

Abstract
Cytochrome c oxidase (CcO) transfers protons from the inner surface of the enzyme to the buried O2 reduction site through two different pathways, termed K and D, and from the outer surface via an undefined route. These proton paths can be inhibited by metals such as zinc or cadmium, but the sites of inhibition have not been established. Anomalous difference Fourier analyses of Rhodobacter sphaeroides CcO crystals, with cadmium added, reveal metal binding sites that include the proposed initial proton donor/acceptor of the K pathway, Glu-101 of subunit II. Mutant forms of CcO that lack Glu-101II (E101A and E101A/H96A) exhibit low activity and eliminate metal binding at this site. Significant activity is restored to E101A and E101A/H96A by adding the lipophilic carboxylic compounds, arachidonic acid and cholic acid, but not by their non-carboxylic analogues. These amphipathic acids likely provide their carboxylic groups as substitute proton donors/acceptors in the absence of Glu-101II, as previously observed for arachidonic acid in mutants that alter Asp-132I of the D pathway. The activity of E101A/H96A is still inhibited by zinc, but this remaining inhibition is nearly eliminated by removal of subunit III, which is known to alter the D pathway. The results identify the Glu-101/His-96 site of subunit II as the site of metal binding that inhibits the uptake of protons into the K pathway and indicate that subunit III contributes to zinc binding and/or inhibition of the D pathway. By removing subunit III from E101A/H96A, thereby eliminating zinc inhibition of the uptake of protons from the inner surface of CcO, we confirm that an external zinc binding site is involved in inhibiting the backflow of protons to the active site.
Oxygen reduction by cytochrome c oxidase (CcO)1 requires the delivery of both electrons and protons to the O2 reduction site, located at the heme a3–CuB center in the transmembrane region of subunit I of the complex. In addition to reducing O2, CcO pumps protons across the protein from the negative (N) to the positive (P) side of the membrane. Two proton uptake routes, the K and D pathways, identified by X-ray crystallography (1, 2) as well as site-directed mutagenesis studies (3–5), lead from the inner surface of CcO toward the heme a3–CuB site.
The K pathway transfers one or two of the protons that are taken up during the reduction of heme a3 and CuB before O2 binds (6–8). Mutagenesis experiments indicate that the K pathway of Rhodobacter sphaeroides begins at Glu-101 (R. sphaeroides numbering) of subunit II (9, 10), although the precise entry point has been questioned on the basis of studies on Paracoccus denitrificans CcO (11). The sequential involvement of Ser-229, Lys-362, Thr-359, the hydroxyl farnesyl of heme a3, and Tyr-288 of subunit I (12) (Figure 1A) is supported by further mutational and computational analyses. Two waters of the K pathway are resolved in the CcO structures (2, 13), and computational studies have suggested positions for more (14).
Figure 1.
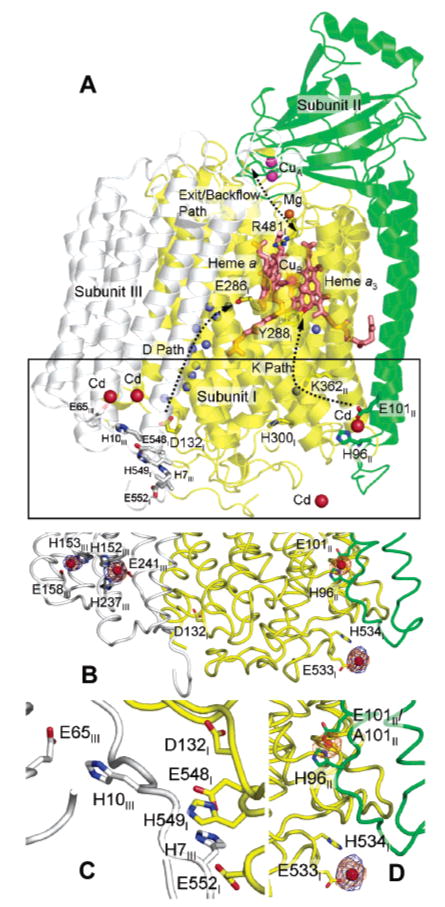
Structure of R. sphaeroides CcO (PDB entry 1M56) showing newly identified sites of cadmium (zinc) binding. For clarity, subunit IV is omitted from the picture. (A) The CcO structure is colored by subunit (yellow for subunit I, green for subunit II, and gray for subunit III). Important residues in the proton pathways are shown as stick models and labeled. The antenna residues in subunits I and III which are proposed to help collect protons into the D pathway are shown in stick models and colored dark gray. Water molecules that are in the important proton uptake pathways are shown as small blue spheres. Heme groups are shown as salmon sticks. Different metals are shown as spheres of different colors (red for Cd, magenta for Cu, salmon for Fe, and orange for Mg). The different proton pathways within the enzyme are shown with dotted curves. In this figure, the conformation of the side chain of H96II is changed so that it forms a ligating interaction with a cadmium ion, an observation made in a high-resolution crystal structure of CcO cocrystallized with cadmium (PDB entry 2GSM). In the original crystal structure of the four-subunit CcO (PDB entry 1M56), the side chain of H96II points away from the presumptive cadmium ion binding site. (B) Boxed area in panel A showing more details of cadmium binding in this region as well as the binding ligands. (C) Antenna residues thought to assist in the uptake of protons into the D path. (D) Details of the cadmium binding ligands. The anomalous difference Fourier density maps contoured at 5σ are colored orange for wild-type CcO and blue for E101A mutant CcO. The difference density peak at the E101II/H96II site is absent in E101A.
The D pathway takes up the remainder of the substrate protons (those destined for O2 reduction), plus all of the pumped protons (those translocated to the outside of the membrane) (15). The D pathway consists of a series of hydrogen-bonded waters, whose positions are highly conserved in X-ray structures, leading from surface-exposed Asp-132 of subunit I to Glu-286, located between hemes a and a3, approximately 26 Å above Asp-132 (2, 13) (Figure 1A). From the top of the D path (Glu-286), protons flow to the heme a3–CuB active site for O2 reduction or to the undefined proton pumping element in the vicinity of the heme a3–CuB center via routes not yet identified. The exit path leading from the pumping element to the outer surface of CcO is also unclear. However, when the uptake of protons via the D pathway is inhibited by mutation or by the presence of a membrane potential, the backflow of protons from the outer surface sustains a slow rate of O2 reduction (3, 16). The backflow proton pathway may be the proton exit pathway functioning in reverse.
Among the tools used to study proton transfer in CcO are the removal of subunit III, the addition of arachidonic acid to rescue proton path mutants, and the inhibition of proton uptake by zinc and other metals. Subunit III removal has a considerable impact on the function of the D pathway, in part because residues at the N-terminus of subunit III neighbor Asp-132 (17, 18) (Figure 1C). Subunit III influences the rate and the pH dependence of proton uptake (17, 19), provides a structural barrier to alternative proton uptake paths (20), and contains residues that may facilitate the delivery of protons to Asp-132 (17). On the other hand, subunit III has no apparent effect on the function of the K pathway (17).
Long-chain, unsaturated fatty acids, such as arachidonic acid, enhance the low activity of mutants that have lost the carboxyl at the entrance of the D pathway (21). The fatty acid increases the rate of proton uptake into the altered D pathway of D132A (20), apparently by providing a substitute carboxyl group as a proton donor/acceptor on the surface of CcO.
