

Abstract
Studies in humans and animal models have demonstrated that acute kidney injury (AKI) has a significant effect on the function of extrarenal organs. The combination of AKI and lung dysfunction is associated with 80% mortality; the lung, because of its extensive capillary network, is a prime target for AKI-induced effects. The study presented here tested the hypothesis that AKI leads to a vigorous inflammatory response and produces distinct genomic signatures in the kidney and lung. In a murine model of ischemic AKI, prominent global transcriptomic changes and histologic injury in both kidney and lung tissues were identified. These changes were evident at both early (6 h) and late (36 h) timepoints after 60-min bilateral kidney ischemia and were more prominent than similar timepoints after sham surgery or 30 min of ischemia. The inflammatory transcriptome (109 genes) of both organs changed with marked similarity, including the innate immunity genes Cd14, Socs3, Saa3, Lcn2, and Il1r2. Functional genomic analysis of these genes suggested that IL-10 and IL-6 signaling was involved in the distant effects of local inflammation, and this was supported by increased serum levels of IL-10 and IL-6 after ischemia-reperfusion. In summary, this is the first comprehensive analysis of concomitant inflammation-associated transcriptional changes in the kidney and a remote organ during AKI. Functional genomic analysis identified potential mediators that connect local and systemic inflammation, suggesting that this type of analysis may be a useful discovery tool for novel biomarkers and therapeutic drug development.
Clinical studies have revealed a strong association between AKI and dysfunction of extrarenal organs, and more recently animal research has shown a significant causal effect of AKI on distant organ dysfunction.1–7 Since the availability of dialysis, AKI-associated distant organ dysfunction constitutes the major cause of death in these patients, with the mortality rate still in the 50% range. Despite this frustrating outcome, little is known about the potential pathophysiological interactions between the kidney and extrarenal organs in critically ill patients. Numerous recent studies have demonstrated that outcomes of AKI are heavily dependent upon the severity of comorbid conditions.8–10 Isolated AKI has a much better prognosis than AKI associated with multiple organ failure,11,12 and the presence of renal insufficiency continues to be a sensitive marker for poor outcome in the hospitalized patient.13 Thus, there is an urgent need to study the systemic effects of AKI, and modern discovery tools have the potential to unveil novel diagnostic and therapeutic targets.
Inflammation is a major component of the initiation and exacerbation of kidney injury during AKI,14,15 and local inflammation of kidney tissues could be a source of the development of inflammation and injury in extrarenal organs.2,16 Given that systemic inflammation typically occurs during AKI,17–24 we hypothesized that inflammation of postischemic kidney tissue coupled with decreased kidney clearance activates inflammatory pathways in distant organs. To test this hypothesis, we utilized an established ischemia reperfusion injury (IRI) model of AKI, which is characterized by both cellular and soluble inflammation.20 We recently compared lung genomic and functional effects of AKI versus nephrectomy on lung tissues combined with a candidate gene approach developed by our group25 to identify multiple biologic responses by lung tissues.7 In the study presented here, we focused on distinct time and severity of injury-specific inflammatory transcriptomic response in the kidney during AKI. We then compared this to an inflammation-specific genomic analysis of lung. Furthermore, we evaluated soluble inflammatory products related to the implicated genes, levels of which were increased in the circulation during AKI and could serve as a link between intrarenal and distant organ dysfunction during AKI.
RESULTS
Mouse Model of AKI-Induced Acute Lung Injury
Development of AKI was demonstrated by a significant rise in serum creatinine concentration at both 6 h (1.68 ± 0.11 mg/dl) and 36 h (2.86 ± 0.12 mg/dl) after 60-min renal ischemia compared with sham (0.78 ± 0.14 mg/dl) (Figure 1) and confirmed by kidney histologic injury demonstrated by development of renal tubular cast formation and extensive tubular injury (Figure 2). Reperfusion was confirmed after clamp removal. Serum creatinine (SCr) after 30-min ischemia was not appreciably different from sham (0.59 ± 0.08 mg/dl) at 6 h (0.81 ± 0.15 mg/dl), but demonstrated a strong trend for elevation at 36 h (2.24 ± 0.67 mg/dl). The lung histologic changes demonstrated modest septal edema and hypercellularity at 6 h, which resolved by 36 h. Meanwhile mice treated with 60-min renal ischemia demonstrated more pronounced septal edema and hypercellularity that persisted at 36 h (Figure 3). The leukocyte-specific Cd11 gene expression pattern demonstrated moderate (10 to 50%) contribution of transcriptional changes of tissue-infiltrating leukocytes to total kidney (Figure 2) and lung (Figure 3) tissue transcriptomics.
Figure 1.
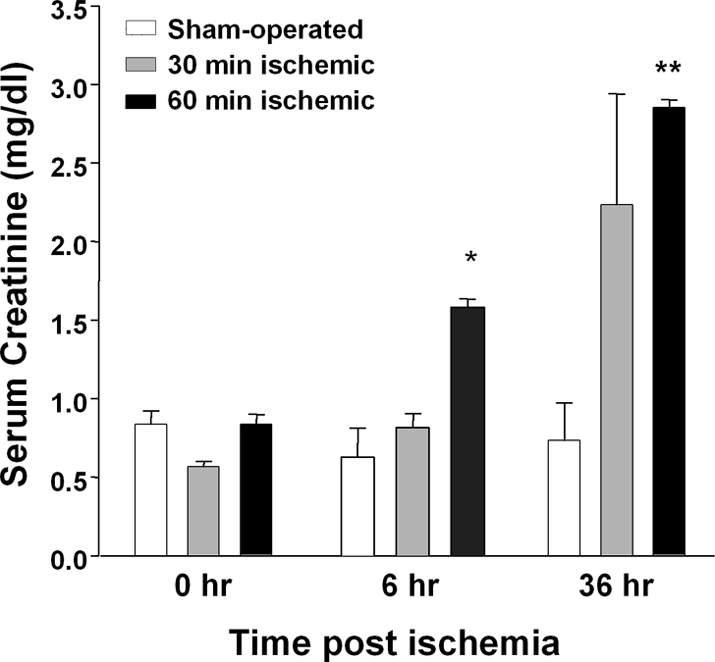
Effect of kidney ischemia-reperfusion injury on renal function. Renal function pre- (0 h) and postischemia reperfusion was evaluated measuring serum creatinine (SCr) concentration. All mice underwent sham operation, 30- or 60-min bilateral renal ischemia followed by reperfusion, and serum creatinine (SCr) was measured at 0 (before), 6, and 36 h after surgery. All three groups of mice had comparable baseline creatinines. Compared with sham, SCr was not different at 6 h after 30 min renal ischemia, and at 36 h approximately half of the tested mice demonstrated an increase in SCr. Sixty-minute renal ischemia led to a significant increase in SCr at 6 h and even higher levels of SCr at 36 h after ischemia. (*P < 0.002 versus corresponding sham-operated mice; **P < 0.0001 versus corresponding sham-operated mice; n = 3 to 5 per group).
