

Abstract
Cytochrome c oxidase is a membrane-bound enzyme that catalyzes the four-electron reduction of oxygen to water. This highly exergonic reaction drives proton pumping across the membrane. One of the key questions associated with the function of cytochrome c oxidase is how the transfer of electrons and protons is coupled and how proton transfer is controlled by the enzyme. In this study we focus on the function of one of the proton transfer pathways of the R. sphaeroides enzyme, the so-called K-proton transfer pathway (containing a highly conserved Lys(I-362) residue), leading from the protein surface to the catalytic site. We have investigated the kinetics of the reaction of the reduced enzyme with oxygen in mutants of the enzyme in which a residue [Ser(I-299)] near the entry point of the pathway was modified with the use of site-directed mutagenesis. The results show that during the initial steps of oxygen reduction, electron transfer to the catalytic site (to form the “peroxy” state, Pr) requires charge compensation through the proton pathway, but no proton uptake from the bulk solution. The charge compensation is proposed to involve a movement of the K(I-362) side chain toward the binuclear center. Thus, in contrast to what has been assumed previously, the results indicate that the K-pathway is used during oxygen reduction and that K(I-362) is charged at pH ≈ 7.5. The movement of the Lys is proposed to regulate proton transfer by “shutting off” the protonic connectivity through the K-pathway after initiation of the O2 reduction chemistry. This “shutoff” prevents a short-circuit of the proton-pumping machinery of the enzyme during the subsequent reaction steps.
Keywords: flow-flash, proton pumping, cytochrome aa3, flash photolysis, gating, R. sphaeroides
Cytochrome c oxidase is a membrane-bound protein complex that catalyzes the reduction of dioxygen to water. The enzyme contains four redox-active metal sites: two copper sites, CuA and CuB, and two heme groups, heme a and heme a3. During the catalytic cycle electrons are first transferred from a water-soluble cytochrome c to CuA, and then consecutively to heme a and the binuclear center heme a3/CuB, where O2 binds and is reduced to water. Part of the free energy released by the oxygen reduction reaction is used to pump protons across the membrane, maintaining an electrochemical gradient that is used for the production of ATP (for reviews, see refs. 1–3).
The reaction cycle of cytochrome c oxidase involves transitions between partly reduced oxygen intermediates, which are built up to detectable concentrations (4). The first intermediate that is formed after binding of O2 to reduced heme a3 is called “peroxy” (P), defined by its optical absorption spectrum, which has a characteristic peak at 607 nm in the difference spectrum of P and the oxidized (O) enzyme (5–8). The intermediate is called “peroxy” because it was originally believed that the reduction level of O2 was at the peroxy level (Fea33+-O−-O−) (for reviews see refs. 2 and 9). However, more recent data indicate that in this first intermediate the O-O bond is broken and an oxo-ferryl state (Fea34+⩵O2−) is formed together with CuB2+-OH− (see, e.g., refs. 10–13). The P species is formed upon reaction of O2 with the two-electron reduced enzyme (heme a3/CuB reduced, and CuA/heme a oxidized, mixed-valence state, m), in which case it is called Pm. Formation of Pm involves oxidation of Fea32+ to Fea34+, CuB+ to CuB2+, and electron transfer to the oxygen intermediate from a tyrosine [Y(I-288)], which forms a radical, Y•(I-288) in the catalytic site (14) (see Fig. 1). The same species (as defined by its optical properties) (6) with a broken O-O bond (13) is formed transiently also upon reaction of the fully reduced enzyme with O2, in which case the intermediate is named Pr. Formation of Pr is associated with simultaneous electron transfer from heme a to rereduce Y•(I-288) (see Fig. 1; CuB2+ is not rereduced; see ref. 13). Thus, there is one more electron at the catalytic site in Pr than in Pm. Because the two intermediates display the same optical absorption spectra (6, 7), their chemical structures are presumably the same. There is no proton uptake from the bulk solution associated with formation of Pm/Pr, and therefore the proton needed to break the O-O bond must be taken internally from the enzyme (15–17), presumably from Y(I-288) (11).
Figure 1.