Micromolar concentrations of divalent metal cations, particularly Zn2+, Cd2+, and Ni2+, inhibit proton transfer pathways in a number of proteins by binding to carboxylate and histidine residues at the entrance or exit points of the pathways. Cytochrome c oxidase activity is inhibited by Zn2+, Cd2+, and Ni2+, and evidence of metal inhibition of the D pathway (22), the K pathway (23), and the proton backflow pathway (24) exists. Single-turnover experiments have shown that zinc slows the uptake of protons into the D pathway up to 30-fold (22). Zinc has also been argued to inhibit the backflow of protons to the active site from the outer surface of CcO because low concentrations of the metal inhibit the catalytic turnover of CcO in phospholipid vesicles (COVs) in the presence of a membrane potential (24). Other proteins containing proton transfer pathways are similarly inhibited. In photosynthetic reaction centers, a Zn or Cd ion binds to two histidines and an aspartic acid residue to inhibit the transfer of protons from the protein surface to the QB quinone binding site (25). Similarly, Zn2+ coordination by a histidine and at least one carboxylate residue inhibits the cytochrome bc1 complex, apparently by slowing the exit of protons from the QO quinone binding site (26).
The sites at which zinc or cadmium bind to inhibit CcO have remained elusive, but a recent crystal structure of subunits I and II of R. sphaeroides CcO at 2.0 Å (13) reveals Cd2+ coordination by Glu-101 and His-96 of subunit II at the proposed entrance of the K pathway (Figure 1A,B). Here we report that mutations altering Glu-101 and His-96 strongly inhibit CcO activity and no longer bind Cd. High rates of activity are restored to these mutant forms by adding arachidonic acid or the detergent cholic acid, both amphipathic molecules that contain single carboxyl groups. The activity of E101A/H96A is still inhibited by zinc, but this remaining inhibition is nearly eliminated by the removal of subunit III from the mutant. The results show that the Glu-101/His-96 site of subunit II is the Cd/Zn binding site that inhibits the uptake of protons into the K pathway and that subunit III is required for zinc inhibition of the D pathway.
Materials and Methods
Materials
Arachidonic acid, methyl arachidonate, horse heart cytochrome c, and soybean phospholipids (asolectin) were from Sigma. Asolectin was recrystallized using the procedure of Sone et al. (27). Dodecyl maltoside, decyl maltoside, sodium cholate, and CHAPS were from Anatrace, Inc. All other chemicals were ACS grade.
Construction of the E101A and E101A/H96A Mutants
The coxII gene was mutated by using the QuikChange (Stratagene) mutagenesis procedure. The primers used were 5′-TCCCCGCTCGCCATCGCCTGGACGA-3′ and 5′-CCAGGCGATGGCGAGCGGGGAATTG-3′ for the E101A mutation, and these primers plus 5′-CGCTTCACCGCCAATTCCCCGCTCG-3′ and 5′-CGAGCGGGGAATTGGCGGTGAAGC-3′ were used for the E101A/H96A double mutant. The mutated plasmids were sequenced by the Michigan State University Genomics Technology Support Facility to confirm the mutations. The mutated coxII genes were ligated back into the subunit II–III operon, and then the mutant subunit II–III operons were ligated into pCH169 (13) which had the wild-type operon removed. The final plasmids were transferred by conjugation into R. sphaeroides strain Δ123 (see below) as described previously (28).
Construction of the Δ123 Strain of R. sphaeroides
An EcoRV site was inserted immediately downstream of the coxI coding region in pJS3 (29) by using PCR. The plasmid was then cut with EcoRV and HinDIII to remove the entire coxI coding sequence and the 272 bp of coxI downstream sequence present in pJS3. This was replaced by 1788 bp of the coxI downstream sequence amplified from R. sphaeroides genomic DNA (Epicentre Fail-Safe PCR kit). The kanamycin resistance cassette was isolated as described previously (13) and inserted into the EcoRV site between the 2016 bp coxI upstream region from pJS3 and the added downstream region. The HinDIII site at the end of the downstream region was then converted to an EcoR1 site by insertion of a linker, and the entire upstream–kanamycin resistance cassette–downstream construct was isolated with EcoRI and cloned into the EcoRI site of suicide vector pSUP202 (30).
This new suicide plasmid was transferred from Escherichia coli strain S17-1 (30) to R. sphaeroides strain YZ200 (31), a strain already lacking the coxII–III operon, by biparental conjugation (28), except that exconjugants were plated on Sistrom's medium plates containing 25 μg/mL kanamycin. Kanamycin-resistant colonies were tested for tetracycline sensitivity to ensure double crossovers. Genomic DNA from kanamycin-resistant, tetracycline-sensitive clones was screened using PCR to confirm replacement in the genome of the coxI coding sequence by the kanamycin resistance cassette. One correct clone was selected as the Δ123 strain.
CcO Isolation and Activity Measurements
Bacterial growth and oxidase purification by Ni affinity chromatography were performed as described by Zhen et al. (31). The removal of subunit III was performed as described by Mills et al. (19). Measurements of the rates of O2 reduction were performed using an oxygen electrode as described previously (32) in assay mixtures containing 50 mM Hepes, MES or CHES, 50 mM KCl, 0.3 mM TMPD, 3.0 mM ascorbate, 0.1% dodecyl maltoside (DDM), 1000 units of catalase, 5–25 pmol of CcO, and 40 μM horse cytochrome c, with other additions as noted. The pH values between 6.2 and 6.8 were obtained using MES-KOH, between 6.8 and 8.2 using Hepes-KOH, and from 8.2 to 9.2 using CHES-KOH. The measurements of the catalytic life span as the CC50, including data evaluation, were performed as described by Mills and Hosler (32).
The protein purification protocol for wild-type and E101A mutant CcO used for crystallization was adapted from an earlier report (28). In short, prior to detergent solubilization, the protein concentration of resuspended membranes was measured by using the BCA protein assay reagent kit (Pierce) supplemented with 0.25% deoxycholate as the detergent. The membranes were diluted to a protein concentration of 10 mg/mL in 10 mM Tris (pH 8.0), 220 mM KCl, and 1 mM imidazole; then dodecyl maltoside was added to a final concentration of 1.0% (w/v), and the membranes were dissolved at 4 °C for 30 min. After centrifugation at 150000g for 30 min to remove unsolubilized material, the supernatant was loaded onto a home-packed Ni-NTA (Qiagen) column (column volume of 18 mL) connected to an AKTA-FPLC system (GE Healthcare). The column was pre-equilibrated with buffer A [10 mM Tris (pH 8.0), 220 mM KCl, 2.5 mM imidazole, and 0.05% dodecyl maltoside]. After the samples had been washed with buffer A, a two-step elution profile was used. First, the concentration of buffer B [10 mM Tris (pH 8.0), 220 mM KCl, 150 mM imidazole, and 0.05% dodecyl maltoside] was increased to 10% (∼15 mM imidazole) and held constant to elute off a major contaminating species. Following this, a linear gradient of buffer B was applied to elute fractions containing CcO. The green fractions were pooled and diluted 4-fold before being loaded onto a 20 mL DEAE-Sepharose FPLC column (Pharmacia) pre-equilibrated with 10 mM Tris (pH 8.0) and 0.05% dodecyl maltoside. A linear gradient of 10 mM Tris (pH 8.0), 0.5 M KCl, and 0.05% dodecyl maltoside over 400 mL was applied at a rate of 2 mL/min. The green fractions were pooled and concentrated, and the buffer was exchanged into 10 mM Tris (pH 8.0), 0.05 M NaCl, and 0.2% decyl maltoside by washing and concentrating two times in a stirred ultrafiltration cell (Millipore) under N2 pressure.
Crystallization, X-ray Data Collection, and Identification of Cadmium Binding Sites
The crystallization conditions were adapted from those described previously (2). A sitting drop vapor diffusion method was used. The reservoir solution (typical volume, 1.0 mL) contained 100 mM MES buffer (pH 6.3–6.6) and 21%–23% PEG400. Typically, 8 μL of a protein solution containing 80–100 μM CcO in 10 mM Tris (pH 8.0), 50 mM NaCl, and 0.2% decyl maltoside was mixed with 4 μL of the reservoir solution and 4 μL of a crystallization additive solution containing 33.3 mM MgCl2, 5% heptanetriol, and 0.026% dodecyl maltoside. The sitting drop was incubated against the reservoir solution at 4 °C, and triangular-shaped CcO crystals appeared after 1 week and continued to grow to their full size in approximately 4 weeks.
