

Abstract
The binding of a cofactor to a protein matrix often involves a reorganization of the polypeptide structure. b Hemoproteins provide multiple examples of this behavior. In this minireview, selected monomeric and single b heme proteins endowed with distinct topological properties are inspected for the extent of induced refolding upon heme binding. To complement the data reported in the literature, original results are presented on a two-on-two globin of cyanobacterial origin (Synechococcus sp. PCC 7002 GlbN) and on the heme-containing module of FixL, an oxygen-sensing protein with the mixed α/β topology of PAS domains. GlbN had a stable apoprotein that was further stabilized and locally refolded by heme binding; in contrast, apoFixLH presented features of a molten globule. Sequence analyses (helicity, disorder, and polarity) and solvent accessibility calculations were performed to identify trends in the architecture of b hemoproteins. In several cases, the primary structure appeared biased toward a partially disordered binding pocket in the absence of the cofactor.
Keywords: globins, cytochromes, b hemoproteins, disorder, stability
1. Introduction
Once enclosed in a polypeptide matrix, a heme cofactor (Fe-protoporphyrin IX, Fig. 1) can be induced to perform a wide range of chemical reactions. At the basis of this exquisite versatility are specific protein-heme interactions that modulate the electronic properties of the iron atom. Axial ligands play a determinant role in fixing the reactivity of the bound heme group. They control the redox potential of the iron as well as its ability to bind exogenous ligands either reversibly or for the purpose of chemical modification. The residues lining the heme binding site also establish essential features: they create water or ligand cavities and hydrogen bond networks; they determine the range and time scale of motions necessary for entry to and exit from; and they distort the heme macrocycle for proper reactivity. The planarity of the heme group and its scant solubility in water at neutral pH impose further restrictions on the properties of the binding site. The architecture of heme cavities therefore fulfills simultaneously multiple and often conflicting requirements.
Fig. 1.
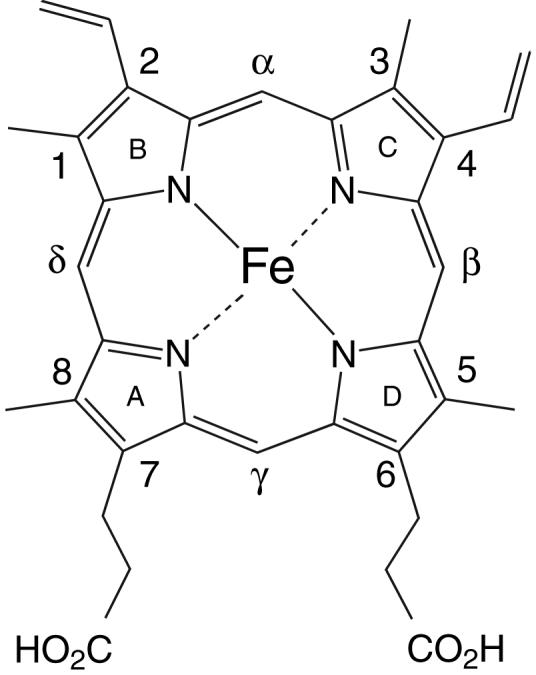
Structure of Fe-protoporphyrin IX. At neutral pH, the Fe(II) complex has a net charge of 2— because of the propionate substituents.
The α helix, with its tubular shape and diameter comparable to the width of the porphyrin ring, provides an adequate motif for interaction with a heme group. As a result, the majority of proteins that bind such a cofactor do so with α helices (Orengo et al., 1997). Notable examples include the globins, many b cytochromes, and some peroxidases (Fig. 2). β Sheets, in contrast to α helices, have a curvature that appears less compatible with strong interactions between protein and heme. Nevertheless, several instances of b heme proteins with a binding site not chiefly composed of α structure have been discovered (Fig. 3). Among them are the oxygen-sensors FixLH (Gong et al., 1998; Taylor and Zhulin, 1999) and DosH (Delgado-Nixon et al., 2000), HasASM, a heme transporter domain from Serratia marcescens (SM) (Arnoux et al., 1999), nitrophorin, a hemipteran nitric oxide (NO) transporter (Weichsel et al., 1998), and hemopexin, a high-affinity heme scavenging glycoprotein (Paoli et al., 1999). Soluble guanylate cyclase and its bacterial homologs (heme-NO and oxygen binding, or H-NOX domains) also enfold their heme group with α and some β structure (Nioche et al., 2004; Pellicena et al., 2004; Poulos, 2006).
Fig. 2.

Ribbon diagrams of the holoproteins discussed in this review, α-binding sites. The heme group and iron axial ligands are shown; the N-terminus is blue and the C-terminus is red. Elements of structure mentioned in sections 3 and 4 are marked. A: Physeter catodon (sperm whale) myoglobin (1JP6), with proximal His93; B: truncated hemoglobin (GlbN) from Synechocystis sp. PCC 6803 (1MWB), with proximal His70 and distal His46; C: Escherichia coli cytochrome b562 (265B), with axial Met7 and His102; D: Bos taurus (bovine) cytochrome b5 (1CYO), with axial His39 and His63; E: Rattus norvegicus (rat) heme oxygenase 1 (1DVE), with axial His25; F: Pseudomonas putida cytochrome P450cam (2A1M), with axial Cys357; and G: Armoracia rustica (horseradish) peroxidase (1ATJ), with axial His170.
Fig. 3.

Ribbon diagrams of the holoproteins discussed in this review, non α-binding sites. The heme group and iron axial ligands are shown; the N-terminus is blue and the C-terminus is red. Elements of structure mentioned in sections 3 and 4 are marked. A: Serratia marcescens hemophore HasA (1B2V) with axial His32 and Tyr75, assisted by His83; B: heme-containing PAS domain of Bradyrhizobium japonicum FixL (1XJ6; residues 153 to 257), with axial His200 and key hydrophobic residue Ile204; C: Escherichia coli direct oxygen sensor (Dos) heme domain (1S66), with axial His77 and Met95; D: Rhodnius prolixus nitrophorin 2 (1EUO), with axial His57.
Removal of the heme group from b hemoproteins can be readily accomplished without damage to the polypeptide. Simple procedures involve the acid denaturation of the protein followed by heme extraction with cold butanone (Teale, 1959) or the precipitation of the protein moiety from cold acidic acetone (Di Iorio, 1981). A gentler alternative suitable for proteins that cannot be efficiently refolded, are difficult to redissolve after the mixed-solvent treatment, or are readily deamidated, relies on the transfer of the heme group to a protein with higher affinity. An alternative to heme extraction is the direct preparation of the apoprotein by recombinant means.
Apoproteins often serve as the starting material for holoprotein studies. Applications include the replacement of Fe-protoporphyrin IX with isotopically-labeled versions of the same, with porphyrins containing other metals, with modified heme cofactors, etc., most often to provide spectroscopic probes, reactivity clues, and new functions, among others. In view of the importance and chemical richness of the holoproteins, comparatively little attention has been devoted to the properties of apoproteins. Among the few studied cases, are there general trends?
When comparing the conformational properties of corresponding holoproteins and apoproteins, it is common to observe some degree of unfolding in the latter (Breslow et al., 1965; Harrison and Blout, 1965; Huntley and Strittmatter, 1972; Yip et al., 1972). This is expected if one considers the number of contacts between heme and protein matrix. In the absence of the heme, intraprotein interactions may not be sufficiently strong to maintain the same geometry as in its presence. Whether or not apoprotein instability and the exposure of protease sites can be tolerated depends upon the fate of the apoprotein in its native environment. The process of holoprotein synthesis can be decomposed into discrete steps such as polypeptide folding, heme binding, and chemical modifications to the protein or heme. Unfortunately, even when the function of the protein is well established, little is known about the detailed mechanism leading to the holoprotein and even less about the lifetime of intermediate species in vivo. As a result, deciphering the primary structure in terms of desired apoprotein features is not a simple task.
Interest in folding questions such as the rate of helix formation, the relationship among sequence, architecture and mechanism, and the determinants of protein stability, has brought light to the behavior of a few selected apoproteins. This minireview examines the literature on monomeric apo b hemoproteins. It focuses on the structural and thermodynamic consequences of heme binding and the information contained in the primary structure. It highlights selected protein families and provides new data on two proteins, a cyanobacterial two-on-two globin and the oxygen sensor FixLH. The compilation reveals experimentally testable trends.
2. Materials and Methods
2.1. Protein expression and purification
The FixLH domain from Bradyrhizobium japonicum (bj) used in this work spanned residues 151 to 256 of the entire bjFixL (Swiss Prot accession number: P23222). The gene was provided by Dr. J. D. Satterlee. Protein overexpression and purification were carried out following published protocols (Suquet et al., 2005) with minor modifications. The apoprotein was obtained from inclusion bodies, and the holoprotein was prepared by reconstitution with hemin. Detailed procedures are provided in the Supplementary Information. The apoprotein from Synechococcus sp. PCC 7002 GlbN (a group I truncated globin, TrEMBL accession number: Q8RT58) was prepared from inclusion bodies according to reported procedures (Scott et al., 2002; Vuletich et al., 2006).
2.2. Thermal denaturation
The response of bjFixLH (holo and apo form) to temperature was monitored with far UV-CD data. Spectra (250-205 nm, 4-s averaging per nm) were collected every 2 °C over the range 17 to 75 °C with an equilibration time of 5 min for each point. The transition was followed at 222 nm. Data were analyzed assuming a two-state denaturation process as described in the Supplementary Information (Pace et al., 1989). The thermal denaturation of apo bjFixLH was also monitored by absorbance measurements. Spectra (250-700 nm) were collected from 19 to 85 °C (every 2 °C) and from 85 to 19 °C (every 3 °C) to inspect reversibility. Equilibration time was 5 min. Data at 280 nm were analyzed as above.
2.3. Chemical denaturation
bjFixLH and Synechococcus apoGlbN were subjected to urea denaturation. The signal was monitored by far-UV CD from 250 to 215 nm with a Jasco J-810 spectropolarimeter equipped with an automatic titrator. The procedure was essentially as described previously (Knappenberger et al., 2006). Met bjFixLH (≤ 5 μM, 1-cm path-length cell) in buffer (20 mM borate, 50 mM NaCl, pH∼8.8) was maintained at 20 °C, and Synechococcus apoGlbN (∼7 μM, 20 mM phosphate, pH ∼7.4, 1-cm path-length cell) was maintained at 25 °C. Each solution was continuously stirred, and equilibration time between points was 5 min. The titration was performed in 0.2-M steps from 0 to 9 M urea. The data were analyzed with a linear free-energy assumption and a two-state model of unfolding (Pace et al., 1989).
2.4. Primary structure analysis
The primary structure of selected proteins was analyzed with the program IUPRED (Dosztanyi et al., 2005a; Dosztanyi et al., 2005b) as available on the IUPRED server (http://iupred.enzim.hu/). Long disorder prediction was used to assess the context-independent likelihood of 30-residue stretches to be disordered; short disorder prediction was used for context-dependent assessment. The results presented in the figures are from long-disorder calculations. Hydropathy profiles were obtained with standard hydrophobicity parameters (Kyte and Doolittle, 1982). In addition, the program AGADIR (Muñoz and Serrano, 1994) (http://www.embl-heidelberg.de/Services/serrano/agadir/agadir-start.html) was used to evaluate the probability of certain protein regions to adopt a helical conformation.
2.5. Solvent accessibility calculations
Solvent accessibility calculations were performed with ACCESS (Lee and Richards, 1971) to determine the percentage of the heme surface area buried upon binding by various proteins. Included in this study are Physeter catodon (sperm whale) myoglobin (PDB ID 1JP6), Synechocystis sp. PCC 6803 GlbN (1MWB), Escherichia coli (Ec) cytochrome b562 (256B), Bos taurus (bovine) cytochrome b5 heme domain (1CYO), Rattus norvegicus (rat) heme oxygenase (1DVE), Pseudomonas putida cytochrome P450cam (2A1M), Armoracia rustica (horseradish) peroxidase C (1ATJ), Serratia marcescens hemophore HasA (1B2V), bjFixLH (1XJ6), Ec direct oxygen sensor (Dos) heme domain (1S66), and Rhodnius prolixus nitrophorin 2 (1EUO). Calculations were performed on a single subunit when a multimer was present in the unit cell and the protein was known to be a monomer in solution. Probe radius was fixed at 1.4 Å. On average, the free heme surface area was 830 Å2.
3. Review and Results
In this section, structural and stability data on several well-studied monomeric b hemoproteins are summarized. The proteins containing a heme pocket constructed from α-helical elements are presented first (Fig. 2, sections 3.1-3.7), followed by those with mixed topologies (Fig. 3, sections 3.8-3.10). Because the three-dimensional structure of hemoproteins depends to some extent on the presence of the heme group, it is of interest to inspect the primary structure of each apoprotein for a predisposition to unfold locally. The program IUPRED (Dosztanyi et al., 2005a; Dosztanyi et al., 2005b) was used to identify sequence regions prone to disorder. This program utilizes pairwise interaction energies to estimate the likelihood of stable structure formation. The database used to derive the interaction and energy matrices does not contain heme proteins. The prominence of α helices in the holoproteins also indicates that an evaluation of secondary structure propensity with a program such as AGADIR (Muñoz and Serrano, 1994) may uncover useful sequence trends.