Figure 2.

Histology of kidney tissues from mice after sham operation, 30- or 60-min renal ischemia and transcriptional contribution of leukocytes to kidney tissue transcriptomics. (A) 30-min sham, (B) 60-min sham, (C) 30-min ischemia and 6-h reperfusion, (D) 60-min ischemia and 6-h reperfusion, (E) 30-min ischemia and 36 h reperfusion, and (F) 60-min ischemia and 36-h reperfusion. Comparing sham kidney with normal histology, both 30- and 60-min ischemic kidneys displayed cast formation and tubular injury at 6- or 36-h postischemia. Sixty-minute ischemic kidney had more injury than 30-min ischemia at each time point. Hemotoxylin and eosin stain. Arrows indicate injured tubules. Open arrows indicate debris and cast formation. (G and H) Transcriptional changes of leukocyte-specific Cd11 gene. Compared with the sham operated controls, the overall contribution of leukocytes to kidney transcriptomics is moderate (5 to 30%). IRI, ischemia reperfursion injury.
Figure 3.

Histology of lung tissues from mice after sham operation, 30- or 60-min renal ischemia, and transcriptional contribution of leukocytes to lung tissue transcriptomics. (A) 30-min sham, (B) 60-min sham, (C) 30-min ischemia and 6-h reperfusion, (D) 60-min ischemia and 6-h reperfusion, (E) 30-min ischemia and 36-h reperfusion, and (F) 60-min ischemia and 36-h reperfusion. Compared with the sham-operated controls, 30-min renal ischemia induced mild lung changes with septal edema and hypercellularity at 6 h, which resolved by 36-h postischemia. Meanwhile, 60-min renal ischemia induced lung changes at 6 h that persisted throughout the 36-h reperfusion period. (G and H) Moderate (10 to 50%) transcriptional changes of Cd11 gene with the highest leukocyte contribution to lung tissue transcriptomics at 36 h after 60 min of kidney ischemia.
Inflammation-Associated Transcriptional Changes Induced by AKI in Kidney and Lung Tissues
Gene ontology analysis of MOE430A GeneChip identified 1035 inflammation-related probes that represented 651 known genes that were tested for association with IRI-induced injury in kidney and lung tissues using Gene Set Enrichment Analysis.26 The generated normalized enrichment scores, which reflect the correlation of inflammatory gene set with the transcriptional effects of AKI demonstrated the highest effect for 60-min ischemia in kidney tissues for both time points (Table 1). The identified NES were significant [false discovery rate (FDR) < 0.25] for seven of eight tested conditions in which 30-min ischemia failed to illicit a significant transcriptional response in lung 6 h after ischemia (Table 1).
Table 1.
Gene set enrichment analysis of inflammatory genes in acute kidney injury (AKI) settings
| Duration of Ischemia | Time of Recovery | Kidney
|
Lung
|
||
|---|---|---|---|---|---|
| NES | FDRa | NES | FDR | ||
| 30 min | 6 h | 1.32 | 0.138 | 0.89 | 1.000 |
| 36 h | 1.20 | 0.216 | 1.28 | 0.131 | |
| 60 min | 6 h | 1.49 | 0.140 | 1.19 | 0.192 |
| 36 h | 1.41 | 0.172 | 1.18 | 0.144 | |
NES, normalized enrichment score; FDR, false discovery rate.
FDR < 0.250 is considered significant.
Genomic Response to Ischemia/Reperfusion in the Kidney
Sixty-minute kidney ischemia resulted in a higher number of kidney inflammatory gene changes at both 6 and 36 h (Figure 4A) compared with 30-min ischemia. At 6 h, Significance Analysis of Microarrays (SAM) identified 55 IRI-affected inflammatory genes after 60 min of ischemia compared with 31 genes after 30 min of ischemia, including 24 genes that were responsive to both mild and severe ischemia. Similarly, at 36 h 54 genes were affected by 60 min of ischemia compared with 28 genes affected by 30 min of ischemia, including 20 genes that were sensitive to both mild and severe ischemia. Sixty-minute ischemia induced sustained inflammation-associated expressional changes in 31 genes (Figure 4A) of which 11 and 20 genes were previously identified as associated with kidney IRI and kidney inflammation, respectively (Table 2).
Figure 4.

Detection of AKI candidate genes affected by both 30- and 60-min ischemia in kidney and lung tissues. Gene expression profiles of IRI-affected kidney tissues (control = 3, IRI = 3) were analyzed using GeneChip Operating Software (GCOS) 1.4 and the Significance Analysis of Microarrays algorithm, and genes with a false discovery rate (FDR) < 1% and ± 2.85 changes in expression were considered significantly affected by IRI. Severity-independent inflammatory candidate genes were selected by crossreferencing significantly IRI-affected genes after 30- and 60-min exposure to ischemia and a recovery period of 6 or 36 h in (A) kidney or (B) lung tissues. Dashed and solid circles represent 30- and 60-min ischemia, respectively.
Table 2.
Renal inflammatory genes that are significantly affected during severe AKI
| Gene Title | Gene Symbol | Fold Change
|
PubMatrix
|
||
|---|---|---|---|---|---|
| 6 h | 36 h | Renal IRI | Kidney Inflammation | ||
| Lipocalin 2 | Lcn2 | 67.91 | 137.43 | 7 | 2 |
| Chemokine (C-X-C motif) ligand 2 | Cxcl2 | 35.31 | 11.44 | 2 | 3 |
| FBJ osteosarcoma oncogene | Fos | 39.02 | 4.69 | 5 | 11 |
| IL-6 | Il6 | 38.25 | 4.65 | 31 | 324 |
| Chemokine (C-X-C motif) ligand 1 | Cxcl1 | 29.58 | 10.71 | 5 | 13 |
| S100 calcium binding protein A8 | S100a8 | 16.36 | 11.49 | 0 | 6 |
| Suppressor of cytokine signaling 3 | Socs3 | 15.68 | 11.25 | 0 | 1 |
| S100 calcium binding protein A9 | S100a9 | 16.08 | 10.65 | 0 | 3 |
| Fos-like antigen 1 | Fosl1 | 13.76 | 11.58 | 0 | 0 |
| Chitinase 3-like 3 | Chi3l3 | 10.87 | 9.03 | 0 | 0 |
| Prostaglandin-endoperoxide synthase 2 | Ptgs2 | 13.08 | 5.08 | 36 | 112 |
| Kruppel-like factor 6 | Klf6 | 6.95 | 10.14 | 0 | 0 |
| Protein C receptor, endothelial | Procr | 11.00 | 4.36 | 0 | 2 |
| Plasminogen activator, urokinase receptor | Plaur | 6.20 | 8.06 | 0 | 3 |
| IL-1 receptor type II | Il1r2 | 7.75 | 6.02 | 0 | 1 |
| Serum amyloid A 3 | Saa3 | 7.35 | 3.92 | 0 | 14 |
| Annexin A1 | Anxa1 | 5.04 | 5.99 | 0 | 3 |
| Nfkb inhibitor, zeta | Nfkbiz | 6.18 | 4.19 | 0 | 0 |
| Coagulation factor III | F3 | 6.61 | 3.66 | 8 | 23 |
| Inhibin beta-B | Inhbb | 6.27 | 3.99 | 0 | 0 |
| CD14 antigen | Cd14 | 5.58 | 4.38 | 0 | 25 |
| Chemokine (C-X-C motif) ligand 7 | Cxcl7 | 5.04 | 4.41 | 0 | 0 |
| Proteolipid protein 2 | Plp2 | 3.26 | 5.97 | 0 | 0 |
| Oncostatin M receptor | Osmr | 3.92 | 3.23 | 2 | 1 |
| Chemokine (C-C motif) ligand 6 | Ccl6 | 3.03 | 4.05 | 0 | 0 |
| Chemokine (C-X-C motif) ligand 4 | Cxcl4 | 2.93 | 3.75 | 1 | 0 |
| Prolactin receptor | Prlr | −2.87 | −5.01 | 0 | 2 |
| Protein C | Proc | −3.26 | −5.15 | 5 | 30 |
| Phosphoenolpyruvate carboxykinase 1 | Pck1 | −2.95 | −6.48 | 0 | 0 |
| Peroxisomal membrane protein 2 | Pxmp2 | −3.25 | −6.57 | 0 | 0 |
| Growth hormone receptor | Ghr | −3.26 | −9.56 | 1 | 1 |
Significance criteria: 2.85-fold change and 1% false discovery rate (see Concise Methods). IRI, ischemia reperfusion injury.