Schematic diagram showing the sequence of events after binding of O2 to reduced heme a3 (intermediate A). Only the binuclear center is shown. In intermediate P, the O-O bond cleavage requires a total of four electrons and one proton. In Pm one of these electrons and a proton are transferred from Y(I-288), which forms a radical (YO•) (τ ≅ 300 μs). Upon reaction of the fully reduced enzyme with O2 an electron from heme a is transferred rapidly to the catalytic site to rereduce Y•(I-288) (Pr state). The Pr state decays into F, which involves proton uptake through the D-pathway. Note that Pr has one more negative charge at the catalytic site as compared with Pm. a3 denotes Fea3. In F, the proton is suggested to bind to OH− at CuB, but it could also bind to other groups.
Upon reaction of the fully reduced enzyme with O2, the decay of Pr is associated with formation of the oxo-ferryl state (F) and coupled to proton uptake from the bulk solution (18), but without additional electron transfer to the binuclear center (see Fig. 1). Upon transfer of a fourth electron to the binuclear center the F state decays, forming the fully oxidized enzyme (O), associated with additional proton uptake from the bulk solution (18).
The enzyme has two proton transfer pathways (called D and K) used for the transfer of pumped protons across the membrane and substrate protons from solution to the catalytic site (19, 20). The K-pathway contains a highly conserved lysine residue K(I-362) and a threonine T(I-359), which presumably can form a protonic connection with Y(I-288) (Fig. 2). Kinetic measurements showed that proton uptake/release, coupled to reduction/oxidation of the binuclear center, was blocked in the KM(I-362) and TA(I-359) mutant enzymes. This observation indicates that at least one of the protons taken up upon reduction of the binuclear center is transferred through the K-pathway (21). Moreover, in KM(I-362) the reduction rate of the oxidized binuclear center is dramatically slowed (22–27), presumably because of impaired proton transfer through the K-pathway that accompanies its reduction (28).
Figure 2.

The structure of the proton-conductive K-pathway leading from the surface of the protein to the binuclear center. Ser(I-299) is located at the protein surface on the proton-input side. It is at hydrogen-bonding distance from a water molecule that in turn is at hydrogen-bonding distance from Lys(I-362). The figure was prepared with visual molecular dynamics software (42) with the R. sphaeroides enzyme structure (Svensson Ek et al., unpublished results).
The role of the K-pathway during reaction of the reduced enzyme with O2 is less clear as all proton-uptake reactions from the bulk solution take place through the D-pathway (25, 29). In addition, in the KM(I-362) and TA(I-359) mutant enzymes the extent and time constant of the overall oxidation of the fully reduced enzyme were not affected (but see this work), although a slow proton uptake that follows the F → O transition was impaired (21).
To elucidate the role of the K-pathway in the different reaction steps of the catalytic reaction of cytochrome c oxidase, we have investigated electron and proton transfer reactions in mutant forms of the Rhodobacter sphaeroides enzyme in which the proton connectivity through the K-pathway was altered by modification of Ser(I-299). The residue is located near the enzyme surface, at a possible entry point of the K-pathway (see also ref. 30), 20–25 Å from the binuclear center (Fig. 2). Consequently, mutations of S(I-299) are expected to leave K(I-362), the key residue of the K-pathway, as well as the catalytic site intact.
Materials and Methods
Mutagenesis, Growth of Bacteria, and Enzyme Purification.
Site-directed mutagenesis of cytochrome c oxidase has been described in detail in ref. 22 (see also ref. 31). Bacteria where grown aerobically in shake incubators. The histidine-tagged enzyme was purified as described (32). The overall activities of the SG(I-299) and SD(I-299) mutant enzymes were ≈100% and ≈40%, respectively, of that of the wild-type enzyme.
Preparation of Fully Reduced Enzyme.
The enzyme stock solution was diluted to a concentration of about 10 μM in a solution of 100 mM Hepes-KOH (pH 7.4), 0.1% dodecyl-β-d-maltoside, and 5 μM phenazine methosulfate in a modified anaerobic cuvette, which was then repetitively evacuated on a vacuum line and flushed with N2. To reduce the enzyme, ascorbate (2 mM) was added to the enzyme solution, followed by replacement of N2 by CO.
Preparation of Mixed-Valence Enzyme.
The buffer in the enzyme solution was replaced with 100 mM Tris⋅HCl (pH 8.5), 0.1% dodecyl-β-d-maltoside with the use of a Centriprep centrifuge filter (Millipore). The enzyme at a concentration of 10 μM was transferred to a modified anaerobic cuvette, and the atmosphere in the cuvette was changed first to N2 and then to CO. Anaerobic incubation in CO results in formation of the mixed-valence state of the enzyme in which heme a3/CuB is reduced and heme a/CuA is oxidized. In cases where fractional reduction of heme a was observed, the enzyme solution was titrated with anaerobic ferricyanide until heme a became reoxidized.