3.1. Vertebrate myoglobins
The first protein for which a structure was determined, sperm whale myoglobin (Mb), revealed an α-helical fold (Kendrew et al., 1958). Of the eight helices, labeled A-H, two enclose the heme group: the proximal (or F) helix, which carries the histidine ligand to the heme iron, and the distal (or E) helix, which contains residues controlling the affinity of the cofactor for oxygen and other ligands (Fig. 2A). The main function of vertebrate Mb is to bind oxygen reversibly for storage, but additional roles, such as cytoprotection against reactive oxygen species and NO scavenging, are now well documented (Wittenberg and Wittenberg, 2003; Merx et al., 2005; Kanatous and Garry, 2006).
Sperm whale, horse, pig and human apomyoglobins (apoMbs) (MW 17 kDa) have been studied the most for their biophysical properties and response to sequence modifications ranging from single amino acid replacements to circular permutation. The sperm whale protein has been targeted for decades, in part because it has sufficient stability in solution and in part because of the availability of the material. Removal of the heme group from the holoprotein results in a decrease in the circular dichroism (CD) signal associated with the helical secondary structure (Harrison and Blout, 1965). Estimates of 15 residues converting from helix to coil have been deduced from the CD data and the known holoprotein structure (Breslow et al., 1965).
The native state of sperm whale apoMb, the final product of the in-vitro folding reaction in the absence of the cofactor, has not been described at high resolution yet. The apoprotein does not crystallize, and, so far, only NMR spectroscopy has provided detailed information about the structure. It appears that a large portion of apoMb is folded as in the holoprotein, with several hydrophobic clusters conserving the same geometry, for example at the A-B-G-H interface (Lecomte et al., 1996). Additional interactions, such as the His24-His119 hydrogen-bonded pair, stabilize the tertiary structure (Cocco et al., 1992). ApoMb also contains regions of relative disorder, mostly on the proximal side of the heme group (Eliezer et al., 1998; Lecomte et al., 1999). These regions, which fluctuate on a time scale inappropriate for NMR observation, refold in the presence of the cofactor.
The IUPRED analysis of apoMb from four sources (pig, human, horse, and sperm whale) is presented in Fig. 4A. Identity levels among these proteins are between 84 and 93%. The disorder threshold is marked by the horizontal line at an index of 0.5. The sperm whale sequence shows a tendency for disorder in the F helix, as observed experimentally. This feature is also present in the pig, horse, and human sequences and reflects the high level (93 or 100%) of identity in this region. When helical propensity is analyzed with AGADIR, all four sequences are found to have a low propensity to form helical structure over the F helix, in the 80-108 span. Thus, both indicators agree that the proximal helix is a region with few to no determinants of the organized structure found in the holoprotein.
Fig. 4.

IUPRED profiles for Mbs (A) and GlbNs (B). (A) The Mb sequences of sperm whale (sw, solid line), human (hu, long dashes), horse (eq, long grey dashes), and pig (pig, short dashes) are shown. The arrow indicates the proximal histidine, and the horizontal bars span the helical regions of the holoproteins. (B) The GlbN sequences of Synechococcus sp. PCC 7002 (solid line), Synechocystis sp. PCC 6803 (short dashes), and Nostoc commune (long grey dashes) are shown. The horizontal bars span the helical regions as defined in the structure of Synechocystis sp. PCC 6803 GlbN (1MWB). Arrows indicate the axial histidines of Synechococcus sp. PCC 7002 and Synechocystis sp. PCC 6803 GlbN, located at positions 46 (distal) and 70 (proximal). Nostoc GlbN appears to use His70 as only axial ligand (Thorsteinsson et al., 1999).
The folded structure of sperm whale apoMb is stable over a useful range of temperature, pH, denaturant and salt concentrations. The protein can form a molten globule intermediate (Haynie and Freire, 1993) and, under certain conditions, can undergo cold denaturation (Nishii et al., 1994). Thus, this particular apoprotein offers a robust background for amino acid replacements and an excellent subject for the exploration of a protein’s energy landscape (Privalov, 1996). Although the population of an equilibrium intermediate in the folding transition complicates the evaluation of energetics (Kirby and Steiner, 1970; Barrick and Baldwin, 1993), it is possible to assess the impact of sequence changes. The difference in standard free energy between fully unfolded and native states (ΔG°U) is 29 kJ mol-1 for the wild-type apoprotein at 25 °C (Hargrove and Olson, 1996). ΔG°U can be increased with substitutions of the distal histidine (His64 at position E7, a residue highly conserved in mammalian globins) with non-polar residues (e.g., Ala, Leu, and Phe). His E7, however, is necessary to bring the O2 affinity within a physiologically useful range and to limit the rate of iron autoxidation (Hargrove et al., 1994; Dou et al., 2002). Thus, the constraints of reactivity conflict with those of stability. Other locations are also sensitive to replacement. The single substitution Leu29Asn (position B10), which makes contact with the distal histidine and other residues in the C helix and the C-terminal end of the E helix, is highly detrimental to the apoprotein native state and results in a compact intermediate-like state under normal conditions of temperature and pH.
The primary structure of Mb has been analyzed for the conservation of residues not directly related to heme and O2 binding (Lesk and Chothia, 1980; Bashford et al., 1987; Ptitsyn and Ting, 1999), and hypotheses regarding the role of individual side chains in the stability of apoMb have been tested with the goal to understand the properties of the protein in its cellular environment (Regis et al., 2005). It is noteworthy that, despite its simplicity, the IUPRED analysis shown in Fig. 4A ranks the sperm whale, horse, human and pig apoMbs in the same order as determined by stability measurements, with the pig protein being the least ordered and least stable. The Leu29Asn and His64Phe variations of sperm whale Mb lead to modified profiles, with disorder increasing in the averaging window of 21 residues for Leu29Asn and decreasing for His64Phe. These effects reflect directly the relative propensities of the substituted residues for pairwise interactions. It is important to note that IUPRED is not expected to predict actual stability (Dosztanyi et al., 2005b); it gives an indication of minimal energy allowed by the composition, but the sequence may prevent the chain from adopting this optimal conformation. For example, exchanging residues (as in the H64V/V68H variant) leaves the composition intact and the overall calculated energy unchanged. Experimentally, however, the double replacement has a strong destabilizing effect on human apoMb (Hargrove et al., 1996).
Holoproteins are typically more stable than apoproteins. In wild-type sperm whale Mb, the contribution of various heme-protein interactions to the structural and thermodynamic properties of the holoprotein have been isolated (Hargrove et al., 1996): coordination to the proximal histidine and specific heme-protein interactions account for 20 kJ mol-1 each, and non-specific heme-protein apolar interactions account for 30 to 40 kJ mol-1. Heme affinity at pH 8 is evaluated at 1014 M-1 (Hargrove et al., 1996), and the difference in ΔG°U for apo and holoproteins should reflect these numbers. The heme group, however, has a strong tendency to interact non-specifically with protein surfaces and to aggregate under most solution conditions (Shen and Hermans, 1972; Robinson et al., 1997). These problems are encountered in all b hemoprotein studies when denaturing strength leads to the release of the heme group from its cavity. Heme adsorption, aggregation, and chemical damage are the main sources of irreversibility in denaturation experiments. Lack of reversibility and access to a fourth thermodynamic state representing a polypeptide with non-specifically bound heme can prevent thermodynamic analysis via comparative apo- and holoprotein denaturation experiments.
Studies of multiple Mb variants have led to several general conclusions. Binding of heme by the apoprotein occurs with a bimolecular rate constant kH nearly independent of the nature of the protein and estimated at 2-8 × 107 M-1 s-1 (Gibson and Antonini, 1960; Hargrove et al., 1996). Heme affinity is therefore dictated principally by the rate of heme dissociation from the protein matrix. This rate, in turn, depends on the nature of the exogenous ligand to the iron, but for a constant ligand, does not vary much among vertebrate globins (k-H ∼ 0.01 h-1 for ferric heme at neutral pH (Hargrove et al., 1996)). This represents a physiological constraint related to the oxidative damage that free hemin could cause to a cell. The stability of apoproteins from various organisms, on the other hand, does vary more widely than either kH or k-H. It therefore appears that the holoprotein stability (i.e., the resistance to lose heme and unfold) is not correlated to apoprotein stability (Hargrove and Olson, 1996).
3.2. Other monomeric globins
Vertebrate globins comprise only a portion of the hemoglobin superfamily (Vinogradov et al., 2006). The non-vertebrate proteins form a large and diverse group, including proteins from a separate lineage and referred to as “truncated” (trHbs) because of the shorter length of the globin domain (MW 14 kDa) (Wittenberg et al., 2002). TrHbs are found in all kingdoms of life, with high representation in bacteria (Vuletich and Lecomte, 2006). Of the characterized trHbs, all can accommodate a heme cofactor that binds exogenous gaseous ligands reversibly. TrHbs differ from vertebrate globins in the topology of their fold, a 2-on-2 helix bundle (Fig. 2B) versus the better known 3-on-3 helix bundle (Fig. 2A) (Pesce et al., 2000). The non-nitrogen-fixing cyanobacteria Synechococcus sp. PCC 7002 and Synechocystis sp. PCC 6803 each contain a truncated globin gene (glbN), coding for products that are 59% identical. In vitro, these two proteins can exist in at least three distinct forms; the holoprotein (a bis-histidine complex) (Couture et al., 2000; Lecomte et al., 2001; Scott et al., 2002), the holoprotein with covalent attachment of the heme group through a third histidine (Scott et al., 2002; Vu et al., 2002), and the apoprotein. As is the case for many of the globins discovered recently in the course of genome sequencing, the function and functional states of these cyanobacterial proteins have not yet been identified, and, a priori, any of the three forms could be of use to the organism.
The apoprotein from Synechocystis GlbN has a significant (∼30%) helical content under native conditions; its NMR characteristics, however, suggest that little tertiary structure is present (Lecomte et al., 2001). In contrast, the apoprotein from Synechococcus GlbN exhibits a helical content of ∼45% and yields NMR spectra that are adequate for a low-resolution characterization of the native state. Backbone amide assignments and sequential nuclear Overhauser effects (which are observed if two 1H nuclei are within ∼5 Å of one another) reveal that stable helical structure is present in portions of all helices found in the holoprotein except helix F, where the proximal His70 resides. Backbone H/D exchange experiments indicate that a core consisting of residues from the B helix, G helix, and the N-terminal portion of the H helix is somewhat protected from interactions with solvent (Vuletich et al., 2006). These slowly exchanging regions of the protein are also the regions with the largest number of long-range interactions. Interestingly, the core locations of the two cyanobacterial proteins contain different residues, with a trend toward bulkier side chains in Synechococcus GlbN.
Here as well, the analysis with IUPRED is enlightening. The profiles for the two cyanobacterial globins, shown in Fig. 4B, are clearly different. Synechococcus GlbN exhibits the lower propensity for disorder at the N-terminus and beyond the F helix. Synechocystis GlbN has lower propensity than Synechococcus GlbN only in the C and E regions. Since the resilient core of Synechococcus GlbN involves B, G, and H residues, the higher disorder index in these regions of Synechocystis GlbN may forecast the lack of a folded core. Also shown in Fig. 4B is the profile for Nostoc GlbN. If there is validity in this approach, the plot indicates that Nostoc apoGlbN should behave more like Synechocystis apoGlbN than Synechococcus apoGlbN.