The supervised hierarchical clustering analysis identified several clusters of genes with similar expression patterns, including a group of genes that were upregulated by both mild and severe injury, and the observed upregulation was sustainable throughout 36 h of reperfusion (Figure 5A) and included the Lcn2 gene, which codes for kidney injury biomarker neutrophil gelatinase-associated lipocalin,19,27 and Cxcl1 and Cxcl2 genes, which code for the keratinocyte-derived cytokine and macrophage inflammatory protein-2 alpha, respectively28,29 [according to nomenclature, mouse gene symbols are italicized, first letter uppercase and all of the rest lowercase; protein symbols are same as the gene symbol, but not italicized and all uppercase (http://www.informatics.jax.org)].
Figure 5.

Hierarchical clustering of inflammatory genes affected by 6- or 36-h exposure to IRI induced by 30- or 60-min of ischemia. (A) The 102 kidney inflammatory genes that were significantly affected by IRI after 30 or 60 min of ischemia were combined and clustered using MeV software. Fold change values (log2) were calculated by subtracting the average of their corresponding controls (n = 3) from individual gene expression value of each biological replicate. The clustering was conducted based on the gene expression pattern rather than amplitude using uncentered Pearson correlation and applying an average linkage algorithm, which identified five major clusters (more than five genes, blue triangles). The expression pattern of the well-known kidney injury-related gene that codes for neutrophil gelatinase-associated lipocalin (Lcn2) is highlighted with a white rectangle and identified in the gene list with a dotted arrow. (B) The 94 lung inflammatory genes that were significantly affected by kidney IRI after 30 or 60 min of ischemia were combined and clustered as described above. The six major clusters (blue triangles) were identified, of which Lcn2-containing clusters (bottom blue rectangle) demonstrated upregulation throughout all conditions. Genes from most representative clusters are listed on the right. Each column represents an experimental condition of ischemia-affected kidney or lung sample and each row represents an expression pattern of a gene throughout given experimental conditions. Red color indicates upregulation and green color indicates downregulation of gene expression relative to corresponding controls, with color intensity corresponding to the fold-change amplitude (fold-change scale shown on the left).
Evaluating Genomic Responses to Ischemic AKI in Lung Tissues
Sixty-minute kidney ischemia activated 30 inflammatory genes in the lung at 6 h, and 22 genes at 36 h (Figure 4B) including 10 genes that were affected at both timepoints (Table 3). Thirty-minute kidney ischemia was insufficient in triggering significant transcriptional changes in the lung compared with sham surgery. PubMatrix analysis of AKI-induced genomic changes in the lung identified 15 genes that are known to be associated with pulmonary inflammation according to the current PubMed database (Table 3). The supervised hierarchical clustering analysis identified two major clusters of genes with similar expression trends elicited by severe kidney injury, including Lcn2 and Cxcl2 genes (Figure 5B).
Table 3.
Lung inflammatory genes that are significantly affected during severe AKI
| Gene Title | Gene Symbol | Fold Change
|
PubMatrix
|
|
|---|---|---|---|---|
| 6 h | 36 h | Lung Inflammation | ||
| Significant at 6-h timepoint | ||||
| chemokine (C-C motif) ligand 9 | Ccl9 | 4.11 | 1.86b | 0 |
| leukocyte immunoglobulin-like receptor B3 | Lilrb3 | 3.63 | 1.80 | 0 |
| thrombospondin 1 | Thbs1 | 3.48 | 1.28 | 3 |
| CD14 antigen | Cd14 | 3.21 | 1.71 | 30 |
| CCAAT/enhancer binding protein (C/EBP), beta | Cebpb | 2.99 | 1.98 | 1 |
| phospholipase A2, group VII | Pla2g7 | 2.91 | 5.60 | 0 |
| Fc receptor, IgE, high affinity I, gamma | Fcer1g | 2.81 | 2.97 | 0 |
| serum amyloid A 2 | Saa2 | 2.59 | 1.32 | 12 |
| fatty acid binding protein 4, adipocytea | Fabp4 | −2.51 | −2.44 | 0 |
| roundabout homolog 1 | Robo1 | −2.69 | −1.45 | 0 |
| Significant at both timepoints | ||||
| serum amyloid A 3 | Saa3 | 15.99 | 6.42 | 3 |
| chemokine (C-X-C motif) ligand 2 | Cxcl2 | 14.67 | 2.69 | 17 |
| IL-1 receptor, type II | Il1r2 | 5.88 | 11.34 | 0 |
| inhibin beta-B | Inhbb | 6.93 | 3.70 | 0 |
| serum amyloid A 1 | Saa1 | 5.91 | 2.45 | 9 |
| suppressor of cytokine signaling 3 | Socs3 | 5.47 | 2.41 | 0 |
| chemokine orphan receptor 1 | Cmkor1 | 3.57 | 3.71 | 1 |
| chemokine (C-C motif) receptor 1 | Ccr1 | 2.73 | 3.76 | 2 |
| formyl peptide receptor, related sequence 2 | Fpr-rs2 | 2.76 | 3.72 | 0 |
| interleukin 4 receptor, alpha | Il4ra | 2.42 | 3.87 | 4 |
| Significant at 36-h timepoint | ||||
| serine/cysteine peptidase inhibitor, clade A 3N | Serpina3n | 2.03 | 7.72 | 0 |
| lipocalin 2 | Lcn2 | 1.64 | 5.81 | 3 |
| FMS-like tyrosine kinase 1 | Flt1 | 1.65 | 4.59 | 0 |
| arachidonate 5-lipoxygenase activating protein | Alox5ap | 2.29 | 3.80 | 0 |
| S100 calcium binding protein A9 | S100a9 | 1.55 | 3.57 | 6 |
| chemokine (C-C motif) ligand 6 | Ccl6 | 2.30 | 3.09 | 0 |
| signal transducer and activator of transcription 3 | Stat3 | 1.99 | 3.04 | 9 |
| peptidoglycan recognition protein 1 | Pglyrp1 | 1.37 | 2.83 | 0 |
| IL-1 receptor-like 1 | Il1rl1 | −1.20 | −2.70 | 3 |
| chemokine (C-X-C motif) ligand 12 | Cxcl12 | −1.82 | −3.57 | 2 |
Significance criteria: 2.38 fold change and 2% false discovery rate (see Concise Methods).