Flow-Flash Experiments.
The enzyme solution was transferred anaerobically to one of the drive syringes (0.5 ml total volume) of a modified flow-flash apparatus (LKS.60, FF.60) from Applied Photophysics (Surrey, UK). The other syringe (2.5 ml total volume) was filled with an oxygen-saturated buffer (for details, see figure legends). A 150-W xenon bulb provided the monitoring beam, which was passed through a monochromator and directed to the detection cell with an optical fiber. Various interference filters (Melles Griot, Irvine, CA) were placed directly after the optical detection cell to block scattered actinic light. The signal was detected with a photomultiplier tube (R928; Hamamatsu, Ichinocho, Japan). It was amplified with a current-to-voltage converter (13AMP005; Melles Griot) and passed through a low-pass filter and a differential amplifier (AM 502; Tegam, Geneva, OH), which was used to subtract an offset voltage, generated from a stabilized power supply (PS 503A; Tegam). The signal was recorded on a digital oscilloscope (Nicolet Pro 90). The reaction was initiated by flash photolysis of the enzyme-CO complex about 100 ms after mixing with a 10-ns, ≈50-mJ laser flash at 532 nm (Brilliant B; Quantel, Les Ulis Cedex, France). Measurements at ≈830 nm were made with a solid-state diode laser (model 56IMS507; Melles Griot), and a photo diode was used instead of the photomultiplier tube to detect changes in absorbance.
Measurements of Internal Electron Transfer.
The cuvette, containing the mixed-valence CO complex, was placed between two monochromators. The second monochromator was used to block the actinic light from the excitation laser, scattered in the sample (instead of the interference filter; cf. above). The remaining part of the experimental set-up was the same as that described above.
Results
Internal Electron Transfer.
To determine whether modification of serine, S(I-299), located close to the cytochrome c oxidase surface on the proton-input side of the K-pathway, had any effect on internal electron transfer between hemes a and a3, we first investigated the reaction in the absence of O2. Flash photolysis of CO from heme a3 in enzyme in which heme a3/CuB is reduced and heme a/CuA is oxidized (mixed-valence state) results in electron transfer from heme a3 to heme a with an observed time constant of ≈3 μs (33). The intramolecular electron transfer was investigated in mutant enzymes in which S(I-299) was replaced by glycin [SG(I-299)] or aspartate [SD(I-299)]. The electron transfer rate was not affected by the mutations (not shown), which indicates that the mutations did not have any effect on the environments of the hemes.
Reaction of the Two-Electron Reduced Enzyme with O2.
Fig. 3 shows absorbance changes associated with reaction of the two-electron reduced (mixed-valence) cytochrome c oxidase with oxygen. At 610 nm, the initial increase in absorbance is due to dissociation of the CO ligand. The following decrease in absorbance is mainly associated with binding of O2 to heme a3, forming the iron-oxo complex (intermediate A). Next, the “peroxy” intermediate (Pm) is formed with a time constant of ≈300 μs (increase in absorbance). Formation of Pm is associated with an internal proton transfer, but there is no proton uptake from the bulk solution (17). The Pm formation rate with the SG(I-299) and SD(I-299) mutant enzymes was the same as with the wild-type enzyme.
Figure 3.

Absorbance changes at 610 nm after flash photolysis of CO from the two-electron reduced (mixed valence) enzyme in the presence of O2. The increase in absorbance with a time constant of ≈300 μs is associated with formation of the Pm intermediate. The same Pm formation rate was obtained with the SD(I-299) (not shown) as with the SG(I-299) enzyme. Experimental conditions: 0.1 M Hepes, pH 7.5, 0.1% dodecyl-β-d-maltoside, ≈2 μM reacting enzyme (the traces have been scaled to 1 μM reacting enzyme), 1 mM O2, 22°C.
Reaction of the Fully Reduced Enzyme with O2.
Absorbance changes associated with reaction with
O2 of the fully reduced wild type and SD(I-299)
and SG(I-299) mutant enzymes were investigated at a number of
wavelengths specific to transitions between oxygen intermediates and
redox changes of the metal cofactors (Fig.
