

Abstract
Hepatitis C virus (HCV) infection is the leading cause of liver cirrhosis and hepatocellular carcinoma and one of the primary indications for liver transplantation. The molecular mechanisms underlying the actions of host factors in HCV replication remain poorly defined. FUSE (far upstream element of the c-myc proto-oncogene) binding protein (FBP) is a cellular factor that we have identified as a binder of HCV 3′ nontranslated region (3′NTR). Mapping of the binding site showed that FBP specifically interacts with the poly(U) tract within the poly(U/UC) region of the 3′NTR. Silencing of FBP expression by small interfering RNA in cells carrying HCV subgenomic replicons severely reduced viral replication, while overexpression of FBP significantly enhanced viral replication. We confirmed these observations by an in vitro HCV replication assay in the cell-free replicative lysate, which suggested that there is a direct correlation between the cellular FBP level and HCV replication. FBP immunoprecipitation coprecipitated HCV nonstructural protein 5A (NS5A), indicating that FBP interacts with HCV NS5A, which is known to function as a link between HCV translation and replication. Although FBP is mainly localized in the nucleus, we found that in MH14 cells a significant level of this protein is colocalized with NS5A in the cytosol, a site of HCV replication. While the mechanism of FBP involvement in HCV replication is yet to be delineated, our findings suggest that it may be an important regulatory component that is essential for efficient replication of HCV.
Hepatitis C virus (HCV), a positive single-stranded RNA virus of the Flaviviridae family, has a 9.6-kb genome (14, 50, 68). The RNA genome contains a single open reading frame that encodes a polyprotein of about 3,000 amino acids that are processed by host and virus proteases into structural proteins (core, E1, and E2) and nonstructural proteins (p7, nonstructural protein 2 [NS2], NS3, NS4A, NS4B, NS5A, and NS5B) (14, 50).
The conserved 5′- and 3′-terminal nontranslated regions (5′NTR and 3′NTR, respectively) of the HCV genomic RNA have multiple regulatory elements essential for viral replication and expression of viral genes. While the 5′NTR of HCV contains the internal ribosomal entry site (IRES) required for cap-independent translation of plus-strand HCV RNA (67), it is also the 3′ region of the minus-strand HCV RNA. It is predicted that both the 3′NTR of the plus strand and the 3′NTR of the minus-strand HCV RNA are highly structured and are the sites for initiation of viral replication (54). In addition to proteins associated with the translational machinery, several cellular proteins have been reported to interact with the 5′NTR; these include polypyrimidine tract binding protein (PTB) (3, 4, 37), La autoantigen (17), nucleolin (38), eukaryotic initiation factor 2B gamma subunit (eIF2Bγ) (43), and the nuclear factors NF90 (nuclear factor 90), NF110, NF45, and RNA helicase A (RHA) (34). Some cellular proteins are also associated with the 3′NTR of HCV, including PTB (13, 29, 36, 37, 66), heterogeneous nuclear ribonucleoprotein C (hnRNP C) (29), La autoantigen (29, 61, 62), Hu antigen receptor protein (HuR) (63), and the nuclear factors (34). In a recent study using an RNA affinity capture system and liquid chromatography followed by tandem mass spectrometry, we identified several cellular proteins that bind and/or interact with the 3′NTR of HCV (31). These proteins include members of the AU-rich element (ARE) binding protein (AREBP), such as FBP, FBP2 (also called K homology type splicing regulatory protein [KSRP]), ARE binding factor 1 (AUF1, also called hnRNP D), HuR, hnRNP C, Y-box binding protein 1 (YB-1), T-cell intracellular antigen 1 (TIA1), and NF90. Several of these AREBPs are involved in regulating the stability of mRNAs. Some, such as AUF1 (58), hnRNP L (59), and KSRP (28), target mRNAs for degradation by binding to AREs within the 3′NTRs of cellular mRNAs. Still others, such as HuR (11, 55), hnRNP C (56), and NF90 (60), increase the stability and longevity of certain mRNAs.
FBP, one of the AREBPs mentioned earlier, activates transcription of the c-myc gene by binding to an element called the far-upstream element (FUSE) (7, 20, 32, 47). FBP not only activates c-myc transcription, but it also, along with PTB, binds the 3′NTR of growth-associated protein 43 (GAP-43) mRNA. These two proteins appear to work in coordination to regulate the stability of the mRNA (33). FBP, which is identical to the DNA helicase V protein, contains powerful ATP-dependent DNA helicase activity (69) and has conserved Arg-Gly-Gly (RGG) motifs, as do many RNA and DNA helicases (18). FBP has been compared with hnRNP K in its ability to target cognate sequences in negatively supercoiled DNA. It also shares some homology with hnRNP K, which has also been shown to bind the CT-element in the c-myc promoter (64). FBP is present only in undifferentiated cells (20). The structure of FBP (K homology domain 3 [KH3] and KH4) bound to a 29-bp fragment from FUSE has been determined by nuclear magnetic resonance (10).
Because of the paucity of information, HCV-host interactions clearly warrant intensive investigation. Several groups, including our own, have begun to define interactions between HCV and human cellular proteins. In this study, we have mapped the FBP interaction site on the HCV 3′NTR and demonstrated a direct correlation between cellular levels of FBP and HCV replication. The results of our investigation imply that FBP may play an important regulatory role in HCV replication and translation processes.
MATERIALS AND METHODS
Plasmids and oligonucleotides.
Plasmids carrying the HCV subgenomic replicon with the luciferase reporter gene (pLMH14 [51]) and without the luciferase reporter gene (pMH14 [49]) were obtained from Makoto Hijikata (Kyoto University, Japan). Plasmid pVP506 was constructed by cloning the PCR-amplified 243-bp fragment of HCV 3′NTR between NdeI and BamHI sites in the pET3b (Novagen) vector 65 nucleotides downstream of the T7 promoter (31). FBP small interfering RNA (siRNA) sense (5′-GGG ACA UCA CUG AAU UCA ATT-3′) and antisense (5′-UUG AAU UCA GUG AUG UCC CTG-3′) were purchased from Ambion (Austin, TX); control siRNA (scrambled) sense (5′-UUC UCC GAA CGU GUC ACG Utt-3′) and antisense (5′-ACG UGA CAC GUU CGG AGA Att-3′) were obtained from Qiagen (Valencia, CA).
The primers for reverse transcription-PCR (RT-PCR) of HCV 5′NTR and actin mRNA and for PCR amplification of full-length HCV 3′NTR (F13) and its variable region (F11), poly(U/UC) region (F22), and 3′ X-tail (F33) were synthesized at the Molecular Resource Facility at UMDNJ. The sequences of these primers are as shown in Table 1. Some of the primers that start with T7 promoter sequences (underlined) were used in preparing the templates for the in vitro transcription assay.
TABLE 1.
Primers used in this study
| Primera | Sequenceb |
|---|---|
| 5′NTR up | 5′-CGG GAG AGC CAT AGT GG-3′ |
| 5′NTR down | 5′-AGT ACC ACA AGG CCT TTC G-3′ |
| Actin up | 5′-CAG GCA CCA GGG CGT GAT GG-3′ |
| Actin down | 5′-AGG CGT ACA GGG ATA GCA CA-3′ |
| F11/F13 up | 5′-TTA ATA CGA CTC ACT ATA GAC CCC GCT GGT TCA TGT GGT GC-3′ |
| F11dn | 5′-AAT GGC CTA TTG GCC TGG A-3′ |
| F33 up | 5′-TTA ATA CGA CTC ACT ATA GTG GCT CCA TCT TAG CCC T-3′ |
| F13/F33 down | 5′-ACA TGA TCT GCA GAG AGG CCA-3′ |
| F22 up | 5′-TTA ATA CGA CTC ACT ATA TCC AGG CCA ATA GGC CAT T-3′ |
| F22 down | 5′-AGG GCT AAG ATG GAG CCA C-3′ |
Primers for reverse transcription-PCR (RT-PCR) of HCV 5′NTR and actin mRNA and for PCR amplification of full-length HCV 3′NTR (F13) and its variable region (F11), poly(U/UC) region (F22), and 3′ X-tail (F33).
T7 promoter sequences are underlined.
Photoaffinity cross-linking of HCV 3′NTR with FBP or cell lysate.
We incubated purified FBP with Cy5-labeled HCV 3′NTR in the presence or absence of RNA competitor. After 20 min of incubation at 37°C, we irradiated the mixture in a UV cross-linker (Spectrolinker XL-1000; Spectronics Corporation) at 300 mJ/cm2 and then treated the sample with RNase A (0.1 μg/μl; Qiagen) for 15 min at 37°C. We resolved the cross-linked RNA-protein complexes on a 8% sodium dodecyl sulfate (SDS)-polyacrylamide gel and visualized them by using a Typhoon scanner (Amersham).
We used the same protocol to carry out photoaffinity cross-linking of 0.5 pmol of Cy5-labeled HCV 3′NTR with 15 μl (15 μg of total protein) cytoplasmic cell lysate of cured MH14 cells. We resolved the cross-linked RNA-protein complexes on a 8% SDS-polyacrylamide gel and visualized the Cy5-labeled RNA protein complexes by using a Typhoon scanner (Amersham). After we transferred the RNA-protein complexes to a nitrocellulose membrane (Schleicher and Schuell Bioscience), we detected the FBP in the RNA-protein complexes by Western blotting using anti-FBP antibody (FBP C-20; Santa Cruz Biotechnology).
Cell culture.
Cured MH14 and MH14 cells were gifts from Makoto Hijikata (Kyoto University, Japan). MH14 cells are a derivative of the Huh7 cell line, which carries stable HCV subgenomic replicons (51). Cured MH14 cells were prepared by treating MH14 cells with 5,000 IU/ml of alpha interferon for 2 weeks. The absence of replicon RNA and viral proteins was checked by Northern blotting, RT-PCR, and Western blotting (51). Huh7 and cured MH14 cells were maintained in Dulbecco's modified Eagle medium (Sigma) supplemented with 10% fetal bovine serum (Sigma), 100 units/ml of nonessential amino acids (Gibco), and 100 μg/ml of penicillin and streptomycin sulfate (Gibco). MH14 cells were cultured in the medium supplemented with 300 μg/ml of G418 (Calbiochem). Cells were grown at 37°C with 5% CO2.
Transfection of FBP siRNA and FBP expression plasmid.
Two sets of MH14 cells were grown in a six-well plate (2 × 105/well) for 24 h and then transfected with FBP siRNA (final concentration, 20 nM per well) or pCIA-CMV-FBP DNA (CMV stands for cytomegalovirus) (4 μg per well) according to the manufacturer's protocol, using Lipofectamine 2000 (Invitrogen) as the transfection reagent. The transfected cells were grown for an additional 48 h. One set of cells was processed for the isolation of total protein; protein concentration was determined by DC protein assay (Bio-Rad). An equal (15-μg) amount of total protein was used for Western blotting. The other set of cells was processed for the isolation of total RNA (TRIzol; Invitrogen) and underwent subsequent RT-PCR analysis (SuperScript II reverse transcriptase [Invitrogen] and Taq DNA polymerase [Invitrogen]) for HCV RNA and actin mRNA. The experiments were performed in triplicate.
Preparation of cell extract and Western blotting.