Significantly affected after 30 min of ischemia (Figure 5A);
failed significance testing.
Discriminating Power of Identified Inflammatory Genes
The ability of AKI-associated inflammatory gene changes to discriminate the severity and stage of kidney and lung tissues was tested using unsupervised hierarchical clustering. The unsupervised hierarchical clustering of 109 genes that were significantly affected by at least one tested condition identified three major clusters that represent sham, 30-min ischemia, and 60-min ischemia samples (Figure 6). Notably, these clusters comprised samples from both tissues demonstrating similar (tissue-independent) response to AKI by inflammatory genes in kidney and lung. The transcriptional changes inflicted by 30 min of kidney injury failed to form an individual cluster in either tissue. The seven subclusters (three or more members) were represented by either sham or severe AKI gene expression profiles, demonstrating that the severely AKI-affected samples from both tissues were efficiently discriminated from their corresponding shams (Figure 6).
Figure 6.

Inflammatory signature identified by hierarchical clustering of kidney and lung gene expression profiles. The 109 inflammatory genes that were significantly affected in kidney, lung, or both tissues were normalized, and unsupervised clustering was performed simultaneously for the kidney and lung samples. In addition to our in-house normalization procedure the original signal intensity values were also processed using conventional Cluster software.51 The “Adjust Data” function was applied to kidney and lung expression profiles using log transformation and mean center normalization of genes and arrays. The normalized gene expression profiles for each tissue were combined; inflammatory genes were extracted and clustered using uncentered Pearson correlation (average linkage). Each row represents an experimental condition including shams and is labeled on the right (K, kidney; L, lung; 30IRI, 30-min ischemia; 60IRI, 60-min ischemia; 1, 2, or 3, biologic replicate). Each column represents the expression pattern of a gene throughout given experimental conditions and the five most representative genes are marked with the arrows at the bottom. Each gene expression value is normalized to the mean column expression value of a given gene throughout all experimental conditions. The resulting values are color-coded as higher than column mean (yellow) or lower than column mean (blue), where color intensity corresponds to the amplitude of deviation from the mean (deviation scale is shown on the left). Three major clusters that represent sham, short exposure to ischemia, and long exposure to ischemia are separated in individual blocks. Seven significant (three or more members) clusters represented by large blue triangles on the sample tree are bracketed and named on the right. The location of candidate genes from Figure 7 is marked with arrows and corresponding gene symbols.
Validation of Genomics Findings
The significant increase in the relative message abundance of Cd14, Socs3, Saa3, Lcn2, and Il1r2 genes was confirmed by real time PCR (rtPCR) in both tissues (Figure 7) and was overall comparable with the corresponding transcriptional changes identified by the microarray technique. Although the sensitivity of these two transcript detecting approaches was interchangeable in kidney tissues, the rtPCR method was more sensitive in lung samples, which is in agreement with our previous findings.30 This higher sensitivity of rtPCR allowed identification of significant changes in the transcript abundance for Lcn2 and Cd14 genes at 6- and 36-h timepoints, respectively (Figure 7). Two downregulated genes in lung tissues, Il1rl1 and Cxcl12 (Table 3), were also successfully validated by rtPCR (data not shown).
Figure 7.

Expression level of inflammatory candidate genes in AKI-affected kidney and lung tissues detected by real-time reverse transcriptase PCR (rtPCR) and Affymetrix GeneChip. The Affymetrix expression values for five inflammatory genes that are listed on the x-axis were generated by hybridization of total mouse RNA isolated from kidney (n = 3, top panel) or lung (n = 3, bottom panel) to MOE430A GeneChip. The transcriptional changes were identified by comparing each gene's expression value to the mean of corresponding controls (n = 3) and expressed as mean of fold changes (open bars) where error bars represent SD. Transcriptional changes in all but two samples were statistically significant (FDR < 1%, FCkidney > 2.85, or FClung > 2.38). The relative message abundance in the same set of samples was evaluated by real-time rtPCR and compared with internal controls (three housekeeping genes) as described in Concise Methods. The fold changes in transcript abundance were computed and expressed as mean average fold change versus corresponding controls (solid bars). The error bars represent SD. Changes in transcript abundance were statistically significant (P < 0.05) in all rtPCR samples. ≠, microarray-identified fold changes failed significance testing according to power-predicted parameters described in Concise Methods. Lcn2, lipocalin 2; Socs3, suppressor of cytokine signaling 3; Saa3, serum amyloid A 3; Il1r2, IL-1 receptor, type II; Cd14, CD14 antigen.
Identification and Validation of Mediators Involved in AKI Inflammatory Signal Propagation
Global functional genomics analysis of validated candidate genes (Cd14, Socs3, Saa3, Lcn2, and Il1r2) indicated that the major AKI-dysregulated canonical pathways were IL-10 signaling (P < 0.000001) and IL-6 signaling (P < 0.0001) (Figure 8). Given that mouse IL-10 is a cytokine synthesis inhibitory factor and inhibits the synthesis of several cytokines, including IFN-γ, interleukin-2 (IL-2), TNFα, and granulocyte colony stimulating factor (G-CSF) produced by activated macrophages and by helper T cells (http://us.expasy.org/cgi-bin/niceprot.pl?P18893), these cytokines were added to a customized Bioplex protein array. The Ccl2 gene that codes for monocyte chemotactic protein-1 (MCP-1) and is coregulated with Il631 was tested as an additional representative of the IL-6 signaling pathway. Although the level of IL-10 protein was significantly higher in serum from mice exposed to AKI than in serum from sham-operated animals, protein levels of IL-10 targets were largely unaffected. The increase in IL-6 and MCP-1 levels in serum from AKI mice was significant and concordant (Figure 9).