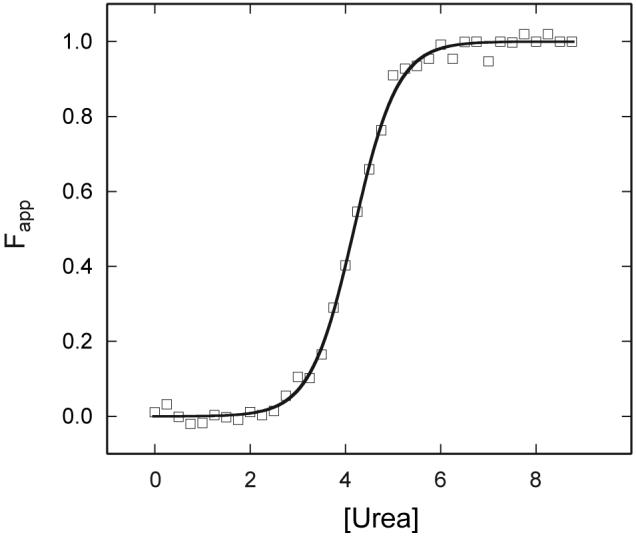
In agreement with the absence of stable tertiary structure in Synechocystis apoGlbN, the thermal denaturation of this protein lacks cooperativity (Lecomte et al., 2001). Also a sign of missing hydrophobic core is a shallow urea denaturation transition, with no native baseline (Knappenberger et al., 2006). In contrast, Synechococcus apoGlbN displays a cooperative denaturation transition (Vuletich et al., 2006). The thermal process has a midpoint of 64 °C near neutral pH, only 10 °C lower than the holoprotein without covalent attachment of the heme group. This process, however, is not completely reversible. Complete reversibility is observed for urea denaturation, and the data (shown in Fig. 5) can be used for thermodynamic purposes. The curve is well modeled with only two states and a midpoint of 3.2 M. The ΔG°U(H2O) is 21.3 ± 0.6 kJ mol-1. In the absence of the covalent linkage of the heme to the protein matrix, the two holoproteins, have measurable stabilities reflected by Tms of 74 °C for Synechocystis GlbN (Lecomte et al., 2001) and 76.4 °C for Synechococcus GlbN (Vuletich et al., 2006). These values demonstrate a limited influence of the apoprotein properties on the behavior of the heme-bound state, or an adaptation of heme-protein interactions to compensate for the apoprotein disparities. It will be necessary to determine the structure of Synechococcus GlbN and to measure its rate of heme loss to understand the factors that level the holoprotein properties. The GlbN studies also show that, even though the length of the truncated globin is reduced by 30 residues compared to the canonical 3-on-3 domain, similar global stabilities can be achieved.
Fig. 5.
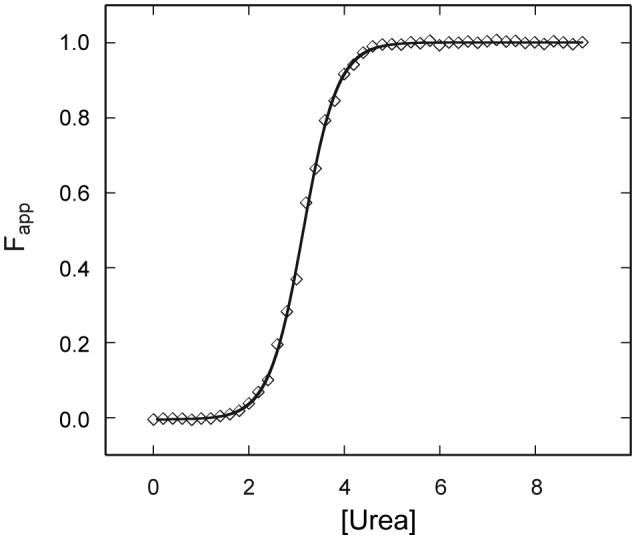
Urea denaturation of Synechococcus sp. PCC 7002 apoGlbN. The apparent fraction folded is shown at pH 7.4 in 20 mM phosphate. The protein concentration was ∼7 μM from a calculated extinction coefficient. The two-state fit is shown by the solid line, and one representative set of data is shown by the symbols.
Single-domain 3-on-3 globins are present in bacteria (Vinogradov et al., 2006) and a recent example illustrates well the adaptability of the fold to different environments. Purified thermoglobin from Aquifex aeolicus, an obligate chemolithautotroph growing at 95 °C, can be heated up to 90 °C and still contains 75% of its room-temperature helical content (Miranda et al., 2005). The secondary structure recovery upon cooling is complete, indicating that the heme is not lost by the protein up to the growth temperature. Both apo and holoproteins are resistant to guanidinium chloride denaturation, the apoprotein showing a midpoint >3.5 M. The fact that this protein has a high stability in the apo and holo states is expected to reflect both sequence and composition. Interestingly, the IUPRED profile (not shown) is entirely below the disorder threshold unlike the Mbs shown in Fig. 4A and thermoglobin appears as an excellent candidate for attempts at apoprotein crystallization.
In addition to providing clues about protein folding in general, apoglobin studies also offer an opportunity to explore the conservation of folding pathways. A comparison of the process as illustrated by sperm whale apoMb and the apoprotein of Glycine max leghemoglobin-a (Lba) (a 3-on-3 globin with ∼22% sequence identity with sperm whale Mb) reinforces that similarity of final native structure does not imply identical mechanisms. The kinetic evaluation shows that the apoLba folding core consists of the E, G, and H helices (Nishimura et al., 2000) whereas the sperm whale apoMb core involves the A, G, and H helices (Nishimura et al., 2005). The slow exchange core of Synechococcus GlbN, if representing the folding core, confirms the importance of the G and H helices in the nucleation process across multiple globins. Although helical propensity varies in the G helix, the H helix seems to be systematically identified by AGADIR in 3-on-3 and 2-on-2 proteins.
3.3. Cytochrome b562
Cytochrome b562 (cyt b562, Fig. 2C) is a ∼100-residue protein present in certain Proteobacteria, including E. coli. It has been extensively studied for its biophysical properties, but despite this, its precise function (likely in electron transport) has yet to be elucidated. Cyt b562 has a four-helix bundle topology (Mathews et al., 1979) with heme axial ligands in the first (Met7) and last (His102) helices. The apoprotein has been characterized by NMR spectroscopy (Feng et al., 1994); these studies show that the consequences of heme removal are limited to a dissipation of regular structure near the heme axial ligands (Feng et al., 1994) and an increase of internal motions at the cofactor site (Fuentes and Wand, 1998; D’Amelio et al., 2002). The IUPRED analysis (not shown) is successful: the first helix has a borderline tendency to be unstructured, and the last helix lies entirely above the 0.5 threshold. Disorder is also anticipated in the loop between the second and third helices. The most ordered part of the protein is the third helix.
The thermal stability of cyt b562 has been measured by differential scanning microcalorimetry. Both holo- and apoproteins undergo a two-state denaturation process (Feng and Sligar, 1991; Fisher, 1991). The apoprotein has a ΔG°U of ∼13 kJ mol-1. This is a decrease of ∼15 kJ mol-1 compared to the ferric holoprotein, the thermal denaturation of which leads to a state with heme non-specifically bound to the protein matrix. Non-specific affinity accounts for a remarkable ∼32 kJ mol-1 (Robinson et al., 1997). Heme affinity is dependent on the state of the iron; it is evaluated at 1 × 108 M-1 for the ferric state (Robinson et al., 1997) and higher in the ferrous state (Fisher, 1991). When the axial Met7 is replaced with an alanine, the stability of the apoprotein is barely affected, but that of the holoprotein decreases significantly (Kamiya et al., 2001).
3.4. Cytochrome b5
The b5 cytochromes (cyt b5, Fig. 2D) form a large family of electron transport proteins. Single domain b5 cytochromes are found as membrane bound proteins (microsomal and outer mitochondrial membrane) or as water soluble entities. The phylogenetic relationship among b5 cytochromes has been recently reviewed (Schenkman and Jansson, 2003). The membrane-bound proteins participate in lipid and steroid metabolism as well as drug catabolism. They serve as electron carriers for oxygenases, including cytochrome P450. The heme-binding domain of b5 cytochromes contains ∼100 residues. The cyt b5 fold is composed of a β-barrel and a four-helix bundle, both of which are small and irregular (Mathews, 1985). The heme cofactor, with iron coordinated by two histidines, is located in the bundle.
In the absence of the cofactor, much of the structure of the α bundle is lost, whereas the β-barrel remains largely intact (Falzone et al., 2001). NMR studies of overexpressed 15N-labeled rat microsomal apocyt b5 in live E. coli cells confirm that the three-dimensional structure of the protein in the crowded cellular environment is similar to that in solution (Bryant et al., 2005). This suggests that the primary structure of the cytochrome heme-binding region encodes few intramolecular interactions unless the cofactor is present and illustrates a balance of specificity and stability (Lattman and Rose, 1993).
IUPRED identifies the region containing the three unfolded helices of apocyt b5 as intrinsically unable to form a large number of stabilizing interactions. Fig. 6 illustrates the prediction for the rat microsomal, rat outer mitochondrial membrane, and bovine microsomal cytochromes. Also shown is the sequence from Ascaris suum cyt b5, a protein with a three-dimensional structure (1X3X) containing an additional helix in the heme binding region (Yokota et al., 2006). This composition-sequence has the potential to fold into a better defined tertiary structure than the other depicted proteins.
Fig. 6.

IUPRED profiles for selected b5 cytochromes. The sequences are for rat microsomal (rat mc, solid line), rat outer mitochondrial membrane (rat om, dash-dot line), bovine microsomal (bovine mc, dotted line) and Ascaris suum (ascaris, grey dashed line). Alignment was performed with BLAST (Altschul et al., 1997) and the residue number reflects the discrepancy in the sequences. The arrows at the bottom indicate the location of the axial histidines; the arrows at the top indicate the limits of the soluble heme domain.
The association of the heme with the apocytochrome has been characterized to some extent. The kH for binding to bovine microsomal apocyt b5 is 4.5 × 107 M-1 s-1, a value within the range of those observed for globins (section 3.1). Combined with the slow dissociation rate constant (k-H ∼ 0.06 h-1 for ferric heme (Vergères et al., 1993)), this indicates an affinity of 3 × 1012 M-1 (Gruenke et al., 1997), intermediate between that of myoglobin and E. coli cyt b562. Rat microsomal apocyt b5 has a ΔG°U of 6 kJ mol-1, a decrease of ∼20 kJ mol-1 compared to the holoprotein (Mukhopadhyay and Lecomte, 2004). Other wild-type forms of the protein are thought to undergo similar, or even greater, decreases in stability upon heme removal (Wang et al., 2006). In the holoprotein form, rat outer mitochondrial membrane cyt b5 is more stable than the bovine microsomal protein (Tms of 86 and 68 °C, respectively). This stability difference is eliminated in the apocytochrome form (ΔG°U = 11-12 kJ mol-1) (Wang et al., 2006).
Apocyt b5 illustrates further architectural principles of b hemoproteins. Because the apoprotein contains a folded core and a largely unfolded heme binding loop, it is possible to make amino acid replacements that affect the holoprotein stability (or heme affinity) without affecting the apparent apoprotein stability. An extreme example parallels that of the Met7 replacement in cyt b562: removal of an axial ligand to the iron from cyt b5 yields a variant with largely unchanged apoprotein properties but displaying much reduced heme affinity (Ihara et al., 2000; Falzone et al., 2001). This is consistent with the observation that rat microsomal apocyt b5 is unable to refold around Zn-PPIX or PPIX (Knappenberger, 2006). In contrast, replacements in the cooperative core of the apoprotein, Pro81Ala in helix 6 for example, can affect both the stability of the apoprotein and that of the holoprotein (Mukhopadhyay and Lecomte, 2004).
3.5. Heme oxygenase
Heme oxygenase (HO, Fig. 2E) is an enzyme found in bacteria, invertebrates, algae, higher plants and mammals (Wilks, 2002; Frankenberg-Dinkel, 2004; Rivera and Zeng, 2005). Unlike the other proteins discussed in this work, HO uses heme both as a cofactor and a substrate. The catalyzed reaction is the regiospecific breakdown of protohemin to produce biliverdin IXa and liberate iron and carbon monoxide (Tenhunen et al., 1968). Depending on the species of origin, HO participates variously in signaling, heme catabolism, and iron acquisition. The mechanism of HO is intricate; it requires three O2 molecules, seven electrons and nine protons, and involves several intermediates (Ortiz de Montellano and Wilks, 2001).
Rat HO isoform 1 (HO-1) is an inducible 32-kDa spleen and liver protein (Sugishima et al., 2002a). The three-dimensional structure of the heme-bound enzyme is an up-down α bundle made of 8 helices (A-H). The protein can be crystallized in the apoprotein state (Sugishima et al., 2002b), a rare property that may be related to the need for a stable free enzyme in the cell. The differences between heme-bound and heme-free structures are few; they resemble those observed when comparing Mb and apoMb. In rat apoHO-1, the N-terminal residues and A helix, which contains the axial His25, are not visible. Localized perturbations are detected in the residues directly following the A helix and on the distal side. Similar observations are made on the human form of the protein (Lad et al., 2003), with variability in the N-terminal region and increased flexibility in the heme pocket. Predictions based on the primary structure are consistent with observation; they show the first 25 residues to have a tendency for disorder in both the rat and the human proteins. In the rat sequence, a short stretch surrounding residue 195 is also above the 0.5 threshold. This stretch is in contact with the A helix.
A comparison of the stability of the protein with and without bound heme is not available. However, the related HO-2, which is found principally in brain and testes, has been studied. HO-2 and HO-1 are 55% identical over the catalytic domain. When heme is bound, the resistance of HO-2 to thermal denaturation increases, with a Tm higher by 10 °C (Rublevskaya and Maines, 1994). Interestingly, the IUPRED profile for HO-2 predicts disorder in the N-terminal region up to the ligating histidine (His44). Thus, this motif of N-terminal disorder is recurring and likely significant.
