

Abstract
The Bcl-2 protein Bax normally resides in the cytosol, but during apoptosis it translocates to mitochondria where it is responsible for releasing apoptogenic factors. Using anoikis as a model, we have shown that Bax translocation does not commit cells to apoptosis, and they can be rescued by reattachment to extracellular matrix within a specific time. Bax undergoes an N-terminal conformational change during apoptosis that has been suggested to regulate conversion from its benign, cytosolic form to the active, membrane bound pore. We now show that the Bax N-terminus regulates commitment and mitochondrial permeabilisation, but not the translocation to mitochondria. We identify Proline 13 within the N-terminus of Bax as critical for this regulation. The subcellular distribution of Proline 13 mutant Bax was identical to wild type Bax in both healthy and apoptotic cells. However, Proline 13 mutant Bax induced rapid progression to commitment, mitochondrial permeabilisation and death. Our data identify changes in Bax controlling commitment to apoptosis that are mechanistically distinct from those controlling its subcellular localisation. Together, they indicate that multiple regulatory steps are required to activate the proapoptotic function of Bax.
Keywords: Bax, anoikis, commitment, mitochondria
Introduction
Bax and Bak forms a requisite gateway for the mitochondrial apoptotic pathway (1), releasing pro-apoptotic factors including cytochrome c and Smac/Diablo (2, 3), activating caspases and bringing about apoptotic cell death. Different BH3-only members of the Bcl-2 family proteins become activated following specific types of cellular stress, but all are dependent on Bax and Bak to induce apoptosis (4-6). Thus, how Bax and Bak are activated is central to understanding apoptosis. Bak is permanently on mitochondria, whereas Bax is predominantly in the cytosol of healthy cells (7-9). During apoptosis, Bax undergoes a number of changes, including translocation to mitochondria, a conformational change exposing cryptic epitopes within the N-terminus, and its assembly into high molecular weight complexes (5, 9-14). The order and significance of these events remain unclear, and they are commonly described as occurring as a single, irrevocable step converting benign, cytosolic Bax into a death inducing pore on mitochondria.
Anoikis is apoptosis following loss of attachment to the extracellular matrix (ECM) (15, 16). Most cells require adhesion to ECM to provide information regarding their position within a tissue, ensuring that they only grow and survive in the correct context. Using this model, we have shown that Bax translocates to mitochondria some considerable time prior to cytochrome c release (13, 17). Furthermore, cells can be rescued if they reattach to ECM before becoming committed to apoptosis, despite Bax translocating to mitochondria (7, 13). Mitochondrial permeabilisation (MMP) and subsequent caspase activation is rapid, taking five minutes from the point at which it is first detected (2, 3, 13, 18). Together, these data imply that following translocation, Bax undergoes a second activation step before inducing MMP, a step yet to be demonstrated mechanistically in intact cells.
Here we show that the N-terminus of Bax regulates this post-translocation step required for full activation. The N-terminus of mitochondrial Bax is not exposed until the moment of MMP. Furthermore, we define Proline13 within the N-terminus as crucial for regulating Bax activation on the OMM. Substitution of Proline13 does not affect the ability of adherent cells to sequester Bax in the cytosol, or its translocation to mitochondria. However, it significantly increases the kinetics of Bax activation and apoptosis commitment following translocation. Our results mechanistically separate two steps in Bax activation, and identify a critical point for apoptosis commitment that is regulated through the conformation of the N-terminus.
Results
Bax translocation to mitochondria does not commit cells to apoptosis
During anoikis Bax translocates to mitochondria a considerable time prior to cytochrome c release and apoptosis (13). This is illustrated in Fig. 1. In adherent epithelial cells, YFP-Bax was distributed throughout the cytoplasm, but following detachment for 30 minutes it colocalised with mtHsp-70 (Fig. 1a). Most cells at this time had not released cytochrome c (Fig. 1b) or displayed apoptotic nuclei (quantified in Fig. 6 d and e). Following detachment for longer periods, progressively more cells underwent mitochondrial permeabilisation and showed apoptotic morphology. YFP alone remained cytosolic (not shown).
Figure 1. Bax translocation occurs rapidly during anoikis, but is reversible.
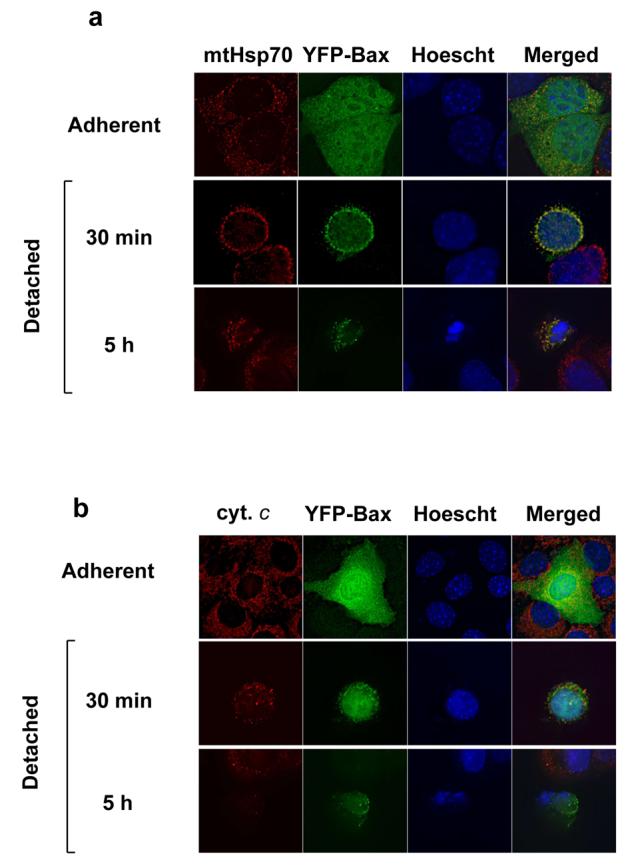

(A) FSK-7 mammary epithelial cells transiently expressing YFP-Bax were either left adherent or detached and plated onto poly-HEMA coated dishes for 30 minutes or 5 hours. Cells were fixed and immunostained for mtHsp70. Nuclei were stained with Hoechst. (B) FSK-7 cells as in (A) immunostained with anti-cytochrome c. (C) FSK-7 cells transiently expressing YFP-Bax were detached for 30 minutes before replating onto ECM coated coverslips for various times. Cells were fixed and immunostained as in (A). (D) FSK-7 cells were either left adherent or detached from ECM for 60 minutes before replating for various times. Cells were then separated into cytosolic and membrane fractions. Fractions were separated by SDS-PAGE and immunoblotted for APAF-1, mtHsp-70 and Bax.
Figure 6. Pro13A mutation does not alter Bax translocation following detachment, but does accelerate anoikis.