Figure 8.

Global functional analysis. The significance value associated with a function in global analysis is a measure for how likely a group of our candidate genes is involved in represented (x-axes) function. The significance is expressed as a P value that is calculated using the right-tailed Fisher's exact test and represented as negative log value (y-axis). The threshold line represents significant P value 0.05 (1.3 in −log units).
Figure 9.

Representative chemokine/cytokine protein levels in mouse serum 6 and 36 h after IRI. Cytokines were measured with a protein multiple Bioplex technique in serum from mice that underwent sham surgery (open bars) or IRI (solid bars) and sacrificed at 6 or 36 h after surgery. (*P < 0.05 and **P < 0.005 versus corresponding sham-operated groups).
DISCUSSION
The study presented here provides novel characterization and disease-oriented bioinformatics analysis of the inflammatory molecular signature associated with both local and distant organ effects of AKI, enabling identification of novel biomarkers and therapeutic targets. Given that there are more than 1000 known inflammatory signaling molecules,28 the global gene expression profiling of lung and kidney tissues performed in these studies facilitated characterization of AKI-associated inflammatory processes involved in an established model of ischemic AKI. We also used a novel bioinformatics approach to correlate kidney and lung genomic changes with biological effects in the tissue samples, combining our data with the extensive information in the public domain (i.e. PubMed, Ingenuity).
The global gene expression profiling of injured mouse kidney revealed significant severity- and time-dependent association of the inflammatory pathway with AKI (Table 1). The injury severity-dependent changes in gene expression were well correlated with the SCr and kidney morphology at early (6 h after injury) and later (36 h after injury) stages of AKI. SCr were significantly increased after 6 h of severe IRI (60-min ischemia) compared with sham and were further elevated after 36 h (Figure 1), whereas 30 min ischemia did not lead to significant change in SCr at 6 h; however, creatinine was significantly up at 36 h. The extended ischemia time of 60 versus 30 min resulted in an increase in the number of inflammation-associated gene changes compared with the shorter exposure at both 6 and 36 h of AKI. A sizable group of these genes responded to both mild and severe ischemic injury and, moreover, was persistently upregulated throughout the course of AKI (Figure 4A). This observation suggests that this particular group of 31 genes could exert continuous inflammatory changes in the kidney as well as distant organs. Many of these genes have already associated with kidney inflammation in prior studies (Table 2).
Proinflammatory genes that were very highly upregulated included lipocalin 2 (fold change (FC) = 137.4), chemokine (C-X-C motif) ligand 2 (FC = 35.3), IL-6 (FC = 38.25), and chemokine (C-X-C motif) ligand 1 (FC = 29.58). These findings generated the hypothesis that products of these actively transcribed genes could leak into circulation and trigger and sustain systemic inflammation. It has already been described that there is an increase of serum levels of CXCL1 and IL-6 after mouse kidney ischemia,6,29 suggesting systemic leakage of proinflammatory cytokines/chemokines released by injured kidney tissues. We used the lung as a biosensor of distant systemic effects of AKI. The histological studies of lungs from mice exposed to 30 min of renal ischemia were either similar to tissues from the sham-operated animals or demonstrated minor changes at 6 h after ischemia, which were totally resolved by 36 h. However, the mice with 60 min renal ischemia developed consistent lung changes with septal edema and hypercellularity at 6 h and these remained evident at 36 h (Figure 3, D and F), similar to our previous report.7 These lung histological findings were consistent with gene expression profiling studies that identified significant inflammatory transcriptional changes in lung tissues after 60, but not 30 min of ischemic kidney injury (Figure 4B). We acknowledge that kidney and lung transcriptional changes could, in part, be attributed to the transcriptomics of infiltrating leukocytes (Figures 2 and 3). Taken together, these findings suggest that the transcriptional inflammatory response in lungs triggered by AKI is dependent on the severity of kidney injury and could underlie lung functional and histological changes.
The analysis of the expression pattern of all identified AKI-associated inflammatory genes in kidney and lung tissues demonstrated a striking similarity in expression changes (Table 2 and Table 3) and revealed a definite inflammatory signature of AKI in both tissues (Figure 6), where the similarity in the inflammatory response to AKI overpowered the tissue-specific differences. We also observed that the inflammatory signatures of severe AKI were time-specific and grouped accordingly to the timepoints (6 or 36 h) rather than to severity of the injury. Notably, the clustering of transcriptional changes inflicted by moderate kidney injury (30 min ischemia) was sporadic for both tissues, which correlated well with the functional (Figure 1) and histological (Figures 2 and 3) findings.
In line with our findings that kidney IRI leads to intrarenal inflammation that promotes distant organ inflammation with similar inflammatory transcriptomics, we selected genes that were affected by AKI in both tissues. Using other selecting criteria such as novelty (were not previously associated with kidney IRI) and solubility (can exist in secretable forms) we selected serum amyloid A3 (Saa3), IL-1 receptor type II (Il1r2), suppressor of cytokine signaling 3 (Socs3), and CD14 antigen (Cd14) (Table 2) as our primary candidates. The established kidney injury marker lipocalin 2 (Lcn2) was chosen as a positive control. Significant upregulation of selected genes was confirmed by rtPCR at both timepoints (Figure 7) and gene ontology analysis identified the innate immunity processes as common to these genes. Our identification of marked changes in the innate immunity Cd14 gene, which codes for the endotoxin lipopolysaccharide (LPS) receptor was in agreement with previous reports on the induction of this gene and association of its soluble product with kidney injury. Previous data support that Cd14 gene expression is a kidney-specific response and attributed to the ability of the intrinsic renal cells to express Cd14 rather than to infiltrating myeloid cells.32,33
The upregulation of another LPS-responsive Saa3 gene that codes for the isoform of serum amyloid A34 in lungs during AKI was recently reported by our group,7 and we now show it is also upregulated in kidney tissues. The next two genes, Il1r2 and Socs3, belong to innate immunity regulators. The Il1r2 gene codes for the IL-1 binding protein, which functions as a decoy receptor and has no direct signaling activity. It sequesters interleukins in the serum and tissues, thus preventing their binding to corresponding functional receptors and interrupting the signaling cascade.35 Another innate immunity regulator, Socs3, belongs to a family of negative-feedback regulators of cytokine signaling. These regulators are induced by their corresponding cytokines, which leads to subsequent shut-down of the respective signaling cascade,36 thus inhibiting effects of cytokines involved in the development of AKI and AKI-associated distant organ injury. Finally, the Lcn2 gene that codes for neutrophil gelatinase-associated lipocalin, a well-known AKI biomarker involved in antibacterial host defense, has been shown to limit bacterial growth by sequestering iron and depriving bacteria of this important element.37 In our studies, Lcn2 was the highest AKI-upregulated gene in kidney tissue (67.9 and 137.4 fold increase at 6 and 36 h after injury, Table 2), which corroborates reports by others.19,27
The global functional analysis that was used to select common pathways for these candidate genes led to the highest association of IL-10 and IL-6 signaling, which was dysregulated by AKI gene expression (Figure 8). The increase in expression of serum IL-10 and concordant inhibition of its targets IFN-γ, IL-2, TNF-α, and G-CSF in serum from mice with severe (60 min of ischemia) AKI was consistent with these findings (Figure 9). Of note, the previously reported inability of mild kidney injury (22 min of ischemia) to affect serum levels IL-106 explains inconsistent inflammatory signature in lungs exposed to mild AKI (30 min of ischemia) observed in our studies and highlights the severity-dependent role of IL-10 in distant injurious signaling. Although the severe AKI increased IL-10 levels in serum, it had no effect on the Il10 gene expression in kidney and lung tissues (Supplement Tables 3 to 4), suggesting that the source of the high levels of IL-10 in serum is not kidney tissues but rather circulating cells like AKI-activated macrophages and helper T cells. This hypothesis is further supported by the previous finding of high levels of IL-10, IL-6, and suppressor of cytokine signaling-3 in rat alveolar macrophages from inflamed lung where production of TNF-α was markedly reduced.38 Given that expression of myeloid cell-specific Il10 gene was undetectable by expression profiling of kidney and lung tissues, our Cd11 expression studies (Figure 2 G and H) were confirmed and support the notion that contribution of infiltrating cells to global transcriptomics of kidney and lung tissues occurs, but is limited.