3.6. Cytochrome P450
The P450 cytochromes (Fig. 2F) are chiefly involved in drug metabolism and steroid biosynthesis (Denisov et al., 2005). They contain approximately 400 residues and their single b heme is the cofactor needed to activate O2 for insertion of one oxygen atom into unactivated C-H bonds. This monooxygenation reaction requires direct interaction of the substrate with activated oxygen and proceeds via a two-electron reduction in a mechanism clearly distinct from that of HO. The structure of cyt P450, including heme distortions (Shelnutt et al., 1998), is well conserved throughout the superfamily. It is mostly α helical (Poulos et al., 1987) and classified as an orthogonal bundle. The heme group is deeply buried in the structure and makes contact with loops and a long helix (helix I). The thermal denaturation of the holoprotein reveals multiple states, an observation in line with the fact that large proteins (> 30 kDa) are typically made of several domains. Little information is available regarding the conformational properties of apocyt P450. A study performed by Pfeil (Pfeil et al., 1993) shows that cyt P450cam, despite its bulk, unfolds partially if the heme is withdrawn. The helical content is lowered, the resistance to proteolysis decreases, and the thermal denaturation, although still cooperative, indicates a net destabilization. Even though the P450 cytochromes play essential biochemical roles, the apoprotein has garnered little attention, and the location of the structure that unfolds is not known.
3.7. Horseradish peroxidase and cytochrome c peroxidase
Horseradish peroxidase (HRP, Fig. 2G), a 44-kDa monomeric protein, is a Class III plant peroxidase. HRP occurs in eight isoenzyme forms. It catalyzes one-electron oxidations via a mechanism that is consistent with contact of the substrate with a heme edge (Ator and Ortiz de Montellano, 1987). HRP contains two domains (Gajhede et al., 1997); it is glycosylated with several N-linked carbohydrates, has disulfide bonds, and binds calcium ions - three characteristics that complicate in vitro studies. Upon heme removal from the C and A1 isozymes, both proteins retain 30 to 40% helicity according to CD data (Strickland et al., 1968). Sequence analysis identifies helix H, located near pyrrole A in domain II, as a candidate for unfolding.
Yeast cytochrome c peroxidase (CCP) is a 34-kDa Class I protein that resembles HRP (Finzel et al., 1984), but does not contain disulfide bridges, carbohydrate moieties or calcium binding site. The thermal denaturation of the holoprotein proceeds with two distinct transitions; the apoprotein exhibits only one, matching the higher temperature transition of the holoprotein. The molecular origin of these transitions, however, does not appear to correspond to the domain structure (Kresheck and Erman, 1988). ApoCCP can form crystals (Yonetani, 1967), and the structure of the iron-free protein has been solved (Su et al., 1994; Bhaskar and Poulos, 2005). IUPRED identifies the stretch (120-145) at the end of the N-terminal portion of domain I as prone to disorder. It is interesting that this region, among others in half of domain I, has elevated B factors (Finzel et al., 1984). A comparison of apoHRP and apoCCP indicates that the latter is the more flexible of the two and suggests it is capable of capturing the heme readily (Tsaprailis et al., 1998).
3.8. HasA, a hemophore, and hemopexin, a heme sponge
Under iron deficient conditions, the Gram-negative bacterium Serratia marcescens secretes a protein endowed with high affinity for b heme (Létoffé et al., 1994). This protein, HasASM (Fig. 3A), sequesters free heme and heme bound to host proteins such as hemoglobin for the purpose of extracting its iron. When heme-loaded, HasASM interacts with and delivers its cargo to a membrane bound receptor, HasR. The affinity for the heme group is evaluated at ∼ 5 × 1010 M-1 at neutral pH and room temperature (Deniau et al., 2003). This is ensured by a distinctive endogenous hexacoordination involving a tyrosine/histidine pair on one side of the heme and a histidine on the other. Heme transfer is thought to occur through a conformational change in HasA upon formation of the HasASM-HasR complex. The resulting alteration of axial ligation in the background of a small number of hydrophobic contacts represents an economical mechanism for modulating the free energy of heme binding (Wolff et al., 2002).
The structure of HasASM in the heme-loaded state has been solved by X-ray crystallography (Arnoux et al., 1999). Both α and β secondary structure are present, and the architecture of the protein is a 2-layer sandwich. The heme group is held via two loops at the edge of the structure. CD data suggest that the heme-free state of HasASM has the same secondary structure content as the heme-bound state (Izadi et al., 1997). The NMR structure of the heme-free state (1YBJ) indicates that the region extending from the axial histidine (His32) over a dozen residues rearranges compared to the heme-loaded structure. The structural perturbations are minor; yet, when the sequences of five of the six hemophore proteins available in Pfam (Bateman et al., 2004) are analyzed with IUPRED, a pattern of higher-than-average tendency for disorder emerges in the 30-48 region of the structure (not shown). The sixth sequence, from Yersinia pseudotuberculosis, differs with a disorder index above the 0.5 threshold over residues 125-149. Helix 5, which borders the heme group, is included in this region.
Hemopexin offers another example of variable heme affinity required by function. This 60-kDa plasma glycoprotein binds heme with record affinity and delivers it, without being degraded, to the liver for disposal (Tolosano and Altruda, 2002). Subpicomolar dissociation constant is achieved with bis-histidine coordination, and as for HasA, affinity modulation is thought to occur via a conformational change triggered by interaction with a specific receptor. The structure of hemopexin is unusual in that it contains two homologous four-bladed β-propeller domains, and the heme binds at their interface with propionates pointing inside the protein rather than toward the solvent (Paoli et al., 1999). The thermodynamics of this system are complicated by the two-domain structure and the ability of the heme to bind to the N-terminal domain as well as at the interface of the two domains (Shipulina et al., 2001).
3.9. FixLH
The protein FixL participates in the nitrogen fixation pathway of rhizobia; along with FixJ, it constitutes a classical two-component system (Monson et al., 1995). bjFixL is 505 residues in length, and consists of three domains: a non-heme-binding Per-Arnt-Sim (PAS) domain, a second PAS domain, and a histidine kinase domain, itself composed of a histidine kinase A subdomain and an HSP90-like ATPase subdomain. The second PAS domain (FixLH, Fig. 3B), which binds heme, serves as an oxygen sensor regulating the kinase activity. The PAS domain is a highly adaptable signal transduction protein module. Thousands of PAS domain genes have been recognized in archaeal, bacterial, and eukaryotic genomes (Bateman et al., 2004; Hefti et al., 2004). In many cases for which a sensing function has been confirmed, the domain resides in the cytoplasm (Taylor and Zhulin, 1999) and makes use of diverse cofactors or prosthetic groups (Ponting and Aravind, 1997). The dynamic properties of the domain, which are supported by a modular, presumably stable, structure (Pandini and Bonati, 2005), are thought to play an essential mechanistic role in signal transduction.
The PAS fold is composed of an anti-parallel β sheet (Fig. 3B, elements Bβ, Aβ, Iβ, Hβ, and Gβ). Strands Bβ and Gβ are separated by the four helices (Cα, Dα, Eα, and Fα) and loops. Although the β-strands of the PAS structures thus far determined are superimposable, the helices and loops are not. One notable aspect of FixLH is a large loop between Fα and Gβ, possibly serving as an on/off switch of the catalytic domain (Gilles-Gonzalez and Gonzalez, 2005; Key and Moffat, 2005). To provide one more example of structural perturbation induced by heme binding, we investigated the effects of urea addition and temperature changes on the holo and apo states of bjFixLH. 1H NMR spectra (not shown) confirm that the ferric form of bjFixLH obtained by reconstitution of apoprotein with hemin exists in a high-spin ferric state; four signals attributable to heme methyl groups are detected between 60 and 95 ppm, in agreement with observations on Sinorhizobium meliloti FixLH (Bertolucci et al., 1996; Rodgers et al., 1996). The 1H NMR spectrum of apo bjFixLH (not shown) contains few resolved resonances outside of the 0-to-10 ppm spectral region. Broad lines and lack of dispersion suggested a limited amount of stable tertiary structure.
Holo bjFixLH is expected to contain 29% of helical structure, 37% of β structure, and 34% β-turns (Key and Moffat, 2005). The CD spectrum of ferric bjFixLH is shown in Fig. 7. A single minimum at 217 nm is observed, consistent with the high β content of the protein. Fig. 7 also contains the spectrum of apo bjFixLH, recorded under the same conditions. The apoprotein spectrum has decreased intensity and shows hints of the double minimum of α helices at 219 and 208 nm. This suggests some degree of β structure unfolding when the heme is removed from the protein.
Fig. 7.
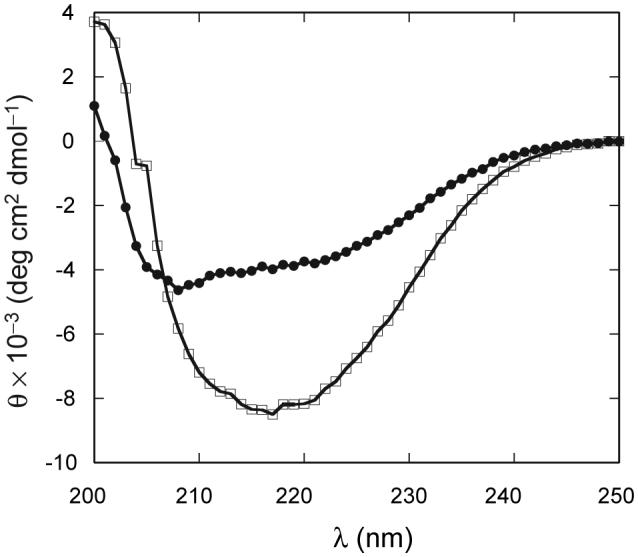
Comparison of far UV-CD spectra of apo (●) and met (□) bjFixLH. Sample conditions were pH 9.0 in 20 mM borate 250 mM NaCl at 25 °C. The spectrum of apo bjFixLH was recorded at a protein concentration of ∼10 μM and that of met bjFixLH at ∼25 μM on a heme basis.
The response of the 222-nm CD signal of ferric bjFixLH to heating was used to construct a denaturation curve. Fig. 8 shows the apparent fraction of denatured protein as a function of temperature. Despite partial irreversibility, the data fit well to a two-state model. The apparent midpoint of the transition is 56.6 ± 0.1 °C at pH ∼8.8 and is obtained reproducibly. The thermal denaturation of apo bjFixLH at pH 9.0 is accompanied with small changes in ellipticity between 200 and 230 nm. The unfolding transition for the apoprotein (Fig. 8) is also irreversible and exhibits a midpoint of 44.3 ± 0.6 °C. The value obtained by absorbance at (43.6 ± 0.9 °C) is within error of the value obtained by CD.
Fig. 8.
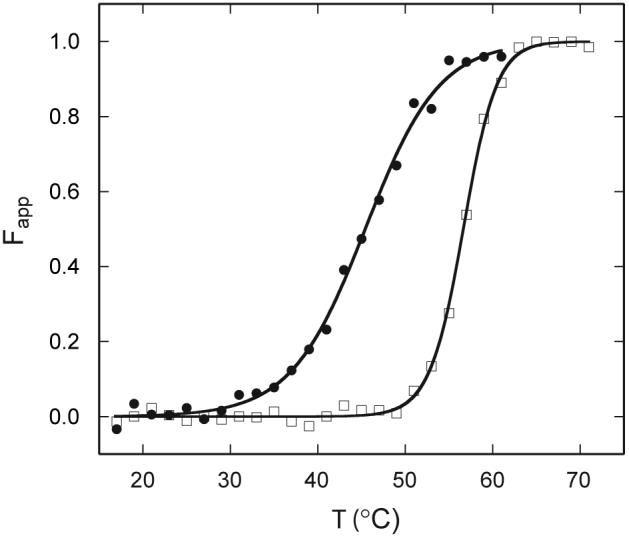
Thermal denaturation of bjFixLH as monitored by far UV-CD spectroscopy (222 nm). The apparent fraction of unfolded protein is presented at pH 9.0 (20 mM borate, 250 mM NaCl). Protein concentrations were ≤ 25 μM (heme basis, met bjFixLH, □) and ≤ 10 μM (apo bjFixLH, ●). Fits are shown by the lines, and one representative set of data is shown by the symbols.
Unlike the thermal denaturation, chemical denaturation of met bjFixLH is completely reversible. Fig. 9 shows the apparent fraction of unfolded protein plotted versus urea concentration, as obtained from the change in ellipticity at 222 nm. The urea concentration at the transition midpoint is 4.2 ± 0.3 M, and ΔG°U(H2O) is 22 ± 1 kJ mol-1. The m value (5.3 ± 0.2 kJ mol-1 M-1) matches the expected 5.6 kJ mol-1 M-1 based on the change in accessible surface area upon unfolding (ΔASA) (Myers et al., 1995). The chemical denaturation of apo bjFixLH (not shown) is reversible, but, as indicated by the breadth of the transition, has a low level of cooperativity. The data did not define a distinct native baseline and could not be used to obtain ΔG°U.