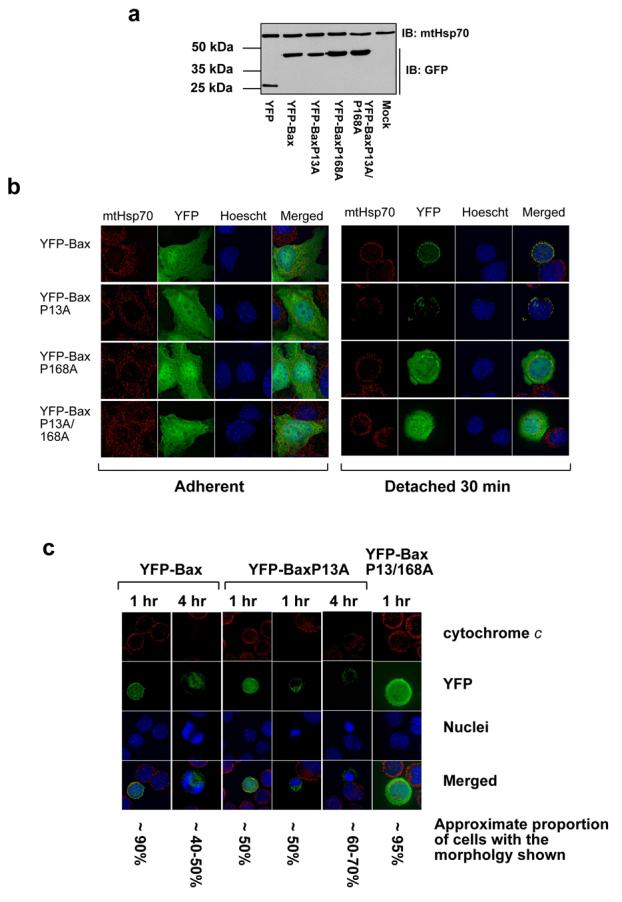

(A) Lysates of FSK-7 cells transiently expressing YFP or YFP-Bax proline mutants were immunoblotted with a polyclonal anti-GFP antibody and a monoclonal anti-mtHsp70 antibody. (B) FSK-7 cells transiently expressing YFP-Bax, YFP-BaxP13A, YFP-BaxP168A or YFP-BaxP13/168A were either left adherent or detached from the ECM and plated onto poly-HEMA coated dishes for 30 minutes. Cells were fixed and stained with anti-mtHsp70 and Hoechst. In adherent cells all YFP-Bax constructs had a cytosolic distribution. After 30 minutes detachment, YFP-Bax and YFP-BaxP13A co-localised with mtHsp70, whereas YFP-BaxP168A and YFP-BaxP13A/P168A remained cytosolic. (C) FSK-7 cells expressing YFP-Bax, YFP-BaxP13A and YFP-BaxP13A/P168A were detached from ECM for 30 minutes or 4 hours before fixing and staining with anti-cytochrome c and Hoechst. The approximate proportions of transfected cells (judged by YFP fluorescence) showing each morphology is indicated. (D) FSK-7 transiently expressing YFP, YFP-Bax, YFP-BaxP13A, YFP-BaxP168A or YFP-BaxP13/168A were left adherent or detached from the ECM for 30 minutes, 1 hour and 4 hours. Apoptosis was quantified by nuclear morphology in YFP-positive cells. Data represents the mean of three independent experiments +/− SEM. (E) FSK-7 transiently expressing YFP, YFP-Bax, YFP-BaxP13A, or YFP-BaxP13/168A were left adherent or detached from the ECM for 30 minutes, 1 hour and 4 hours. Cells were fixed and immunostained with anti-cytochrome c. The percentage of YFP-positive cells that had released cytochrome c was determined. Data represents the mean of three independent experiments +/− SEM. (F) MEFs transiently expressing YFP-Bax or YFP-BaxP13A were left adherent or detached from the ECM for various times. Apoptosis was quantified by nuclear morphology in YFP-positive cells. Data represents the mean of three independent experiments +/− SEM.
Prior to mitochondrial permeabilisation Bax translocation is reversible (13). Cells were detached for 30 minutes before replating onto ECM for various times. Prior to replating, there was colocalisation between YFP-Bax and mitochondria (Fig. 1c). Following reattachment for 1 hour colocalisation between YFP-Bax and mtHsp-70 was lost. This became more apparent when cells had reattached for longer. Loss of endogenous Bax from mitochondria during reattachment was demonstrated biochemically. Cells were detached and replated for various times, and separated into cytosol and membrane fractions. These were immunoblotted for Bax, APAF-1 (cytosol) and mtHsp-70 (mitochondria) (Fig. 1d). Detachment resulted in accumulation of Bax in the membrane fraction, which was lost upon reattachment. At present we cannot distinguish if this is due to Bax relocalisation back to the cytosol or degradation on the OMM.
Anoikis is a stochastic process and cells were either alive or apoptotic. The inability to find cells with an intermediate morphology indicates a rapid transition between these two states. The kinetics of MMP following staurosporine or UV irradiation are rapid (2, 3). These represent irreversible stimuli, whereas epithelial cells require prolonged detachment before committing to apoptosis. To determine if the differences between stimuli were due to different kinetics of MMP, FSK-7 cells expressing H2B-CFP (nuclear marker) and SMAC-YFP were detached from ECM and imaged in suspension every 3 minutes (Fig 2b). Although the time between detachment and MMP varied between cells, the kinetics of SMAC-YFP release were identical to those published for STS and UV treated cells (3-6 minutes), immediately followed by cell death.
Figure 2. OMM permeabilisation and cell death is rapid and invariant during anoikis.

Live cell imaging of detached FSK-7 cells transiently expressing SMAC-YFP and H2B-CFP. Images were captured every 3 minutes. In the panel on the left, the sequence shown starts 240 minutes following detachment. Note at 240 min, SMAC-YFP is punctate, but has become diffuse by 243 min. Membrane blebbing is seen in the phase image at 246 min, and the nucleus is clearly condensed at 252 min. In the panel on the right, the sequence starts 129 minutes following detachment. SMAC release is first apparent at 132 minutes, and is complete by 135 minutes when membrane blebbing is seen.
These data show Bax translocation and MMP are distinct events. Furthermore, the transition between these two stages during physiological apoptosis occurs rapidly, and is not affected by the reversible nature of the apoptotic stimulus.
Exposure of the Bax N-terminus is associated with MMP and not translocation
As Bax translocation has been suggested to be regulated through the N-terminal conformational change (19, 20), we asked if exposure of the N-terminus occurred following translocation but prior to MMP. Cells expressing YFP-Bax were detached for various times and immunostained with an N-terminal conformation dependent antibody (anti-NT) (Fig. 3a). In adherent cells YFP-Bax was cytosolic and not anti-NT reactive. Following detachment for 1 hour, YFP-Bax colocalised with mitochondria, but was still not immunoreactive. Anti-NT immunoreactivity was only seen in cells with apoptotic nuclei (see the cell detached for 5 hr). Identical data were obtained with another N-terminal antibody, monoclonal 6A7, and in cells treated with staurosporine (data not shown).
Figure 3. Bax undergoes an N-terminal conformational change concurrent with OMM permeabilisation.