Cells were washed twice with phosphate-buffered saline (PBS) and then lysed in warmed lysis buffer containing 1% SDS, 10% glycerol, 1 mM dithiothreitol (DTT), and 10 mM Tris HCl (pH 6.8). The lysed cells were boiled for 5 min, cooled to room temperature, and centrifuged at 13,000 rpm for 10 min at 4°C. We saved the supernatant and determined the protein concentration (DC protein assay; Bio-Rad) of each sample. An aliquot of the supernatant equivalent to 15 μg of protein was resolved on a 8% SDS-polyacrylamide gel and then transferred to a nitrocellulose membrane (Schleicher and Schuell Bioscience). The membrane was first blocked for 1 h at room temperature by 5% nonfat dry milk in PBS buffer containing 0.5% Tween 20. The membrane was then washed four times with the same buffer and treated with FBP primary antibody (FBP C-20; Santa Cruz Biotechnology) (diluted 1:500) overnight at 4°C. The membrane was washed again with the same buffer four times, then treated with a 1:5,000 dilution of horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature, and washed thoroughly. The membrane was incubated with horseradish peroxidase substrate (Western Lightning chemiluminescence reagent; PerkinElmer Life Sciences) for 1 min, exposed on Kodak X-Omat AR X-ray film (Eastman Kodak), and developed.
Preparation of nuclear and cytoplasmic extracts for Western blotting.
We prepared cytoplasmic and nuclear extracts from cured MH14 and MH14 cells using NE-PER nuclear and cytoplasmic extract reagent (Pierce) according to the manufacturer's protocol. Three percent volume of each of the extracts was subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting for FBP and NS5A. The samples were also Western blotted for poly(ADP-ribose) polymerase (PARP) and α-tubulin as markers for the nucleus and cytosol, respectively.
Preparation of replicative cell lysate and cell-free HCV replication assay.
MH14 cells were cultured as described above in 10-cm-diameter petri dishes. We prepared the replicative cytoplasmic fractions from the cells following the protocol described previously (3, 71). Cells were washed with cold buffer containing 150 mM sucrose, 30 mM HEPES (pH 7.4), 33 mM ammonium chloride, 7 mM KCl, and 4.5 mM magnesium acetate. We then treated the cells with lysolecithin solution (250 μg/ml) in the washing buffer for 1 min and then added 3 ml of washing buffer to each dish. We removed the buffer and collected the cells by scraping them into 120 μl of replication buffer containing 100 mM HEPES (pH 7.4); 50 mM ammonium chloride; 7 mM KCl; 1 mM spermidine; 1 mM (each) of ATP, GTP, CTP, and UTP; 1 mM DTT; and 10% glycerol. We transferred the cells to a new tube, lysed them gently by pipetting up and down several times, and then centrifuged the lysed cells at 1,600 rpm for 5 min at 4°C. The supernatant fraction (replicative lysate) was divided into aliquots and stored at −80°C until use. For the cell-free HCV replication assay, we incubated 50 μl of replicative lysate for 0 h or 3 h at 30°C with or without FBP. We terminated the reaction by adding 0.5% SDS in STE buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 150 mM NaCl) and extracted total RNA twice with phenol-chloroform-isoamyl alcohol (25:24:1) and twice with water-saturated ether. We precipitated the RNA by ethanol and dissolved it in diethyl pyrocarbonate (DEPC)-treated water. The total RNA underwent subsequent RT-PCR analysis (SuperScript II reverse transcriptase [Invitrogen] and Taq DNA polymerase [Invitrogen]) for HCV RNA and actin mRNA. The PCR products were visualized by agarose gel electrophoresis. The experiments were performed in triplicate.
Detection and quantification of HCV RNA in cell lysate.
We isolated total RNA from the cells with TRIzol reagent (Invitrogen) according to the manufacturer's protocol. The RNA was dissolved in DEPC-treated water, quantified by NanoDrop (ND1000; NanoDrop Technologies), divided into aliquots, and stored at −80°C. For RT-PCR, 1 μg of total RNA was taken in a 20-μl reaction mixture containing 250 ng of each primer pair and 1 mM (each) of the four deoxynucleoside triphosphates (Invitrogen). The mixture was heated to 65°C for 5 min, chilled on ice, and supplemented with 4 μl of 5× first-strand buffer (Invitrogen), 2 μl of 0.1 M DTT, and 1 μl (40 units) of RNaseOUT (Invitrogen). The mixture was incubated at 42°C for 2 min; the reaction was started by the addition of 1 μl (200 units) of SuperScript II reverse transcriptase (Invitrogen). After 50 min of incubation at 42°C, the reaction was terminated by heating at 70°C for 15 min. For PCR, we took 2 μl of synthesized cDNA in a total volume of 50 μl of a PCR mixture containing 40 mM Tris HCl (pH 8.4), 50 mM KCl, 2 mM MgCl2, 0.2 mM (each) of the four deoxynucleoside triphosphates, 0.2 μM of each primer pair, and 2 units of Taq DNA polymerase (Invitrogen). PCR was done for 15 to 35 cycles, with 1 cycle consisting of 1 min at 95°C, 1 min at 55°C, and 1 min at 72°C. The product was analyzed by agarose gel electrophoresis. The experiments were performed in triplicate.
In vitro transcription of the full-length HCV subgenomic replicon, HCV 3′NTR RNA, and its fragments.
For in vitro transcription of full-length HCV subgenomic replicon RNA, we used the MEGAscript T7 kit (Ambion). For in vitro transcription of RNA corresponding to the full-length HCV 3′NTR (F13) and fragments corresponding to the variable region (F11), the poly(U/UC) region (F22), and conserved 3′ X-tail (F33) of HCV 3′NTR, we used T7 RNA polymerase (Roche Applied Science). To synthesize Cy5-labeled RNA, we added Cy5-labeled UTP (Amersham) in the reaction solution. Reactions were carried out according to the manufacturer's protocols. The transcripts were purified by phenol-chloroform extraction and ethanol precipitation, dissolved in DEPC-treated water, and stored at −80°C.
Quantification of gel image.
All gel images (Western blotting and RT-PCR) were quantified by QuantityOne software (version 4.4.1; Bio-Rad). All analyses were based on triplicate experiments.
Luciferase assay.
Cured MH14 cells seeded on a 24-well plate (3 × 104/well) were transfected with FBP siRNA (final concentration, 20 nM per well) or with FBP-overexpressing plasmid pCIA-CMV-FBP (4 μg per well) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were grown for another 24 h and then transfected with the luciferase HCV replicon RNA (Luc-HCV RNA) (0.5 μg per well), using DMRIE-C reagent (Invitrogen) according to the manufacturer's instructions. After 24 h of incubation, total proteins were extracted with the lysis buffer supplied in the luciferase reporter assay system kit (Promega). The luciferase activity was measured according to the manufacturer's protocol. Assays were performed in four parallel sets.
Immunoprecipitation.
We washed the MH14 cells (5 × 106 cells per assay) twice with warmed 1× PBS and then lysed the cells in cold buffer containing 150 mM NaCl, 50 mM Tris HCl (pH 7.4), 1% Triton X-100, and a 1× protease inhibitor cocktail (Roche Applied Science). We also treated the lysate with 60 μg of RNase A to avoid nonspecific RNA binding proteins that may be captured in FBP immunoprecipitation (FBP-IP) via RNA bridging. We incubated the cell lysate with 2 μg of anti-FBP antibody (FBP C-20; Santa Cruz Biotechnology) for 1 h at 4°C and then added 20 μl of protein A/G Plus agarose beads (Santa Cruz Biotechnology). The mixture was then incubated overnight at 4°C. The immunoprecipitates were collected by centrifugation at 2,500 rpm for 5 min at 4°C. After we washed the pellets four times with lysis buffer, we resuspended the immunoprecipitates in 1× Laemmli gel loading buffer. Samples were boiled and centrifuged to pellet the agarose beads. The supernatant was subjected to 8% SDS-PAGE analysis. The coprecipitated proteins were identified by Western blotting with corresponding antibodies.
Immunofluorescence.
We put one coverslip in every well of the six-well plate, split the cured MH14 cells or MH14 cells into the wells (1.5 × 105 cells per well), and grew the cells for 24 h. We took out the coverslips, washed the coverslip with PBS two times, then fixed the cells with 3% paraformaldehyde in PBS for 30 min, and permeabilized them with 0.1% Triton X-100 in PBS for 5 min. We incubated the permeabilized cells with 50 mM glycine in PBS for 10 min and stained them with anti-FBP antibody (FBP C-20; Santa Cruz Biotechnology) or anti-NS5A antibody (Biodesign) for 1 h at a 1:100 dilution in PBS buffer containing 0.1% gelatin and then with Cy3- or Alexa Fluor 488-labeled secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) at a 1:100 dilution in the same buffer for another 1 h. After we washed the coverslip with PBS four times, we incubated the coverslips in 0.1 mg/ml DAPI (4′,6′-diamidino-2-phenyindole; Sigma Chemical Co.) for 5 min and then washed the coverslips with PBS for four times. We allowed the coverslips to air dry and then mounted them with mounting medium (ProLong antifade kit; Molecular Probes, Eugene, OR). We analyzed the cells under a TE-300 Nikon fluorescence microscope (Nikon, Japan). Pictures were taken and merged using Spot Advanced software (Diagnostic Instruments, Sterling Heights, MI).
Coomassie blue staining.
After we wetted the membrane in 100% methanol, we stained it with 0.1% Coomassie brilliant blue R (Sigma-Aldrich) in 50% methanol and 7% acetic acid for 2 min. We rinsed the membrane with Milli-Q water and destained it with solution I (50% methanol, 7% acetic acid) for 10 min and then with solution II (90% methanol, 10% acetic acid) for 10 min.
RESULTS
Interaction between FBP and HCV 3′NTR.
In a previous study, using an RNA affinity capture system and liquid chromatography followed by tandem mass spectrometry, we identified FBP as one of the HCV 3′NTR binding proteins (31). In this study, to verify the interaction between FBP and HCV 3′NTR, we carried out photoaffinity cross-linking between the cytoplasmic cell lysate of cured MH14 cells, which does not harbor the HCV replicon, and Cy5-labeled HCV 3′NTR RNA. We added Cy5-labeled, in vitro-transcribed HCV 3′NTR RNA to the cytoplasmic cell lysate of cured MH14 cells, incubated the mixture for 20 min at 37°C, and then irradiated the incubation mixture in a UV cross-linker at 300 mJ/cm2. We digested the cross-linked samples with RNase A (0.1 μg/μl) to remove free and uncross-linked RNA and then resolved protein-RNA cross-linked complexes on a 8% SDS-polyacrylamide gel, visualizing the complexes with a Typhoon scanner (Fig. 1, lane 1). We found a number of cellular proteins that bind to HCV 3′NTR RNA, including one that migrated to the 74-kDa position. We transferred the protein-RNA complexes on the gel to a nitrocellulose membrane and subjected it to Western blotting with anti-FBP antibody and found that the protein-RNA complexes at the 74-kDa position contained FBP protein (Fig. 1, lane 2). This indicated that FBP binds to HCV 3′NTR RNA.
FIG. 1.
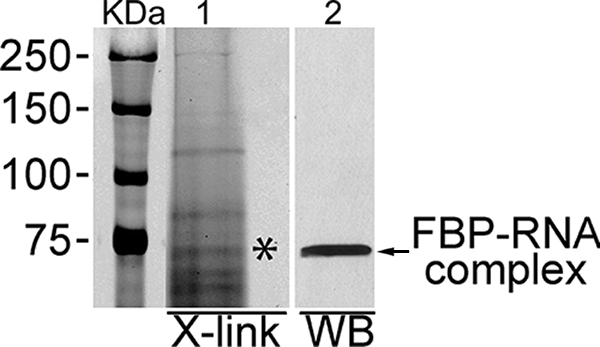
Interaction between cytoplasmic FBP and Cy5-labeled HCV 3′NTR. Cy5-labeled, in vitro-transcribed HCV 3′NTR RNA was incubated with cytoplasmic extract from cured MH14 cells for 20 min at 37°C and then UV irradiated. The sample was then treated with RNase A and resolved by SDS-PAGE. The cross-linked (X-link) protein-RNA complexes were detected by using a Typhoon scanner (lane 1). We then transferred the RNA-protein complexes to the nitrocellulose membrane and Western blotted (WB) using anti-FBP antibody (lane 2). The positions of molecular mass protein markers (in kilodaltons) are shown to the left of the gel.