In contrast to Il10, the expression of Il6 was highly upregulated (>38-fold change versus sham) in kidney tissues (Table 2), thus directly linking increased Il6 gene expression and increased IL-6 serum concentration. The kidney expression of Ccl2 (>3-fold change, Supplement Table 3) was also concordant with the increase of its product MCP-1 in serum. These findings suggest that distant effects of AKI can be conducted by both mechanisms: via AKI-activated leukocytes and by direct leakage of proinflammatory signaling molecules from injured kidney into circulation.
The study presented here is, to our knowledge, the first comprehensive inflammation-based genomic map of kidney and lung tissues during AKI. By using bioinformatics tools for both more refined analysis and linking to published data, we have developed a hypothesis about kidney injury-releasing inflammatory mediators into the circulation. Our study has also identified novel AKI-associated genes representing the inflammatory component of injury in both organs. These data offer a new opportunity for understanding the mechanisms involved in local and distant effects of AKI, and identify new diagnostic and therapeutic targets.
CONCISE METHODS
Animal Model
All animal protocols were approved by the Johns Hopkins Animal Care and Use Committee. Kidney IRI was performed in male C57BL6/J mice (6 to 8 wk old, Jackson Laboratory, Bar Harbor, Maine) as described previously.7 The ischemia duration was 30 or 60 min with a recovery time of 6 or 36 h.
Evaluating Renal Function
Blood samples were collected from each animal before ischemia and at sacrifice, and centrifuged for 10 min at 8000 rpm to obtain serum. SCr levels were measured as a marker of renal function, using a 557A Creatinine kit that uses a kinetic modification of the Jaffe reaction based on the alkaline picric acid method (Sigma Diagnostics, St. Louis, Missouri) and analyzed on a Cobas Mira S Plus automated analyzer (Roche Diagnostics Corp. Indianapolis, Indiana).
Histological Studies
For assessment of kidney morphologic injury, hematoxylin and eosin (H&E) staining was performed as described previously.20 At 6 or 36 h, animals were exsanguinated to remove the blood from organs, kidneys and lungs were harvested (lungs were preinflated with 5% agarose gel through the trachea) and fixed with formalin, embedded with paraffin, and stained with H&E reagents for histological examination.
Statistical and power prediction for minimum array requirements analyses
The measurements of SCr were analyzed with one-way ANOVA. Individual group means were then compared with a Tukey multiple-comparison test. P values less than 0.05 were considered significant.
The microarray sample size determination for class comparisons39 was calculated as described previously.7 Identified SD for control kidney (σ = 0.274) and lung (σ = 0.271) tissues were submitted to the microarray sample size identifying formula40 with power (1 − β) = 90% and 1% FDR (significance level α = 0.01) for kidney and 2% for more heterogeneous lung tissue.41 The power.t.test function of R2.3.1 program (www.r-project.org) identified fold change for kidney log2(Δ) = 1.513 (numerical 2.85) and lung log2(Δ) = 1.249 (numerical 2.38).
Transcript Profiling with Affymetrix Oligonucleotide Arrays
Affymetrix GeneChip profiling was performed at the Johns Hopkins Lowe Family Genomics Core. The total kidney or lung mRNA was isolated from parenchymal tissues (capsules and pedicles were removed upon organ collection). The processed RNA (quality monitored on an Agilent 2100 Bioanalyzer) was hybridized to Affymetrix GeneChip MG-430A 2.0 (MOE430A 22,626 transcripts) and hybridization signals measured by a Agilent Gene Array Scanner as described previously.7 The resulting digitized matrix (CEL files) was processed as described in Figure 10.
Figure 10.

Flow chart of Affymetrix gene expression data processing (electronic masking of oligonucleotide probes, background adjustment, and chip intensity normalization). The rectangular boxes represent data sets generated and used by the algorithm. Oval shapes depict steps at which manipulations with the data were conducted. Rhomboids represent the steps at which decision is made. The digitized hybridization signals were analyzed on a probe level using Bioconductor (www.bioconductor.org) affy package.48 Probes that produced detectable hybridization signal in less than 5% of kidney or lung sample hybridizations were considered nonfunctional and combined into tissue specific mask (*.MSK) files. The subsequent electronic masking of nonfunctional probes was conducted during GCOS 1.4 expression data re-evaluation as described previously.49 The process of identification of the background hybridization signal was modified from a previously reported approach.50 Briefly, the quartiles of highest hybridization signals among all “Absent” calls (P > 0.04) from each chip were averaged and considered a nonspecific (background) hybridization signal of a given chip. The average of all “Absent” values was considered an array brightness coefficient for chip normalization. The expression data were stratified by experimental conditions (n = 3) and hybridization of each transcript was evaluated. The transcripts that were called “Present” by the GCOS 1.4 algorithm and produced signal at least twice as high as that of background in at least two of three hybridizations in any given cluster were considered tissue-specific. The signal intensity values of these transcripts from each chip were increased by corresponding to a given chip background value (background adjustment) and divided by a chip brightness coefficient (normalization). All of the data manipulation processes were written in Python 2.2 (www.python.org) and scripts are available upon request.