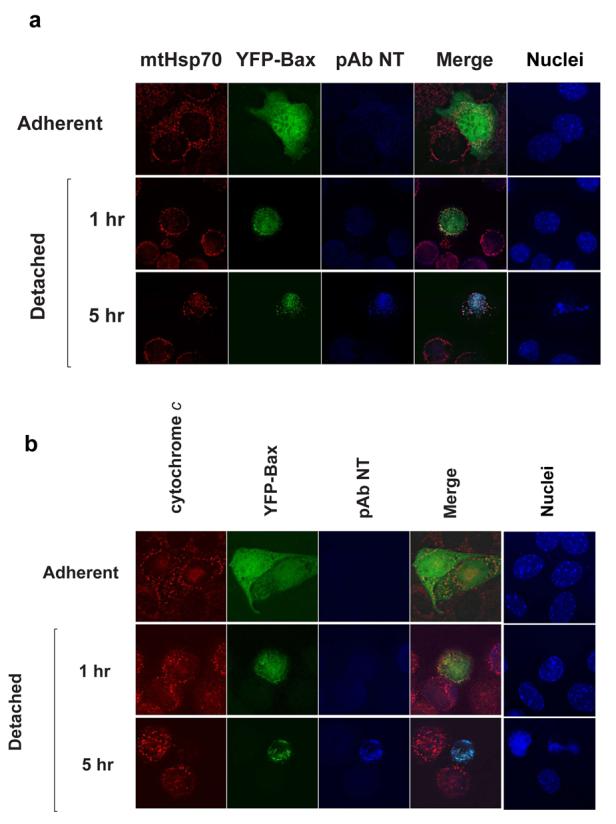
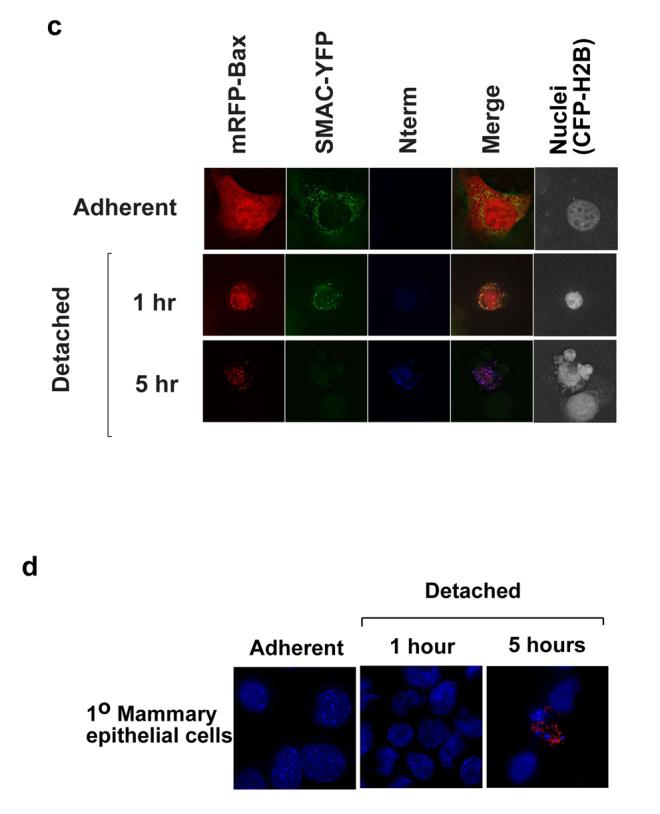
(A) FSK-7 cells expressing YFP-Bax were either kept adherent, or detached for 1 hour and 5 hours. Cells were fixed and stained with anti-Bax NT, anti-mtHsp70 and Hoechst. Note in cells detached for 1 hour YFP-Bax colocalised with mtHsp70, but there was little anti-NT immunoreactivity. In cells detached for 5 hours, Bax NT reactivity is seen in cells with apoptotic nuclei. (B) FSK-7 cells transiently expressing YFP-Bax were treated as in (A), and immunostained for anti-NT and cytochrome c. Anti-NT reactivity was only seen in cells that had lost mitochondrial cytochrome c. (C) FSK-7 cells transiently expressing mRFP-Bax, SMAC-YFP and H2B-CFP were left adherent or detached for 1 hour and 5 hours. Cells were fixed and immunostained for the N-terminal epitope. Note significant N-terminal exposure is only seen in the cell detached for 5 hours that shows diffuse SMAC-YFP and a fragmented nucleus. (D) Primary mouse mammary epithelial cells were left adherent or detached from ECM for the indicated times. Cells were fixed and immunostained for the N-terminal epitope of endogenous Bax. Note that only the cell with apoptotic morpholgy is positive for the N-terminal Bax epitope.
We next examined the relationship between N-terminal exposure and cytochrome c release (Fig. 3b). Following detachment, although YFP-Bax was mitochondrial, N-terminal exposure was only seen in cells that no longer had punctate cytochrome c. These all had apoptotic nuclei. Identical results were obtained with cells expressing mRFP-Bax and SMAC-YFP (Fig. 3c). In adherent cells, mRFP-Bax and SMAC-YFP did not colocalise, and there was no immunoreactivity. Following detachment for 1 hr, colocalisation was seen between mRFP-Bax and SMAC-YFP. Exposure of the N-terminus was only seen in cells that no longer had a punctate SMAC-YFP distribution. We also examined Bax epitope exposure in primary mammary epithelial cells. As with FSK-7 cells, anti-NT immunoreactivity was only seen in cells that had apoptotic nuclear morphology (Fig. 3d).
We examined N-terminal epitope exposure of endogenous Bax biochemically. Cytosolic Bax can be activated by exposure to detergents (21). This is shown in Fig. 4a, where cytosolic fractions from adherent FSK-7 cells were analysed by immunoprecipitating with anti-Bax antibodies in the presence or absence of Triton-X100. Two conformation dependent N-terminal anti-Bax antibodies, polyclonal anti-NT and monoclonal 6A7, only immunoprecipitated Bax in the presence of Triton-X100. In contrast, monoclonal 5B7 and a polyclonal raised against Bax residues 1-170 (SC) precipitated Bax in the presence or absence of detergent. This detergent sensitivity necessitates specific extraction conditions to maintain the native N-terminal conformation of membrane associated Bax. Membrane fractions from cells detached for 1 hour were extracted in CHAPS or RIPA, and immunoprecipitated with anti-Bax antibodies NT, SC and 6A7 (Fig. 4b). Anti-Bax SC precipitated Bax from the CHAPS and RIPA extracted membranes. In contrast, anti-NT and 6A7 only precipitated Bax in RIPA, despite a considerable amount of Bax being present in the CHAPS fraction (the total lane contains 10% of the CHAPS extract used in the immunoprecipitation). Thus, endogenous Bax on the OMM in cells detached for 1 hour does not have an exposed N-terminus.
Figure 4. Endogenous Bax does not show an exposed N-terminus following translocation to the OMM.