4
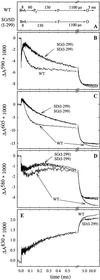 ). The initial rapid
change in absorbance at t = 0 in the traces shown in
Fig. 4 is due to CO dissociation. With the wild-type enzyme, at 590 nm
(Fig. 4B), the initial increase in absorbance (after the CO
dissociation change) is associated with binding of
O2 to reduced heme
a3, forming the iron-oxo complex (τ
≅ 8 μs). The subsequent decrease in absorbance (τ = 60
± 10 μs) is associated with formation of the
Pr intermediate, concomitant with oxidation of
heme a, which is also seen as a decrease in absorbance at
605 nm (Fig. 4C). The next intermediate, F, is formed with a
time constant of ≈130 μs, seen most clearly at 580 nm as an
absorbance increase (Fig. 4D). At the same time the electron
on CuA equilibrates with heme a, seen
as an increase in absorbance at 830 nm (net oxidation of
CuA, Fig. 4E). In the next step the
electron on the CuA/heme a
equilibrium is transferred to the binuclear center (τ ≅ 1.1 ms),
which is seen as a further increase in absorbance at 830 nm (oxidation
of CuA) and an absorbance decrease at 605 nm
(oxidation of heme a) and at 580/590 nm.
). The initial rapid
change in absorbance at t = 0 in the traces shown in
Fig. 4 is due to CO dissociation. With the wild-type enzyme, at 590 nm
(Fig. 4B), the initial increase in absorbance (after the CO
dissociation change) is associated with binding of
O2 to reduced heme
a3, forming the iron-oxo complex (τ
≅ 8 μs). The subsequent decrease in absorbance (τ = 60
± 10 μs) is associated with formation of the
Pr intermediate, concomitant with oxidation of
heme a, which is also seen as a decrease in absorbance at
605 nm (Fig. 4C). The next intermediate, F, is formed with a
time constant of ≈130 μs, seen most clearly at 580 nm as an
absorbance increase (Fig. 4D). At the same time the electron
on CuA equilibrates with heme a, seen
as an increase in absorbance at 830 nm (net oxidation of
CuA, Fig. 4E). In the next step the
electron on the CuA/heme a
equilibrium is transferred to the binuclear center (τ ≅ 1.1 ms),
which is seen as a further increase in absorbance at 830 nm (oxidation
of CuA) and an absorbance decrease at 605 nm
(oxidation of heme a) and at 580/590 nm.
Figure 4.
Absorbance changes after flash photolysis of CO from the fully reduced enzyme in the presence of O2. (A) Sequence of events in the wild-type (WT) and mutant enzymes. The length of the arrows has been adjusted to approximately fit the time course of the absorbance changes in B–E. At 590 nm (B) the increase in absorbance with τ ≅ 8 μ s is associated with binding of O2 to reduced heme a3 (intermediate A). The subsequent decrease in absorbance with τ ≅ 60 μs is associated with the formation of Pr. This kinetic phase is not observed with the SD(I-299)/SG(I-299) mutant enzymes. At 605 nm (C) the redox state, primarily of heme a, is monitored. In both mutant enzymes, heme a is oxidized more slowly than in the wild-type enzyme, and the oxidation coincides with formation of F (increase in absorbance at 580 nm, D). Changes in the redox level of CuA contribute significantly at 830 nm (E), where an increase in absorbance corresponds to CuA oxidation. The noise level is larger at short times because the traces were recorded on a logarithmic scale. Experimental conditions: 0.1 M Hepes, pH 7.4, 0.1% dodecyl-β-d-maltoside, ≈2 μM reacting enzyme (all traces have been normalized to 1 μM reacting enzyme), 1 mM O2, 22°C.
With both the SD(I-299) and SG(I-299) mutant enzymes the iron-oxo complex (intermediate A) was formed at the same rate as the wild-type enzyme (τ ≅ 8 μs; see Fig. 4B). However, the subsequent oxidation of heme a (Fig. 4C) and formation of the Pr intermediate (Fig. 4B) (τ ≅ 60 μs with the wild-type enzyme) were not observed with these mutant enzymes. Instead, the A intermediate decayed directly to F at about the same rate as in the wild-type enzyme (τ ≅ 130 μs). The subsequent F → O transition was unaffected by the mutations (τ ≅ 1.1 ms).
In the wild-type enzyme, the 1.1-ms phase is followed by a slower reaction phase with a time constant of ≈5 ms (see Fig. 4), associated with proton uptake from the bulk solution (18, 21). This kinetic phase was not observed with the SG(I-299) and SD(I-299) mutant enzymes (most clearly seen in Fig. 4D).