Identification of Inflammatory Genes and Gene Set Enrichment Analysis
Gene ontology information for each probe set of MOE430A GeneChip was obtained from NetAffx (http://www.affymetrix.com/analysis/index.affx), enriched by cross-referencing with their human orthologues, and queried for [inflamm], [cytokine], [IL], [chemokine], and [chemotaxis] terms. The resulting gene list was used as an inflammatory gene set database for Gene Set Enrichment Analysis.26 Genes that were classified as “Present” by GeneChip Operating Software (GCOS 1.4) were used as an expression matrix and 1000 permutations were performed for each condition. FDR <0.25 was considered significant.
Computational Identification of AKI-Associated Candidate Genes
SAM 2.2042 was conducted using full permutation of three control and three IRI samples (720 permutations) without application of arbitrary restrictions.43 Genes with 2.85-fold change and 1% FDR (kidney tissues) and 2.38-fold change and 2% FDR (lung tissues) were considered significantly affected by AKI. The contribution of tissue infiltrating cells to kidney and lung transcriptomics was evaluated using leukocyte-specific gene coding for CD11 antigen (1455733_at probe).
Genomic Clustering and Signature Analyses
Hierarchical clustering was performed using the MeV (MultiExperiment Viewer) component (http://www.tm4.org/mev.html) of TM4 system for microarray data management and analysis.44 The crosstissue and inflammatory signature analyses were conducted using supervised or unsupervised clustering, respectively, with application of uncentered Pearson correlation and average linkage algorithm.
In Vitro and In Silico Validation of Gene Expression Data
rtPCR validation of selected candidate genes was conducted as described previously.45 Briefly, transcript levels of selected candidate genes were identified (n = 3 per condition, randomly selected from 5 samples) by ABI Prism 7700 Sequence Detector Systems using premanufactured probes and primers (Perkin-Elmer/Applied Biosystems). The ΔCt value for each sample was normalized using three endogenous control genes (Gapdh, Actb, and Pgk1). All changes in the gene expression between sham-operated (n = 3) and injured (n = 3) groups were evaluated with unpaired t test and were significant (P < 0.05). AKI-affected transcripts were matched against “kidney inflammation,” “lung inflammation,” and “renal ischemia reperfusion injury” terms in PubMed using the PubMatrix automated literature search engine.46
Global Functional Analysis
The functional analysis that identifies the biological functions that were significantly associated with identified candidate genes was conducted using the Ingenuity Pathways Knowledge Base tool (http://www.ingenuity.com). Fischer's exact test was used to calculate a P value determining the probability that each biological function assigned to our five candidate genes (tested set: 1450826_a_at, 1419532_at, 1416576_at, 1417268_at, 1427747_a_at) was due to chance alone.47
Cytokine Measurement in Mouse Serum
The custom cytokine protein array was manufactured as multiplex cytokine kit (Bio-Rad, Hercules, California) based on the Luminex technology and included IL-2, IL-6, IL-10, TNF, MCP-1, and G-CSF antibodies. The cytokine array plate layout consisted of eight standards in duplicate (32,000 to 1.95 pg/ml), two blank wells (for background fluorescence subtraction), and each sample in triplicate wells. The 6- and 36-h serum samples from mice with severe (60 min of ischemia) AKI were processed for Bioplex analysis according to manufacturer's protocol and reported signal for each cytokine was converted to concentration values using Bioplex Manager 3.0 software (Bio-Rad).
DISCLOSURES
None.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Acute Lung Injury SCCOR HL-073994 to K.C.B. and H.R.; and in part by the Mary Beryl Patch Turnbull Scholar Program (K.C.B.) and National Kidney Foundation (D.N.G.). A portion of this work was presented at the 39th annual meeting of the American Society of Nephrology, November 14 to 19, San Diego, California.
Published online ahead of print. Publication date available at www.jasn.org.
D.N.G. and M.L. contributed equally to this work.
REFERENCES
- 1.Levy EM, Viscoli CM, Horwitz RI: The effect of acute renal failure on mortality. A cohort analysis. JAMA 275: 1489–1494, 1996 [PubMed] [Google Scholar]
- 2.Kramer AA, Postler G, Salhab KF, Mendez C, Carey LC, Rabb H: Renal ischemia/reperfusion leads to macrophage-mediated increase in pulmonary vascular permeability. Kidney Int 55: 2362–2367, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Kelly KJ: Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol 14: 1549–1558, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Rabb H, Wang Z, Postler G, Soleimani M: Possible molecular basis for changes in potassium handling in acute renal failure. Am J Kidney Dis 35: 871–877, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Zarbock A, Schmolke M, Spieker T, Jurk K, Van Aken H, Singbartl K: Acute uremia but not renal inflammation attenuates aseptic acute lung injury: a critical role for uremic neutrophils. J Am Soc Nephrol 17: 3124–3131, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Hoke TS, Douglas IS, Klein CL, He Z, Fang W, Thurman JM, Tao Y, Dursun B, Voelkel NF, Edelstein CL, Faubel S: Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol 18: 155–164, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Hassoun H, Grigoryev DN, Lie M, Liu M, Cheadle C, Tuder RM, Rabb H: Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol 293: F30–F40, 2007 [DOI] [PubMed] [Google Scholar]
- 8.McCarthy JT: Prognosis of patients with acute renal failure in the intensive-care unit: a tale of two eras. Mayo Clin Proc 71: 117–126, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Bagshaw SM, Laupland KB, Doig CJ, Mortis G, Fick GH, Mucenski M, Godinez-Luna T, Svenson LW, Rosenal T: Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care 9: R700–R709, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen YC, Fang JT, Tien YC, Chang MY, Huang CC: Organ system failures predict prognosis in critically ill patients with acute renal failure requiring dialysis. Chang Gung Med J 23: 8–13, 2000 [PubMed] [Google Scholar]
- 11.Liano F, Junco E, Pascual J, Madero R, Verde E: The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. The Madrid Acute Renal Failure Study Group. Kidney Int Suppl 66: S16–S24, 1998 [PubMed] [Google Scholar]
- 12.Liano F, Pascual J: Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int 50: 811–818, 1996 [DOI] [PubMed] [Google Scholar]
- 13.Clermont G, Acker CG, Angus DC, Sirio CA, Pinsky MR, Johnson JP: Renal failure in the ICU: Comparison of the impact of acute renal failure and end-stage renal disease on ICU outcomes. Kidney Int 62: 986–996, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Lu CY, Hartono J, Senitko M, Chen J: The inflammatory response to ischemic acute kidney injury: A result of the ‘right stuff’ in the ‘wrong place’? Curr Opin Nephrol Hypertens 16: 83–89, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Thurman JM: Triggers of inflammation after renal ischemia/reperfusion. Clin Immunol 123: 7–13, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maitra R, Grigoryev DN, Bera TK, Pastan IH, Lee B: Cloning, molecular characterization, and expression analysis of Copine 8. Biochem Biophys Res Commun 303: 842–847, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Goes N, Urmson J, Ramassar V, Halloran PF: Ischemic acute tubular necrosis induces an extensive local cytokine response. Evidence for induction of interferon-gamma, transforming growth factor-beta 1, granulocyte-macrophage colony-stimulating factor, interleukin-2, and interleukin-10. Transplantation 59: 565–572, 1995 [PubMed] [Google Scholar]