Cytosolic and membrane fractions from FSK-7 cells were immunoprecipitated (IP) and immunoblotted (WB) with various antibodies to determine N-terminal epitope exposure. (A) Cytosolic fractions prepared from adherent FSK-7 cells in the absence of detergent. Bax was immunoprecipitated from 400 μg of lysate with the indicated antibodies either in the absence of detergent or following addition of 0.1% Triton-X100, followed by immunoblotti ng. Total represents 40 μg of cell lysate. (B) Membrane fractions isolated from FSK-7 cells following detachment from ECM for 1 hour. Membrane fractions were extracted in either 1% CHAPS or RIPA, and 400 μg were immunoprecipitated with the indicated antibodies, followed by immunoblotting. Total represents 40 μg of the CHAPS extracted membranes. Note that CHAPS extracted Bax is not immunoprecipitated with either anti-NT or mAb 6A7. (C) Membrane fractions from FSK-7 cells detached from ECM for 5 hours were extracted sequentially in CHAPS, RIPA and Urea. The CHAPS and RIPA fractions were immunoprecipitated with the indicated antibodies. Total extracts and the immunoprecipiates were immunoblotted with anti-Bax mAb 5B7. Note that very little Bax is immunoprecipitated from the CHAPS extract, and there are significant amounts of Bax in the RIPA and Urea extracts. (D) FSK-7 cells treated with DMSO or staurosporine (STS) for 18 hours were separated into cytosolic and membrane fractions. The membrane fraction was extracted in CHAPS, and the CHAPS insoluble material solubilised in Urea. Note that the majority of Bax in the STS treated cells is not soluble in CHAPS.
When cells were detached for 5 hours, anti-NT could precipiate some Bax from CHAPS extracted membranes, but it was considerably less than that precipitated with SC (Fig. 4c). As cells apoptose, Bax becomes resistant to detergent extraction (13). We suspected that CHAPS was selectively extracting Bax from cells that had not undergone MMP. To address this, the CHAPS insoluble material was sequentially extracted in RIPA, and the RIPA insoluble fraction solubilised in urea. Both anti-NT and SC immunoprecipitated Bax from the RIPA extract, suggesting cells that had been detached for prolonged periods had a significant proportion of Bax that was not extracted in CHAPS. It was noteworthy that a significant amount of Bax was only extracted in urea. Indeed, when we examined a population of cells in which 100% apoptosis had been induced with staurosporine, almost all the Bax was present in a CHAPS insoluble fraction that was only solubilised in urea (Fig. 4d).
These data indicate that following translocation to mitochondria, endogenous Bax does not display an exposed N-terminus. Furthermore, it is difficult to examine biochemically the N-terminal conformation of Bax following MMP as it becomes resistant to extraction using conditions that maintain its native structure. The N-terminal conformational switch seen concomitant with MMP using YFP-Bax correlates with this profound change in its biochemical properties.
Proline 13 forms part of the Bax N-terminal epitope
The kinetics of MMP are too rapid to image Bax N-terminal exposure during permeabilisation. We therefore introduced mutations within Bax that might affect this transition. Comparison of the sequence of human, mouse and rat Bax indicated a conserved proline at position 13, just before α1 helix (Fig. 5a). The context of Proline13 was similar to Proline168, previously shown to control Bax translocation (19). We made a Proline13 to alanine substitution (BaxP13A), along with the previously described BaxP168A, which prevents translocation, and the double BaxP13/168A. Along with wildtype (WT) YFP-Bax, these were transiently expressed in MEFs. Immunoblotting with anti-GFP indicated all were expressed as full-length proteins at similar levels (Fig. 5b).
Figure 5. Proline 13 is required for immunoreactivity with the N-terminal Bax antibody.
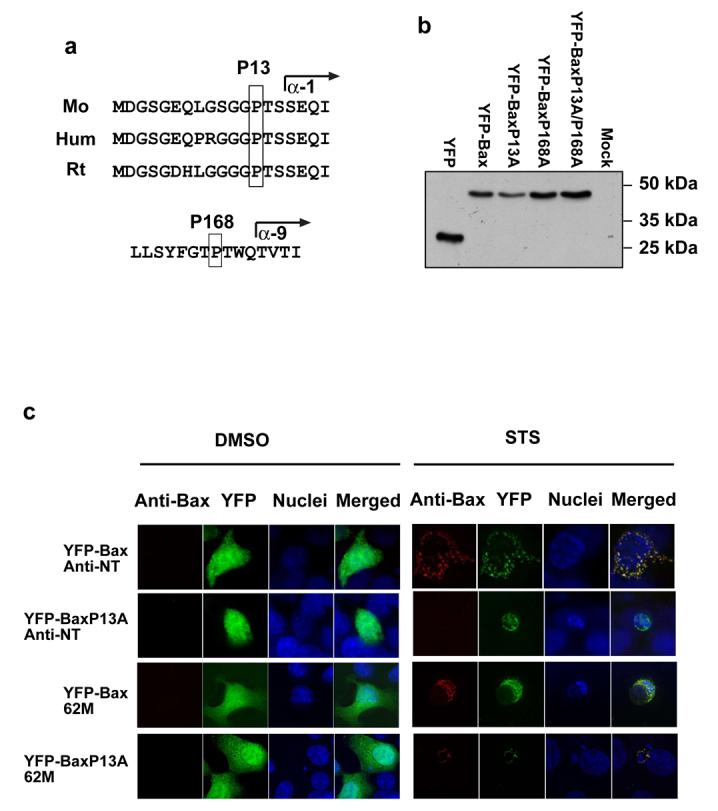

(A) Proline 13 is conserved in mouse, human and rat Bax. The residue forms part of the anti-NT epitope (amino acids 1-21) and precedes helix α-1. Proline 168 is conserved in mouse and human Bax, and precedes helix α-9. (B) MEFs transiently expressing YFP or the YFP-Bax proline mutants were lysed as described in the methods. Lysates were separated by SDS-PAGE and immunoblotted with a polyclonal anti-GFP antibody. (C) Bax deficient HCT116 cells transiently expressing YFP-Bax or YFPBaxP13A were either treated with DMSO or 10 μM staurosporine in DMSO for 4 hours. Following treatment cells were fixed and stained with anti-NT or 62M. After treatment, YFP-Bax and YFP-BaxP13A had a punctate distribution, and nuclei had apoptotic morphology. YFP-Bax was reactive with both anti-Bax antibodies. YFP-BaxP13A was only reactive with anti-Bax 62M, and not with anti-NT. (D) Bax−/−/Bak−/− MEFs were transiently transfected with YFP or the YFP-Bax proline mutants and treated with 10μM staurosporine for various times before fixing. Apoptosis was quantified in transfected cells (identified by YFP fluorescence) by nuclear morphology. Data represents 3 independent experiments +/− SEM.
We asked if the P13A substitution had affected the ability of Bax to fold correctly or localise to mitochondria. Bax-deficient HCT116 cells expressing YFP-Bax or YFP-BaxP13A were treated with DMSO or staurosporine, and immunostained with anti-NT or 62M (Fig. 5c). Anti-Bax 62M recognises a conformation dependent epitope proximal to the BH3-domain (13). With DMSO, YFP-Bax and YFP-BaxP13A were cytosolic and not reactive with anti-NT or 62M. Following staurosporine treatment, YFP-Bax had a punctate distribution which immunostained with both antibodies. Staurosporine also induced punctate YFP-BaxP13A in apoptotic cells, but this was only reactive with 62M. We expressed the YFP-Bax fusions in Bax/Bak double knock out MEFs (DKO-MEFs). These were treated with staurosporine and apoptosis quantified (Fig. 5d). DKO-MEFs expressing YFP or YFP-BaxP168A were resistant to staurosporine. In contrast, YFP-Bax and YFP-BaxP13A both sensitised DKO-MEFs to apoptosis (p<0.001). There was no difference in apoptosis between YFP-Bax or YFP-BaxP13A expressing DKO-MEFs. YFP-BaxP13/168A did not sensitise DKO-MEFs to staurosporine.
