholar]
- Kadler, K.E., Hojima, Y., Prockop, D.J. Assembly of type I collagen fibrils de novo. Between 37 and 41 degrees C the process is limited by micro-unfolding of monomers. J. Biol. Chem. 1988;263:10517–10523. [PubMed] [Google Scholar]
- Kar, K., Amin, P., Bryan, M.A., Persikov, A.V., Mohs, A., Wang, Y.H., Brodsky, B. Self-association of collagen triple helic peptides into higher order structures. J. Biol. Chem. 2006;281:33283–33290. doi: 10.1074/jbc.M605747200. [DOI] [PubMed] [Google Scholar]
- Kielty, C.M., Grant, M.E. The collagen family: Structure assembly and organization in the extracellular matrix. In: Royce P.M., Steinmann B., editors. Connective tissue and its heritable disorders. Wiley-Liss; New York: 2002. pp. 159–222. [Google Scholar]
- Kishimoto, T., Morihara, Y., Osanai, M., Ogata, S., Kamitakahara, M., Ohtsuki, C., Tanihara, M. Synthesis of poly(Pro-Hyp-Gly)n by direct polycondensation of (Pro-Hyp-Gly)n, where n = 1, 5, and 10, and stability of the triple-helical structure. Biopolymers. 2005;79:163–172. doi: 10.1002/bip.20348. [DOI] [PubMed] [Google Scholar]
- Knupp, C., Squire, J.M. A new twist in the collagen story—the type VI segmented supercoil. EMBO J. 2001;20:372–376. doi: 10.1093/emboj/20.3.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotch, F.W., Raines, R.T. Self-assembly of synthetic collagen triple helices. Proc. Natl. Acad. Sci. 2006;103:3028–3033. doi: 10.1073/pnas.0508783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, R.Z., Venugopal, M.G., Bella, J., Mayville, P., Brodsky, B., Berman, H.M. Staggered molecular packing in crystals of a collagen-like peptide with a single charged pair. J. Mol. Biol. 2000;301:1191–1205. doi: 10.1006/jmbi.2000.4017. [DOI] [PubMed] [Google Scholar]
- Kramer, R.Z., Bella, J., Brodsky, B., Berman, H.M. The crystal and molecular structure of a collagen-like peptide with a biologically relevant sequence. J. Mol. Biol. 2001;311:131–147. doi: 10.1006/jmbi.2001.4849. [DOI] [PubMed] [Google Scholar]
- Kuznetsova, N., Leikin, S. Does the triple helical domain of type I collagen encode molecular recognition and fiber assembly while telopeptides serve as catalytic domains? Effect of proteolytic cleavage on fibrillogenesis and on collagen–collagen interaction in fibers. J. Biol. Chem. 1999;274:36083–36088. doi: 10.1074/jbc.274.51.36083. [DOI] [PubMed] [Google Scholar]
- Kuznetsova, N., Rau, D.C., Parsegian, V.A., Leikin, S. Solvent hydrogen-bond network in protein self-assembly: Solvation of collagen triple helices in nonaqueous solvents. Biophys. J. 1997;72:353–362. doi: 10.1016/S0006-3495(97)78674-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikin, S., Rau, D.C., Parsegian, V.A. Temperature-favoured assembly of collagen is driven by hydrophilic not hydrophobic interactions. Nat. Struct. Biol. 1995;2:205–210. doi: 10.1038/nsb0395-205. [DOI] [PubMed] [Google Scholar]
- Leikin, S., Parsegian, V.A., Yang, W., Walrafen, G.E. Raman spectral evidence for hydration forces between collagen triple helices. Proc. Natl. Acad. Sci. 1997;94:11312–11317. doi: 10.1073/pnas.94.21.11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikina, E., Mertts, M.V., Kuznetsova, N., Leikin, S. Type I collagen is thermally unstable at body temperature. Proc. Natl. Acad. Sci. 2002;99:1314–1318. doi: 10.1073/pnas.032307099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., Brodsky, B., Baum, J. NMR shows hydrophobic interactions replace glycine packing in the triple helix at a natural break in the (Gly-X-Y)n repeat. J. Biol. Chem. 2007;282:22699–22706. doi: 10.1074/jbc.M702910200. [DOI] [PubMed] [Google Scholar]