To confirm that the binding of FBP to HCV 3′NTR is specific, we carried out photoaffinity cross-linking between purified FBP and Cy5-labeled HCV 3′NTR RNA in the presence or absence of competitor RNA. When we incubated a fixed concentration of Cy5-labeled HCV 3′NTR RNA (0.5 pmol) with increasing concentrations of purified FBP (0.25, 0.5, and 1.0 pmol), we found corresponding increases in the cross-linking as a function of FBP concentration (Fig. 2A). Similarly, when we incubated a fixed concentration of purified FBP (0.5 pmol) with decreasing concentrations of Cy5-labeled HCV 3′NTR RNA (0.5, 0.25, and 0.125 pmol), we found corresponding decreases in the cross-linking as a function of the concentration of Cy5-labeled HCV 3′NTR RNA (Fig. 2B, lanes 1 to 3). Decreases in cross-linking also occurred in the presence of increasing concentrations of cold HCV 3′NTR RNA (0.5, 2.5, and 5 pmol) (Fig. 2B, lanes 4 to 6), indicating that Cy5-labeled HCV 3′NTR RNA and cold HCV 3′NTR RNA compete for binding to FBP. In contrast, when we incubated a fixed concentration of purified FBP (0.5 pmol) with a fixed concentration of Cy5-labeled HCV 3′NTR RNA (0.5 pmol) in the presence of a 10-fold excess of a non-NTR RNA corresponding to human immunodeficiency virus type 1 (HIV-1) transactivation-responsive element (TAR), we found no reduction in cross-linking, indicating that FBP interaction with HCV 3′NTR RNA is highly specific and cannot be outcompeted by RNA other than HCV 3′NTR (Fig. 2C). We also examined the ability of two RNA homopolymers, poly(rU) and poly(rC), to compete with HCV 3′NTR for binding to FBP (Fig. 2D). Interestingly, poly(rU) completely outcompeted Cy5-labeled HCV 3′NTR (lane 2), while poly(rC) was ineffective (lane 3). These results suggest the possibility of FBP binding selectively to the poly(U)-rich region of the HCV 3′NTR.
FIG. 2.
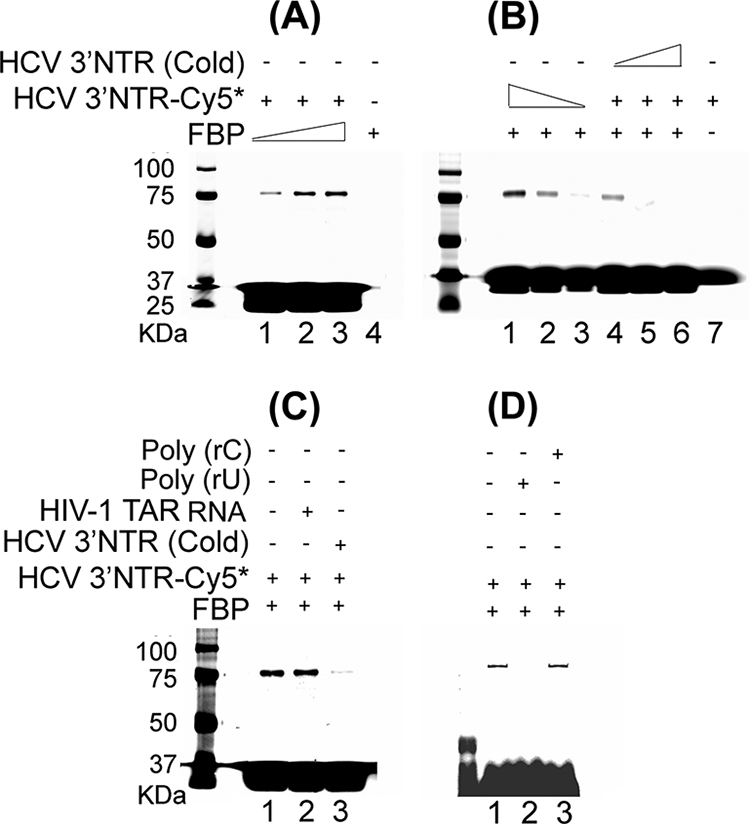
Specificity of interaction between FBP and HCV 3′NTR. (A) Cross-linking of Cy5-labeled HCV 3′NTR (HCV 3′NTR-Cy5) with FBP as a function of increasing FBP concentration. Cy5-labeled 3′NTR (0.5 pmol) was incubated with 0.25, 0.5, and 1.0 pmol of purified FBP (lanes 1 through 3, respectively). Lane 4 contained 0.5 pmol of purified FBP only. The samples were UV irradiated, then treated with RNase A, and resolved by SDS-PAGE. The cross-linked RNA-protein complexes were detected with a Typhoon scanner. (B) FBP cross-linking with 3′NTR as a function of the concentration of Cy5-labeled HCV 3′NTR and competition by increasing concentrations of cold HCV 3′NTR. In lanes 1 through 3, a fixed concentration of FBP (0.5 pmol) was cross-linked with 0.5, 0.25, and 0.125 pmol of Cy5-labeled HCV 3′NTR, respectively. In lanes 4 though 6, a fixed concentration of FBP (0.5 pmol) was cross-linked with a fixed concentration of Cy5-labeled HCV 3′NTR (0.5 pmol) in the presence of 0.5, 2.5, and 5 pmol of cold HCV 3′NTR, respectively. Lane 7 contained Cy5-labeled HCV 3′NTR (0.05 pmol) only. (C) FBP cross-linking with HCV 3′NTR in the presence of nonspecific heteropolymeric HIV-1 TAR RNA as the competitor. Lane 1 contains no competitor. Lane 2 contains 5 pmol of cold HIV-1 TAR RNA. Lane 3 contains 5 pmol of cold HCV 3′NTR. (D) FBP cross-linking with Cy5-labeled HCV 3′NTR in the presence of poly(rU) or poly(rC) as the competitor. Lane 1 contains no competitor, while lanes 2 and 3 contain poly(rU) and poly(rC) as the competitor, respectively. The positions of molecular mass markers (in kilodaltons) are shown to the left of the gels.
Mapping of FBP binding sites within HCV 3′NTR.
After establishing that FBP specifically binds to HCV 3′NTR, we mapped the binding site to identify the region of the HCV 3′NTR with which FBP specifically interacts. We carried out photoaffinity cross-linking between FBP and Cy5-labeled, in vitro-transcribed HCV 3′NTR RNA (F13) in the absence or presence of in vitro-transcribed cold RNA corresponding to the variable region (F11), the poly(U/UC) region (F22), or the conserved 3′ X-tail (F33) of HCV 3′NTR as the competitor (Fig. 3A). We incubated 0.5 pmol purified FBP with 0.5 pmol Cy5-labeled full-length HCV 3′NTR (F13) in the presence of 10-fold excess of these competitors. The RNA fragment corresponding to the poly(U/UC) region (F22) of the 3′NTR was the only one that outcompeted FBP binding to the full-length Cy5-labeled HCV 3′NTR (F13) (Fig. 3B, lane 4). Thus, the poly(U/UC) region of HCV 3′NTR is the specific site for FBP interaction.
FIG. 3.

FBP binds to the poly(U/UC) region of HCV 3′NTR. (A) Schematic representation of PCR amplification and in vitro transcription of full-length HCV 3′NTR (F13) and its fragments corresponding to the variable region (F11), poly(U/UC) region (F22), and conserved 3′ X-tail (F33). (B) Cross-linking of FBP with Cy5-labeled full-length HCV 3′NTR (3′NTR-Cy5) (F13) in the absence (−) and presence (+) of in vitro-transcribed cold RNA fragments corresponding to F11, F22, and F33. Lane 1, 0.5 pmol of Cy5-labeled full-length HCV 3′NTR RNA (F13) alone without FBP; lane 2, 0.5 pmol of purified FBP cross-linked with 0.5 pmol of Cy5-labeled HCV 3′NTR in the absence of competitor. Lanes 3 through 5 show cross-linking of FBP with 0.5 pmol of Cy5-labeled HCV 3′NTR (F13) in the presence of a 10-fold excess of competitor RNA corresponding to F11, F22, and F33, respectively. The positions of molecular mass markers (in kilodaltons) are shown to the left of the gel. (C) Direct cross-linking of FBP with Cy5-labeled F11, F22, F33, or F13. Purified FBP (0.5 pmol) was cross-linked with 0.5 pmol of Cy5-labeled RNA fragments corresponding to F11, F22, F33, and full-length HCV 3′NTR (F13). The individual Cy5-labeled input RNA probe is shown in the bottom blot.
We then performed direct cross-linking of FBP with Cy5-labeled, in vitro-transcribed RNA fragments corresponding to the variable region (F11), poly(U/UC) region (F22), conserved 3′ X-tail (F33), and full-length 3′NTR (F13). FBP specifically cross-linked to the poly(U/UC) region (F22) of HCV 3′NTR, but not to the other RNA fragments (Fig. 3C). These results clearly established that FBP specifically interacts with the poly(U/UC) track of HCV 3′NTR, which is known to be crucial for HCV replication.
Knocking down of FBP expression severely impairs HCV replication.
It is known that the poly(U/UC) tract and the highly conserved 3′ X-tail in the HCV 3′NTR are required for viral infectivity (72) and viral replication in host cells (25, 42, 73). FBP, which belongs to the family of AREBPs, activates transcription of the c-myc gene by binding to the FUSE. It also binds the 3′NTR of the GAP-43 mRNA and regulates stability of the mRNA in mammalian cells (33). Since our RNA affinity capture experiments (31) and the protein-RNA cross-linking experiment showed that FBP binds to HCV 3′NTR, we examined whether FBP has an important function in HCV replication in host cells. In designing our experimental strategy, we considered a primary question. The question was would HCV replication be affected if FBP expression in the host cells were decreased or increased? To address this question, we first induced the down-regulation of FBP in MH14 cells, an HCV replicon-containing cell line, using FBP siRNA. Cured MH14 cells devoid of HCV replicons were used as negative controls. After transfection with FBP siRNA, we collected the total protein from one set of cells and total RNA from the other set of cells and then examined FBP and viral RNA levels in the cells by Western blotting and RT-PCR, respectively. Western blotting (Fig. 4A) showed that at 48 h after transfection with 20 nM FBP siRNA, the amount of FBP was significantly decreased (lane 4) than the amount in untransfected MH14 cells (lane 1), MH14 cells incubated with transfection reagent alone (lane 2), or MH14 cells transfected with scrambled control siRNA (lane 3). FBP expression in MH14 cells transfected with FBP siRNA was reduced by 74% compared to that in the control (Fig. 4C), while the levels of actin protein, a housekeeping gene, remained unchanged. This demonstrated that FBP siRNA selectively reduced FBP expression in MH14 cells without significantly influencing cell growth.
FIG. 4.