To obtain a satisfactory global (simultaneously at 445, 580, 590, 605, and 830 nm) fit of the mutant-enzyme data with a sum of exponential functions, we had to include a component with a time constant of ≈30 μs. This component is clearly seen at 580 nm (decrease in absorbance, Fig. 4D) and at 590 nm (increase in absorbance, Fig. 4B). The amplitude of this component was zero at 605 nm, i.e., there was no oxidation of heme a on this time scale (heme a was oxidized with τ ≅ 130 μs).
Discussion
Results from other studies indicate that the D-pathway is used for the transfer of both pumped and substrate protons during reaction of the fully reduced cytochrome c oxidase with O2 (25, 29). The role of the K-pathway is less clear. It is evidently needed to allow rapid reduction of the binuclear center (21–25) as one of the two protons taken up upon reduction of the binuclear center is transferred through the K-pathway (21, 26, 27). In addition, the pathway may be used for proton uptake immediately after oxidation of the fully reduced enzyme (21, 34). The K-pathway has also been suggested to be used for the release of hydroxide (instead of proton uptake) or to provide a “dielectric well” used to neutralize changes in charge at the binuclear center (23) (see below).
We have previously shown that upon reaction of the mixed-valence enzyme with oxygen, formation of the Pm intermediate involves an internal proton transfer but no proton uptake from the bulk solution (17). As indicated in the Introduction, this proton may be taken from Y(I-288) (see ref. 15). The time constant and extent of Pm formation were the same in SD(I-299) and SG(I-299) as in the wild-type enzyme (see Fig. 3), which indicates that the internal proton transfer takes place over a short distance and does not involve the “lower” part of the K-pathway.
During reaction of the fully reduced SD(I-299) and SG(I-299) mutant enzymes with oxygen, formation of the Pr intermediate and the accompanying oxidation of heme a were impaired. The same result was obtained previously with the KM(I-362) and TA(I-359) mutant enzymes (see figure 1B in ref. 21), although this fact was not discussed in the earlier publication. Because the rate of the internal electron transfer between heme a and the binuclear center (after flash photolysis of CO from the mixed-valence enzyme) and the formation of P (Pm) per se are not impaired in the SD(I-299) and SG(I-299) mutant enzymes, the results indicate that the transfer of the additional negative charge to the binuclear center during Pr formation requires charge compensation through the K-pathway (see Figs. 1 and 5). Because formation of Pr is not associated with proton uptake from the bulk solution, this charge compensation does not involve proton transfer all of the way through the K-pathway. In other words, even though formation of both Pm and Pr involves a localized, internal proton transfer, in the latter case the excess negative charge at the catalytic site requires charge compensation over a longer distance through the K-pathway.
Figure 5.

A schematic model explaining the experimental data in terms of structural events. In the wild-type enzyme, the protonated, charged (+) K(I-362) forms a hydrogen bond with a water molecule, which is hydrogen bonded to S(I-299). Formation of Pr involves the transfer of an additional electron to the binuclear center. To compensate for this negative charge, the K(I-362) side chain has to move toward the binuclear center.
According to the model in Fig. 5, in the reduced enzyme, K(I-362) is protonated and points in a direction down toward the input side of the proton transfer pathway. Upon binding of O2, electron transfer from heme a is charge compensated by a shift of the position of the positively charged side chain of K(I-362) toward the binuclear center. In other words, the movement of K(I-362) increases the redox potential at the catalytic site. In the mutant enzymes the water molecule, which in the wild-type enzyme is hydrogen bonded to S(I-299) (see Figs. 2 and 5), is displaced. This displacement destabilizes the protonated state of K(I-362) and puts the side chain in a position closer to the binuclear center where the unprotonated K(I-362) cannot pick up a proton to compensate for the excess charge at the binuclear center on the time scale of Pr formation. Alternatively, in SD(I-299) the negative charge of the aspartate may lock the protonated K(I-362) in a position pointing toward D(I-299), hindering the movement of K(I-362).
Electrostatic calculations (35) and visual analysis of the Paracoccus denitrificans cytochrome c oxidase structure (19) suggested that K(I-362) is neutral at pH 7–8. However, in the calculations the water molecule found near K(I-362) in the R. sphaeroides structure was not explicitly included, which is likely to alter the calculated pKa of the group.