Fig. 9.
Urea denaturation of met bjFixLH as monitored by far UV-CD spectroscopy (222 nm). The apparent fraction of unfolded protein is presented at pH 9.0 (20 mM borate, 250 mM NaCl). Final protein concentration was ∼5 μM. The fit is shown by the line, and one representative set of data is shown by the symbols.
Although it was not possible to determine the location of the lost structure by NMR spectroscopy, IUPRED provides hints regarding weak determinants of folded structure. As shown in Fig. 10 (solid line), the region encompassing the histidine ligand to the iron (Eα-Fα) has a disorder index above the 0.5 threshold. Likewise, a portion of Hβ, which lines the opposite side of the heme group, qualifies as disordered. Helix Fα (196-210) contains a single hydrophobic residue, Ile204, which serves as a contact with the Aβ-Bβ turn and belongs to the set of hydrophobic residues defining the PAS domain (Perutz et al., 1999). Secondary structure prediction (Muñoz and Serrano, 1994) identifies Fα as a weak helix; only a low propensity turn is anticipated at 203-206. Thus, it is possible that Ile204 cannot form the stabilizing contact if Fα is disordered.
Fig. 10.

IUPRED profiles for bjFixLH (solid line) and EcDosH (dotted line). The two sequences were aligned with BLAST (Altschul et al., 1997). The numbering and elements of secondary structure shown with horizontal bar pertain to bjFixLH. The arrow at position 200 indicates the axial His of both bjFixLH and EcDosH. The second arrow (position 218) indicates the axial Met of EcDosH.
3.10. DosH and nitrophorins
DosH (Fig. 3C), like FixLH, is a heme-binding PAS domain functioning as an oxygen sensor (Delgado-Nixon et al., 2000). EcDosH has low amino acid sequence identity to bjFixLH (27% over a 99-residue length), but the fold is conserved as shown by the recently solved X-ray structure of the domain (Park et al., 2004). Per chance, both O2-bound and O2-free molecules were present in the EcDosH unit cell, which provided clues as to the signaling mechanism. In the absence of O2, Met95 coordinates the iron. The region affected by the change in ligand extends over the FG turn, Gβ, and the HI turn. Once more, IUPRED performs well (Fig. 10, dotted line), identifying the 80-100 region overlapping with Fβ as prone to disorder, and therefore likely to undergo conformational changes. Extension of this observation suggests that this portion of the structure unfolds in the apoprotein state. The C-terminal portion of the EcDosH sequence has a lower disorder index than bjFixLH; this raises the hope that it will be possible to characterize the structural properties of the EcDosH apoprotein.
Nitrophorins (NPs) are 20-kDa saliva b hemoproteins used by the blood-sucking insect Rhodnius prolixus to induce vasodilation. They do so by delivering NO to the wound (Montfort et al., 2000). NPs (Fig. 3D) have the lipocalin fold (Andersen et al., 1998; Weichsel et al., 1998; Andersen and Montfort, 2000) and encapsulate the heme cofactor between β strands and loops. NO binding occurs in the ferric state and causes minor structural rearrangements of loops on the distal side (Weichsel et al., 2000), leading to the protection of NO and heme from solvent. The IUPRED profiles (not shown) of NP-1 and NP-4 (88% identity) are similar, and so are those of NP-2 and NP-3 (81% identity). Of the four proteins, NP-4 has the lowest overall disorder index, with the entire sequence having values below 0.5. The short stretches that are affected by NO binding do not stand out. As for EcDosH, a study of the apoprotein will provide further information on the intrinsic flexibility of the rearranging loops in the absence of cofactor.
4. Discussion
The set of proteins presented above is disparate and, a priori, one expects no unifying feature to emerge. Yet, the survey reveals properties recurring regardless of the purpose of the heme, whether cofactor, substrate, chemosensor, or cargo. In the discussion, we set aside the details that condition heme reactivity and focus on structural and thermodynamic relationships.
4.1. Heme accessibility
The strength of interaction between a heme group and a polypeptide host depends on van der Waals contacts with cavity residues, among other factors (section 3.1 (Hargrove et al., 1996)). The number of contacts is in turn related to the ASA buried upon binding. The extent of burial of the cofactor in the holoprotein therefore deserves inspection. Are proteins with low heme ASA comparatively more affected by heme removal than those with high heme ASA? Artificial proteins that are modified in a region remote from the heme group and have unchanged holoprotein structure (for example, the P81A variant of rat microsomal cyt b5) highlight that the ability of parts of the protein not involved in cofactor contacts to adopt a long-lived tertiary structure autonomously is influential. Also key is the recognition that iron ligation, which involves only one or two residues, can contribute large amounts of free energy. With this in mind, the wild-type proteins covered in section 3 can be inspected.
Solvent ASAs of the heme group are listed in Table 1. The values range from 1% (HRP) to 25% (cyt b5). In some cases, the degree of exposure makes functional sense. For example, FixLH requires a buried cofactor to control properly its redox potential, heme plane geometry, and O2 affinity, and eventually to couple ligand binding to a sensitive switch. Studies of a R220H variant of bjFixLH reveal that the replacement allows water to bind in the heme pocket in a redox- and pH-dependent fashion, a property that is not physiologically desirable (Balland et al., 2006; Jasaitis et al., 2006). EcDosH (Fig. 3C) also contains a hydrophobic pocket with no water molecule (Park et al., 2004). That apo bjFixLH has low tertiary structure content and stability may be related to the number of contacts severed by heme removal. Likewise, cyt P450cam (section 3.6) and HRP (section 3.7) have a buried heme. Access to heme edge (HRP) or distal cavity (cyt P450), however, is required for function. Both peroxidases must therefore exhibit some degree of flexibility that is not captured in the static X-ray structure. The disppearance of a significant amount of structure in the apoprotein state may reflect this intrinsic flexibility as well as lost intermolecular interactions.
Table 1.
Solvent accessibility of the heme group in various b heme proteins.
| Protein | PDB ID | Fig. | MW (kDa) | Iron ligation | Heme ASA (%) |
|---|---|---|---|---|---|
| horseradish peroxidase Ca | 1ATJ | 2G | 33.7 | His | < 1% |
| cytochrome P450camb | 2A1M | 2F | 46.6 | Cys | 3% |
| FixLHc | 1XJ6 | 3B | 13.4 | His | 6% |
| DosHd | 1S66 | 3C | 13.6 | His/Met | 10% |
| GlbNe | 1MWB | 2B | 13.7 | His/His | 12% |
| cytochrome b562f | 256B | 2C | 11.8 | His/Met | 14% |
| myoglobing | 1JP6 | 2A | 17.2 | His | 18% |
| heme oxygenaseh | 1DVE | 2E | 30.6 | His | 20% |
| nitrophorin 2i | 1EUO | 3D | 20.1 | His | 22% |
| HasAsmj | 1B2V | 3A | 19.3 | His/Tyr | 22% |
| cytochrome b5k | 1CYO | 2D | 10.6 | His/His | 25% |
From Armoracia rusticana
from Pseudomonas putida, with camphor bound
from Bradyrhizobium japonicum
from Escherichia coli, with no oxygen bound
truncated globin from Synechocystis sp. PCC 6803
from Escherichia coli
from Physeter catodon
from Rattus norvegicus
from Rhodnius prolixus
hemophore from Serratia marcescens
soluble domain from Bos taurus microsomal protein.
In contrast, HasASM, which operates in heme scavenging and delivery, has a relatively high cofactor exposure (22%). Less stringent control of heme properties may be required for transport purposes and, in this case, heme accessibility is tolerated, perhaps to facilitate delivery. Heme removal disrupts a smaller number of stabilizing interactions and the apoprotein is well folded. Likewise, high accessibility of the heme group and localized unfolding upon its removal is observed for HO-1 (section 3.5), a protein that also has a functional apoprotein.
Cyt b5 has the greatest heme exposure of the set. This may be because solvent accessibility plays a part in regulating the redox potential of the iron (Rivera et al., 1998). Nevertheless, cyt b5 achieves high heme affinity (section 3.4) with two histidines as axial ligands, and unfolding upon heme removal is extensive despite the relatively low number of intramolecular contacts involved. This protein illustrates diverse factors at work: number and nature of axial ligands, number and nature of contacts with the heme group, composition of the heme site, and the stability of the hydrophobic core remote from the heme binding site. In general, the number of possible local interactions with the heme group is finite and so is the free energy derived from these interactions. The balance of this energy and the structural core energy provided by the rest of the protein as well as the efficiency of the coupling between the local and remote regions make heme ASA a weak determinant of apoprotein behavior.
4.2. Where is the structure lost?
Where structural information is available, it appears that binding the heme group enhances the global stability of the protein and refolds specific elements of secondary structure. Unless the protein is thoroughly destabilized in the absence of the heme, (1) the loss of structure is limited to the binding site, often encompassing a 20- to 30-residue stretch that contains one axial ligand to the heme iron; (2) the apoprotein contains a structured cooperative core that is extended by heme binding; and (3) IUPRED meets with a measure of success at identifying the regions of weak order propensity.
The implication of the first two points is that the primary structure codes for a decoupling of elements of the binding site from the rest of the three-dimensional structure. The third point suggests that certain families of sequences, in particular those that contain α-helices and therefore are rich in short-range interactions, may be subjects for meaningful prediction. For example, Mb, cyt b562, and cyt b5 display trends by which the experimentally determined location of disorder is recognized. Furthermore, the overall disorder index for apoMb appears to correlate with stability. As a counterexample, apoLba has a lower disorder tendency than sperm whale apoMb, but is known to be less stable to urea denaturation (Nishimura et al., 2000). As mentioned for the H64V/V68H double replacement in section 3.1, IUPRED only returns a potential for stability, and its results are most reliable at the structural level. The secondary structure content of apoLba is similar to that of sperm whale apoMb (Nishimura et al., 2000). Unfortunately, the status of the E helix (disorder index above 0.3) and F helix (disorder index below 0.3) is not known in this protein. Perhaps more importantly, point (3) suggests that the primary structures are distinctly biased toward local “low foldability” near the heme. This property may be encoded so that the dismantling of stable structure is not required prior to heme binding in the biosynthesis or function of holoproteins.
From a practical point of view, the ability to anticipate which parts of a protein may be relatively more flexible and less prone to folding than the rest would be useful, especially in view of the large number of primary structures becoming available through genome sequencing. Tests of the prediction would be to determine the structure and in-vitro kH constant of Ascaris suum apocyt b5 (Fig. 6) and of thermoglobin at room temperature. A plausible expectation is that heme association would be slower for a tightly collapsed state and reflect a rate limiting step involving the protein’s conformational rearrangement rather than heme desolvation.
4.3. Relationship to hydropathy
The conspicuous success of IUPRED raises two questions: what are the characteristics of the disordered regions and why are they necessary? The algorithm evaluates pairwise interaction energies, with hydrophobic residues contributing the most to order. A simple hydropathy analysis with the Kyte and Doolittle scale (Kyte and Doolittle, 1982) and a window of 21 residues returns profiles that parallel the IUPRED profiles; the disordered regions tend to have negative hydropathic indices, i.e., high polarity. The demarcation between regions, however, does not appear as clearly through hydropathy as it does with disorder prediction. A combination of the two plots is likely to be a useful tool for anticipating the location of weak conformational determinants in apoproteins.
The necessity of polar regions in the heme site may stem from intertwined functional and folding considerations. In practically all cases, chemistry cannot be supported solely by hydrophobic residues. For example, hydrogen bonding side chains are often required to control axial and exogenous ligand binding, and charged residues at the heme edge participate in the recognition of electron transfer partners. The distribution of polar groups along the sequence, however, appears systematically biased to favor disorder, and it is not clear that this is the only solution to the chemistry problem. The concentration of polar residues in certain stretches of the protein may prevent the hydrophobic collapse mentioned above and allow conformational fluctuations (section 4.6).
4.4. Folding considerations
The insolubility of the heme group and its tendency to aggregate create practical problems for holoprotein kinetic as well as thermodynamic studies. In the few reported instances of rapid mixing experiments, the bis-cyanide complex of ferric heme, hemin-(CN)2 is utilized because of its comparatively favorable properties (Kawamura-Konishi et al., 1988; Crespin et al., 2005). One problem, however, is that the cyanide ligand(s) has to be decoordinated prior to ligation with protein residues, and this process constitute an additional kinetic step (Yee and Peyton, 1991). Furthermore, it is unclear how these studies relate to the process in the cell. In general, for soluble proteins, it is not known whether the heme group binds to a nascent protein chain, the fully folded apoprotein, or an intermediate species. It is also possible that other cellular components, specific factors or non-specific surfaces, play a role in the assembly, in particular for membrane-bound proteins that cannot travel to the site of heme biosynthesis (Senjo et al., 1985; Takami, 1993).