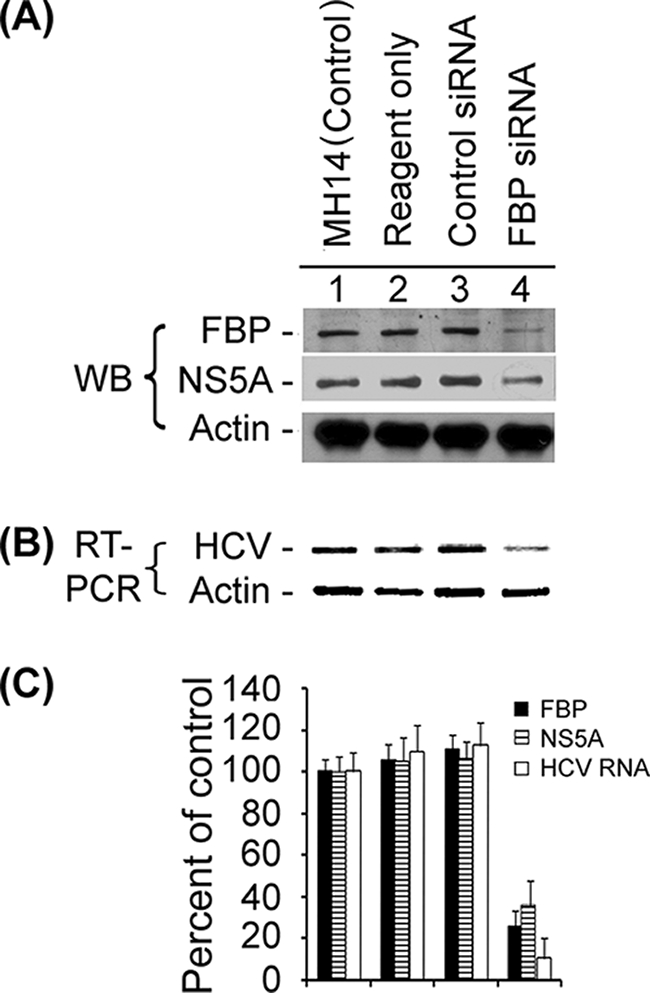
Effect of siRNA-mediated reduced expression of FBP on HCV replication. MH14 cells harboring replicating subgenomic HCV replicons were transfected with FBP siRNA. At 48 h posttransfection, the cells were harvested, lysed, and Western blotted (WB) for FBP, NS5A, and actin (A) and subjected to RT-PCR for HCV RNA and actin mRNA (B). (C) Quantification of WB and RT-PCR results.
In assessing HCV replicon RNA from total RNA, we used a pair of primers targeted to the 5′NTR of HCV replicon RNA in MH14 cells to detect the amount of HCV replicon RNA in the cells by RT-PCR. At 48 h posttransfection with 20 nM FBP siRNA, the amount of HCV replicon RNA in MH14 cells was significantly decreased (Fig. 4B, lane 4) compared to the amount in untransfected MH14 cells (lane 1), MH14 cells incubated with the transfection reagent alone (lane 2), or MH14 cells transfected with scrambled control siRNA (lane 3). Quantification showed that the HCV replicon RNA in MH14 cells transfected with FBP siRNA was reduced by 89% compared to that in control cells (Fig. 4C). This corresponded well with the reduced level of FBP in the cells. On the basis of the results obtained from both Western blotting of FBP and RT-PCR of HCV RNA, we concluded that HCV replication was reduced when FBP expression was diminished or inhibited in MH14 cells.
Knocking down of FBP expression also impairs HCV translation.
Since HCV replication was inhibited upon reduced expression of FBP in the cells, we decided to determine what effect this reduction would have on the translation of HCV. We detected the viral protein NS5A in the cells by Western blotting. The results of Western blotting of NS5A (Fig. 4A) showed that 48 h after transfection with FBP siRNA, the amount of NS5A was significantly decreased (lane 4) than the amount in untransfected MH14 cells (lane 1) or in MH14 cells incubated with transfection reagent alone (lane 2) or transfected with scrambled control siRNA (lane 3). We determined that NS5A expression in MH14 cells transfected with FBP siRNA was reduced by 64% compared to that in control cells (Fig. 4C). Thus, while HCV replication was decreased upon down-regulation of FBP expression in the cells, the reduced translation of HCV protein correlated well with the reduced level of HCV positive-stranded RNA template.
Another HCV replicon construct, pLMH14, with the luciferase reporter gene was also used to determine the effect of FBP siRNA on HCV translation (51). We set up a highly efficient and sensitive HCV replicon system with this HCV replicon construct in cured MH14 cells. We used XbaI to linearize the pLMH14 plasmid DNA and used it as a template to make in vitro-transcribed full-length transcripts of Luc-HCV RNA. We had two parallel sets of cured MH14 cells transfected with Luc-HCV RNA. We collected total protein from one set of cells and detected FBP by Western blotting using the anti-FBP antibody (Fig. 5A, lanes 1 to 6). At 48 h posttransfection with FBP siRNA, the amount of FBP was decreased in cured MH14 cells (lane 6) compared to the amount in untransfected cells (lane 1), cells incubated with the transfection reagent alone (lane 2), cells transfected with scrambled control siRNA (lane 3), cells transfected with Luc-HCV RNA (lane 4), or cells cotransfected with Luc-HCV RNA and scrambled control siRNA (lane 5). As in our earlier siRNA experiment with MH14 cells, the levels of FBP in FBP siRNA-transfected cured MH14 cells was reduced by 75% compared to the control (Fig. 5B, lanes 1 to 6), indicating that FBP siRNA reduced FBP levels in the cured MH14 cells. We collected the total protein from the other set of cells with a special lysis buffer provided by the luciferase reporter assay system kit (Promega) and then measured luciferase activity using the manufacturer's protocol (Fig. 5C, lanes 1 to 6). Cured MH14 cells transfected with Luc-HCV RNA exhibited a very high level of luciferase activity at 24 h posttransfection (Fig. 5C, lane 4, positive control), indicating the presence of active translation of HCV replicons in the cells. Cured MH14 cells transfected with both Luc-HCV RNA and scrambled control siRNA (Fig. 5C, lane 5) also exhibited a high level of luciferase activity, indicating that scrambled control siRNA did not affect the translation of HCV replicon in the cells. In contrast, in cured MH14 cells transfected with both Luc-HCV RNA and FBP siRNA (lane 6), the detectable luciferase activity was reduced by 79% compared to that in the positive control shown in lane 4. As expected, cured MH14 cells (lane 1), cured MH14 cells incubated with either transfection reagent alone (lane 2) or with scrambled control siRNA (lane 3) did not show any luciferase activity due to the lack of Luc-HCV RNA in the cells. Collectively, these results indicated that the translation of Luc-HCV RNA was severely impaired when FBP expression was reduced by the delivery of FBP siRNA into the cells. Combined with the results from Western blotting, RT-PCR, and the luciferase reporter assay in MH14 cells and cured MH14 cells, these findings demonstrated that normal levels of cellular FBP are required for efficient HCV replication in host cells.
FIG. 5.
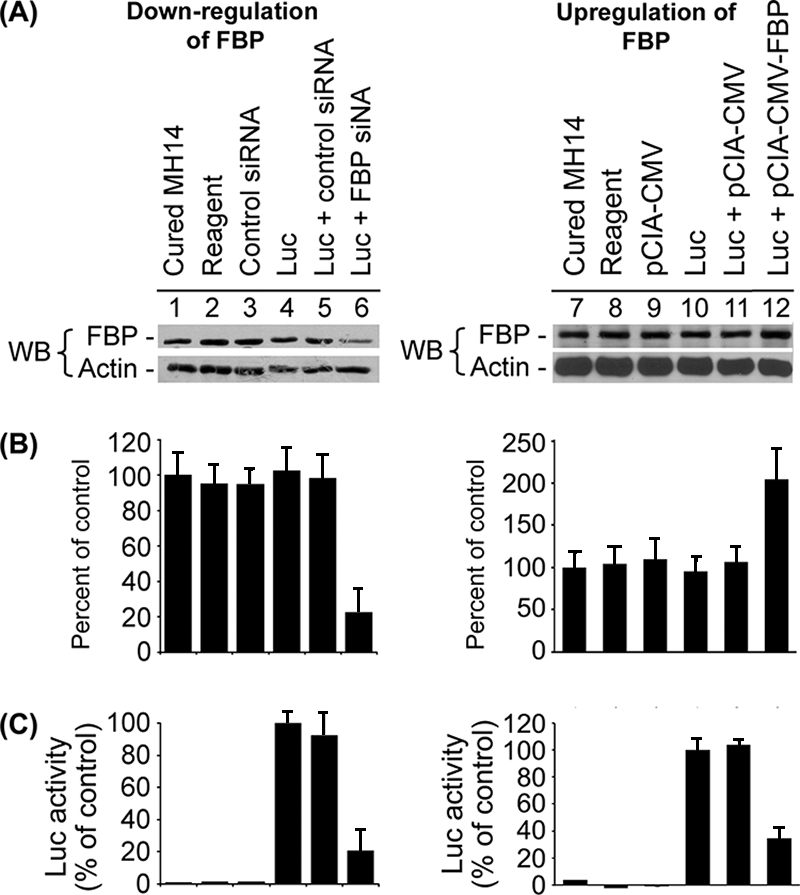
Effects of down-regulation and up-regulation of FBP on the translation of Luc-HCV RNA replicon in cured MH14 cells. Cured MH14 cells transfected with either FBP siRNA or pCIA-CMV-FBP were grown for 24 h and then transfected with Luc-HCV RNA containing the luciferase reporter gene (Luc). At 24 h posttransfection, cells were harvested, lysed, and Western blotted (WB) for FBP and actin (A) and the results were quantified (B). An aliquot of the lysate was assayed for luciferase (Luc) activity (C).
Up-regulation of FBP expression enhanced HCV replication.
Having found that reduction of FBP expression in cells severely impaired HCV replication, we examined the effect of increased expression of FBP on viral replication. We overexpressed FBP by transfecting the FBP expression construct pCIA-CMV-FBP into MH14 cells and then investigated its effect on HCV replication using Western blot analysis to detect subsequent FBP levels of expression in cells (Fig. 6A). Expression of FBP in MH14 cells transfected with FBP expression plasmid was about 2.7-fold higher (Fig. 6C, lane 4) than that in untransfected MH14 cells (lane 1) or MH14 cells either incubated with transfection reagent alone (lane 2) or transfected with pCIA-CMV vector (lane 3), thus confirming that transfection with pCIA-CMV-FBP increased FBP in MH14 cells.
FIG. 6.
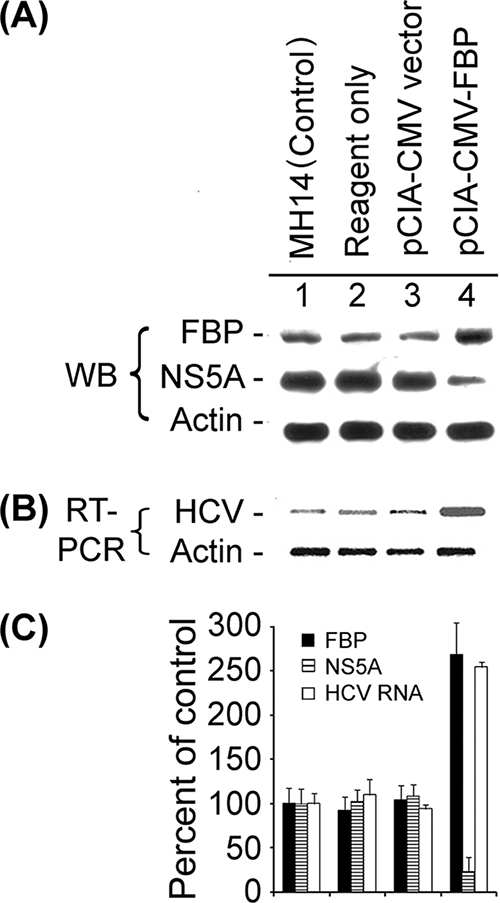
Effect of overexpression of FBP on HCV replication in MH14 cells. After 48 h of transfection with the FBP-expressing plasmid pCIA-CMV-FBP, MH14 cells were harvested, lysed, and Western blotted (WB) for FBP, NS5A, and actin (A) and then subjected to RT-PCR for HCV RNA and actin mRNA (B). (C) Quantification of WB and RT-PCR results.