Prior to X-ray data collection, the crystals were soaked in a stabilizing solution containing CdCl2. The soaking solution was prepared as 91 mM MES, from a stock solution at pH 6.6, 18 mM Tris, from a stock solution at pH 8.0, and 30 mM MgCl2, 2.5% heptanetriol, 0.16% decyl maltoside, 0.016% dodecyl maltoside, 21–23% PEG400, and 2 mM CdCl2. The crystals were soaked in this solution for approximately 6 h to allow binding of cadmium ions to CcO. Afterward, the soaking solution was exchanged stepwise to a cryoprotectant solution which contained the same ingredients as the soaking solution, including CdCl2, except for a higher (32%) concentration of PEG400. The crystals were picked up from the cryoprotectant solution and flash-cooled in liquid N2.
The anomalous diffraction data were collected at station 23ID-D, GM/CA-CAT, APS, Argonne National Laboratory (Argonne, IL). The wavelength of the X-ray was set at 1.984 Å, where cadmium has a strong anomalous signal (f″ = 7.1 e). As the crystals diffracted only to ∼3.5 Å, the anomalous data for both wild-type CcO and E101A were processed only to 3.9 Å to ensure acceptable data quality. The anomalous data were processed as the rhombohedral crystal form with a space group of R3 (a = b = 341.0 Å and c = 89.9 Å for wild-type CcO; a = b = 341.6 Å and c = 91.6 Å for the E101A mutant) using HKL2000 (33). Molecular replacement phasing using Phaser (34) and the published structure of the four-subunit R. sphaeroides CcO (PDB entry 1M56) as the search model was performed first; this was followed by a rigid body refinement against the anomalous data. The anomalous difference Fourier map was calculated with the CCP4 program suite (35) using the phases of the rigid-body refined structure. All metals in the structure were removed from the phase calculation to minimize bias. Strong anomalous difference density peaks were found, including some that correctly indicated the locations of Ca and sulfur atoms in the structure; this was expected as they too have anomalous signals at the selected X-ray wavelength. The strongest peaks were assigned as cadmium binding sites (Figure 1B). In a control experiment, during which the crystals were not subjected to Cd soaking prior to flash-cooling and data collection, the anomalous difference Fourier map showed no peaks at the locations shown in Figure 1B, as expected. Although Figure 1B shows only one R. sphaeroides CcO molecule with the observed cadmium binding sites, there are two R. sphaeroides CcO molecules in the asymmetric unit, and the observed cadmium binding sites were found in both molecules.
Results
Identification of Cadmium Binding Sites
Cadmium binding sites were identified on the surface of the four-subunit aa3-type CcO of R. sphaeroides by anomalous difference Fourier analyses using the anomalous data obtained from crystals soaked in cadmium and the phases from a previous structure of the complex (2) (see Materials and Methods for details). Figure 1 shows the observed cadmium binding sites (orange electron density) in WT CcO, which are also proposed binding sites for zinc. The Glu-101/His-96 metal binding site on subunit II was also observed in a high-resolution structure (2.0 Å) of the enzyme containing only subunits I and II (13), making the assignment of metal binding at this site highly reliable. Additionally, there are three other cadmium or zinc binding sites on the inner surface of R. sphaeroides CcO. Their respective binding ligands are His-152, His-237, and Glu-241 of subunit III, His-153 and Glu-158 of subunit III, and Glu-533 and His-534 of subunit I. The latter is at a crystal contact interface.
Characterization of the Activity of Mutants that Eliminate Binding of Metal at the Entrance of the K Pathway
Mutants E101A and E101A/H96A of R. sphaeroides CcO were engineered to eliminate the metal binding site created by these residues. The abolishment of metal binding in E101A was confirmed in a crystal of the mutant by the absence of the intense peak at this site in the anomalous difference Fourier map, as shown in Figure 1D. In the work of Tomson et al. (10) that identified Glu-101 as the initial proton acceptor/donor of the K pathway of R. sphaeroides CcO, the alteration of Glu-101 to alanine inhibited CcO activity by approximately 92%. This is confirmed here (Table 1), and the same level of inhibition was observed for the double mutant E101A/H96A. Neither mutation affected the proton pumping efficiency of CcO (data not shown), consistent with previous results using mutants that affect K pathway activity (3).
Table 1.
Vmax Values of O2 Reduction by Wild-Type CcO, E101A, and E101A/H96A at pH 6.5
| Vmax (electrons/s)a | percent remaining activity | |
|---|---|---|
| wild type | 1500 ± 32 | 100 |
| E101A | 120 ± 3 | 8 |
| E101A/H96A | 110 ± 2 | 7 |
No cholic acid, arachidonic acid, or soybean phospholipid was added to the assays. The error is the standard error of the fit of the data to the Michaelis–Menten equation.
The Activity of the K Pathway Mutants Is Rescued by Arachidonic Acid and Cholic Acid
Arachidonic acid (Aa), a 20-carbon fatty acid with four double bonds (Figure 2), is capable of effecting a partial chemical rescue of mutants that have lost the carboxyl (Asp-132) at the entrance of the D pathway, apparently by providing an alternative proton donor/acceptor (20, 21). The activity of D132A at pH 7.0 increases from 34 to 250 e−/s with the addition of 250 μM Aa (Table 2). The activity of the K pathway mutant, E101A/H96A, increases from 46 to >1000 e−/s upon addition of 250 μM Aa (Figure 3), while the activity of wild-type CcO is little altered by the addition of Aa. The addition of the methyl ester of Aa (250 μM) leads to no increase in the activity of E101A/H96A, indicating that the carboxyl group of Aa is required for the stimulation of activity (Figure 3). The data indicate that Aa restores a high level of K pathway activity when Glu-101 is altered, by supplying a substitute carboxyl group. Unlike most chemical rescue experiments using soluble compounds (36, 37), relatively low concentrations of Aa are required, implying tighter and more specific binding in the vicinity of the missing carboxyl.
Figure 2.
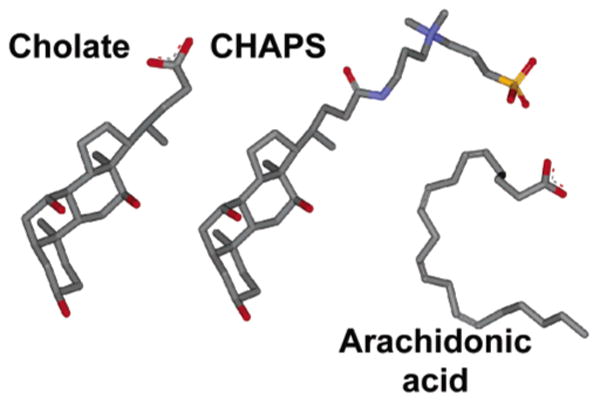
Molecular structures of cholic acid, CHAPS, and arachidonic acid. The structures are colored by atom type (gray for C, blue for N, red for O, and orange for S).
Table 2.
Effect of Cholic Acid and Arachidonic Acid on the Activity of the D Pathway Mutant D132A
| activity (electrons/s)a | ||
|---|---|---|
| addition | D132A III(+) | D132A III(−) |
| none | 34 ± 1 | 530 ± 35b |
| 1.37 mM cholic acid | 37 ± 3 | 630 ± 36 |
| 250 μM arachidonic acid | 250 ± 14 | 1300 ± 48 |
The error is the standard deviation of multiple measurements at pH 7.0.
The removal of subunit III increases the activity of D132A by allowing a bound water to function as an alternative proton donor/acceptor for the D pathway (20).
Figure 3.