These data indicate that substitution of Proline13 to alanine does not inhibit Bax function. Although Proline13 is required for recognition by anti-NT, exposure of the BH3-proximal 62M epitope indicates the gross folding of YFP-BaxP13A and its activation during apoptosis are unaffected.
Proline 13 regulates commitment to apoptosis in epithelial cells during anoikis
We next asked if BaxP13A affected progression of anoikis in epithelial cells. Similar levels of expression of the YFP-Bax fusion proteins were observed in FSK-7 cells (Fig. 6a). In adherent cells, all the YFP-Bax fusions were cytosolic (Fig. 6b). Following detachment for 30 minutes, YFP-Bax and YFP-BaxP13A redistributed to mitochondria (Fig. 6b and c). YFP-BaxP168A and YFP-BaxP13/168A remained cytosolic. Anoikis requires prolonged detachment from ECM before MMP occurs, but we noted that following short times many of the cells expressing YFP-BaxP13A were apoptotic and had released cytochrome c (Fig. 6c). We quantified the ability of the YFP-Bax mutants to sensitise cells to anoikis following detachment for various times, measuring nuclear morphology (Fig. 6d) and cytochrome c release (Fig. 6e). In adherent cells, there was no induction of apoptosis by any YFP-Bax fusions compared with YFP (p>0.05). After 30 min and 1 hr detachment there was no significant difference between YFP-Bax and YFP (p>0.05). YFP-Bax cells showed significant apoptosis after 4 hours (p<0.001). In contrast, cells expressing YFP-BaxP13A showed significant apoptosis after just 30 minutes detachment (p<0.001). After 4 hours there was no difference between YFP-Bax and YFP-BaxP13A (p>0.05). YFP-BaxP168A and YFP-BaxP13/168A did not sensitise cells to anoikis. We examined the ability of BaxP13A to induce anoikis in mouse embryonic fibroblasts (MEFs). MEFs are more resistant to anoikis than epithelial cells, but can be sensitised by expression of YFP-Bax (Fig. 6f). As with epithelial cells, YFP-BaxP13A significantly accelerated the progression of anoikis in MEFs. These data indicate that YFP-BaxP13A initiates apoptosis more rapidly following translocation to mitochondria through accelerated progression to MMP.
We asked if YFP-BaxP13A could integrate into the OMM and oligomerise. Cells expressing YFP, YFP-Bax or YFP-BaxP13A treated with staurosporine for 1 hour. Cytosol and mitochondria were isolated, and the mitochondria extracted in alkali. Fractions were immunoblotted for YFP (Fig. 7a). YFP was exclusively in the cytosol. Both YFP-Bax and YFP-BaxP13A were distributed between the cytosol and the alkali resistant mitochondrial fraction, indicating that both integrated into the OMM. To examine the oligomerisation, membrane fractions isolated from cells that had been detached from ECM for 1 hour were analysed by 2-dimensional blue-native PAGE, and immunoblotted for Bax (Fig. 7b). Both YFP-Bax and YFP-BaxP13A were present in oligomeric complexes of approximately 200kDa, as was endogenous Bax. mtHsp70 was seen at molecular weights approximate to known complexes with Tim44 and Tim17/23 (22). Together, these show no obvious difference between YFP-Bax and YFP-BaxP13A in both oligomerisation and membrane integration.
Figure 7. BaxP13A integrates into the OMM and oligomerises.

(A) FSK-7 cells transiently expressing YFP, YFP-Bax or YFP-BaxP13A were treated with staurosporine for 1 hour. Cytosolic and mitochondrial fractions were prepared as described in the methods, and the latter extracted in 0.1M Na2CO3. The cytosolic, Na2CO3 soluble (extracted) and insoluble (integrated) fractions were separated, along with a total cell lysate, by SDS-PAGE and immunoblotted for YFP. (B) FSK-7 cells transiently expressing YFP-Bax or YFP-BaxP13A were detached from ECM for 1 hour. Mitochondrial fractions isolated from cells were subjected to 2D-BN-PAGE as described in the methods. The 1st dimension BN-PAGE gel was cut into 16 slices which were extracted in SDS-PAGE sample buffer. These were then separated by conventional SDS-PAGE, and immunoblotted for Bax, YFP and mtHsp70.
We asked if YFP-BaxP13A showed altered regulation by other Bcl-2 proteins. We expressed mRFP-Bax or mRFP-BaxP13A alone or with GFP-Bcl-XL. Cells were detached from ECM for various times. mRFP-Bax and the mRFP-BaxP13A sensitised cells to anoikis in an identical manner as YFP-tagged forms, and co-expression of Bcl-XL inhibited anoikis induced by both (Fig. 8a). Bcl-XL inhibited translocation of mRFP-Bax and mRFP-BaxP13A following detachment (data not shown). We asked if BaxP13A was activated by BH3-only proteins. YFP, YFP-Bax or YFP-BaxP13A were expressed with truncated Bid (tBid) in DKO MEFs. YFP-Bax and YFP-BaxP13A did not induce significant apoptosis when expressed alone, but did when co-expressed with tBid (Fig. 8b). These data indicate that the proline 13 mutation has not uncoupled it from the regulatory functions of other Bcl-2 family proteins.
Figure 8. YFP-BaxP13A accerlerates cell commitment to apoptosis.
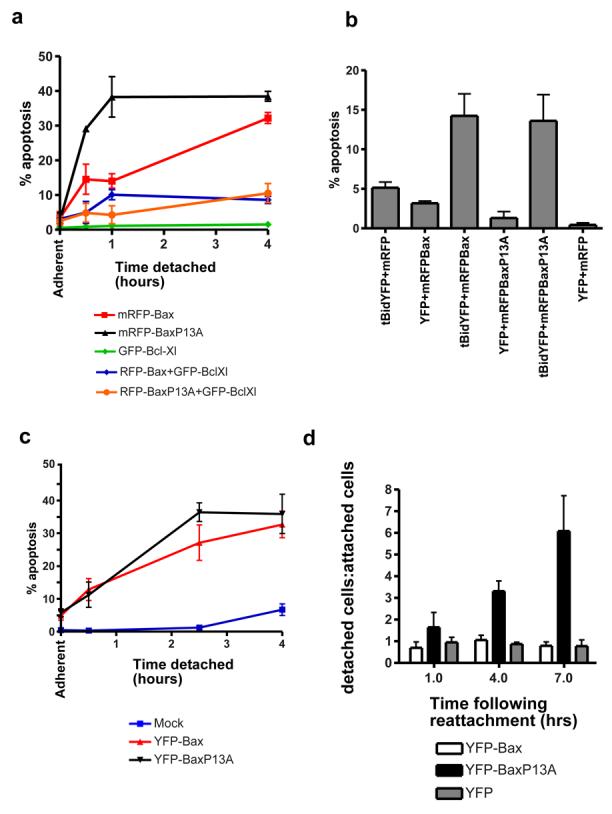
(A) FSK-7 cells transiently expressing the indicated fusion proteins were detached from ECM for various times, and apoptosis quantified by nuclear morphology. mRFPBax or mRFP-BaxP13A sensitised cells to anoikis in an identical manner as seen with YFP-fusions (compare with Fig. 6d and e). Co-expression of GFP-Bcl-XL inhibited anoikis mediated by both WT Bax, and the P13A mutant. (B) DKO-MEFs were transiently transfected with combinations of mRFP, mRFP-Bax, mRFP-BaxP13A, YFP and tBid-YFP, as indicated. 24 hours post transfection, cells were harvested and apoptosis quantified in the transfected cells by nuclear morphology. Co-expression of tBid and either WT Bax or the P13A mutant resulted in an increase in apoptosis. Note that the increase in apoptosis induced by co-expression of Bax and tBid occurs to a similar level as previously reported in Bax/Bak DKO-MEFs by Wei et al. (1). (C) FSK-7 cells expressing YFP-Bax or YFP-BaxP13A were detached from ECM in the presence of the Jnk inhibitor SP600125. Apoptosis was quantified by nuclear morphology. (D) FSK-7 cells transiently expressing YFP, YFP-Bax or YFP-BaxP13A detached from ECM for 30 minutes. After 30 minutes, cells were allowed to reattach to ECM coated coverslips for 1 hour, 4 hours or 7 hours. At each time point cells in the supernatant were collected by cytospinning, and cells on the coverslip were fixed. Cells were stained with Hoechst. Quantification of YFP-positive cells in the supernatant (detached) and on the coverslip (attached). Results are expressed as the ratio of cells in each fraction (detached/attached). Note that over time, there is no increase in the ratio for YFP or YFP-Bax expressing cells, indicating that those cells that were able to reattach were rescued from an apoptotic fate. In contrast, the proportion of detached YFP-BaxP13A cells increases significantly over time, indicating that cells that had initially reattached were not rescued and still underwent apoptosis. Data represents the mean of three independent experiments +/− SEM.