- Marini, J.C., Forlino, A., Cabral, W.A., Barnes, A.M., San Antonio, J.D., Milgrom, S., Hyland, J.C., Korkko, J., Prockop, D.J., De Paepe, A., et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllyharju, J., Kivirikko, K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Nagarajan, V., Kamitori, S., Okuyama, K. Structure analysis of a collagen-model peptide with a (Pro-Hyp-Gly) sequence repeat. J. Biochem. 1999;125:310–318. doi: 10.1093/oxfordjournals.jbchem.a022288. [DOI] [PubMed] [Google Scholar]
- Okuyama, K., Narita, H., Kawaguchi, T., Noguchi, K., Tanaka, Y., Nishino, N. Unique side chain conformation of a Leu residue in a triple-helical structure. Biopolymers. 2007;86:212–221. doi: 10.1002/bip.20724. [DOI] [PubMed] [Google Scholar]
- Persikov, A.V., Pillitteri, R.J., Amin, P., Schwarze, U., Byers, P.H., Brodsky, B. Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum. Mutat. 2004a;24:330–337. doi: 10.1002/humu.20091. [DOI] [PubMed] [Google Scholar]
- Persikov, A.V., Xu, Y., Brodsky, B. Equilibrium thermal transitions of collagen model peptides. Protein Sci. 2004b;13:893–902. doi: 10.1110/ps.03501704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persikov, A.V., Ramshaw, J.A., Brodsky, B. Prediction of collagen stability from amino acid sequence. J. Biol. Chem. 2005;280:19343–19349. doi: 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- Prockop, D.J., Fertala, A. Inhibition of the self-assembly of collagen I into fibrils with synthetic peptides. Demonstration that assembly is driven by specific binding sites on the monomers. J. Biol. Chem. 1998;273:15598–15604. doi: 10.1074/jbc.273.25.15598. [DOI] [PubMed] [Google Scholar]
- Ramachandran, G.N., Kartha, G. Structure of collagen. Nature. 1955;176:593–595. doi: 10.1038/176593a0. [DOI] [PubMed] [Google Scholar]
- Rele, S., Song, Y., Apkarian, R.P., Qu, Z., Conticello, V.P., Chaikof, E.L. D-periodic collagen-mimetic microfibers. J. Am. Chem. Soc. 2007;129:14780–14787. doi: 10.1021/ja0758990. [DOI] [PubMed] [Google Scholar]
- Rich, A., Crick, F.H. The molecular structure of collagen. J. Mol. Biol. 1961;3:483–506. doi: 10.1016/s0022-2836(61)80016-8. [DOI] [PubMed] [Google Scholar]
- Stephan, S., Sherratt, M.J., Hodson, N., Shuttleworth, C.A., Kielty, C.M. Expression and supramolecular assembly of recombinant α1(VIII) and α(VIII) collagen homotrimers. J. Biol. Chem. 2004;279:21469–21477. doi: 10.1074/jbc.M305805200. [DOI] [PubMed] [Google Scholar]
- Vitagliano, L., Berisio, R., Mazzarella, L., Zagari, A. Structural bases of collagen stabilization induced by proline hydroxylation. Biopolymers. 2001;58:459–464. doi: 10.1002/1097-0282(20010415)58:5<459::AID-BIP1021>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Yurchenco, P.D., Ruben, G.C. Basement membrane structure in situ: Evidence for lateral associations in the type IV collagen network. J. Cell Biol. 1987;105:2559–2568. doi: 10.1083/jcb.105.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenco, P.D., Ruben, G.C. Type IV collagen lateral associations in the EHS tumor matrix. Comparison with amniotic and in vitro networks. Am. J. Pathol. 1988;132:278–291. [PMC free article] [PubMed] [Google Scholar]
- Yurchenco, P.D., Schittny, J.C. Molecular architecture of basement membranes. FASEB J. 1990;4:1577–1590. doi: 10.1096/fasebj.4.6.2180767. [DOI] [PubMed] [Google Scholar]