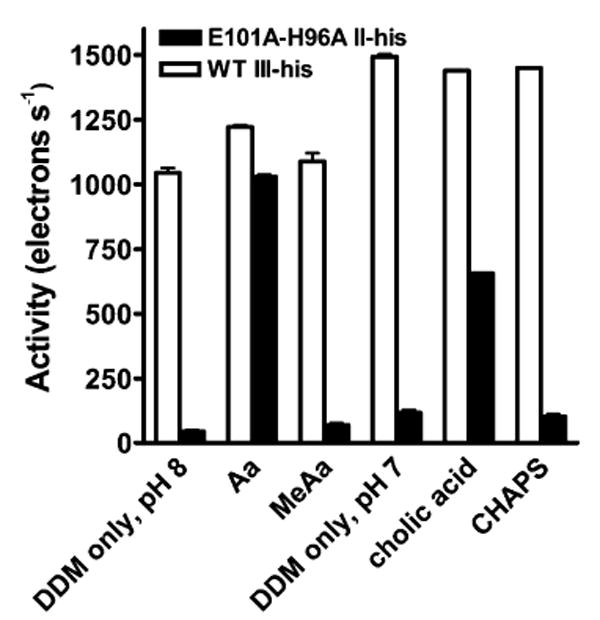
Effect of arachidonic acid, cholic acid, and their non-carboxylic analogues on the O2 reduction activity of wild-type CcO (white bars) and E101A/H96A (black bars). Rates of O2 reduction activity were measured as described in Materials and Methods. Arachidonic acid (Aa) and methyl arachidonate (MeAa) were added to a final concentration of 250 μM; these experiments were performed at pH 8.0. Cholic acid and CHAPS were added to a final concentration of 1.4 mM; these experiments were performed at pH 7.0. All experiments included 2 mM (0.1%) dodecyl maltoside (DDM). The activity of wild-type CcO in the presence of only DDM declines with pH (18); hence, it is greater at pH 7 than at pH 8. The error is the standard deviation of two or more determinations.
Like Aa, cholic acid binds to the surface of membrane proteins and contains a single carboxyl group (Figure 2). Cholic acid also stimulates the activity of E101A/H96A (Figure 3) to a level of ∼750 e−/s at pH 6.2 (Figure 4) with half-maximum stimulation at 500 μM cholic acid (data not shown). In this concentration range, cholic acid has no effect on the activity of wild-type CcO (Figure 3). The detergent CHAPS contains the same hydroxylated ring system of cholic acid, but the carboxyl group is replaced with a zwitterion of a quaternary amine and a sulfate group (Figure 2). The addition of similar levels of CHAPS failed to stimulate the activity of E101A/H96A or wild-type CcO (Figure 3). Like Aa, then, cholic acid apparently restores activity to the altered K pathway of E101A/H96A by supplying its carboxyl group as a substitute for Glu-101.
Figure 4.
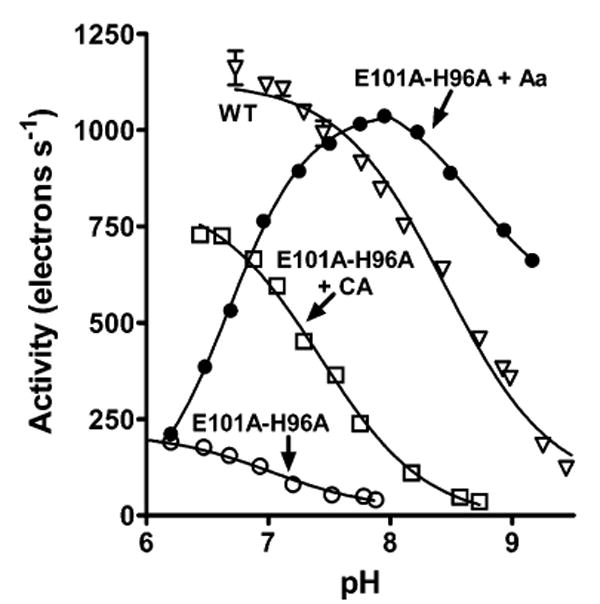
pH dependence of the O2 reduction activity of E101A/H96A in the presence of cholic acid or arachidonic acid. Rates of O2 reduction were measured at different pH values for E101A/H96A in the presence of 250 μM arachidonic acid [Aa (●)] or 1.4 mM cholic acid [CA (□)]. The pH dependencies of wild-type CcO [from ref 18 (▽)] and E101A/H96A in the absence of Aa and CA (○) are shown for comparison. With the exception noted below, the data were fit to the equation Y = TNmin + (TNmax − TNmin)/(1 + 10pH−pKa), where TN is the turnover number in electrons per second. The fits yielded pKa values of 7.4 for E101A/H96A with CA, 7.0 for E101A/H96A in the absence of either CA or Aa, and 8.5 for wild-type CcO (WT). The data for the descending portion of the data of E101A/H96A activity in the presence of arachidonic acid were fit to the above equation, while the data for the ascending portion were fit to the equation Y = TNmin + (TNmax − TNmin)/(1 + 10pKa−pH), yielding a pKa value of 6.7.
The activity of the E101A/H96A mutant is lower than that of wild-type CcO even in the presence of cholic acid or Aa, indicating that the rate of proton uptake into the K pathway is limiting catalytic turnover. The estimated pKa of cholic acid-supported steady-state activity by E101A/H96A is 7.4 (Figure 4). This value is considerably shifted from the apparent pKa of 8.5 measured for the activity of wild-type CcO (Figure 4), where the K pathway activity may not be limiting (17, 19). The estimated pKa value of 7.4 is reasonably close to the value of 6.6 attributed to the cognate of Glu-101 in bovine CcO (38, 39), suggesting that the carboxyl groups of Glu-101 in wild-type CcO and cholic acid in E101A/H96A occupy similar sites.
The pH dependence of E101A/H96A activity in the presence of Aa is more complex, showing a strong maximum at pH 8 (Figure 4) with a pKa of activation of 6.7. This pKa likely relates to that of the Aa bound to the K pathway site. The declining activity with higher pH may reflect the intrinsic loss of CcO activity.
Cholic Acid Has Little Effect on the Uptake of Protons into the D Pathway
Arachidonic acid restores activity to mutants of both the D and K pathways; might cholic acid do the same? Table 2 shows Aa stimulation of the D pathway mutant D132A, in agreement with previous results (20, 21), but cholic acid fails to stimulate this mutant. Removal of subunit III from D132A [creating D132A III(−)] makes it even more sensitive to Aa stimulation (Table 2), but cholic acid still has little effect. The results suggest that there is specificity to the positioning of these two chemical rescue agents and that cholic acid does not find a suitable binding site close to the D path, only the K path.
Inhibition of the D Pathway by Zinc and the Role of Subunit III
The activities of E101A and E101A/H96A, with (Figure 5) or without cholic acid (data not shown), were found still to be inhibited by zinc even though the metal binding site at the entrance of the K path has been completely removed. Assuming that this zinc sensitivity reflects inhibition of proton uptake, zinc could be inhibiting the uptake of protons into the D pathway, as shown in single-turnover experiments (40). On the other hand, zinc could be inhibiting an alternative entry route into the K pathway, such as that facilitated by cholic acid. One way to discriminate between these possibilities is to eliminate or reduce the ability of zinc to inhibit D pathway activity. Since subunit III impacts the region of entry of protons into the D pathway (18, 20), the ability of zinc to inhibit E101A/H96A in the absence of subunit III was tested. The data of Figure 5 show that E101A/H96A III(−) is only weakly inhibited by micromolar concentrations of zinc. This result indicates that the presence of subunit III is, in fact, required for strong inhibition of the D pathway by zinc.
Figure 5.