We have previously demonstrated that Bax activation during anoikis is regulated by kinase signalling pathways (7). c-Jun N-terminal kinase (Jnk) has been implicated in Bax activation and anoikis (23-25). We asked if the N-terminal proline mutation had made Bax more sensitive to a pro-apoptotic signal from Jnk by treating cells expressing YFP-Bax or YFP-BaxP13A with SP600125. Interestingly, although anoikis was not inhibited, the rapid progression to MMP seen with YFP-BaxP13A was, with no statistical difference between WT and mutant Bax (p>0.05).
Jnk activity increases with prolonged detachment from ECM and might contribute to apoptosis commitment (Zouq and Gilmore, unpublished data). Increased sensitivity to a commitment signal by BaxP13A may affect the ability to rescue cells by reattachment to ECM. To measure commitment, cells expressing YFP, YFP-Bax or YFP-BaxP13A were detached for 30 minutes and then allowed to reattach to ECM. Identical numbers of transfected cells were replated for 1, 4 or 7 hrs. At each time, we measured the proportion of transfected cells that reattached with those in the supernatant, presented as a ratio (Fig. 6d). The ratio for YFP and YFP-Bax expressing cells remained constant, indicating that cells that reattached to ECM were not committed to apoptosis and were rescued. YFP-BaxP13A expressing cells showed increasing number of detached cells at 4 and 7 hours, indicating that although YFP-BaxP13A expressing cells initially reattached to ECM, these were already committed to apoptosis and still underwent cell death.
Together, these data indicate that the N-terminus of Bax is important for regulating cell commitment to apoptosis, but is not important in the initial targeting to mitochondria. Furthermore, they have mechanistically separated Bax translocation and OMM permeabilisation.
Discussion
Although Bax plays a critical role in apoptosis, our understanding of its regulation is far from complete. We now provide an important insight into this process. We demonstrate that translocation of Bax to mitochondria is not associated with changes in its N-terminal conformation. Instead, changes in the Bax N-terminus are associated with MMP. Mutating Proline13 within the Bax N-terminus influences MMP but not translocation, possibly by sensitising it to pro-apoptotic kinase signalling. Our data show that the N-terminus of Bax controls a molecular switch regulating commitment to apoptosis.
Several seminal papers have contributed to current models of Bax regulation. Bax, a cytosolic monomer in healthy cells, has a cryptic N-terminal epitope which is exposed by detergents (9, 12, 21). This conformational change occurs during apoptosis when Bax translocates to mitochondria and oligomerises (5, 10, 12, 14). However, several lines of evidence suggest Bax activation requires distinct regulatory steps, and translocation per se does not induce apoptosis. Healthy cells can have an appreciable amount of mitochondrial Bax (5, 7, 8, 19). Bax translocation can occur hours before MMP (7, 13, 17). A mutation within the Bax mitochondrial targeting sequence induced constitutive localisation to mitochondria, but not apoptosis or N-terminal epitope exposure (26). Bax translocation but not activation can be seen in myc deficient cells (27). These studies indicate that following Bax translocation a second event is required before MMP.
Bax, Bak and Bcl-XL are C-terminal anchored mitochondrial proteins (19, 28). The C-terminus of monomeric Bax is hidden in a hydrophobic pocket, and it has been suggested that the N-terminus regulates reorientation of this sequence to allow translocation (19, 20, 26). Although the structure of Bax shows no interaction between N- and C-termini, allosteric effects cannot be discounted. We show that the N-terminal conformational change is not concomitant with translocation, and N-terminal exposure only occurs when MMP is apparent. Thus, reorientation of the C-terminus probably occurs without detectable N-terminal changes.
The nature of the N-terminal conformational switch is unclear, as only one Bax structure is known. The change may be localised, with the epitope adopting a specific conformation recognised by the antibody, and may regulate Bax association with mitochondrial proteins (10, 13). Alternatively, Bax may undergo global unfolding during integration into mitochondria. In vitro studies suggest that when Bax forms a pore, its hydrophobic helices-5 and -6 become exposed (29, 30). We have previously shown that anti-Bax 62M did not significantly detect mitochondrial Bax until MMP, and now show exposure of the N-terminus at the same time (13). Concomitant exposure of spatially distinct epitopes suggests global unfolding. This correlates with endogenous Bax becoming resistant to detergent extraction, and may account for the large clusters described in apoptotic cells (31, 32).
Cells expressing BaxP13A show altered regulation of progression to MMP and commitment. BaxP13A was retained in the cytosol in adherent cells, accumulated on mitochondria following detachment like WT-Bax, and is not constitutively apoptotic in adherent cells. Instead, the P13A substitution may alter interactions with regulatory factors on mitochondria. Bax complexes on mitochondria have been proposed to represent the apoptotic pore (10, 13). However, it is as likely that interactions with mitochodnrial factors keep Bax inactive prior to commitment. Bak is regulated this way, binding VDAC2 in healthy cells (33). Possible Bax binding proteins, including components of the permeability transition pore, are discussed elsewhere, (34). Although it is not yet known if endogenous mitochondrial proteins keep Bax inactive, the principle is sound, shown by the anti-apoptotic viral proteins, M11L and vMIA (35-37). These bind Bax on mitochondria, preventing N-terminal epitope exposure and cytochrome c release. BH3-only proteins are strong candidates for interacting with Bax and Bak to activate them. Activated Bid displaces Bak from VDAC2, activating it (33). BaxP13A can be activated by tBid in DKO-MEFs to the same extent as wildtype Bax, and is supressed by Bcl-XL, suggesting that it is not completely uncoupled from this regulatory pathway. However, the P13A substitution may have introduced some flexibility into the N-terminus that might reduce the energy required for activation, or sensitise it to modification. In either case, this only occurs in the context of the mitochondrial membrane, an idea which is supported by in vitro studies (38).