One extreme scenario has the apoprotein associating with the heme before assuming its native three-dimensional structure. This “fly-casting” mechanism, by which the unfolded protein captures the cofactor, may accelerate the coupled events of folding and binding (Shoemaker et al., 2000). Thus far, there is little evidence that fly-casting is at work for apoMb in vitro (Kawamura-Konishi et al., 1988; Crespin et al., 2005). The folding of cyt b5 is a complex process that depends on the presence of the heme group (Manyusa et al., 1999). Studies of apocyt b5 variants suggest that there is a kinetic advantage in having a partially folded pocket (Ihara et al., 2000). In contrast, apoHRP is thought to refold before binding the heme group, whereas apoCCP, which can exist in the yeast cell (Djavadi-Ohaniance et al., 1978), does less so (Tsaprailis et al., 1998). In the absence of definite information regarding the lifetime of apoproteins in the cell and the mechanism for free heme presentation, the constraints on the folding process cannot be evaluated.
4.5. Extent of destabilization
Thermodynamic stability is only one of the many properties that must be controlled for optimal function. The variety of heme ligation motifs and heme pocket structures renders a correlation between heme affinity and apoprotein stability unlikely across different families of protein. Within a family, however, some insight has been achieved. An apoprotein that must refold extensively upon cofactor binding may be expected, at least for entropic reasons, to have a lower affinity for the cofactor than an apoprotein with fully preformed structure. A relationship between stability of residual structure in the apoprotein and affinity for the heme group has been sought in cytochrome b5 (Mukhopadhyay and Lecomte, 2004; Cowley et al., 2005) and the globins (Hargrove et al., 1994; Hargrove and Olson, 1996). Because the folding core of the apoprotein behaves as a cooperative unit on its own, and much of the structure contacting the heme group is dissipated in its absence, affinity and apoprotein stability can be decoupled. This decoupling is less effective when the apoprotein becomes marginally stable, as captured in the equations derived for the population of the native holoprotein state during denaturation (Hargrove and Olson, 1996).
4.6. Are certain folds intrinsically incapable of stability?
Is it possible to construct a stable apoprotein that has a structure nearly identical to that of the holoprotein, or is it necessary to include a flexible region despite its susceptibility to the cellular degradation machinery? Although four-helix bundles, as presented by cyt b562, need not contain a cofactor, it is not clear that more complex b hemoprotein folds have architectural properties that permit high rigidity and stability without the aid of the large heme group.
The PAS fold (sections 3.9 and 3.10) can serve to explore this point. Several such domains need to survive in the cellular environment while in the free state, and indeed, cofactor binding is not a strict requirement for attaining a PAS topology. This is illustrated by the N-terminal domain of the human ether-a-go-go-related gene-encoded voltage sensor (HERG), which is well folded under physiological conditions (MacKinnon et al., 1998) and has an IUPRED disorder tendency reaching 0.2 at most. Likewise, human PAS kinase (hPASK) is obtainable in the free state, which assumes a stable structure with a Tm of 84 °C (Amezcua et al., 2002). Small molecule screening has revealed that the binding site matches that of FixLH, but is not fully preorganized to accommodate ligands. This “deformability” is reflected in a rise in disorder tendency in the Fα region. Interestingly, the ligands that bind more tightly to the protein tend to bury themselves more deeply into it, highlighting that it is the iron ligation capability of the heme group and the fine-tuning of reactivity that obscure the relationship between ASA and apoprotein properties mentioned in section 4.1.
The helical bundle of globins also has geometrical characteristics that can be adapted to the absence of cofactor. RsbR, a protein belonging to the Bacillus subtilis stressosome and adopting the globin fold (2BNL), shows that it is feasible to occlude the heme pocket completely (Murray et al., 2005) and extend the stable core over the whole structure. The propensity for helix formation in the E and F regions of RsbR, as measured by the program AGADIR, is strong, and hydrophobic contacts and electrostatic interactions contribute to closing the heme cavity. PAS units share these traits with the globins: a solid domain can be produced, as evidenced by HERG, and the proteins that bind heme do so by altering the dynamic properties of the regions involved in the association (Pandini and Bonati, 2005).
The preservation of a well-defined, crystallizable fold in the absence of the heme and regardless of its final ASA may be an indication that the apoprotein has a specific role to play in the cell. It should be noted, however, that apocyt b5 has been implicated as a stimulator of several P450 cytochromes (Schenkman and Jansson, 2003). If this function is confirmed, it would indicate that a partially disordered apoprotein can fulfill specific tasks unrelated to a heme group. Thus, although a well-folded apoprotein may signify a functional role as in HasASM and HO, a partially folded one would offer no reliable clue as to its uses in the cell.
5. Conclusions
From the accumulated data on various b hemoproteins emerges a qualitative pattern by which the wild-type proteins did not evolve toward a compact and maximally ordered apoprotein structure. Certain sections of sequence, detectable in primary structure analyses, appear to resist folding until the cofactor provides a significant set of interactions. The control of conformational dynamics in regions surrounding the heme holds the key to achieving suitable cofactor, substrate, and ligand association and dissociation rates. In view of the hydropathy and disorder profiles, it appears that polar stretches are generally observed not only because they encode the chemical properties of the heme, but also possibly because they decrease the probability of forming kinetic traps and intraprotein interactions that would compete with heme-protein interactions. Knowledge of the function of a protein gives insight into the evolution of the heme binding sites, whereas information about the expression of the gene and presentation of the heme to the polypeptide is required to understand the evolution of other aspects of the proteins. Further studies to determine the structure of apoproteins within different classes of b hemoproteins and, ideally, to elucidate the folding pathway in the cell will be needed to decipher the significance of sequence trends.
Supplementary Material
Acknowledgments
The authors thank Nancy Scott and Dr. Jane Knappenberger for assistance with protein preparation and Dr. James D. Satterlee for the gift of the FixLH plasmid. Figs. 2 and 3 were made with MOLSCRIPT (Kraulis, 1991). This work was supported by Grant GM-054217 from the National Institutes of Health (GM), Grant MCB-0349409 of the National Science Foundation, and Grant NNG04GN33H of the National Aeronautic and Space Administration to DAV.
Abbreviations
- apoMb
apomyoglobin
- ASA
accessible surface area
- bj
Bradyrhizobium japonicum
- CCP
cytochrome c peroxidase
- CD
circular dichroism
- cyt b5
cytochrome b5
- cyt b562
cytochrome b562
- ΔG°U
apparent standard free energy of unfolding
- Dos
direct oxygen sensor
- Ec or E. coli
Escherichia coli
- GlbN
cyanobacterial truncated hemoglobin N (group I trHb or two-on-two globin)
- Hb
hemoglobin
- HERG
human ether-a-go-go-related gene-encoded voltage sensor
- HO
heme oxygenase
- HRP
horseradish peroxidase
- Lba
leghemoglobin-a
- Mb
myoglobin
- NMR
nuclear magnetic resonance
- NO
nitric oxide
- NP
nitrophorin
- PAS
Per-Arnt-Sim
- SM
Serratia marcescens
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amezcua CA, Harper SM, Rutter J, Gardner KH. Structure and interactions of PAS kinase N-terminal PAS domain: Model for intramolecular kinase regulation. Structure. 2002;10:1349–1361. doi: 10.1016/s0969-2126(02)00857-2. [DOI] [PubMed] [Google Scholar]
- Andersen JF, Montfort WR. The crystal structure of nitrophorin 2. A trifunctional antihemostatic protein from the saliva of Rhodnius prolixus. J. Biol. Chem. 2000;275:30496–30503. doi: 10.1074/jbc.M002857200. [DOI] [PubMed] [Google Scholar]
- Andersen JF, Weichsel A, Balfour CA, Champagne DE, Montfort WR. The crystal structure of nitrophorin 4 at 1.5 Å resolution: transport of nitric oxide by a lipocalin-based heme protein. Structure. 1998;6:1315–1327. doi: 10.1016/s0969-2126(98)00131-2. [DOI] [PubMed] [Google Scholar]
- Arnoux P, et al. The crystal structure of HasA, a hemophore secreted by Serratia marcescens. Nat. Struct. Biol. 1999;6:516–520. doi: 10.1038/9281. [DOI] [PubMed] [Google Scholar]
- Ator MA, Ortiz de Montellano PR. Protein control of prosthetic heme reactivity. Reaction of substrates with the heme edge of horseradish peroxidase. J. Biol. Chem. 1987;262:1542–1551. [PubMed] [Google Scholar]
- Balland V, et al. Functional implications of the propionate 7-arginine 220 interaction in the FixLH oxygen sensor from Bradyrhizobium japonicum. Biochemistry. 2006;45:2072–2084. doi: 10.1021/bi051696h. [DOI] [PubMed] [Google Scholar]
- Barrick D, Baldwin RL. Three-state analysis of sperm whale apomyoglobin folding. Biochemistry. 1993;32:3790–3796. doi: 10.1021/bi00065a035. [DOI] [PubMed] [Google Scholar]
- Bashford D, Chothia C, Lesk AM. Determinants of a protein fold. Unique features of the globin amino acid sequences. J. Mol. Biol. 1987;196:199–216. doi: 10.1016/0022-2836(87)90521-3. [DOI] [PubMed] [Google Scholar]
- Bateman A, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolucci C, Ming L, Gonzalez G, Gilles-Gonzalez MA. Assignment of the hyperfine-shifted 1H-NMR signals of the heme in the oxygen sensor FixL from Rhizobium meliloti. Chem. Biol. 1996;3:561–566. doi: 10.1016/s1074-5521(96)90147-7. [DOI] [PubMed] [Google Scholar]
- Bhaskar B, Poulos TL. The 1.13-Å structure of iron-free cytochrome c peroxidase. J. Biol. Inorg. Chem. 2005;10:425–430. doi: 10.1007/s00775-005-0654-4. [DOI] [PubMed] [Google Scholar]
- Breslow E, Beychok S, Hardman KD, Gurd FRN. Relative conformations of sperm whale metmyoglobin and apomyoglobin in solution. J. Biol. Chem. 1965;240:304–309. [PubMed] [Google Scholar]
- Bryant JE, Lecomte JTJ, Lee AL, Young GB, Pielak GJ. Protein dynamics in living cells. Biochemistry. 2005;44:9275–9279. doi: 10.1021/bi050786j. [DOI] [PubMed] [Google Scholar]
- Cocco MJ, Kao YH, Phillips AT, Lecomte JTJ. Structural comparison of apomyoglobin and metaquomyoglobin: pH titration of histidines by NMR spectroscopy. Biochemistry. 1992;31:6481–6491. doi: 10.1021/bi00143a018. [DOI] [PubMed] [Google Scholar]
- Couture M, et al. Structural investigations of the hemoglobin of the cyanobacterium Synechocystis PCC 6803 reveal a unique distal heme pocket. Eur. J. Biochem. 2000;267:4770–4780. doi: 10.1046/j.1432-1327.2000.01531.x. [DOI] [PubMed] [Google Scholar]
- Cowley AB, Sun N, Rivera M, Benson DR. Divergence in nonspecific hydrophobic packing interactions in the apo state, and its possible role in functional specialization of mitochondrial and microsomal cytochrome b5. Biochemistry. 2005;44:14606–14615. doi: 10.1021/bi051337m. [DOI] [PubMed] [Google Scholar]
- Crespin MO, Boys BL, Konermann L. The reconstitution of unfolded myoglobin with hemin dicyanide is not accelerated by fly-casting. FEBS Lett. 2005;579:271–274. doi: 10.1016/j.febslet.2004.11.088. [DOI] [PubMed] [Google Scholar]
- D’Amelio N, Bonvin AM, Czisch M, Barker P, Kaptein R. The C terminus of apocytochrome b562 undergoes fast motions and slow exchange among ordered conformations resembling the folded state. Biochemistry. 2002;41:5505–5514. doi: 10.1021/bi011863n. [DOI] [PubMed] [Google Scholar]