The model in Fig. 5 is consistent with the earlier proposed function of the K-pathway as a “dielectric well” that could neutralize changes in charge at the binuclear center (23). However, it should be noted that we propose this role for the pathway only during formation of the Pr intermediate (where the charge movement also shuts off the proton transfer capability of the pathway). The pathway is used for proton transfer during reduction of the binuclear center and possibly also immediately after oxidation of the fully reduced enzyme (36, 37).
The 5-ms phase that follows the 1.1-ms F → O transition during oxidation of the fully reduced enzyme was not observed with the SG(I-299) and SD(I-299) mutant enzymes. The same result was obtained previously with the KM(I-362) mutant enzyme, and it was suggested that the 5-ms phase is associated with protonation or the release of OH− formed at the binuclear center during reduction of O2 (21, 34). This scenario is further supported by the results from the present study.
An absorbance change with a time constant of ≈30 μs was observed with the mutant enzymes. This event may reflect a fractional formation of a peroxy state with an intact O-O bond (see ref. 17) before oxidation of heme a and Pr formation. The time constant of the event is likely to depend on the reduction state of heme a, which explains why it is not observed during reaction of the mixed-valence enzyme with O2. Alternatively, it may be masked by the slower O2 binding to the mixed-valence enzyme (17). The absence of the 30-μs phase with the wild-type enzyme does not exclude the possibility that the corresponding event takes place, because it may be masked by the absorbance changes associated with Pr formation (τ ≅ 60 μs).
Two important questions related to this study are discussed in the context of the results presented above:
1. Why are the optical absorbance spectra of Pm and Pr the same despite the charge difference (see, e.g., ref. 11)? In Pr the excess negative charge appears on Y(I-288) (see Fig. 1; CuB remains oxidized) (13). As shown in the model in Fig. 5 the excess negative charge results in the movement of the positively charged K(I-362) side chain toward the binuclear center. Effectively, this shift moves the negative charge density away from the catalytic site. Thus, a possible (electronically induced) spectral difference as a result of the charge difference at Y(I-288) is attenuated, which explains why the same spectra are found for Pm as for Pr. Alternatively, the proton at K(I-362) could be transferred to Y·(I-288).
2. Is the same amount of free energy available for proton pumping when starting with state Pm as with state Pr? This question has been brought up by several authors also at a time when the chemical structure of Pm/Pr was not known. For example, it was noted that a too rapid electron transfer from heme a to the binuclear center (i.e., formation of Pr) may result in uncoupling of electron transfer and proton pumping. As discussed above, in Pm a Tyr radical is formed. In photosystem II the YZ•/YZ couple has a midpoint potential of ≈1 V (see ref. 38). If the same applies to Y(I-288) in cytochrome c oxidase, the electron transfer from heme a (Em ≅ 0.4 V in R. sphaeroides cytochrome aa3) (39) to Y•(I-288) would be highly exergonic. Thus, if the third electron is transferred too rapidly to the catalytic site (i.e., before F formation and the transfer of a “pumped proton”), this energy would be lost (see ref. 17). However, the results from this study indicate that the driving force for the electron transfer from heme a to the catalytic site (to form Pr) is relatively modest because the electron cannot be transferred unless the charge is compensated for by movement of K(I-362). Consequently, the midpoint potential of Y•/Y(I-288) is probably relatively low (similar to that of heme a), and thus the free-energy difference between Pm and Pr is small (for a discussion on the number of protons pumped during the different steps of the reaction cycle, see refs. 40 and 41).
Conclusions
We show that the K-pathway is involved during reaction of the fully reduced enzyme with O2 upon electron transfer from heme a to the catalytic site. Even though this reaction step does not involve proton uptake from the bulk solution, it requires compensation of the excess charge at the catalytic site (cf. ref. 28). We propose that this charge compensation is achieved by switching the positively charged K(I-362) side chain to a position pointing toward the binuclear center. This structural rearrangement closes the K-pathway for further proton transfer during oxygen reduction, which ensures that during the subsequent, highly exergonic steps, no protons are transferred directly to the catalytic site through the K-pathway. Instead the transfer of the remaining substrate protons takes place through the D-pathway, controlled by the uptake of pumped protons. This mechanism prevents a short-circuit of the pumping machinery by preventing reduction of P without the uptake of pumped protons.