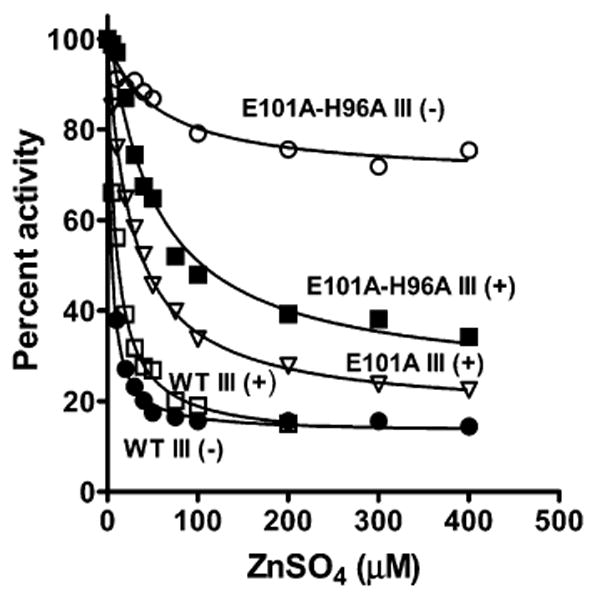
Inhibition of steady-state O2 reduction by zinc for wild-type CcO and different forms of the K pathway mutants in the presence of cholic acid. The activity of CcO was measured as described in Materials and Methods, in the presence of ZnSO4 at pH 7.4. All reactions included 1.1 mM cholic acid and 1 mg/mL soybean phospholipid (asolectin). The zinc inhibition constants were determined by nonlinear least-squares fitting as described by Mills et al. (24). Symbols and Ki values are as follows: WT III(−), 3.8 ± 0.2 μM (●); WT III(+), 9.3 ± 0.6 μM (□); E101A III(+), 28 ± 1.5 μM (▽); E101A/H96A III(+), 50 ± 9 μM (■); and E101A/H96A III(−), 56 ± 18 μM (○). The data for WT III(−) were taken from ref 19.
The requirement for the presence of subunit III for zinc inhibition of the D pathway was confirmed through measurements of the catalytic life span of CcO. Previous studies have shown that slow uptake of protons into the D pathway (but not the K pathway) increases the probability of irreversible inactivation, or suicide inactivation, of CcO during turnover (32). The probability of suicide inactivation increases, and thus the catalytic life span decreases, as the rate of delivery of protons to the active site via the D pathway declines. Catalytic life span is measured in terms of the average number of catalytic cycles required to inactivate the enzyme (one cycle = 1O2 + 4e− + 4H+ → 2H2O). The catalytic life span of dodecyl maltoside-solubilized wild-type CcO at pH 7.4 was measured as ∼1.6 × 106 catalytic cycles (Table 3). In the presence of 400 μM zinc, the catalytic life span of wild-type III(+) drops 16-fold (Table 3), consistent with significant inhibition of the uptake of protons into the D pathway by zinc. In contrast, the addition of 400 μM zinc to WT III(−) fails to decrease its catalytic life span (Table 3), confirming that the D pathway of CcO is not inhibited by zinc in the absence of subunit III. Note that the catalytic life span of WT III(−) is already much shorter than that of WT III(+). This is because the removal of subunit III, by itself, increases the probability of suicide inactivation by slowing the uptake of protons into the D pathway at pH >7.0 (41). Nevertheless, the catalytic life span of WT III(−) is highly sensitive to factors that further slow the uptake of protons into the D pathway, such as increases in pH, the mutation of Asp-132, or the presence of a transmembrane voltage gradient (32). Therefore, if zinc was slowing the uptake of protons into the D pathway of WT III(−) to any significant extent, a decrease in the catalytic life span would be evident.
Table 3.
Effect of Zinc on the Catalytic Life Span of Wild-Type CcO Containing Subunit III and CcO Lacking Subunit III
| average number of catalytic cycles required to inactivate the enzymea | ||
|---|---|---|
| addition | WT III(+) | WT III(−) |
| none | ∼1.6 × 106 | (6.2 ± 0.9) × 103 |
| 400 μM ZnSO4 | (105 ± 4) × 103 | (8.3 ± 0.4) × 103 |
No cholic acid, arachidonic acid, or soybean phospholipid was added to the assays. One catalytic cycle = 1O2 + 4e− + 4H+ → 2H2O.
Proton Backflow Is Inhibited by Zinc in a CcO Form that Lacks Zn Inhibition of the K and D Pathways
Once CcO is incorporated into phospholipid vesicles (cytochrome oxidase vesicles or COVs), the consumption of protons from the interior of the vesicle rapidly generates a transmembrane electrical potential that inhibits the uptake of protons from the inner surface of the CcO complex (24, 42). Under these “controlled” turnover conditions, low activity is supported by the remaining uptake of protons from the inner surface plus the backflow of protons from the outer surface of CcO to the active site (24, 42). Micromolar concentrations of zinc inhibit proton backflow (24). Measurements of the rate of cytochrome c oxidation by COVs containing E101A/H96A III(−) show that its rate of controlled turnover is substantially inhibited by low concentrations of zinc, although the inhibition profile differs somewhat from that of wild-type CcO lacking subunit III (Figure 6). Thus, the zinc binding site required for the inhibition of proton backflow is retained in a CcO form in which zinc inhibition of the D and K pathways is largely eliminated.
Figure 6.
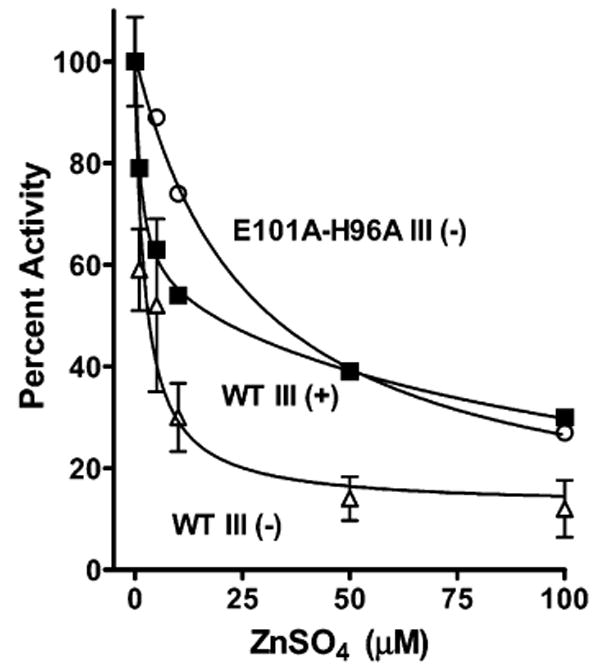
Zinc inhibits on the outside of COVs in the presence of a membrane potential even when zinc inhibition of the uptake of protons from the interior of the COVs is largely eliminated by the H96A/E101A mutant and the removal of subunit III. The preparation of cytochrome c oxidase vesicles (COVs) was like that described by Qian et al. (47), and the rate of horse cytochrome c oxidation was measured at 550 nm in an OLIS-rsm stopped-flow spectrophotometer with increasing concentrations of zinc added to the outside of the COVs. No ionophores were added to diminish the membrane potential (i.e., controlled state) (24): WT III(−) (△), WT III(+) (■), and E101A/H96A III(−) (○). The Ki values measured from fitting (as in Figure 5) were 1.3 and 116 μM for WT III(+), 2.6 μM for WT III(−), and 27 μM for E101A/H96A III(−).
Discussion
Chemical Rescue of Proton Pathway Activity in CcO by Arachidonic Acid and Cholic Acid
The long-chain, poly-unsaturated fatty acid arachidonic acid has been shown to increase the rate of uptake of protons into the D pathway in the absence of Asp-132 (20, 43). A corresponding increase in the rate of O2 reduction results (21). Apparently, the fatty acid provides a substitute carboxyl group in the absence of Asp-132 since arachidonol, a non-carboxylic analogue of Aa, has no effect (21).
Here, we show that in the absence of the initial proton donor/acceptor of the K pathway, Glu-101 of subunit II, the addition of Aa allows near-normal activity at pH 8.0. Since methyl arachidonate, also a non-carboxylic analogue of Aa, does not stimulate activity (Figure 3), the results indicate that Aa reconstitutes K pathway activity by supplying a carboxyl group as a substitute for that of Glu-101, similar to the effect of Aa on the D pathway of Asp-132 mutants.