- Delgado-Nixon VM, Gonzalez G, Gilles-Gonzalez MA. Dos, a heme-binding PAS protein from Escherichia coli, is a direct oxygen sensor. Biochemistry. 2000;39:2685–2691. doi: 10.1021/bi991911s. [DOI] [PubMed] [Google Scholar]
- Deniau C, et al. Thermodynamics of heme binding to the HasASM hemophore: Effect of mutations at three key residues for heme uptake. Biochemistry. 2003;42:10627–10633. doi: 10.1021/bi030015k. [DOI] [PubMed] [Google Scholar]
- Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- Di Iorio EE. Preparation of derivatives of ferrous and ferric hemoglobin. Methods Enzymol. 1981;76:57–72. doi: 10.1016/0076-6879(81)76114-7. [DOI] [PubMed] [Google Scholar]
- Djavadi-Ohaniance L, Rudin Y, Schatz G. Identification of enzymically inactive apocytochrome c peroxidase in anaerobically grown Saccharomyces cerevisiae. J. Biol. Chem. 1978;253:4402–4407. [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005a;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005b;347:827–839. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- Dou Y, Maillett DH, Eich RF, Olson JS. Myoglobin as a model system for designing heme protein based blood substitutes. Biophys. Chem. 2002;98:127–148. doi: 10.1016/s0301-4622(02)00090-x. [DOI] [PubMed] [Google Scholar]
- Eliezer D, Yao J, Dyson HJ, Wright PE. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat. Struct. Biol. 1998;5:148–155. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- Falzone CJ, Wang Y, Vu BC, Scott NL, Bhattacharya S, Lecomte JTJ. Structural and dynamic perturbations induced by heme binding in cytochrome b5. Biochemistry. 2001;40:4879–4891. doi: 10.1021/bi002681g. [DOI] [PubMed] [Google Scholar]
- Feng Y, Sligar SG, Wand AJ. Solution structure of apocytochrome b562. Nat. Struct. Biol. 1994;1:30–35. doi: 10.1038/nsb0194-30. [DOI] [PubMed] [Google Scholar]
- Feng YQ, Sligar SG. Effect of heme binding on the structure and stability of Escherichia coli apocytochrome b562. Biochemistry. 1991;30:10150–10155. doi: 10.1021/bi00106a011. [DOI] [PubMed] [Google Scholar]
- Finzel BC, Poulos TL, Kraut J. Crystal structure of yeast cytochrome c peroxidase refined at 1.7-Å resolution. J. Biol. Chem. 1984;259:13027–13036. [PubMed] [Google Scholar]
- Fisher MT. Differences in thermal stability between reduced and oxidized cytochrome b562 from Escherichia coli. Biochemistry. 1991;30:10012–10018. doi: 10.1021/bi00105a028. [DOI] [PubMed] [Google Scholar]
- Frankenberg-Dinkel N. Bacterial heme oxygenases. Antioxid. Redox Signal. 2004;6:825–834. doi: 10.1089/ars.2004.6.825. [DOI] [PubMed] [Google Scholar]
- Fuentes EJ, Wand AJ. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry. 1998;37:9877–9883. doi: 10.1021/bi980894o. [DOI] [PubMed] [Google Scholar]
- Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Crystal structure of horseradish peroxidase C at 2.15 Å resolution. Nat. Struct. Biol. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- Gibson QH, Antonini E. Kinetic studies on the reaction between native globin and haem derivatives. Biochem. J. 1960;77:328–341. doi: 10.1042/bj0770328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles-Gonzalez MA, Gonzalez G. Heme-based sensors: defining characteristics, recent developments, and regulatory hypotheses. J. Inorg. Biochem. 2005;99:1–22. doi: 10.1016/j.jinorgbio.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Gong W, Hao B, Mansy SS, Gonzalez G, Gilles-Gonzalez MA. Structure of a biological oxygen sensor: A new mechanism for heme-driven signal transduction. Proc. Natl. Acad. Sci. U. S. A. 1998;95:15177–15182. doi: 10.1073/pnas.95.26.15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenke LD, Sun J, Loehr TM, Waskell L. Resonance Raman spectral properties and stability of manganese protoporphyrin IX cytochrome b5. Biochemistry. 1997;36:7114–7125. doi: 10.1021/bi970407p. [DOI] [PubMed] [Google Scholar]
- Hargrove MS, Barrick D, Olson JS. The association rate constant for heme binding to globin is independent of protein structure. Biochemistry. 1996;35:11293–11299. doi: 10.1021/bi960371l. [DOI] [PubMed] [Google Scholar]
- Hargrove MS, Krzywda S, Wilkinson AJ, Dou Y, Ikeda-Saito M, Olson JS. Stability of myoglobin: a model for the folding of heme proteins. Biochemistry. 1994;33:11767–11775. doi: 10.1021/bi00205a012. [DOI] [PubMed] [Google Scholar]
- Hargrove MS, Olson JS. The stability of holomyoglobin is determined by heme affinity. Biochemistry. 1996;35:11310–11318. doi: 10.1021/bi9603736. [DOI] [PubMed] [Google Scholar]
- Harrison SC, Blout ER. Reversible conformational changes of myoglobin and apomyoglobin. J. Biol. Chem. 1965;240:299–303. [PubMed] [Google Scholar]
- Haynie DT, Freire E. Structural energetics of the molten globule state. Proteins. 1993;16:115–140. doi: 10.1002/prot.340160202. [DOI] [PubMed] [Google Scholar]
- Hefti MH, Françoijs K-J, de Vries SC, Dixon R, Vervoort J. The PAS fold: A redefinition of the PAS domain based upon structural prediction. Eur. J. Biochem. 2004;271:1198–1208. doi: 10.1111/j.1432-1033.2004.04023.x. [DOI] [PubMed] [Google Scholar]
- Huntley TE, Strittmatter P. The effect of heme binding on the tryptophan residue and the protein conformation of cytochrome b5. J. Biol. Chem. 1972;247:4641–4647. [PubMed] [Google Scholar]
- Ihara M, Takahashi S, Ishimori K, Morishima I. Functions of fluctuation in the heme-binding loops of cytochrome b5 revealed in the process of heme incorporation. Biochemistry. 2000;39:5961–5970. doi: 10.1021/bi9922289. [DOI] [PubMed] [Google Scholar]
- Izadi N, et al. Purification and characterization of an extracellular heme-binding protein, HasA, involved in heme iron acquisition. Biochemistry. 1997;36:7050–7057. doi: 10.1021/bi962577s. [DOI] [PubMed] [Google Scholar]
- Jasaitis A, et al. Role of distal arginine in early sensing intermediates in the heme domain of the oxygen sensor FixL. Biochemistry. 2006;45:6018–6026. doi: 10.1021/bi060012i. [DOI] [PubMed] [Google Scholar]
- Kamiya N, et al. How does heme axial ligand deletion affect the structure and the function of cytochrome b562? Protein Eng. 2001;14:415–419. doi: 10.1093/protein/14.6.415. [DOI] [PubMed] [Google Scholar]
- Kanatous SB, Garry DJ. Gene deletional strategies reveal novel physiological roles for myoglobin in striated muscle. Respir. Physiol. Neurobiol. 2006;151:151–158. doi: 10.1016/j.resp.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Kawamura-Konishi Y, Kihara H, Suzuki H. Reconstitution of myoglobin from apoprotein and heme, monitored by stopped-flow absorption, fluorescence and circular dichroism. Eur. J. Biochem. 1988;170:589–595. doi: 10.1111/j.1432-1033.1988.tb13738.x. [DOI] [PubMed] [Google Scholar]
- Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC. A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature. 1958;181:662–666. doi: 10.1038/181662a0. [DOI] [PubMed] [Google Scholar]
- Key J, Moffat K. Crystal structures of deoxy and CO-bound bjFixLH reveal details of ligand recognition and signalling. Biochemistry. 2005;44:4627–4635. doi: 10.1021/bi047942r. [DOI] [PubMed] [Google Scholar]
- Kirby EP, Steiner RF. The tryptophan microenvironments in apomyoglobin. J. Biol. Chem. 1970;245:6300–6306. [PubMed] [Google Scholar]
- Knappenberger JA. Ph.D. thesis. The Pennsylavania State University, University Park; PA, U. S. A.: 2006. Modulation of thermodynamic and kinetic properties in b hemoproteins: characterization of a family of PsaE-cytochrome b5 chimeras and Synechocystis sp. PCC 6803 hemoglobin. [Google Scholar]
- Knappenberger JA, Kuriakose SA, Vu BC, Nothnagel HJ, Vuletich DA, Lecomte JTJ. Proximal influences in two-on-two globins: Effect of the Ala69Ser replacement on Synechocystis sp. PCC 6803 hemoglobin. Biochemistry. 2006;45:11401–11413. doi: 10.1021/bi060691x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- Kresheck GC, Erman JE. Calorimetric studies of the thermal denaturation of cytochrome c peroxidase. Biochemistry. 1988;27:2490–2496. doi: 10.1021/bi00407a035. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lad L, et al. Comparison of the heme-free and -bound crystal structures of human heme oxygenase-1. J. Biol. Chem. 2003;278:7834–7843. doi: 10.1074/jbc.M211450200. [DOI] [PubMed] [Google Scholar]
- Lattman EE, Rose GD. Protein folding— what’s the question? Proc. Natl. Acad. Sci. U. S. A. 1993;90:439–441. doi: 10.1073/pnas.90.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecomte JTJ, Kao YH, Cocco MJ. The native state of apomyoglobin described by proton NMR spectroscopy: the A-B-G-H interface of wild-type sperm whale apomyoglobin. Proteins. 1996;25:267–285. doi: 10.1002/(SICI)1097-0134(199607)25:3<267::AID-PROT1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Lecomte JTJ, Sukits SF, Bhattacharya S, Falzone CJ. Conformational properties of native sperm whale apomyoglobin in solution. Protein Sci. 1999;8:1484–1491. doi: 10.1110/ps.8.7.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecomte JTJ, Scott NL, Vu BC, Falzone CJ. Binding of ferric heme by the recombinant globin from the cyanobacterium Synechocystis sp. PCC 6803. Biochemistry. 2001;40:6541–6552. doi: 10.1021/bi010226u. [DOI] [PubMed] [Google Scholar]
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Lesk AM, Chothia C. How different amino acid sequences determine similar protein structures: the structure and evolutionary dynamics of the globins. J. Mol. Biol. 1980;136:225–230. doi: 10.1016/0022-2836(80)90373-3. [DOI] [PubMed] [Google Scholar]
- Létoffé S, Ghigo JM, Wandersman C. Iron acquisition from heme and hemoglobin by a Serratia marcescens extracellular protein. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9876–9880. doi: 10.1073/pnas.91.21.9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R, Cohen SL, Kuo A, Lee A, Chait BT. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 1998;280:106–109. doi: 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- Manyusa S, Mortuza G, Whitford D. Analysis of folding and unfolding reactions of cytochrome b5. Biochemistry. 1999;38:14352–14362. doi: 10.1021/bi991413j. [DOI] [PubMed] [Google Scholar]
- Mathews FS. The structure, function and evolution of cytochromes. Prog. Biophys. Mol. Biol. 1985;45:1–56. doi: 10.1016/0079-6107(85)90004-5. [DOI] [PubMed] [Google Scholar]
- Mathews FS, Bethge PH, Czerwinski EW. The structure of cytochrome b562 from Escherichia coli at 2.5 Å resolution. J. Biol. Chem. 1979;254:1699–1706. [PubMed] [Google Scholar]
- Merx MW, Gödecke A, Flögel U, Schrader J. Oxygen supply and nitric oxide scavenging by myoglobin contribute to exercise endurance and cardiac function. FASEB J. 2005;19:1015–1017. doi: 10.1096/fj.04-2886fje. [DOI] [PubMed] [Google Scholar]
- Miranda JJ, Maillett DH, Soman J, Olson JS. Thermoglobin, oxygen-avid hemoglobin in a bacterial hyperthermophile. J. Biol. Chem. 2005;280:36754–36761. doi: 10.1074/jbc.M505918200. [DOI] [PubMed] [Google Scholar]
- Monson EK, Ditta GS, Helinski DR. The oxygen sensor protein, FixL, of Rhizobium meliloti. Role of histidine residues in heme binding, phosphorylation, and signal transduction. J. Biol. Chem. 1995;270:5243–5250. doi: 10.1074/jbc.270.10.5243. [DOI] [PubMed] [Google Scholar]
- Montfort WR, Weichsel A, Andersen JF. Nitrophorins and related antihemostatic lipocalins from Rhodnius prolixus and other blood-sucking arthropods. Biochim. Biophys. Acta. 2000;1482:110–118. doi: 10.1016/s0167-4838(00)00165-5. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay K, Lecomte JTJ. A relationship between heme binding and protein stability in cytochrome b5. Biochemistry. 2004;43:12227–12236. doi: 10.1021/bi0488956. [DOI] [PubMed] [Google Scholar]
- Muñoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1994;1:399–409. doi: 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- Murray JW, Delumeau O, Lewis RJ. Structure of a nonheme globin in environmental stress signaling. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17320–17325. doi: 10.1073/pnas.0506599102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nioche P, Berka V, Vipond J, Minton N, Tsai AL, Raman CS. Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science. 2004;306:1550–1553. doi: 10.1126/science.1103596. [DOI] [PubMed] [Google Scholar]