Acknowledgments
We thank Sophie Plantard for experimental assistance, Jon Hosler and Catherine Pecoraro for help with site-directed mutagenesis, and Michael Carey (Applied Photophysics Ltd.) for technical assistance. These studies were supported by grants from the Swedish Foundation for International Cooperation in Research and Higher Education and the Swedish Natural Science Research Council. In memory of Prof. Gerald T. Babcock (Jerry), who died on December 22, 2000. A short time before Jerry passed away he carefully read the manuscript of this article and raised a number of important questions. We address his comments and questions in the discussion section. Jerry was a great scientist and a dear friend. We will miss him a lot.
Abbreviations
- WT
wild type
- CuA
copper A
- CuB
copper B
- τ
time constant (exp(−t/τ))
- binuclear center
heme a3 and CuB
- catalytic site
the binuclear center and redox-active/protonatable groups in its immediate vicinity
- Pm and Pr
the peroxy intermediates formed at the binuclear center upon reaction of the two-electron reduced (mixed-valence, “m”) and fully reduced (“r”) cytochrome c oxidase, respectively, with O2 (in Pr there is one more electron at the catalytic site as compared with Pm): F, oxo-ferryl intermediate
- O
fully oxidized enzyme. Mutant-enzyme nomenclature: S(I-299), serine of subunit I at position 299
- SD(I-299)
replacement of S(I-299) by aspartic acid. If not otherwise indicated, amino acid residues are numbered according to the R. sphaeroides cytochrome aa3 sequence
References
- 1.Ferguson-Miller S, Babcock G T. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Babcock G T, Wikström M. Nature (London) 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 3.Zaslavsky D, Gennis R B. Biochim Biophys Acta. 2000;1458:164–179. doi: 10.1016/s0005-2728(00)00066-9. [DOI] [PubMed] [Google Scholar]
- 4.Varotsis C, Zhang Y, Appelman E H, Babcock G T. Proc Natl Acad Sci USA. 1993;90:237–241. doi: 10.1073/pnas.90.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vygodina T V, Konstantinov A A. Ann NY Acad Sci. 1988;550:124–138. doi: 10.1111/j.1749-6632.1988.tb35329.x. [DOI] [PubMed] [Google Scholar]
- 6.Morgan J E, Verkhovsky M I, Wikström M. Biochemistry. 1996;35:12235–12240. doi: 10.1021/bi961634e. [DOI] [PubMed] [Google Scholar]
- 7.Sucheta A, Georgiadis K E, Einarsdóttir Ó. Biochemistry. 1997;36:554–565. doi: 10.1021/bi962422k. [DOI] [PubMed] [Google Scholar]
- 8.Wikström M, Morgan J E. J Biol Chem. 1992;267:10266–10273. [PubMed] [Google Scholar]
- 9.Babcock G T. Proc Natl Acad Sci USA. 1999;96:12971–12973. doi: 10.1073/pnas.96.23.12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabian M, Wong W W, Gennis R B, Palmer G. Proc Natl Acad Sci USA. 1999;96:13114–13117. doi: 10.1073/pnas.96.23.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Proshlyakov D A, Pressler M A, Babcock G T. Proc Natl Acad Sci USA. 1998;95:8020–8025. doi: 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Proshlyakov D A, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. Biochemistry. 1996;35:76–82. doi: 10.1021/bi9511705. [DOI] [PubMed] [Google Scholar]
- 13.Hansson Ö, Karlsson B, Aasa R, Vänngård T, Malmström B G. EMBO J. 1982;1:1295–1297. doi: 10.1002/j.1460-2075.1982.tb01313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Proshlyakov D A, Pressler M A, DeMaso C, Leykam J F, DeWitt D L, Babcock G T. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 15.Blomberg M R A, Siegbahn P E M, Babcock G T, Wikström M. J Inorg Biochem. 2000;80:261–269. doi: 10.1016/s0162-0134(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 16.Karpefors M, Ädelroth P, Aagaard A, Smirnova I A, Brzezinski P. Isr J Chem. 1999;39:427–437. [Google Scholar]
- 17.Karpefors M, Ädelroth P, Namslauer A, Zhen Y J, Brzezinski P. Biochemistry. 2000;39:14664–14669. doi: 10.1021/bi0013748. [DOI] [PubMed] [Google Scholar]