The level of restoration of K pathway activity by Aa is similar to its restoration of D pathway activity in the absence of subunit III. Arachidonic acid increases the activity of D132A to 250 e−/s in the presence of subunit III but to 1300 e−/s in its absence (Table 2). Once subunit III is removed, part of a ring of residues surrounding Asp-132 is lost and a new hydrophobic face of subunit I is exposed. This apparently gives Aa greater access to the D pathway, since the increase in activity correlates with an increase in the rate of uptake of protons into the pathway (20). In fact, once subunit III is removed, Aa becomes the dominant proton donor/acceptor for the D pathway regardless of whether Asp-132 is present (19, 20).
Cholic acid also stimulates the activity of E101A/H96A, although not to an extent as great as that with Aa. Like arachidonic acid, cholic acid is a molecule that contains a single carboxyl group and can bind to the hydrophobic surface of CcO. The evidence that it rescues the activity of Glu-101 mutants by supplying its carboxyl group as an alternative proton donor/acceptor derives from the finding that the non-carboxylated analogue of cholic acid, CHAPS, fails to stimulate the activity of E101A/H96A. Since the ring structures of cholic acid and CHAPS are identical, both molecules should bind in a similar manner on the surface of CcO.
Although cholic acid stimulates K pathway activity in the absence of Glu-101, it has only a weak effect on the D pathway in the absence of Asp-132, the normal proton donor/acceptor of the D pathway. Presumably, the detergent does not bind in such a way to present its carboxyl group in an appropriate position to donate protons into the D pathway. Arachidonic acid is more successful, perhaps because a flexible, long-chain fatty acid can adopt more configurations on the protein surface than the ring system of cholic acid, allowing it to occupy existing phospholipid binding sites (13). A small stimulation of the D pathway by cholic acid is observed in the absence of subunit III (Table 2), most likely because the removal of the subunit allows the detergent greater access to the entry of the D pathway.
Identification of the Inhibitory Zinc Binding Site of the K Pathway
The inhibition of proton pathways by zinc or cadmium is well-documented for protein complexes that transfer protons to and from buried substrate binding sites, including cytochrome c oxidase, the bc1 complex, and the photosynthetic reaction center (25, 26). For the latter two complexes, metal binding sites composed of histidine and carboxylate residues have been previously identified by the positions of Cd or Zn ions in crystal structures (25, 26). The cadmium binding site of Glu-101 and His-96 of subunit II, seen in a 2.0 Å resolution structure of subunits I and II of R. sphaeroides CcO (13) as well as in the anomalous difference Fourier map of the cadmium-soaked four-subunit crystal presented here, should be the site that binds zinc to inhibit the uptake of protons into the K pathway. However, elimination of zinc inhibition of the D pathway was required to prove this (see below). The glutamate–histidine zinc binding motif conforms with that of the other membrane protein complexes, as well as with a survey of surface binding sites for zinc on soluble proteins that shows a strong preference for sites composed of histidine–glutamate or histidine–aspartate pairs (44).
Crystal structures of CcO have yet to reveal a metal binding site at or near Asp-132, the initial proton donor/acceptor of the D pathway. Nevertheless, single-turnover experiments have shown that micromolar concentrations of zinc inhibit the transfer of a proton from bulk solvent through Asp-132 to the active site (40, 45). The protein surface surrounding Asp-132 is composed of residues of both subunits I and III, and the removal of subunit III significantly alters the activity of the D pathway, but not the K pathway (19). The removal of subunit III from E101A and E101A/H96A created CcO forms showing little inhibition of their activity by zinc. This finding leads to four conclusions. (1) Subunit III is required for zinc inhibition of the uptake of protons into the D pathway. (2) The K pathway activity restored by the addition of cholic acid is not inhibited by zinc. This leads directly to conclusion 3, that the zinc binding site formed by Glu-101 and His-96 does, in fact, confer the sensitivity of the normal K pathway to zinc. (4) Finally, the zinc sensitivity of O2 reduction activity that remains in E101A and E101A/H96A reflects inhibition of the D pathway.
Using this last conclusion, the estimated Ki values for zinc inhibition of the D pathway [28–50 μM (Figure 5)] are greater than that measured for both pathways [∼9 μM (Figure 5) (19, 24)]. This suggests that zinc binds more tightly to the Glu-101/His-96 site than to the undefined zinc binding site that inhibits the D pathway. Consistent with this, detergent-solubilized WT III(−), in which only the K pathway should be inhibited by zinc, has previously been shown to have a lower Ki for zinc, ∼4 μM, under the same experimental conditions as this study (ref 19 and Figure 5). This value provides an estimate of the Kd for Zn2+ binding at the Glu-101/His-96 site. (Since metal binding is strongly influenced by interactions with other molecules, it should be noted that all of the Ki values reported here were obtained in the presence of cholic acid and soybean phospholipids at pH 7.4, as specified in Figure 5.)
Zinc Inhibition of the D Pathway
The requirement of subunit III for zinc inhibition of steady-state uptake of protons into the D pathway could indicate a single site of zinc binding on subunit III or a single site on subunit I [as has recently been proposed (46)] that may be altered by the removal of subunit III. However, inspection of the structure of CcO suggests a third possibility. The area around Asp-132 includes several conserved histidine and carboxylate residues that have been proposed to function as a proton antenna for the D pathway (15, 20). These include His-549, Glu-548, and Glu-552 of subunit I, with His-7, His-10, and Glu-65 of subunit III (Figure 1C). These antenna residues are also reasonably positioned to form multiple zinc binding sites composed of histidine–carboxylate residue pairs. Zinc binding at these sites could inhibit D pathway proton uptake by inhibiting the activity of the proton antenna and/or by placing positive charges in the vicinity of Asp-132. Thus, the requirement for the presence of subunit III for zinc sensitivity could reflect a requirement for zinc binding at multiple sites to effect significant metal inhibition of uptake of protons into the D pathway.
It is also possible that the removal of subunit III affects only the ability of zinc bound on subunit I to inhibit proton uptake. In single-turnover experiments at pH 7.5, 100 μM Zn2+ inhibits the rate of uptake of protons into the D pathway more than 20-fold (22). Inhibition may result from the direct binding of Zn2+ by Asp-132 or by increased stabilization of the anionic form of Asp-132 if Zn2+ binds to nearby residues. In the latter case, Zn2+ binding would lower the pKa of Asp-132. At pH 7.4, the removal of subunit III inhibits uptake of protons into the D pathway to the same extent as the addition of Zn2+ (17). The similar result likely arises from a similar effect on the pKa of Asp-132. The removal of subunit III has been argued to lower the pKa of Asp-132, apparently because the residue is more exposed to bulk water in the absence of subunit III which increases the stability of its anionic (deprotonated) form (17, 20). The electrostatic influence of Zn2+ on the pKa of Asp-132 may be minimized by the increased level of exposure to solvent water to this region upon removal of subunit III. Thus, there could be a single Zn2+ binding site on subunit I that remains in the absence of subunit III, but its inhibitory effect is lost.
The Ki values (28–50 μM) estimated here for zinc inhibition of the D pathway of E101A/H96A III(+) during steady-state turnover are significantly higher than that measured for zinc inhibition of the uptake of a single proton into the D pathway of wild-type CcO [∼1 μM (40)]. In addition to other experimental differences, it may be that protons compete more successfully for the zinc binding site(s) around the entry to the D pathway during continuous proton uptake.
The cadmium binding sites seen on the inner surface of subunit III in the four-subunit CcO structure are more than 23 Å from Asp-132 of subunit I. It is possible, but unlikely, that zinc binding at these sites would inhibit the uptake of protons into the D pathway. However, the presence of cadmium at these sites, as well as the cadmium ion bound to His-534 and Glu-533 of subunit I, is consistent with the propensity of histidine–carboxylate residue pairs to bind metal.