When we used RT-PCR to detect the HCV replicon RNA in FBP-overexpressing MH14 cells, the HCV replicon RNA level also increased by 2.5-fold (Fig. 6B and C, lanes 4) over that in untransfected MH14 cells (lanes 1), MH14 cells incubated with transfection reagent alone (lanes 2), or MH14 cells transfected with pCIA-CMV vector (lanes 3). These results strongly suggest that increased FBP expression significantly stimulates replication of the HCV replicon RNA in MH14 cells.
Up-regulation of FBP expression inhibited HCV translation.
To study the effect of up-regulation of FBP expression on HCV translation, we examined the viral protein NS5A in FBP-overexpressing MH14 cells by Western blotting. We found that with the increased expression level of FBP in the cells, the expression level of NS5A was significantly decreased (Fig. 6, lane 4) compared to that in untransfected MH14 cells (lane 1), MH14 cells incubated with transfection reagent alone (lane 2), or MH14 cells transfected with pCIA-CMV vector without the FBP coding region (lane 3). We also quantified the expression level of NS5A in the FBP-overexpressing MH14 cells (Fig. 6C). As shown in Fig. 6, the expression of NS5A was reduced by 76% from that in control cells, suggesting that HCV translation was strongly inhibited upon increased expression of FBP in the cells.
We also used a HCV subgenomic replicon-containing luciferase reporter (Luc-HCV RNA) (51) to determine the effect of FBP overexpression on HCV translation. We had two parallel sets of cured MH14 cells transfected with Luc-HCV RNA. One set was used for Western blot analysis, while the other set was used for detection of luciferase activity. We collected total protein from one set of cells and detected FBP by Western blotting using the anti-FBP antibody (Fig. 5A, lanes 7 to 12). We observed that at 48 h posttransfection with pCIA-CMV-FBP, the expression level of FBP was significantly increased in the transfected cells (lane 12) than in the untransfected cells (lane 7), cells incubated with transfection reagent alone (lane 8), cells transfected with pCIA-CMV vector only (lane 9), cells transfected with Luc-HCV RNA (lane 10), or cells cotransfected with Luc-HCV RNA and pCIA-CMV vector (lane 11). We noted that the levels of FBP expression in the cured MH14 cells transfected with pCIA-CMV-FBP was approximately twofold higher than that in the control (Fig. 5B, lanes 7 to 12), indicating that transfection with the FBP-overexpressing plasmid increased the FBP expression level in the cells. We collected cells from the other set of experiments and lysed them with special lysis buffer provided by the luciferase reporter assay system kit (Promega). We then measured luciferase activity in the cell lysate using the manufacturer's protocol. The results are depicted in Fig. 5C (lanes 7 to 12). As shown in Fig. 5C, cured MH14 cells transfected with Luc-HCV RNA exhibited a very high level of luciferase activity at 24 h posttransfection (lane 10, positive control), indicating a high level of HCV translation in the cells. Similarly, cured MH14 cells cotransfected with Luc-HCV RNA and pCIA-CMV vector without FBP (lane 11) also exhibited a high level of luciferase activity. In contrast, luciferase activity in the cured MH14 cells cotransfected with Luc-HCV RNA and FBP-overexpressing plasmid was drastically reduced by 66% (lane 12) compared to that in the positive control (lane 10). The observed reduction in the luciferase activity clearly indicated that overexpression of FBP in the cells strongly inhibits HCV translation. As expected, the luciferase activity was not detected in untransfected cured MH14 cells (lane 7) or in cells incubated with transfection reagent (lane 8) or transfected with the vector alone (lane 9).
Exogenous FBP stimulates in vitro HCV replication in replicative lysate of MH14 cells.
Having found that FBP significantly stimulates HCV replication in MH14 cells, we postulated that FBP would have a stimulatory effect on HCV replication in a cell-free replication system. To test this hypothesis, we prepared a cell-free replicative lysate from MH14 cells and applied it to a cell-free HCV replication assay with or without exogenous FBP. After 3 h of incubation, we isolated the total RNA and examined the level of HCV RNA by RT-PCR, which reflected the activity of HCV replicative complexes in the replicative lysate. After incubation, the level of HCV RNA in the absence of exogenous FBP was only 44% higher due to newly synthesized HCV RNA (Fig. 7, lane 2) compared to the level at zero time (lane 1), which is the basal replication activity in the replicative lysate. In the presence of exogenous FBP, the level of newly synthesized HCV RNA was 100% higher at 0.25 pmol and 200% higher at 0.5 pmol of exogenous FBP concentrations (lanes 3 and 4) than the level at basal replication activity (lane 1). This result confirmed that FBP is an important modulator of HCV replication.
FIG. 7.
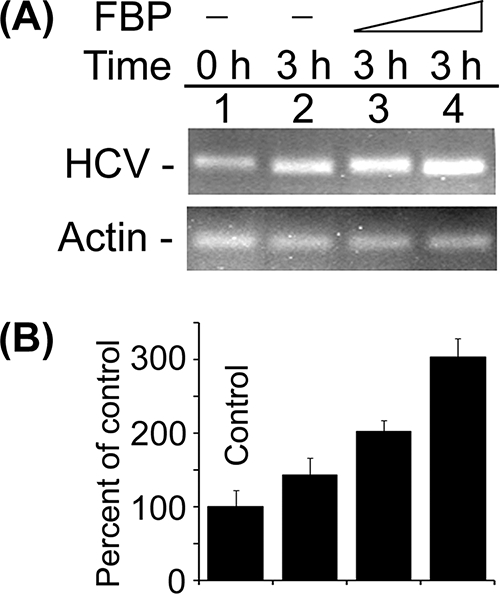
Stimulation of in vitro HCV replication in cell-free replicative lysate of MH14 cells by the addition of purified FBP. An aliquot of cell-free replicative lysate of MH14 cells was incubated in the absence (−) and presence of 0.25 and 0.5 pmol of FBP under standard reaction conditions. At the end of incubation, RT-PCR for HCV RNA (A) was done. (B) Quantification of HCV RNA results.
Coimmunoprecipitation of FBP and HCV NS5A from MH14 cell lysate.
Since FBP binds HCV 3′NTR and modulates HCV replication, we examined whether this protein also interacts with HCV proteins. We first incubated MH14 cell lysate with RNase A and then with FBP antibody and protein A/G Plus agarose beads. The RNase A treatment of the lysate was carried out to avoid capturing nonspecific proteins along with FBP-IP via RNA bridging. We found that incubation of MH14 cell lysate with RNase A under the experimental conditions efficiently degraded cellular RNAs as judged by the complete disappearance of both 28S and 18S RNAs in the treated samples (Fig. 8C) and HCV RNA as judged by RT-PCR of HCV RNA (Fig. 8D). We analyzed the precipitated proteins by SDS-PAGE followed by Western blotting with anti-NS5A antibody and Coomassie blue staining. With Coomassie blue staining, we noted FBP and NS5A bands at 74- and 58-kDa positions (Fig. 8B, lane 2), which were missing in beads only (control) (Fig. 8B, lane 3). After Western blotting with anti-NS5A antibody, we detected NS5A protein in the FBP-IP fraction (Fig. 8A, lane 1) that migrated to the same position as NS5A from the control MH14 cell lysate did (Fig. 8A, lane 3). Another band seen below NS5A could be immunoglobulin G heavy chain (Fig. 8A, lane 1), which could also be seen with Coomassie blue staining (Fig. 8B, lane 2). The treatment of the lysate with RNase A ascertained that coimmunoprecipitation of NS5A with FBP is not via RNA bridging. It also indicated that FBP may modulate HCV replication by interacting with viral protein NS5A in the cells.
FIG. 8.

Coimmunoprecipitation of FBP and NS5A. (A) MH14 cell lysate was treated with RNase A and then incubated with anti-FBP antibody and protein A/G Plus agarose beads overnight at 4°C to immobilize the FBP-FBP antibody complex on the beads. The beads were washed several times, and the bound protein complex was resolved by SDS-PAGE, transferred onto the membrane, and Western blotted (WB) for NS5A using anti-NS5A antibody (lane 1). The lane with beads only (without FBP antibody) was used as a negative control (lane 2). The crude cell lysate from MH14 cell was used as a positive control (lane 3). FBP IP, FBP immunoprecipitate. (B) The protein bands in the immunoprecipitates were resolved on SDS-polyacrylamide gels and visualized by Coomassie blue staining. The positions of molecular mass markers (M) (in kilodaltons) are shown to the left of the gels in panels A and B. IgG, immunoglobulin G. RNase A-mediated degradation of cellular 28S and 18S RNAs and HCV RNA in MH14 cell lysate under the experimental conditions is shown in panels C and D, respectively.
Cellular distribution and localization of FBP and NS5A.
FBP is a transcription activator of the c-myc proto-oncogene and is localized in the nucleus, while HCV replication occurs in the cytoplasm. We also found that FBP-IP coimmunoprecipitated HCV viral protein NS5A from the cell lysate of MH14 cells. It is not clear how FBP in the nucleus could associate with HCV NS5A and stimulate HCV replication in the cytosol. To probe the possibility that some FBP is localized in the cytosol and affects HCV replication, we used Western blotting of cytoplasmic and nuclear extracts, as well as an immunofluorescence experiment to detect the distribution of both FBP and NS5A in MH14 cells. In this experiment, we used cured MH14 cells as controls. Western blotting of cytoplasmic and nuclear extracts of MH14 cells and cured MH14 cells showed that although FBP is mainly localized in the nucleus, a small but significant portion is in the cytosol (Fig. 9).
FIG. 9.
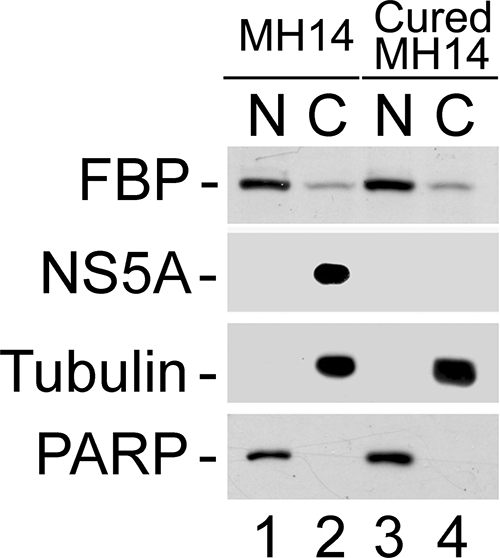
Cellular distribution of FBP and NS5A in MH14 cells. Nuclear (N) and cytoplasmic (C) extracts from MH14 and cured MH14 cells were prepared and Western blotted for FBP and NS5A. PARP and α-tubulin were also Western blotted as the specific nuclear and cytosolic markers, respectively.
NS5A is a component of the intracellular membrane-associated viral replication complex (46). It is also a major viral protein that affects the rate of HCV replication (9). Having found that FBP is also able to modulate HCV replication in MH14 cells, we addressed the question of whether these two factors colocalize in the cells and interact with each other. First, to detect the distribution of NS5A in cured MH14 and MH14 cells, we carried out Western blotting of cell fractions, finding that NS5A was detected only in MH14 cells harboring the HCV replicon. As expected, NS5A was found to be localized only in the cytosol (Fig. 9). In order to further ascertain that the observed results were not due to contamination of cytoplasmic extract with the nuclear extract and vice versa, we also Western blotted for PARP and α-tubulin as the specific nuclear and cytoplasmic markers, respectively. These observations strongly suggested that FBP in the cytoplasm interacts with HCV 3′NTR and viral protein NS5A at the site of viral replication in the cytosol. Similar patterns of distribution of FBP and NS5A were observed in immunofluorescence images (Fig. 10A and B). When immunofluorescence showed FBP and NS5A in the same cell, we found that portions of the FBP and NS5A were colocalized in the cytosol (Fig. 10C).
FIG. 10.