Some models of apoptosis imply that Bcl-2 family proteins are the key arbiters of MMP (4). However, it is likely that other regulatory proteins are also important, possibly facilitating Bcl-2 family interactions via kinase signalling. In vitro studies isolate Bax from the signalling pathways that regulate it in vivo, and we noted that in contrast to anoikis, apoptosis induced by staurosporine, a broad range kinase inhibitor, showed no difference between wildtype Bax or BaxP13A. The rapid transition to MMP induced by BaxP13A was also attenuated by inhibiting Jnk. A recent paper has suggested that Jnk and p38MAPK can phosphorylate Bax and activate it (24). We have no evidence that Bax is directly phosphorylated during anoikis, but Jnk activity does appear to regulate activation of BaxP13A on mitochondria. Using a physiological model of cell death might therefore distinguish important points of apoptosis regulation not seen in other models.
Our data indicate that survival signals regulate Bax at multiple levels, and have identified a mechanistic switch between its inactive and active states on mitochondria. Identification of what controls this switch may identify critical points at which this process can be targeted in the treatment of disease.
Materials and methods
Cells
Fsk-7 mouse mammary epithelial cells and Bax deficient HCT116 human colon carcinoma cells (kindly provided by B. Vogelstein) were cultured as previously described (13, 39). SV40 transformed bax−/− /bak−/− mouse embryonic fibroblasts (DKO MEFs) were kindly provided by Nika Danial (Dana Faber, Boston), and were grown as described (1).
Antibodies
Polyclonal anti-Bax NT (06-499) was from Upstate Biotechnology. Monoclonal anti-Bax 6A7 (B8429) and 5B7 (B9054) were from Sigma. Polyclonal anti-Bax 62M has been described (7). Polyclonal anti-Bax aa1-171 (sc-6236) was from Sant Cruz. Monoclonal anti-cytochrome c (556432) was from BD Biosciences. Polyclonal anti-GFP (A11122) was from Molecular Probes. Monoclonal mtHsp 70 (MA3-028) was from Affinity Bioreagents. Anti-APAF-1 (AAP-300) was from Stressgen.
Plasmids and transfections
YFP-Bax has been previously described (13). Monomeric RFP (mRFP) was a generous gift from Roger Tsien (University of California, San Diego). The coding sequence for mRFP was cloned into pEYFP-C1-Bax using the restriction enzymes Age I and Bgl II, replacing the EYFP coding sequence and generating pmRFPC1-Bax. Smac-YFP was generated by PCR amplification of the complete coding sequence of murine SMAC, which was cloned into pEYFP-N1 (Clontech). The vector expressing Histone 2B (H2B)-CFP was a gift from Dr. Peter March (Manchester). Bax proline mutants were generated using PCR amplification with Pfu polymerase (Promega), and were ligated into pEYFP-C1 or pmRFPC1. All mutations were confirmed by sequencing. Cells were plated onto coverslips or 60 mm dishes at 80% confluence and transfected as previously described (13). After 24 hours cells were harvested for either immunoblotting, anoikis assays, live cell imaging or immunofluorescence.
Cell fractionation and immunoblotting
Cytosol and membrane fractions were prepared as previously described (13). For analysis of protein expression, cells were lysed in NET buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1% NP-40) with a protease inhibitor cocktail (Calbiochem). 10 μg of protein was separated on a conventional 12.5% reducing SDS-PAGE gel and electroblotted onto nitrocellulose (BioRad). Following 1° antibody incubations, detection was performed using peroxidase conjugated 2° antibodies (Jackson ImmunoResearch) and chemiluminesence (Pierce). To determine protein integration into the OMM, adherent cells were scraped into hypotonic lysis buffer (10mM Tris.Cl, pH 7.5, 10mM NaCl, 1.5 mM MgCl2) containing protease inhibitors and allowed to swell on ice for 5 min prior to homogenising with a glass dounce homogeniser. MS buffer (2.5X; 525 mM mannitol, 175mM sucrose, 12.5 mM Tris.Cl, pH 7.5, 2.5 mM EDTA) was added to 1X. The homogenates were centrifuged at 1300g for 10 min at 4°C to pellet nuclei and unbroken cells. The supernatant was centrifuged as before. The subsequent supernatant was centrifuged at 17000g (S17) for 15 min at 4°C to obtain the mitochondrial pellet. The S17 was centrifuged at 100,000g for 30 min at 4°C to produce the cytosolic fraction. The mitochondrial fraction was washed once with 1X MS buffer and then solubilised in 0.1M Na2CO3 for 30 min on ice. After centrifugation at 100,000g for 30 min at 4°C, the supernatant (carbonate soluble fraction) was saved and the pellet (carbonate insoluble fraction) solubilised in SDS sample buffer. Blue native (BN)-PAGE was carried out essentially as described by Brookes et al.(40), following extraction of membranes in 1% CHAPS, 10% glycerol, 0.5 M aminocaproic acid in 50 mM Bis/Tris, pH 7.0. The 1st dimension BN-PAGE was then resolved in the 2nd dimension by conventional SDS-PAGE, and immunoblotted.
Immunofluorescence and imaging
Anoikis was induced in FSK-7 cells by trypsinising them and plating them onto dishes coated with polyhydroxyethylmethacrylate (poly-HEMA) in complete growth media. To image detached FSK-7 cells, they were cytospun onto polysine slides as previously described (13). Apoptosis was induced in HCT116 Bax deficient cells and SV40 bax−/− /bak−/− MEFs via treatment with 10μM staurosporine (Calbiochem) in DMSO for the time indicated. Cells were fixed in 4% formaldehyde in PBS and permeabilised with 0.5% Triton X100/PBS. 1° antibodies were incubated in PBS/0.1% Triton-X100/0.1% horse serum (1 hour, 37°C). Following washing in PBS, 2° goat anti-mouse or goat anti-rabbit Cy5, Cy2 or RhodamineRx conjugates were incubated in above buffer (30 minutes, 37°C). Where indicated, nuclei were stained with 1 μg/ml Hoechst 33258. Images were collected on an Olympus IX70 microscope, equipped with a Deltavision imaging system, using a 100 × PLAN-APO 1.4NA objective. Images were processed by constrained iterative deconvolution using softWoRx™ (Applied Precision). For images showing relative immunostaining with the Bax N-terminal antibody, identical exposures and brightness/contrast adjustments were made. Apoptosis was quantified by assessing nuclear morphology in transfected cells using an Axioplan2 microscope (Carl Zeiss MicroImaging, Inc.). Transfected cells were identified by YFP-fluorescence, and nuclear morphology assessed following staining with Hoechst. Time course apoptosis assays were analysed with two-way ANOVA with Bonferronis post-test to obtain p values, using Prism 4 software (GraphPad).
Live cell imaging
To image cells undergoing anoikis, we utilised a novel hydrogel which allows immobilisation of non-adherent cells under physiological conditions. Transfected cells were trypsinised and resuspended in growth media with 50% CyGel (Biostatus) on ice. The cell suspension was placed in a 30 mm MatTek dish and covered with a glass coverslip. CyGel was set by transferring cells to 37°C for 5 minutes. Images were captured every 3 minutes with a Leica AS-MDW (37°C, 5% CO2) with a HCX PL APO 63x/1.3NA glycerol objective.