Zinc Inhibition of the Proton Backflow Pathway
When the uptake of protons into the D pathway is inhibited, as in the presence of a membrane potential or in mutants such as D132A, proton backflow from the outer surface of CcO partially or largely substitutes for the D pathway to maintain a slow rate of O2 reduction (24, 42). One purpose for proton backflow in vivo may be to keep protons flowing to the active site during transient periods of a high membrane potential. This would lower the probability of suicide inactivation at the active site by shortening the lifetime of reactive intermediates (32) and limit the accumulation of inactive CcO. While proton backflow does occur in the absence of a membrane potential, such as in the mutant D132A (32), zinc inhibits proton backflow only in the presence of a membrane potential (24). One explanation for this is that the binding site for zinc on the outer surface of CcO may only form in the presence of a membrane potential. An alternative, albeit less likely, explanation is that the membrane potential drives the rapid uptake of Zn2+ into the COVs and the observed inhibition of cytochrome c oxidation results from inhibition of the uptake of protons into the D and K pathways. The subunit III-depleted K pathway mutants studied here refute the latter hypothesis, since low concentrations of zinc have little effect on the uptake of protons from the inner surface of CcO, while 100 μM zinc inhibits the controlled turnover of reconstituted E101A/H96A III(−) by ∼70%, similar to that seen for wild-type CcO and WT III(−) (Figure 6). Because this inhibition cannot be that of the D or K pathway in this CcO form, the result confirms the previous finding that binding of zinc to the outer surface of CcO slows proton backflow in the presence of a membrane potential (24).
Conclusions
Zinc inhibits all three proton transfer pathways of CcO, the K pathway, the D pathway, and the proton backflow pathway. The site of inhibitory metal binding for the K pathway is firmly established by crystallographic analysis as Glu-101 and His-96 of subunit II. This binding/inhibitory site is consistent with studies implicating Glu-101 as the initial proton donor/acceptor of the K pathway. The site that binds zinc to inhibit the D pathway remains undefined; however, subunit III is required for zinc inhibition, and multiple zinc binding sites within the “antenna” residues for the D pathway are possible. Zinc inhibition of the backflow of protons from the outer surface of CcO in the presence of a transmembrane voltage gradient is confirmed. Arachidonic acid functions as a carboxyl group donor to restore considerable activity to either the K pathway or the D pathway in mutants that alter the initial proton donor/acceptor of these pathways. Cholic acid does the same for the K pathway, but only weakly for the D pathway. Characterization of these lipidic carboxylates extends their usefulness in studies of the proton pathways of CcO, and possibly other proteins containing long-range proton transfer pathways.
Footnotes
Supported by National Institutes of Health Grants GM56824 (J.H.), GM26916 (S.F.-M.), and HL56773 (R.M.G.).
Abbreviations: Aa, arachidonic acid; BCA, bicinchoninic acid; CcO, cytochrome c oxidase; CA, cholic acid; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; COV, cytochrome c oxidase vesicle; DDM, dodecyl maltoside; MeAa, methyl arachidonate; Ni-NTA, Ni2+-nitriloacetic acid agarose; PCR, polymerase chain reaction; PEG, polyethylene glycol; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine; WT, wild-type CcO; WT III(−), wild-type CcO lacking subunit III.
References
- 1.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 2.Svensson-Ek M, Abramson J, Larsson G, Tornroth S, Brzezinski P, Iwata S. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 3.Fetter JR, Qian J, Shapleigh J, Thomas JW, Garcia-Horsman A, Schmidt E, Hosler J, Babcock GT, Gennis RB, Ferguson-Miller S. Possible proton relay pathways in cytochrome c oxidase. Proc Natl Acad Sci USA. 1995;92:1604–1608. doi: 10.1073/pnas.92.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Horsman JA, Puustinen A, Gennis RB, Wikström M. Proton transfer in cytochrome bo3 ubiquinol oxidase of Escherichia coli: Second-site mutations in subunit I that restore proton pumping in the mutant Asp135→Asn. Biochemistry. 1995;34:4428–4433. doi: 10.1021/bi00013a035. [DOI] [PubMed] [Google Scholar]
- 5.Hosler JP, Shapleigh JP, Mitchell DM, Kim Y, Pressler MA, Georgiou C, Babcock GT, Alben JO, Ferguson-Miller S, Gennis RB. Polar residues in helix VIII of subunit I of cytochrome c oxidase influence the activity and the structure of the active site. Biochemistry. 1996;35:10776–10783. doi: 10.1021/bi9606511. [DOI] [PubMed] [Google Scholar]
- 6.Junemann S, Meunier B, Gennis RB, Rich PR. Effects of mutation of the conserved lysine-362 in cytochrome c oxidase from Rhodobacter sphaeroides. Biochemistry. 1997;36:14456–14464. doi: 10.1021/bi971458p. [DOI] [PubMed] [Google Scholar]
- 7.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc Natl Acad Sci USA. 1997;94:9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ädelroth P, Gennis RB, Brzezinski P. Role of the pathway through K(I-362) in proton transfer in cytochrome c oxidase from R. sphaeroides. Biochemistry. 1998;37:2470–2476. doi: 10.1021/bi971813b. [DOI] [PubMed] [Google Scholar]
- 9.Branden M, Tomson F, Gennis RB, Brzezinski P. The entry point of the K-proton-transfer pathway in cytochrome c oxidase. Biochemistry. 2002;41:10794–10798. doi: 10.1021/bi026093+. [DOI] [PubMed] [Google Scholar]
- 10.Tomson FL, Morgan JE, Gu G, Barquera B, Vygodina TV, Gennis RB. Substitutions for glutamate 101 in subunit II of cytochrome c oxidase from Rhodobacter sphaeroides result in blocking the proton-conducting K-channel. Biochemistry. 2003;42:1711–1717. doi: 10.1021/bi026750y. [DOI] [PubMed] [Google Scholar]
- 11.Richter OM, Durr KL, Kannt A, Ludwig B, Scandurra FM, Giuffre A, Sarti P, Hellwig P. Probing the access of protons to the K pathway in the Paracoccus denitrificans cytochrome c oxidase. FEBS J. 2005;272:404–412. doi: 10.1111/j.1742-4658.2004.04480.x. [DOI] [PubMed] [Google Scholar]
- 12.Branden M, Sigurdson H, Namslauer A, Gennis RB, Adelroth P, Brzezinski P. On the role of the K-proton transfer pathway in cytochrome c oxidase. Proc Natl Acad Sci USA. 2001;98:5013–5018. doi: 10.1073/pnas.081088398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cukier RI. A molecular dynamics study of water chain formation in the proton-conducting K channel of cytochrome c oxidase. Biochim Biophys Acta. 2005;1706:134–146. doi: 10.1016/j.bbabio.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Branden G, Gennis RB, Brzezinski P. Transmembrane proton translocation by cytochrome c oxidase. Biochim Biophys Acta. 2006;1757:1052–1063. doi: 10.1016/j.bbabio.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 16.Mills DA, Geren L, Hiser C, Schmidt B, Durham B, Millett F, Ferguson-Miller S. An arginine to lysine mutation in the vicinity of the heme propionates affects the redox potentials of the hemes and associated electron and proton transfer in cytochrome c oxidase. Biochemistry. 2005;44:10457–10465. doi: 10.1021/bi050283d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilderson G, Salomonsson L, Aagaard A, Gray J, Brzezinski P, Hosler J. Subunit III of cytochrome c oxidase of Rhodobacter sphaeroides is required to maintain rapid proton uptake through the D pathway at physiologic pH. Biochemistry. 2003;42:7400–7409. doi: 10.1021/bi0341298. [DOI] [PubMed] [Google Scholar]