Cellular localization of FBP and NS5A in MH14 cells. (A) Localization of FBP in MH14 cells and cured MH14 cells. Cells were grown on a coverslip for 24 h, fixed, treated with anti-FBP antibody, and then treated with Cy3-labeled secondary antibody. DAPI was used to stain the nuclei. (B) Localization of HCV NS5A in MH14 cells and cured MH14 cells (negative control). Cells were fixed and treated with anti-NS5A antibody and then treated with Alexa Fluor 488 (Alex488)-labeled secondary antibody. DAPI was used to stain the nuclei. (C) Colocalization of FBP and NS5A in cytosol in MH14 cells. MH14 cells were fixed, treated with anti-FBP antibody and Cy3-labeled secondary antibody, and then treated with anti-NS5A antibody and Alexa Fluor 488-labeled secondary antibody. DAPI was used to stain the nuclei. They were then individually observed for FBP and NS5A localization. The same slide and the same field in different channels were used to observe the DAPI-stained nucleus, Cy3-labeled FBP, and Alexa 488-labeled NS5A. Pictures were taken and merged using Spot advanced software (Diagnostic Instruments, Sterling Heights, MI).
Relationship among FBP, c-myc, and HCV infection.
It has been reported that FBP binds the pyrimidine-rich FUSE of the human c-myc proto-oncogene and activates c-myc transcription (7, 20, 32, 47). The c-myc transcription factor targets approximately 10% of transcribed genes and coordinates many essential cellular processes, including proliferation, growth, and differentiation (44, 45). It has been proposed that the sequence-specific FBP and its antagonist, the FBP-interacting repressor (FIR), impose tight regulation on c-myc transcription (12, 44). In this study, we also detected the regulation of c-myc by FBP in MH14 cells harboring the HCV replicon. In the experiments involving up- and down-regulation of FBP to determine the effects on HCV replication, we also detected cellular levels of c-myc, finding that those levels changed in coordination with the cellular level of FBP. In MH14 cells transfected with FBP siRNA and in cells showing decreased levels of expression of FBP (31% of the control), the c-myc level decreased to 46% of that in control cells (Fig. 11A and B, lanes 1 to 4). With the transfection of FBP-expressing plasmid and the increase in FBP expression level (1.7-fold of control), the c-myc level in MH14 cells increased about twofold over the level in control cells (Fig. 11A and B, lanes 5 to 8). Moreover, the cellular FBP levels in MH14 cells harboring HCV replicons were significantly higher than those in parental Huh7 cells or cured MH14 cells (Fig. 12). Similarly, c-myc levels in both MH14 cells and cured MH14 cells were higher than those in Huh7 cells (Fig. 12). These findings, in spite of the fact that the HCV replicon has been totally removed from cured MH14 cells (51), indicate the possibility that elevated levels of these two proteins may be essential for efficient replication of HCV replicons in this cell line.
FIG. 11.
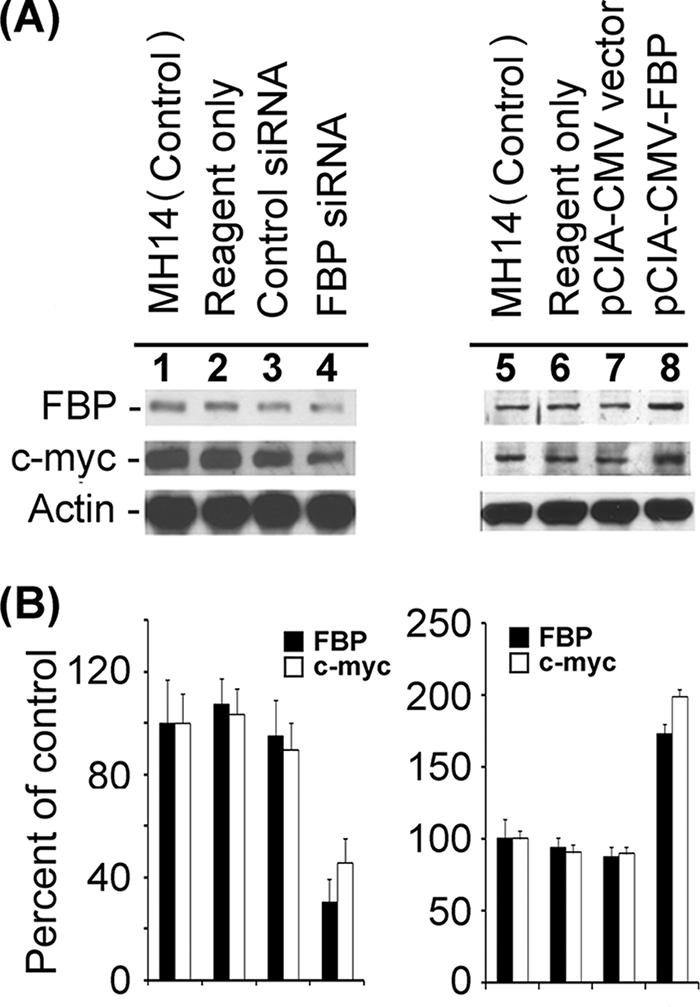
Effect of down- or up-regulation of FBP on c-myc expression in MH14 cells. (A) FBP expression in MH14 cells was either reduced by transfection of FBP siRNA (lanes 1 to 4) or enhanced by transfection of pCIA-CMV-FBP (lanes 5 to 8). At 48 h posttransfection, the cells were lysed and Western blotted for FBP and c-myc. (B) Quantification of the Western blot of FBP and c-myc proteins.
FIG. 12.
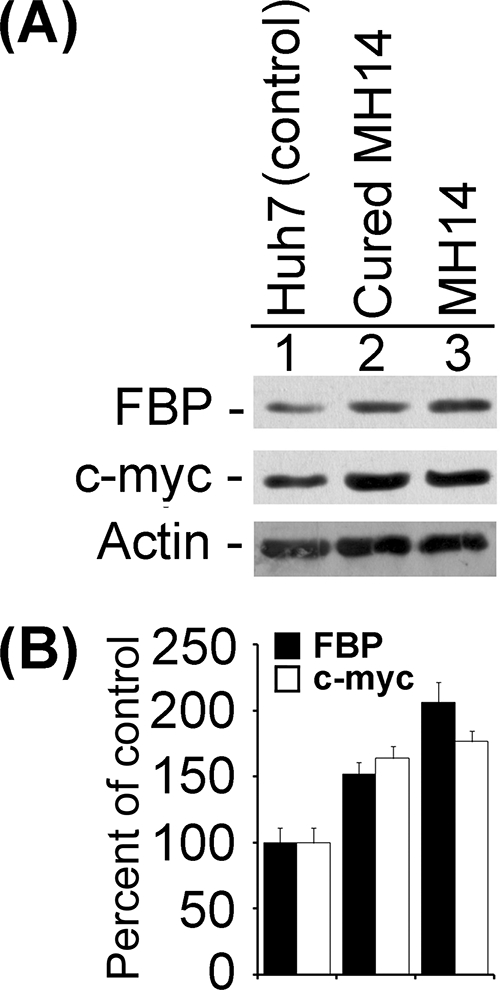
Expression levels of FBP and c-myc proteins in MH14 cells and cured MH14 cells and their parental Huh7 cells. The equivalent protein from cell lysates of MH14 cells, cured MH14 cells, and Huh7 cells were Western blotted for FBP and c-myc proteins (A), and the results were quantified (B).
DISCUSSION
FBP is a 644-amino-acid protein consisting of three domains. Its activation and repression domains are located at the C- and N-terminal regions, respectively; the central domain, containing four KH motifs, binds to the noncoding strand of the FUSE and destabilizes the nucleic acid duplex (8, 20). FBP was first discovered as a single-stranded DNA binding protein (20). Subsequent studies of a known human DNA helicase V revealed that FBP and human DNA helicase V are, in fact, the same protein (69). FBP moves in a 3′-to-5′ direction along the bound strand and is able to unwind both DNA/DNA and RNA/RNA duplexes (69).
It has been reported that FBP binds the pyrimidine-rich FUSE of the human c-myc proto-oncogene and activates c-myc transcription (7, 20, 32, 47). Interestingly, HCV 3′NTR also contains a poly(U/UC) region, located in the middle region of the HCV 3′NTR and upstream of the HCV conserved 3′ X-tail (41, 65). Both the poly(U/UC) region and the conserved 3′ X-tail are essential for HCV infectivity (72) and viral replication (25, 42, 73). It is our laboratory that first reported that FBP binds to the highly structured HCV 3′NTR by RNA affinity capture experiments (31). Since FBP is a transcription factor, it is expected to be located at the site of its function, in the nucleus. How does a nucleus-localized host protein interact with HCV RNA in the cytosol and modulate viral replication? Using Western blotting of cytoplasmic and nuclear extracts, as well as immunofluorescence experiments, we have found that a significant portion of FBP is also present in the cytosol (Fig. 9 and 10A and C), where it can interact with HCV RNA. Many other proteins that are primarily localized in the nucleus also interact with HCV RNA in the cytosol. Various nuclear localization cellular factors, such as PTB (13, 29, 31, 36, 37, 66), hnRNP C (29, 52, 63), hnRNP L (30, 31), and nuclear factors (NF90, NF110, NF45, and RHA) (31, 34, 35), interact with HCV RNA and influence HCV replication and/or translation. Nucleocytoplasmic shuttling proteins, such as HuR (6, 23, 24, 31, 55, 63), shuttle between the nucleus and cytosol and, depending on cell conditions, can interact with HCV RNA in the cytosol. The interaction between the nucleus-localized host cellular factors and HCV RNA suggests two important aspects of host-HCV interaction on HCV infection. First, HCV infection may affect the cellular distribution and/or expression pattern of many host proteins, such as HuR (63) and the nuclear factors (NF90, NF110, NF45 and RHA) (34). Second, some of these affected host proteins, such as PTB (2, 4, 37) and the nuclear factors (34), may have important, active functions in the viral life cycle.
In this study, we demonstrated that the binding of FBP to HCV 3′NTR is not fortuitous or nonspecific. Its binding to Cy5-labeled HCV 3′NTR is not competitively eliminated by nonspecific RNA, such as HIV-1 TAR RNA (Fig. 2C, lane 2), but is competitively eliminated by cold unlabeled HCV 3′NTR in a concentration-dependent manner (Fig. 2B, lanes 4 to 6). We also demonstrated that the poly(U) tract within the poly(U/UC) region of HCV 3′NTR is the binding site of FBP, since binding of FBP to HCV 3′NTR is completely outcompeted by poly(rU) but not by poly(rC) (Fig. 2D) or by RNA fragments corresponding to the variable region or the conserved 3′ X-tail of HCV 3′NTR (Fig. 3B, lanes 3 and 5). However, an RNA fragment corresponding to the poly(U/UC) region of HCV 3′NTR effectively outcompeted the binding of full-length HCV 3′NTR to FBP (Fig. 3B, lane 4). Direct binding studies with Cy5-labeled RNA fragments corresponding to the variable region, conserved 3′ X-tail, and poly(U/UC) region of HCV 3′NTR clearly established that FBP specifically interacts with the poly(U/UC) region, since other RNA fragments were not recognized for binding (Fig. 3C).
FBP binding to the polypyrimidine-rich region of the HCV 3′NTR is analogous to its binding to the pyrimidine-rich FUSE of c-myc. Since FBP binding to the pyrimidine-rich FUSE results in activation of transcription of the c-myc gene, we hypothesized that FBP binding to the polypyrimidine region of HCV 3′NTR may have a similar function in activating the transcription (viral replication) of the HCV genome. The poly(U/UC) region in the HCV 3′NTR is a regulatory region required for HCV replication (25, 42, 73) and infectivity (72). Our finding that reducing the FBP level in MH14 cells by transfection with siRNA dramatically reduced HCV replication (Fig. 4) supports our contention that FBP is involved in the regulation of HCV replication. In contrast, enhancing the level of FBP expression in MH14 cells strongly stimulated HCV replication (Fig. 6). It was still possible that the effects of siRNA and overexpression of FBP on HCV replication might occur indirectly via unrelated changes in cell physiology. To confirm our result, we used an in vitro cell-free HCV replication system to evaluate the effect of FBP on HCV replication. We found that HCV replication activity in the replicative cell lysate of MH14 cells was significantly stimulated by the addition of exogenous FBP (Fig. 7). This result clearly demonstrated that FBP promotes HCV replication.