- 18.Ädelroth P, Ek M, Brzezinski P. Biochim Biophys Acta. 1998;1367:107–117. doi: 10.1016/s0005-2728(98)00142-x. [DOI] [PubMed] [Google Scholar]
- 19.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature (London) 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 20.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 21.Ädelroth P, Gennis R B, Brzezinski P. Biochemistry. 1998;37:2470–2476. doi: 10.1021/bi971813b. [DOI] [PubMed] [Google Scholar]
- 22.Hosler J P, Shapleigh J P, Mitchell D M, Kim Y, Pressler M A, Georgiou C, Babcock G T, Alben J O, Ferguson-Miller S, Gennis R B. Biochemistry. 1996;35:10776–10783. doi: 10.1021/bi9606511. [DOI] [PubMed] [Google Scholar]
- 23.Jünemann S, Meunier B, Gennis R B, Rich P R. Biochemistry. 1997;36:14456–14464. doi: 10.1021/bi971458p. [DOI] [PubMed] [Google Scholar]
- 24.Vygodina T V, Pecoraro C, Mitchell D, Gennis R, Konstantinov A A. Biochemistry. 1998;37:3053–3061. doi: 10.1021/bi971876u. [DOI] [PubMed] [Google Scholar]
- 25.Konstantinov A A, Siletsky S, Mitchell D, Kaulen A, Gennis R B. Proc Natl Acad Sci USA. 1997;94:9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruitenberg M, Kannt A, Bamberg E, Ludwig B, Michel H, Fendler K. Proc Natl Acad Sci USA. 2000;97:4632–4636. doi: 10.1073/pnas.080079097. . (First Published April 18, 2000; 10.1073/pnas.080079097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wikström M, Jasaitis A, Backgren C, Puustinen A, Verkhovsky M I. Biochim Biophys Acta. 2000;1459:514–520. doi: 10.1016/s0005-2728(00)00191-2. [DOI] [PubMed] [Google Scholar]
- 28.Mitchell R, Rich P R. Biochim Biophys Acta. 1994;1186:19–26. doi: 10.1016/0005-2728(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 29.Ädelroth P, Svensson Ek M, Mitchell D M, Gennis R B, Brzezinski P. Biochemistry. 1997;36:13824–13829. doi: 10.1021/bi9629079. [DOI] [PubMed] [Google Scholar]
- 30.Ma J X, Tsatsos P H, Zaslavsky D, Barquera B, Thomas J W, Katsonouri A, Puustinen A, Wikström M, Brzezinski P, Alben J O, et al. Biochemistry. 1999;38:15150–15156. doi: 10.1021/bi991764y. [DOI] [PubMed] [Google Scholar]
- 31.Aagaard A, Gilderson G, Mills D A, Ferguson-Miller S, Brzezinski P. Biochemistry. 2000;39:15847–15850. doi: 10.1021/bi0012641. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell D M, Gennis R B. FEBS Lett. 1995;368:148–150. doi: 10.1016/0014-5793(95)00626-k. [DOI] [PubMed] [Google Scholar]
- 33.Ädelroth P, Brzezinski P, Malmström B G. Biochemistry. 1995;34:2844–2849. doi: 10.1021/bi00009a014. [DOI] [PubMed] [Google Scholar]
- 34.Brzezinski P, Ädelroth P. J Bioenerg Biomembr. 1998;30:99–107. doi: 10.1023/a:1020567729941. [DOI] [PubMed] [Google Scholar]
- 35.Kannt A, Roy C, Lancaster D, Michel H. Biophys J. 1998;74:708–721. doi: 10.1016/S0006-3495(98)73996-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karpefors M, Ädelroth P, Aagaard A, Sigurdson H, Svensson Ek M, Brzezinski P. Biochim Biophys Acta. 1998;1365:159–169. doi: 10.1016/s0005-2728(98)00058-9. [DOI] [PubMed] [Google Scholar]
- 37.Ädelroth P, Sigurdson H, Hallén S, Brzezinski P. Proc Natl Acad Sci USA. 1996;93:12292–12297. doi: 10.1073/pnas.93.22.12292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tommos C, Babcock G T. Biochim Biophys Acta. 2000;1458:199–219. doi: 10.1016/s0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- 39.Ädelroth P, Mitchell D M, Gennis R B, Brzezinski P. Biochemistry. 1997;36:11787–11796. doi: 10.1021/bi962824s. [DOI] [PubMed] [Google Scholar]
- 40.Verkhovsky M I, Jasaitis A, Verkhovskaya M L, Morgan J E, Wikström M. Nature (London) 1999;400:480–483. doi: 10.1038/22813. [DOI] [PubMed] [Google Scholar]
- 41.Michel H. Proc Natl Acad Sci USA. 1998;95:12819–12824. doi: 10.1073/pnas.95.22.12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]