- 18.Hosler JP. The influence of subunit III of cytochrome c oxidase on the D pathway, the proton exit pathway and mechanism-based inactivation in subunit I. Biochim Biophys Acta. 2004;1655:332–339. doi: 10.1016/j.bbabio.2003.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Mills DA, Tan Z, Ferguson-Miller S, Hosler J. A role for subunit III in proton uptake into the D pathway and a possible proton exit pathway in Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 2003;42:7410–7417. doi: 10.1021/bi0341307. [DOI] [PubMed] [Google Scholar]
- 20.Ädelroth P, Hosler JP. Surface proton donors for the D-pathway of cytochrome c oxidase in the absence of subunit III. Biochemistry. 2006;45:8308–8318. doi: 10.1021/bi0605843. [DOI] [PubMed] [Google Scholar]
- 21.Fetter J, Sharpe M, Qian J, Mills D, Ferguson-Miller S, Nicholls P. Fatty acids stimulate activity and restore respiratory control in a proton channel mutant of cytochrome c oxidase. FEBS Lett. 1996;393:155–160. doi: 10.1016/0014-5793(96)00874-5. [DOI] [PubMed] [Google Scholar]
- 22.Aagaard A, Namslauer A, Brzezinski P. Inhibition of proton transfer in cytochrome c oxidase by zinc ions: Delayed proton uptake during oxygen reduction. Biochim Biophys Acta. 2002;1555:133–139. doi: 10.1016/s0005-2728(02)00268-2. [DOI] [PubMed] [Google Scholar]
- 23.Kannt A, Ostermann T, Muller H, Ruitenberg M. Zn2+ binding to the cytoplasmic side of Paracoccus denitrificans cytochrome c oxidase selectively uncouples electron transfer and proton translocation. FEBS Lett. 2001;503:142–146. doi: 10.1016/s0014-5793(01)02719-3. [DOI] [PubMed] [Google Scholar]
- 24.Mills DA, Schmidt B, Hiser C, Westley E, Ferguson-Miller S. Membrane potential-controlled inhibition of cytochrome c oxidase by zinc. J Biol Chem. 2002;277:14894–14901. doi: 10.1074/jbc.M111922200. [DOI] [PubMed] [Google Scholar]
- 25.Axelrod HL, Abresch EC, Paddock ML, Okamura MY, Feher G. Determination of the binding sites of the proton transfer inhibitors Cd2+ and Zn2+ in bacterial reaction centers. Proc Natl Acad Sci USA. 2000;97:1542–1547. doi: 10.1073/pnas.97.4.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berry E, Zhang Z, Bellamy H, Huang L. Crystallographic location of two Zn2+ binding sites in the avian cytochrome bc1 complex. Biochim Biophys Acta. 2000;1459:440–448. doi: 10.1016/s0005-2728(00)00182-1. [DOI] [PubMed] [Google Scholar]
- 27.Sone N, Yoshida M, Hirata H, Kagawa Y. Reconstitution of vesicles capable of energy transformation from phospholipids and adenosine triphosphatase of a thermophilic bacterium. J Biochem. 1977;81:519–528. doi: 10.1093/oxfordjournals.jbchem.a131485. [DOI] [PubMed] [Google Scholar]
- 28.Hiser C, Mills DA, Schall M, Ferguson-Miller S. C-Terminal truncation and histidine-tagging of cytochrome c oxidase subunit II reveals the native processing site, shows involvement of the C-terminus in cytochrome c binding, and improves the assay for proton pumping. Biochemistry. 2001;40:1606–1615. doi: 10.1021/bi0018988. [DOI] [PubMed] [Google Scholar]
- 29.Shapleigh JP, Gennis RB. Cloning, sequencing, and deletion from the chromosome of the gene encoding subunit I of the aa3-type cytochrome c oxidase of Rhodobacter sphaeroides. Mol Microbiol. 1992;6:635–642. doi: 10.1111/j.1365-2958.1992.tb01511.x. [DOI] [PubMed] [Google Scholar]
- 30.Simon R, Priefer U, Pühler A. A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in gram negative bacteria. Bio/Technology. 1983;1:784–791. [Google Scholar]
- 31.Zhen Y, Qian J, Follmann K, Hayward T, Nilsson T, Dahn M, Hilmi Y, Hamer AG, Hosler JP, Ferguson-Miller S. Overexpression and purification of cytochrome c oxidase from Rhodobacter sphaeroides. Protein Expression Purif. 1998;13:326–336. doi: 10.1006/prep.1998.0903. [DOI] [PubMed] [Google Scholar]
- 32.Mills DA, Hosler JP. Slow proton transfer through the pathways for pumped protons in cytochrome c oxidase induces suicide inactivation of the enzyme. Biochemistry. 2005;44:4656–4566. doi: 10.1021/bi0475774. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. 2004;D60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 35.Bailey S. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi E, Wraight CA. Small weak acids reactivate proton transfer in reaction centers from Rhodobacter sphaeroides mutated at AspL210 and AspM17. J Biol Chem. 2006;281:4413–4422. doi: 10.1074/jbc.M511359200. [DOI] [PubMed] [Google Scholar]
- 37.Ädelroth P, Paddock ML, Tehrani A, Beatty JT, Feher G, Okamura MY. Identification of the proton pathway in bacterial reaction centers: Decrease of proton transfer rate by mutation of surface histidines at H126 and H128 and chemical rescue by imidazole identifies the initial proton donors. Biochemistry. 2001;40:14538–14546. doi: 10.1021/bi011585s. [DOI] [PubMed] [Google Scholar]
- 38.Parul D, Palmer G, Fabian M. Proton interactions with hemes a and a3 in bovine heart cytochrome c oxidase. Biochemistry. 2005;44:4562–4571. doi: 10.1021/bi048435c. [DOI] [PubMed] [Google Scholar]
- 39.Antalik M, Jancura D, Palmer G, Fabian M. A role for the protein in internal electron transfer to the catalytic center of cytochrome c oxidase. Biochemistry. 2005;44:14881–14889. doi: 10.1021/bi050824z. [DOI] [PubMed] [Google Scholar]
- 40.Faxen K, Salomonsson L, Ädelroth P, Brzezinski P. Inhibition of proton pumping by zinc ions during specific reaction steps in cytochrome c oxidase. Biochim Biophys Acta. 2006;1757:388–394. doi: 10.1016/j.bbabio.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Bratton MR, Pressler MA, Hosler JP. Suicide inactivation of cytochrome c oxidase: Catalytic turnover in the absence of subunit III alters the active site. Biochemistry. 1999;38:16236–16245. doi: 10.1021/bi9914107. [DOI] [PubMed] [Google Scholar]
- 42.Mills DA, Ferguson-Miller S. Influence of structure, pH and membrane potential on proton movement in cytochrome oxidase. Biochim Biophys Acta. 2002;1555:96–100. doi: 10.1016/s0005-2728(02)00261-x. [DOI] [PubMed] [Google Scholar]
- 43.Smirnova IA, Ädelroth P, Gennis RB, Brzezinski P. Aspartate-132 in cytochrome c oxidase from Rhodobacter sphaeroides is involved in a two-step proton transfer during oxoferryl formation. Biochemistry. 1999;38:6826–6833. doi: 10.1021/bi982865j. [DOI] [PubMed] [Google Scholar]
- 44.Christianson DW, Alexander RS. Carboxylate-histidine-zinc interactions in protein structure and function. J Am Chem Soc. 1989;111:6412–6419. [Google Scholar]
- 45.Aagaard A, Brzezinski P. Zinc ions inhibit oxidation of cytochrome c oxidase by oxygen. FEBS Lett. 2001;494:157–160. doi: 10.1016/s0014-5793(01)02299-2. [DOI] [PubMed] [Google Scholar]
- 46.Francesco F, Giachini L, Boscherini F, Venturoli G, Capitanio G, Martino PL, Papa S. The inhibitory binding site(s) of Zn2+ in cytochrome c oxidase. FEBS Lett. 2007;581:611–616. doi: 10.1016/j.febslet.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 47.Qian J, Mills DA, Geren L, Wang K, Hoganson CW, Schmidt B, Hiser C, Babcock GT, Durham B, Millett F, Ferguson-Miller S. Role of the conserved arginine pair in proton and electron transfer in cytochrome c oxidase. Biochemistry. 2004;43:5748–5756. doi: 10.1021/bi036279o. [DOI] [PubMed] [Google Scholar]