It has been suggested that the replication and translation processes of positive-stranded RNA viruses are mutually exclusive (27). We therefore examined whether alteration of FBP has an effect on the translation of HCV. While we determined the effects of decreased and increased cellular levels of FBP on HCV replication, we also carried out Western blotting for HCV NS5A to compare the levels of translation of viral proteins. Since HCV genomic RNA is first translated into a polyprotein that is then cleaved into individual viral proteins by a combination of viral and cellular proteases (14), we presumed that detection of one of the viral proteins, NS5A, might reflect the overall viral translational activity in the cells. We carried out another experiment involving transfection of Luc-HCV RNA in cured MH14 cells to determine the modulation of HCV translation under the condition of decreased or increased FBP expression. Since expression of luciferase activity is under the control of the HCV IRES, its expression level may directly reflect the level of HCV translation in the cell.
When the levels of FBP expression were either decreased or increased in transfected MH14 cells, we found that the level of NS5A was uniformly reduced compared to that in control cells (Fig. 4 and 6). We also obtained similar results when Luc-HCV RNA was transfected in cured MH14 cells (Fig. 5). Because siRNA-mediated down-regulation of FBP expression reduced HCV replication, the observed reduction of NS5A in MH14 cells and the reduction of luciferase activity in cured MH14 cells may reflect low translational activity due to the reduced number of HCV genomic RNA templates in the cells (Fig. 4 and 5). However, the NS5A level was decreased even when HCV genomic RNA templates were increased due to elevated HCV replication caused by increased FBP expression in the cells (Fig. 6). Luciferase activity was also decreased when FBP was overexpressed in Luc-HCV RNA-transfected cured MH14 cells (Fig. 5).
One explanation of this observation is that, as we have shown, HCV is a positive-stranded RNA virus (14); also, its genomic RNA is used as the template for both translation and replication. The translation and replication complexes move in opposite directions on the genomic RNA template. Thus, these two processes must be mutually exclusive, which means they cannot operate at the same time on the same template. In poliovirus, however, a switch from translation to replication and vice versa has been demonstrated (27). It is therefore possible that FBP may be a key factor in regulating this switch from translation to HCV replication. This may well explain the low level of NS5A observed in MH14 cells even though the level of HCV genomic message was high due to increased FBP expression in the cells. An alternative explanation is that FBP may subtly reduce or turn down the IRES-mediated translation machinery by interacting with other parts of the HCV genome or with viral proteins, such as NS5A. The results of our immunoprecipitation experiments indicated that NS5A was coimmunoprecipitated with FBP, suggesting that FBP interacts with NS5A in the cells, although it is still not clear if FBP interacts with NS5A directly or through another viral or cellular protein(s). There is indeed the possibility of involvement of other cellular or viral factors that may interact with both FBP and NS5A and thus may provide a bridging effect. HCV NS5A has been shown to be an important regulatory link between HCV translation and replication (5, 22, 48, 53). Indeed, many studies have demonstrated that the hyperphosphorylated form of NS5A (p58) favors translation and inhibits HCV replication (5, 22, 53). Neddermann et al. (53) demonstrated that inhibition of HCV replication in cells in the presence of an elevated level of hyperphosphorylated NS5A (p58) can be reversed by kinase inhibitors. McCormick et al. (48) reported that the hyperphosphorylation of NS5A as a consequence of reduced HCV polymerase activity enhanced translation of HCV protein. Appel et al. (5) reported that alanine substitutions for serine residues in NS5A reduced hyperphosphorylation of NS5A and enhanced HCV replication. We found that NS5A (39) coimmunoprecipitated with FBP (Fig. 8). It is possible, therefore, that FBP either inhibits hyperphosphorylation or sequesters the hyperphosphorylated form of NS5A, which, in turn, facilitates the switch from HCV translation to replication. There could be other possibilities that may explain observed reduction of HCV translation by increased FBP expression in the cells. These possibilities may include the following. (i) FBP interaction with NS5A may prevent NS5A being used as a substrate for hyperphosphorylation. (ii) FBP may be inhibitory to specific cellular protein kinases responsible for hyperphosphorylation of NS5A. (iii) The FBP-NS5A complex may interact with HCV IRES in the 5′NTR, either directly or through other viral or cellular factors and inhibit IRES-dependent HCV translation.
FBP is an important transcription activator of the c-myc proto-oncogene (20, 32, 47). The c-myc transcription factor targets approximately 10% of transcribed genes and coordinates many essential cellular processes, including proliferation, growth, and differentiation (44, 45). The dysfunction of c-myc gene regulation has been associated with the genesis of many tumors. Clinical studies have found that chronic HCV infection can progressively develop into liver cirrhosis (LC) and hepatocellular carcinoma (HCC) (19, 57). Approximately half of HCC patients have a history of HCV infection (19, 40). In patients with chronic hepatitis C (CHC), LC, and HCC, constant higher levels of expression of c-myc than occurs in healthy liver tissues have been reported (21, 26). It has been reported that during the progression of CHC to LC and then to HCC, the expression level of c-myc occurs in the following order: healthy liver tissue < CHC < LC < HCC (21, 26). The higher expression level of c-myc in HCC has been correlated with a worse prognosis (1, 15, 16, 70).
In the present study, we found that the cellular level of c-myc protein was correlated with the cellular level of FBP. Decreased or increased expression of FBP led to decreased or increased expression of c-myc in transfected MH14 cells, respectively (Fig. 11). Interestingly, the expression levels of FBP and c-myc proteins in MH14 cells and cured MH14 cells were consistently higher than the levels in their parental cell line, Huh7 (Fig. 12). It is possible that HCV infection triggers the expression and alters the cellular distribution of FBP in host cells by an unknown mechanism, which, in turn, up-regulates the expression of c-myc in host cells. Alternatively, high expression of FBP in MH14 cells may favor efficient replication of the HCV subgenomic replicon, since cured MH14 cells also had elevated levels of FBP, even after the virus has been eliminated from the cells. This could explain the consistently high expression of c-myc in patients with LC and HCC caused by chronic HCV infection. The progression of CHC to LC and HCC usually takes several years (19). Although high expression of c-myc in HCV-induced CHC, LC, and HCC has been confirmed (21, 26), it is still not known whether FBP expression is also elevated in patients having these diseases. We plan to test the hypothesis that there is a relationship among HCV infection, high level of expression of FBP/c-myc, and the subsequent development of CHC, LC, and HCC.
Acknowledgments
We thank Makoto Hijikata (Kyoto University, Japan) for providing MH14 cells, cured MH14 cells, and plasmids pMH14 and pLMH14. We thank David L. Levens (National Cancer Institute, Bethesda, MD) for providing plasmid pET28a-FBP.
Footnotes
Published ahead of print on 9 April 2008.
REFERENCES
- 1.Abou-Elella, A., T. Gramlich, C. Fritsch, and T. Gansler. 1996. c-myc amplification in hepatocellular carcinoma predicts unfavorable prognosis. Mod. Pathol. 995-98. [PubMed] [Google Scholar]
- 2.Ali, N., and A. Siddiqui. 1995. Interaction of polypyrimidine tract-binding protein with the 5′ noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J. Virol. 696367-6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali, N., K. D. Tardif, and A. Siddiqui. 2002. Cell-free replication of the hepatitis C virus subgenomic replicon. J. Virol. 7612001-12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anwar, A., N. Ali, R. Tanveer, and A. Siddiqui. 2000. Demonstration of functional requirement of polypyrimidine tract-binding protein by SELEX RNA during hepatitis C virus internal ribosome entry site-mediated translation initiation. J. Biol. Chem. 27534231-34235. [DOI] [PubMed] [Google Scholar]
- 5.Appel, N., T. Pietschmann, and R. Bartenschlager. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 793187-3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atasoy, U., J. Watson, D. Patel, and J. D. Keene. 1998. ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is upregulated during serum stimulation and T cell activation. J. Cell Sci. 1113145-3156. [DOI] [PubMed] [Google Scholar]
- 7.Avigan, M. I., B. Strober, and D. Levens. 1990. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J. Biol. Chem. 26518538-18545. [PubMed] [Google Scholar]
- 8.Bazar, L., D. Meighen, V. Harris, R. Duncan, D. Levens, and M. Avigan. 1995. Targeted melting and binding of a DNA regulatory element by a transactivator of c-myc. J. Biol. Chem. 2708241-8248. [DOI] [PubMed] [Google Scholar]
- 9.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 2901972-1974. [DOI] [PubMed] [Google Scholar]
- 10.Braddock, D. T., J. M. Louis, J. L. Baber, D. Levens, and G. M. Clore. 2002. Structure and dynamics of KH domains from FBP bound to single-stranded DNA. Nature 4151051-1056. [DOI] [PubMed] [Google Scholar]
- 11.Brennan, C. M., and J. A. Steitz. 2001. HuR and mRNA stability. Cell. Mol. Life Sci. 58266-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang, P. C., C. W. Chi, G. Y. Chau, F. Y. Li, Y. H. Tsai, J. C. Wu, and Y. H. Wu Lee. 2006. DDX3, a DEAD box RNA helicase, is deregulated in hepatitis virus-associated hepatocellular carcinoma and is involved in cell growth control. Oncogene 251991-2003. [DOI] [PubMed] [Google Scholar]
- 13.Chung, R. T., and L. M. Kaplan. 1999. Heterogeneous nuclear ribonucleoprotein I (hnRNP-I/PTB) selectively binds the conserved 3′ terminus of hepatitis C viral RNA. Biochem. Biophys. Res. Commun. 254351-362. [DOI] [PubMed] [Google Scholar]
- 14.Clarke, B. 1997. Molecular virology of hepatitis C virus. J. Gen. Virol. 782397-2410. [DOI] [PubMed] [Google Scholar]
- 15.Cui, J., B. W. Dong, P. Liang, X. L. Yu, and D. J. Yu. 2005. Construction and clinical significance of a predictive system for prognosis of hepatocellular carcinoma. World J. Gastroenterol. 113027-3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui, J., B. W. Dong, P. Liang, X. L. Yu, and D. J. Yu. 2004. Effect of c-myc, Ki-67, MMP-2 and VEGF expression on prognosis of hepatocellular carcinoma patients undergoing tumor resection. World J. Gastroenterol. 101533-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das, S., M. Ott, A. Yamane, W. Tsai, M. Gromeier, F. Lahser, S. Gupta, and A. Dasgupta. 1998. A small yeast RNA blocks hepatitis C virus internal ribosome entry site (HCV IRES)-mediated translation and inhibits replication of a chimeric poliovirus under translational control of the HCV IRES element. J. Virol. 725638-5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis-Smyth, T., R. C. Duncan, T. Zheng, G. Michelotti, and D. Levens. 1996. The far upstream element-binding proteins comprise an ancient family of single-strand DNA-binding transactivators. J. Biol. Chem. 27131679-31687. [DOI] [PubMed] [Google Scholar]
- 19.Di Bisceglie, A. M. 1997. Hepatitis C and hepatocellular carcinoma. Hepatology 2634S-38S. [DOI] [PubMed] [Google Scholar]