Apoptosis commitment assay
Transiently transfected FSK-7 cells were trypsinised 24 hr post transfection and plated onto poly-HEMA coated dishes for 30 minutes. Identical numbers of transfected cells were then replated in triplicate onto glass coverslips in a 12 well plate precoated with ECM proteins (foetal calf serum, 1 hr at 37°C). For each time point, one coverslip and its supernatant were analysed. YFP-positive cells were quantified in each and the ratio of detached to attached transfected cells determined. The data presented represents three independent experiments.
Acknowledgements
This work was funded by The Wellcome Trust. J-P. U. was supported by a studentship from the BBSRC. We thank Dr. Jennefer Lyndsay for critical reading of the manuscript.
Abbreviations used in this paper
- OMM
Outer mitochondrial membrane
- MMP
mitochondrial membrane permeabilisation
- ECM
extracellular matrix
- GFP
green fluorescent protein
- YFP
yellow fluorescent protein
- CFP
cyan fluorescent protein
- mRFP
monomeric red fluorescent protein
- mtHsp70
mitochondrial heat shock protein 70
- SMAC
second mitochondrial activator of caspases
- APAF-1
apoptotic peptidase activating factor 1
- H2B
histone 2B
- CHAPS
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate
- RIPA buffer
Radioimmunoprecipitation Buffer
- BN-PAGE
blue native polyacrylamide gel electrophoresis
- STS
staurosporine
- Jnk
c-Jun N-terminal kinase
- MEFs
mouse embryonic fibroblasts
- tBid
truncated Bid
References
- 1.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2(3):156–62. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 3.Rehm M, Dussmann H, Prehn JH. Real-time single cell analysis of Smac/DIABLO release during apoptosis. J Cell Biol. 2003;162(6):1031–43. doi: 10.1083/jcb.200303123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8(3):705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 5.Desagher S, Osen Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111(3):331–42. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 7.Gilmore AP, Metcalfe AM, Romer LH, Streuli CH. Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J. Cell Biol. 2000;149:431–445. doi: 10.1083/jcb.149.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, et al. Regulated targeting of BAX to mitochondria. J. Cell Biol. 1998;143:207–215. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. Journal of Biological Chemistry. 1998;273:10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- 10.Antonsson B, Montessuit S, Sanchez B, Martinou JC. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem. 2001;276(15):11615–23. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- 11.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20(3):929–35. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valentijn AJ, Metcalfe AD, Kott J, Streuli CH, Gilmore AP. Spatial and temporal changes in Bax subcellular localization during anoikis. J Cell Biol. 2003;162(4):599–612. doi: 10.1083/jcb.200302154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139(5):1281–92. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilmore AP. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–7. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- 16.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. Journal Of Cell Biology. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang P, Valentijn AJ, Gilmore AP, Streuli CH. Early events in the anoikis program occur in the absence of caspase activation. J Biol Chem. 2003;278(22):19917–25. doi: 10.1074/jbc.M210337200. [DOI] [PubMed] [Google Scholar]
- 18.Rehm M, Dussmann H, Janicke RU, Tavare JM, Kogel D, Prehn JH. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process. Role of caspase-3. J Biol Chem. 2002;277(27):24506–14. doi: 10.1074/jbc.M110789200. [DOI] [PubMed] [Google Scholar]
- 19.Schinzel A, Kaufmann T, Schuler M, Martinalbo J, Grubb D, Borner C. Conformational control of Bax localization and apoptotic activity by Pro168. J Cell Biol. 2004;164(7):1021–32. doi: 10.1083/jcb.200309013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103(4):645–54. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 21.Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. Journal Of Biological Chemistry. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 22.Moro F, Sirrenberg C, Schneider HC, Neupert W, Brunner M. The TIM17.23 preprotein translocase of mitochondria: composition and function in protein transport into the matrix. Embo J. 1999;18(13):3667–75. doi: 10.1093/emboj/18.13.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frisch SM, Vuori K, Kelaita D, Sicks S. A Role For Jun-N-Terminal Kinase In Anoikis - Suppression By Bcl-2 and Crma. Journal Of Cell Biology. 1996;135(n5):1377–1382. doi: 10.1083/jcb.135.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281(30):21256–65. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 25.Papadakis ES, Finegan KG, Wang X, Robinson AC, Guo C, Kayahara M, et al. The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett. 2006;580(5):1320–6. doi: 10.1016/j.febslet.2006.01.053. [DOI] [PubMed] [Google Scholar]
- 26.Nechushtan A, Smith CL, Hsu YT, Youle RJ. Conformation of the Bax C-terminus regulates subcellular location and cell death. Embo Journal. 1999;18:2330–2341. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soucie EL, Annis MG, Sedivy J, Filmus J, Leber B, Andrews DW, et al. Myc potentiates apoptosis by stimulating Bax activity at the mitochondria. Mol Cell Biol. 2001;21(14):4725–36. doi: 10.1128/MCB.21.14.4725-4736.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C. Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol. 2003;160(1):53–64. doi: 10.1083/jcb.200210084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Saez AJ, Mingarro I, Perez-Paya E, Salgado J. Membrane-insertion fragments of Bcl-xL, Bax, and Bid. Biochemistry. 2004;43(34):10930–43. doi: 10.1021/bi036044c. [DOI] [PubMed] [Google Scholar]
- 30.Heimlich G, McKinnon AD, Bernardo K, Brdiczka D, Reed JC, Kain R, et al. Bax-induced cytochrome c release from mitochondria depends on alpha-helices-5 and -6. Biochem J. 2004;378(Pt 1):247–55. doi: 10.1042/BJ20031152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. 2001;153(6):1265–76. doi: 10.1083/jcb.153.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capano M, Crompton M. Biphasic translocation of Bax to mitochondria. Biochem J. 2002;367(Pt 1):169–78. doi: 10.1042/BJ20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301(5632):513–7. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 34.Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2(1):63–7. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 35.Su J, Wang G, Barrett JW, Irvine TS, Gao X, McFadden G. Myxoma virus M11L blocks apoptosis through inhibition of conformational activation of Bax at the mitochondria. J Virol. 2006;80(3):1140–51. doi: 10.1128/JVI.80.3.1140-1151.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poncet D, Larochette N, Pauleau AL, Boya P, Jalil AA, Cartron PF, et al. An anti-apoptotic viral protein that recruits Bax to mitochondria. J Biol Chem. 2004;279(21):22605–14. doi: 10.1074/jbc.M308408200. [DOI] [PubMed] [Google Scholar]
- 37.Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, et al. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci U S A. 2004;101(21):7988–93. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem. 2003;278(49):48935–41. doi: 10.1074/jbc.M306289200. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290(5493):989–92. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 40.Brookes PS, Pinner A, Ramachandran A, Coward L, Barnes S, Kim H, et al. High throughput two-dimensional blue-native electrophoresis: a tool for functional proteomics of mitochondria and signaling complexes. Proteomics. 2002;2(8):969–77. doi: 10.1002/1615-9861(200208)2:8<969::AID-PROT969>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]