- 18.Lemay S, Rabb H, Postler G, Singh AK: Prominent and sustained up-regulation of gp130-signaling cytokines and the chemokine MIP-2 in murine renal ischemia-reperfusion injury. Transplantation 69: 959–963, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, Ruff SM, Zahedi K, Shao M, Bean J, Mori K, Barasch J, Devarajan P: Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 365: 1231–1238, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Rabb H, Daniels F, O'Donnell M, Haq M, Saba SR, Keane W, Tang WW: Pathophysiological role of T lymphocytes in renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 279: F525–F531, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Woodward VK, Shelton JM, Richardson JA, Zhou XJ, Link D, Kielar ML, Jeyarajah DR, Lu CY: Ischemia-reperfusion induces G-CSF gene expression by renal medullary thick ascending limb cells in vivo and in vitro. Am J Physiol Renal Physiol 286: F1193–F1201, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Sheridan AM, Bonventre JV: Cell biology and molecular mechanisms of injury in ischemic acute renal failure. Curr Opin Nephrol Hypertens 9: 427–434, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Kitada H, Sugitani A, Yamamoto H, Otomo N, Okabe Y, Inoue S, Nishiyama K, Morisaki T, Tanaka M: Attenuation of renal ischemia-reperfusion injury by FR167653 in dogs. Surgery 131: 654–662, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Subramanian S, Bowyer MW, Egan JC, Knolmayer TJ: Attenuation of renal ischemia-reperfusion injury with selectin inhibition in a rabbit model. Am J Surg 178: 573–576, 1999 [DOI] [PubMed] [Google Scholar]
- 25.Grigoryev DN, Finigan JH, Hassoun P, Garcia JG: Science review: Searching for gene candidates in acute lung injury. Crit Care 8: 440–447, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP: Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt-Ott KM, Mori K, Li JY, Kalandadze A, Cohen DJ, Devarajan P, Barasch J: Dual action of neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol 18: 407–413, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Ibelgauft H: COPE: Cytokines and cells online pathfinder encyclopaedia. Version 15.8 (January 2006), http://www.copewithcytokines.de/cope.cgi, accessed October 2006
- 29.Molls RR, Savransky V, Liu M, Bevans S, Mehta T, Tuder RM, King LS, Rabb H: Keratinocyte-derived chemokine is an early biomarker of ischemic acute kidney injury. Am J Physiol Renal Physiol 290: F1187–F1193, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Grigoryev DN, Ma SF, Shimoda LA, Johns RA, Lee B, Garcia JG: Exon-based mapping of microarray probes: Recovering differential gene expression signal in underpowered hypoxia experiment. Mol Cell Probes 21: 134–139, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ: The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol 26: 2490–2496, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Bussolati B, David S, Cambi V, Tobias PS, Camussi G: Urinary soluble CD14 mediates human proximal tubular epithelial cell injury induced by LPS. Int J Mol Med 10: 441–449, 2002 [PubMed] [Google Scholar]
- 33.Morrissey J, Guo G, McCracken R, Tolley T, Klahr S: Induction of CD14 in tubular epithelial cells during kidney disease. J Am Soc Nephrol 11: 1681–1690, 2000 [DOI] [PubMed] [Google Scholar]
- 34.Wilson TC, Bachurski CJ, Ikegami M, Jobe AH, Kallapur SG: Pulmonary and systemic induction of SAA3 after ventilation and endotoxin in preterm lambs. Pediatr Res 58: 1204–1209, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Mantovani A, Bonecchi R, Martinez FO, Galliera E, Perrier P, Allavena P, Locati M: Tuning of innate immunity and polarized responses by decoy receptors. Int Arch Allergy Immunol 132: 109–115, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Heeg K, Dalpke A: TLR-induced negative regulatory circuits: Role of suppressor of cytokine signaling (SOCS) proteins in innate immunity. Vaccine 21[Suppl 2]: S61–S67, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A: Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432: 917–921, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Garn H, Siese A, Stumpf S, Barth PJ, Muller B, Gemsa D: Shift toward an alternatively activated macrophage response in lungs of NO2-exposed rats. Am J Respir Cell Mol Biol 28: 386–396, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Dobbin K, Simon R: Sample size determination in microarray experiments for class comparison and prognostic classification. Biostatistics 6: 27–38, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Wei C, Li J, Bumgarner RE: Sample size for detecting differentially expressed genes in microarray experiments. BMC Genomics 5: 87, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon BA, Easley RB, Grigoryev DN, Ma SF, Ye SQ, Lavoie T, Tuder RM, Garcia JG: Microarray analysis of regional cellular responses to local mechanical stress in acute lung injury. Am J Physiol Lung Cell Mol Physiol 291: L851–L861, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Tusher VG, Tibshirani R, Chu G: Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98: 5116–5121, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsson O, Wahlestedt C, Timmons JA: Considerations when using the significance analysis of microarrays (SAM) algorithm. BMC Bioinformatics 6: 129, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J: TM4 microarray software suite. Methods Enzymol 411: 134–193, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Barnes KC, Grant A, Gao P, Baltadjieva D, Berg T, Chi P, Zhang S, Zambelli-Weiner A, Ehrlich E, Zardkoohi O, Brummet ME, Stockton M, Watkins T, Gao L, Gittens M, Wills-Karp M, Cheadle C, Beck LA, Beaty TH, Becker KG, Garcia JG, Mathias RA: Polymorphisms in the novel gene acyloxyacyl hydroxylase (AOAH) are associated with asthma and associated phenotypes. J Allergy Clin Immunol 118: 70–77, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Becker KG, Hosack DA, Dennis G, Jr, Lempicki RA, Bright TJ, Cheadle C, Engel J: PubMatrix: A tool for multiplex literature mining. BMC Bioinformatics 4: 61, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li CJ, Li RW, Wang YH, Elsasser TH: Pathway analysis identifies perturbation of genetic networks induced by butyrate in a bovine kidney epithelial cell line. Funct Integr Genomics 7: 193–205, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Irizarry R, Gautier L, Cope L: An R package for analyses of Affymetrix oligonucleotide arrays. The Analysis of Gene Expression Data: Methods and Software, edited by Zeger SL, New York, Springer, 2003
- 49.Grigoryev DN, Ma SF, Simon BA, Irizarry RA, Ye SQ, Garcia JG: In vitro identification and in silico utilization of interspecies sequence similarities using GeneChip technology. BMC Genomics 6: 62, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grigoryev DN, Ma SF, Irizarry RA, Ye SQ, Quackenbush J, Garcia JG: Orthologous gene-expression profiling in multi-species models: Search for candidate genes. Genome Biol 5: R34, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eisen MB, Spellman PT, Brown PO, Botstein D: Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95: 14863–14868, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.