- 20.Duncan, R., L. Bazar, G. Michelotti, T. Tomonaga, H. Krutzsch, M. Avigan, and D. Levens. 1994. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 8465-480. [DOI] [PubMed] [Google Scholar]
- 21.El-Bassiouni, A., M. Nosseir, M. Zoheiry, E. El-Ahwany, A. Ghali, and N. El-Bassiouni. 2006. Immunohistochemical expression of CD95 (Fas), c-myc and epidermal growth factor receptor in hepatitis C virus infection, cirrhotic liver disease and hepatocellular carcinoma. APMIS 114420-427. [DOI] [PubMed] [Google Scholar]
- 22.Evans, M. J., C. M. Rice, and S. P. Goff. 2004. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. USA 10113038-13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan, X. C., and J. A. Steitz. 1998. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc. Natl. Acad. Sci. USA 9515293-15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan, X. C., and J. A. Steitz. 1998. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 173448-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friebe, P., and R. Bartenschlager. 2002. Genetic analysis of sequences in the 3′ nontranslated region of hepatitis C virus that are important for RNA replication. J. Virol. 765326-5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu, X. M., Q. X. Yang, C. K. Shao, and Z. Y. Feng. 2006. Expressions of h-TERT, c-myc, PCNA and cell apoptosis in liver carcinogenesis. Nan Fang Yi Ke Da Xue Xue Bao 26821-823. (In Chinese.) [PubMed] [Google Scholar]
- 27.Gamarnik, A. V., and R. Andino. 1998. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 122293-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gherzi, R., K. Y. Lee, P. Briata, D. Wegmuller, C. Moroni, M. Karin, and C. Y. Chen. 2004. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol. Cell 14571-583. [DOI] [PubMed] [Google Scholar]
- 29.Gontarek, R. R., L. L. Gutshall, K. M. Herold, J. Tsai, G. M. Sathe, J. Mao, C. Prescott, and A. M. Del Vecchio. 1999. hnRNP C and polypyrimidine tract-binding protein specifically interact with the pyrimidine-rich region within the 3′NTR of the HCV RNA genome. Nucleic Acids Res. 271457-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hahm, B., Y. K. Kim, J. H. Kim, T. Y. Kim, and S. K. Jang. 1998. Heterogeneous nuclear ribonucleoprotein L interacts with the 3′ border of the internal ribosomal entry site of hepatitis C virus. J. Virol. 728782-8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris, D., Z. Zhang, B. Chaubey, and V. N. Pandey. 2006. Identification of cellular factors associated with the 3′-nontranslated region of the hepatitis C virus genome. Mol. Cell Proteomics 51006-1018. [DOI] [PubMed] [Google Scholar]
- 32.He, L., J. Liu, I. Collins, S. Sanford, B. O'Connell, C. J. Benham, and D. Levens. 2000. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 191034-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irwin, N., V. Baekelandt, L. Goritchenko, and L. I. Benowitz. 1997. Identification of two proteins that bind to a pyrimidine-rich sequence in the 3′-untranslated region of GAP-43 mRNA. Nucleic Acids Res. 251281-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isken, O., M. Baroth, C. W. Grassmann, S. Weinlich, D. H. Ostareck, A. Ostareck-Lederer, and S. E. Behrens. 2007. Nuclear factors are involved in hepatitis C virus RNA replication. RNA 131675-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isken, O., C. W. Grassmann, R. T. Sarisky, M. Kann, S. Zhang, F. Grosse, P. N. Kao, and S. E. Behrens. 2003. Members of the NF90/NFAR protein group are involved in the life cycle of a positive-strand RNA virus. EMBO J. 225655-5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ito, T., and M. M. Lai. 1997. Determination of the secondary structure of and cellular protein binding to the 3′-untranslated region of the hepatitis C virus RNA genome. J. Virol. 718698-8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito, T., and M. M. Lai. 1999. An internal polypyrimidine-tract-binding protein-binding site in the hepatitis C virus RNA attenuates translation, which is relieved by the 3′-untranslated sequence. Virology 254288-296. [DOI] [PubMed] [Google Scholar]
- 38.Izumi, R. E., B. Valdez, R. Banerjee, M. Srivastava, and A. Dasgupta. 2001. Nucleolin stimulates viral internal ribosome entry site-mediated translation. Virus Res. 7617-29. [DOI] [PubMed] [Google Scholar]
- 39.Kaneko, T., Y. Tanji, S. Satoh, M. Hijikata, S. Asabe, K. Kimura, and K. Shimotohno. 1994. Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem. Biophys. Res. Commun. 205320-326. [DOI] [PubMed] [Google Scholar]
- 40.Katoh, H., H. Ojima, A. Kokubu, S. Saito, T. Kondo, T. Kosuge, F. Hosoda, I. Imoto, J. Inazawa, S. Hirohashi, and T. Shibata. 2007. Genetically distinct and clinically relevant classification of hepatocellular carcinoma: putative therapeutic targets. Gastroenterology 1331475-1486. [DOI] [PubMed] [Google Scholar]
- 41.Kolykhalov, A. A., S. M. Feinstone, and C. M. Rice. 1996. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 703363-3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolykhalov, A. A., K. Mihalik, S. M. Feinstone, and C. M. Rice. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 742046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kruger, M., C. Beger, Q. X. Li, P. J. Welch, R. Tritz, M. Leavitt, J. R. Barber, and F. Wong-Staal. 2000. Identification of eIF2Bgamma and eIF2gamma as cofactors of hepatitis C virus internal ribosome entry site-mediated translation using a functional genomics approach. Proc. Natl. Acad. Sci. USA 978566-8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levens, D. 2002. Disentangling the MYC web. Proc. Natl. Acad. Sci. USA 995757-5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levens, D. L. 2003. Reconstructing MYC. Genes Dev. 171071-1077. [DOI] [PubMed] [Google Scholar]
- 46.Lindenbach, B. D., and C. M. Rice. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436933-938. [DOI] [PubMed] [Google Scholar]
- 47.Liu, J., F. Kouzine, Z. Nie, H. J. Chung, Z. Elisha-Feil, A. Weber, K. Zhao, and D. Levens. 2006. The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO J. 252119-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCormick, C. J., D. Brown, S. Griffin, L. Challinor, D. J. Rowlands, and M. Harris. 2006. A link between translation of the hepatitis C virus polyprotein and polymerase function: possible consequences for hyperphosphorylation of NS5A. J. Gen. Virol. 8793-102. [DOI] [PubMed] [Google Scholar]
- 49.Miyanari, Y., M. Hijikata, M. Yamaji, M. Hosaka, H. Takahashi, and K. Shimotohno. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 27850301-50308. [DOI] [PubMed] [Google Scholar]
- 50.Moradpour, D., F. Penin, and C. M. Rice. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5453-463. [DOI] [PubMed] [Google Scholar]
- 51.Murata, T., T. Ohshima, M. Yamaji, M. Hosaka, Y. Miyanari, M. Hijikata, and K. Shimotohno. 2005. Suppression of hepatitis C virus replicon by TGF-beta. Virology 331407-417. [DOI] [PubMed] [Google Scholar]
- 52.Nakielny, S., and G. Dreyfuss. 1996. The hnRNP C proteins contain a nuclear retention sequence that can override nuclear export signals. J. Cell Biol. 1341365-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neddermann, P., M. Quintavalle, C. Di Pietro, A. Clementi, M. Cerretani, S. Altamura, L. Bartholomew, and R. De Francesco. 2004. Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J. Virol. 7813306-13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oh, J. W., G. T. Sheu, and M. M. Lai. 2000. Template requirement and initiation site selection by hepatitis C virus polymerase on a minimal viral RNA template. J. Biol. Chem. 27517710-17717. [DOI] [PubMed] [Google Scholar]
- 55.Peng, S. S., C. Y. Chen, N. Xu, and A. B. Shyu. 1998. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 173461-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajagopalan, L. E., C. J. Westmark, J. A. Jarzembowski, and J. S. Malter. 1998. hnRNP C increases amyloid precursor protein (APP) production by stabilizing APP mRNA. Nucleic Acids Res. 263418-3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saito, I., T. Miyamura, A. Ohbayashi, H. Harada, T. Katayama, S. Kikuchi, T. Y. Watanabe, S. Koi, M Onji, Y. Ohta, Q.-L.Choo, M. Houghton, and G. Kuo. 1990. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 876547-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarkar, B., Q. Xi, C. He, and R. J. Schneider. 2003. Selective degradation of AU-rich mRNAs promoted by the p37 AUF1 protein isoform. Mol. Cell. Biol. 236685-6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shih, S. C., and K. P. Claffey. 1999. Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein L. J. Biol. Chem. 2741359-1365. [DOI] [PubMed] [Google Scholar]
- 60.Shim, J., H. Lim, J. R. Yates, and M. Karin. 2002. Nuclear export of NF90 is required for interleukin-2 mRNA stabilization. Mol. Cell 101331-1344. [DOI] [PubMed] [Google Scholar]
- 61.Spangberg, K., L. Goobar-Larsson, M. Wahren-Herlenius, and S. Schwartz. 1999. The La protein from human liver cells interacts specifically with the U-rich region in the hepatitis C virus 3′ untranslated region. J. Hum. Virol. 2296-307. [PubMed] [Google Scholar]
- 62.Spangberg, K., L. Wiklund, and S. Schwartz. 2001. Binding of the La autoantigen to the hepatitis C virus 3′ untranslated region protects the RNA from rapid degradation in vitro. J. Gen. Virol. 82113-120. [DOI] [PubMed] [Google Scholar]
- 63.Spangberg, K., L. Wiklund, and S. Schwartz. 2000. HuR, a protein implicated in oncogene and growth factor mRNA decay, binds to the 3′ ends of hepatitis C virus RNA of both polarities. Virology 274378-390. [DOI] [PubMed] [Google Scholar]
- 64.Takimoto, M., T. Tomonaga, M. Matunis, M. Avigan, H. Krutzsch, G. Dreyfuss, and D. Levens. 1993. Specific binding of heterogeneous ribonucleoprotein particle protein K to the human c-myc promoter, in vitro. J. Biol. Chem. 26818249-18258. [PubMed] [Google Scholar]
- 65.Tanaka, T., N. Kato, M. J. Cho, K. Sugiyama, and K. Shimotohno. 1996. Structure of the 3′ terminus of the hepatitis C virus genome. J. Virol. 703307-3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsuchihara, K., T. Tanaka, M. Hijikata, S. Kuge, H. Toyoda, A. Nomoto, N. Yamamoto, and K. Shimotohno. 1997. Specific interaction of polypyrimidine tract-binding protein with the extreme 3′-terminal structure of the hepatitis C virus genome, the 3′X. J. Virol. 716720-6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsukiyama-Kohara, K., N. Iizuka, M. Kohara, and A. Nomoto. 1992. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 661476-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Doorn, L. J. 1994. Review: molecular biology of the hepatitis C virus. J. Med. Virol. 43345-356. [DOI] [PubMed] [Google Scholar]
- 69.Vindigni, A., A. Ochem, G. Triolo, and A. Falaschi. 2001. Identification of human DNA helicase V with the far upstream element-binding protein. Nucleic Acids Res. 291061-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang, Y., M. C. Wu, J. S. Sham, W. Zhang, W. Q. Wu, and X. Y. Guan. 2002. Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer 952346-2352. [DOI] [PubMed] [Google Scholar]
- 71.Waris, G., S. Sarker, and A. Siddiqui. 2004. Two-step affinity purification of the hepatitis C virus ribonucleoprotein complex. RNA 10321-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yanagi, M., M. St. Claire, S. U. Emerson, R. H. Purcell, and J. Bukh. 1999. In vivo analysis of the 3′ untranslated region of the hepatitis C virus after in vitro mutagenesis of an infectious cDNA clone. Proc. Natl. Acad. Sci. USA 962291-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yi, M., and S. M. Lemon. 2003. 3′ nontranslated RNA signals required for replication of hepatitis C virus RNA. J. Virol. 773557-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]