- Nishii I, Kataoka M, Tokunaga F, Goto Y. Cold denaturation of the molten globule states of apomyoglobin and a profile for protein folding. Biochemistry. 1994;33:4903–4909. doi: 10.1021/bi00182a019. [DOI] [PubMed] [Google Scholar]
- Nishimura C, Lietzow MA, Dyson HJ, Wright PE. Sequence determinants of a protein folding pathway. J. Mol. Biol. 2005;351:383–392. doi: 10.1016/j.jmb.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Nishimura C, Prytulla S, Jane Dyson H, Wright PE. Conservation of folding pathways in evolutionarily distant globin sequences. Nat. Struct. Biol. 2000;7:679–686. doi: 10.1038/77985. [DOI] [PubMed] [Google Scholar]
- Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH-a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Wilks A. Heme oxygenase structure and mechanism. Adv. Inorg. Chem. 2001;51:359–407. [Google Scholar]
- Pace CN, Shirley BA, Thomson JA. Protein structure: a practical approach. IRL Press, Oxford; New York: 1989. [Google Scholar]
- Pandini A, Bonati L. Conservation and specialization in PAS domain dynamics. Protein Eng. Des. Sel. 2005;18:127–137. doi: 10.1093/protein/gzi017. [DOI] [PubMed] [Google Scholar]
- Paoli M, Anderson BF, Baker HM, Morgan WT, Smith A, Baker EN. Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two β-propeller domains. Nat. Struct. Biol. 1999;6:926–931. doi: 10.1038/13294. [DOI] [PubMed] [Google Scholar]
- Park H, Suquet C, Satterlee JD, Kang C. Insights into signal transduction involving PAS domain oxygen-sensing heme proteins from the X-ray crystal structure of Escherichia coli Dos heme domain (Ec DosH) Biochemistry. 2004;43:2738–2746. doi: 10.1021/bi035980p. [DOI] [PubMed] [Google Scholar]
- Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz MF, Paoli M, Lesk AM. Fix L, a haemoglobin that acts as an oxygen sensor: signalling mechanism and structural basis of its homology with PAS domains. Chem. Biol. 1999;6:R291–R297. doi: 10.1016/s1074-5521(99)80121-5. [DOI] [PubMed] [Google Scholar]
- Pesce A, et al. A novel two-over-two α-helical sandwich fold is characteristic of the truncated hemoglobin family. EMBO J. 2000;19:2424–2434. doi: 10.1093/emboj/19.11.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeil W, Nolting BO, Jung C. Apocytochrome P450cam is a native protein with some intermediate-like properties. Biochemistry. 1993;32:8856–8862. doi: 10.1021/bi00085a017. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Aravind L. PAS: a multifunctional domain family comes to light. Curr. Biol. 1997;7:R674–R677. doi: 10.1016/s0960-9822(06)00352-6. [DOI] [PubMed] [Google Scholar]
- Poulos TL. Soluble guanylate cyclase. Curr. Opin. Struct. Biol. 2006;16:736–743. doi: 10.1016/j.sbi.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Poulos TL, Finzel BC, Howard AJ. High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- Privalov PL. Intermediate states in protein folding. J. Mol. Biol. 1996;258:707–725. doi: 10.1006/jmbi.1996.0280. [DOI] [PubMed] [Google Scholar]
- Ptitsyn OB, Ting KL. Non-functional conserved residues in globins and their possible role as a folding nucleus. J. Mol. Biol. 1999;291:671–682. doi: 10.1006/jmbi.1999.2920. [DOI] [PubMed] [Google Scholar]
- Regis WC, Fattori J, Santoro MM, Jamin M, Ramos CH. On the difference in stability between horse and sperm whale myoglobins. Arch. Biochem. Biophys. 2005;436:168–177. doi: 10.1016/j.abb.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Rivera M, et al. The reduction potential of cytochrome b5 is modulated by its exposed heme edge. Biochemistry. 1998;37:1485–1494. doi: 10.1021/bi972390g. [DOI] [PubMed] [Google Scholar]
- Rivera M, Zeng Y. Heme oxygenase, steering dioxygen activation toward heme hydroxylation. J. Inorg. Biochem. 2005;99:337–354. doi: 10.1016/j.jinorgbio.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Robinson CR, Liu Y, Thomson JA, Sturtevant JM, Sligar SG. Energetics of heme binding to native and denatured states of cytochrome b562. Biochemistry. 1997;36:16141–16146. doi: 10.1021/bi971470h. [DOI] [PubMed] [Google Scholar]
- Rodgers KR, Lukat-Rodgers GS, Barron JA. Structural basis for ligand discrimination and response initiation in the heme-based oxygen sensor FixL. Biochemistry. 1996;35:9539–9548. doi: 10.1021/bi9530853. [DOI] [PubMed] [Google Scholar]
- Rublevskaya I, Maines MD. Interaction of Fe-protoporphyrin IX and heme analogues with purified recombinant heme oxygenase-2, the constitutive isozyme of the brain and testes. J. Biol. Chem. 1994;269:26390–26395. [PubMed] [Google Scholar]
- Schenkman JB, Jansson I. The many roles of cytochrome b5. Pharmacol. Ther. 2003;97:139–152. doi: 10.1016/s0163-7258(02)00327-3. [DOI] [PubMed] [Google Scholar]
- Scott NL, Falzone CJ, Vuletich DA, Zhao J, Bryant DA, Lecomte JTJ. The hemoglobin of the cyanobacterium Synechococcus sp. PCC 7002: evidence for hexacoordination and covalent adduct formation in the ferric recombinant protein. Biochemistry. 2002;41:6902–6910. doi: 10.1021/bi025609m. [DOI] [PubMed] [Google Scholar]
- Senjo M, Ishibashi T, Imai Y. Purification and characterization of cytosolic liver protein facilitating heme transport into apocytochrome b5 from mitochondria. Evidence for identifying the heme transfer protein as belonging to a group of glutathione S-transferases. J. Biol. Chem. 1985;260:9191–9196. [PubMed] [Google Scholar]
- Shelnutt JA, Song X-Z, Ma J-G, Jia SL, Jentzen W, Medforth CJ. Nonplanar porphyrins and their significance in proteins. Chem. Soc. Rev. 1998;27:31–42. [Google Scholar]
- Shen LL, Hermans J., Jr. Kinetics of conformation change of sperm-whale myoglobin. II. Characterization of the rapidly and slowly formed denatured species (D and D*) Biochemistry. 1972;11:1842–1844. doi: 10.1021/bi00760a017. [DOI] [PubMed] [Google Scholar]
- Shipulina NV, Smith A, Morgan WT. Effects of reduction and ligation of heme iron on the thermal stability of heme-hemopexin complexes. J. Protein Chem. 2001;20:145–154. doi: 10.1023/a:1011033625009. [DOI] [PubMed] [Google Scholar]
- Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. U. S. A. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland EH, Kay E, Shannon LM, Horwitz J. Peroxidase isoenzymes from horseradish roots. 3. Circular dichroism of isoenzymes and apoisoenzymes. J. Biol. Chem. 1968;243:3560–3565. [PubMed] [Google Scholar]
- Su XD, Yonetani T, Skoglund U. The crystal structure of the iron-free cytochrome c peroxidase and its implication for the enzymatic mechanism. FEBS Lett. 1994;351:437–442. doi: 10.1016/0014-5793(94)00910-4. [DOI] [PubMed] [Google Scholar]
- Sugishima M, et al. Crystal structure of rat heme oxygenase-1 in complex with heme bound to azide. Implication for regiospecific hydroxylation of heme at the α-meso carbon. J. Biol. Chem. 2002a;277:45086–45090. doi: 10.1074/jbc.M207267200. [DOI] [PubMed] [Google Scholar]
- Sugishima M, et al. Crystal structure of rat apo-heme oxygenase-1 (HO-1): mechanism of heme binding in HO-1 inferred from structural comparison of the apo and heme complex forms. Biochemistry. 2002b;41:7293–7300. doi: 10.1021/bi025662a. [DOI] [PubMed] [Google Scholar]
- Suquet C, Savenkova M, Satterlee JD. Recombinant PAS-heme domains of oxygen sensing proteins: high level production and physical characterization. Protein Expr. Purif. 2005;42:182–193. doi: 10.1016/j.pep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Takami M. Catabolism of heme moiety of hemoglobin.haptoglobin in rat liver cells in vivo. J. Biol. Chem. 1993;268:20335–20342. [PubMed] [Google Scholar]
- Taylor BL, Zhulin IB. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 1999;63:479–506. doi: 10.1128/mmbr.63.2.479-506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teale FWJ. Cleavage of heme-protein link by acid methylethylketone. Biochim. Biophys. Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. U. S. A. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsteinsson MV, et al. A cyanobacterial hemoglobin with unusual ligand binding kinetics and stability properties. Biochemistry. 1999;38:2117–2126. doi: 10.1021/bi9819172. [DOI] [PubMed] [Google Scholar]
- Tolosano E, Altruda F. Hemopexin: structure, function, and regulation. DNA Cell Biol. 2002;21:297–306. doi: 10.1089/104454902753759717. [DOI] [PubMed] [Google Scholar]
- Tsaprailis G, Chan DW, English AM. Conformational states in denaturants of cytochrome c and horseradish peroxidases examined by fluorescence and circular dichroism. Biochemistry. 1998;37:2004–2016. doi: 10.1021/bi971032a. [DOI] [PubMed] [Google Scholar]
- Vergères G, Chen DY, Wu FF, Waskell L. The function of tyrosine 74 of cytochrome b5. Arch. Biochem. Biophys. 1993;305:231–241. doi: 10.1006/abbi.1993.1416. [DOI] [PubMed] [Google Scholar]
- Vinogradov SN, et al. A phylogenomic profile of globins. BMC Evol. Biol. 2006;6:31. doi: 10.1186/1471-2148-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu BC, Jones AD, Lecomte JTJ. Novel histidine-heme covalent linkage in a hemoglobin. J. Am. Chem. Soc. 2002;124:8544–8545. doi: 10.1021/ja026569c. [DOI] [PubMed] [Google Scholar]
- Vuletich DA, Falzone CJ, Lecomte JTJ. Structural and dynamic repercussions of heme binding and heme-protein cross-linking in Synechococcus sp. PCC 7002 hemoglobin. Biochemistry. 2006;45:14075–14084. doi: 10.1021/bi061532g. [DOI] [PubMed] [Google Scholar]
- Vuletich DA, Lecomte JTJ. A phylogenetic and structural analysis of truncated hemoglobins. J. Mol. Evol. 2006;62:196–210. doi: 10.1007/s00239-005-0077-4. [DOI] [PubMed] [Google Scholar]
- Wang L, Sun N, Terzyan S, Zhang X, Benson DR. A histidine/tryptophan π-stacking interaction stabilizes the heme-independent folding core of microsomal apocytochrome b5 relative to that of mitochondrial apocytochrome b5. Biochemistry. 2006;45:13750–13759. doi: 10.1021/bi0615689. [DOI] [PubMed] [Google Scholar]
- Weichsel A, Andersen JF, Champagne DE, Walker FA, Montfort WR. Crystal structures of a nitric oxide transport protein from a blood-sucking insect. Nat. Struct. Biol. 1998;5:304–309. doi: 10.1038/nsb0498-304. [DOI] [PubMed] [Google Scholar]
- Weichsel A, Andersen JF, Roberts SA, Montfort WR. Nitric oxide binding to nitrophorin 4 induces complete distal pocket burial. Nat. Struct. Biol. 2000;7:551–554. doi: 10.1038/76769. [DOI] [PubMed] [Google Scholar]
- Wilks A. Heme oxygenase: evolution, structure, and mechanism. Antioxid. Redox Signal. 2002;4:603–614. doi: 10.1089/15230860260220102. [DOI] [PubMed] [Google Scholar]
- Wittenberg JB, Bolognesi M, Wittenberg BA, Guertin M. Truncated hemoglobins: A new family of hemoglobins widely distributed in bacteria, unicellular eukaryotes and plants. J. Biol. Chem. 2002;277:871–874. doi: 10.1074/jbc.R100058200. [DOI] [PubMed] [Google Scholar]
- Wittenberg JB, Wittenberg BA. Myoglobin function reassessed. J. Exp. Biol. 2003;206:2011–2020. doi: 10.1242/jeb.00243. [DOI] [PubMed] [Google Scholar]
- Wolff N, et al. Histidine pKa shifts and changes of tautomeric states induced by the binding of gallium-protoporphyrin IX in the hemophore HasASM. Protein Sci. 2002;11:757–765. doi: 10.1110/ps.3630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee S, Peyton DH. Proton NMR investigation of the reconstitution of equine myoglobin with hemin dicyanide. Evidence for late formation of the proximal His93F8-iron bond. FEBS Lett. 1991;290:119–122. doi: 10.1016/0014-5793(91)81240-9. [DOI] [PubMed] [Google Scholar]
- Yip YK, Waks M, Beychok S. Influence of prosthetic groups on protein folding and subunit assembly. I. Conformational differences between separated human α- and β-globins. J. Biol. Chem. 1972;247:7237–7244. [PubMed] [Google Scholar]
- Yokota T, Nakajima Y, Yamakura F, Sugio S, Hashimoto M, Takamiya S. Unique structure of Ascaris suum b5-type cytochrome: an additional α-helix and positively charged residues on the surface domain interact with redox partners. Biochem. J. 2006;394:437–447. doi: 10.1042/BJ20051308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonetani T. Studies on cytochrome c peroxidase. X. Crystalline apo- and reconstituted holoenzymes. J. Biol. Chem. 1967;242:5008–5013. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.