

Abstract
The active sites of metalloenzymes are often deeply buried inside a hydrophobic protein sheath, which protects them from undesirable hydrolysis and polymerization reactions, allowing them to achieve their normal functions. In order to mimic the hydrophobic environment of the active sites in bacterial monooxygenases, diiron(II) compounds of the general formula [Fe2([G-3]COO)4(4-RPy)2] were prepared, where [G-3]COO− is a third-generation dendrimer-appended terphenyl carboxylate ligand and 4-RPy is a pyridine derivative. The dendrimer environment provides excellent protection for the diiron center, reducing its reactivity toward dioxygen by about 300-fold compared with analogous complexes of terphenyl carboxylate ([G-1]COO−) ligands. An FeIIFeIII intermediate was characterized by electronic, electron paramagnetic resonance, Mössbauer, and X-ray absorption spectroscopic analyses following the oxygenation of [Fe2−([G-3]COO)4(4-PPy)2], where 4-PPy is 4-pyrrolidinopyridine. The results are consistent with the formation of a superoxo species. This diiron compound, in the presence of dioxygen, can oxidize external substrates.
Introduction
The active sites of metalloenzymes are often surrounded by a hydrophobic sheath of amino acids that shields them from undesirable hydrolysis and polymerization reactions and facilitates their normal functions. Dendrimers are highly branched molecules with well-defined 3-D structures. As in metalloenzymes, a site-isolated environment can be provided by dendrons arranged to protect a core moiety from unwanted side reactions.1 This distinctive property of dendrimers makes them excellent candidates to mimic a hydrophobic metalloprotein core framework. Dendritically functionalized iron(II) porphyrins are able to bind O2 reversibly and model the chemistry of heme proteins.2 In addition, a porphyrin compound with a dendrimer at its periphery exhibited size and shape selectivity due to cavities formed within the dendrons.3 A dendrimer-derived mimic of a non-heme diiron protein was achieved when modified triazacyclononanes were used as ligands to prepare diiron(III) complexes, which could be reduced to their diiron(II) forms upon irradiation.4 The fourth-generation dendrimer diiron(III) compound used in this work was highly robust against alkaline hydrolysis, demonstrating that the dendrons stabilize the active site just as do hydrophobic protein frames.
Terphenyl carboxylate ligands recently developed in our laboratory5,6 and elsewhere7 form diiron(II) complexes that closely resemble the active site of the hydroxylase of soluble methane monooxygenase (MMOH) in its reduced state. In the presence of dioxygen, several of these complexes convert to di(μ-hydroxo)diiron(III) species that mimic the oxidized state of MMOH.6,8–12 In some of this chemistry, the formation of peroxodiiron(III) and/or di(μ-oxo)diiron(IV) intermediates was suggested, although no direct evidence for such species was obtained.6
In the present study we have modified the terphenyl carboxylate ligand framework to incorporate a dendrimer sheath by utilizing, in particular, a poly(benzylether) carboxylic acid [G-3]-COOH (7). Because of the dendritic polybenzylether units and terminal long-chain alkyl groups of 7, we anticipated that its diiron complexes would be highly soluble in a variety of organic solvents and that the dimetallic center would be well protected. Our previous research on diiron model compounds revealed that oxygenated intermediates could often be observed at low temperatures, T < –78 °C. With the dendritic shield, we envisaged increased thermal stability, since solvent and substrate access is more restricted, and that we would more readily be able to investigate the structure and reactivity of these intermediates.
Experimental Section
General Considerations
All reagents were purchased from commercial sources and used as received except for Fe(OTf)2·2MeCN,13 2,6-dibromo-3,5-dihydroxybenzoic acid,14 [FeII2(μ-O2CArTol)2(O2CArTol)2−(Py)2] (A),5 [FeII2(μ-O2CArTol)4(t-Bu-Py)2] (B),8 and [FeIII2(μ-OH)2(μ-O2CArTol)2(O2CArTol)2(t-Bu-Py)2](C),8 which were prepared according to literature procedures. All solvents were saturated with nitrogen and purified by passage through activated Al2O3 columns under nitrogen. Air-sensitive manipulations were performed under nitrogen in an MBraun glovebox.
Physical Measurements
1H NMR spectra were recorded on Inova 300 MHz and Bruker 500 MHz DRX spectrometers. Chemical shifts were referenced to residual deuterated solvent peaks. FT-IR spectra were recorded on an Avatar 360 FT-IR instrument, and samples were prepared as KBr pellets. UV–vis spectra were recorded on a Hewlett-Packard 8453 diode array spectrophotometer. ESI-MS spectrometry was performed with an Agilent 1100 Series LC/MSD system. MALDI-TOF mass spectrometry was performed on a Voyager-DE instrument from PerSeptive Biosystems using α-cyanohydroxycinnamic acid as a matrix. FAB and EI mass spectrometry was performed by the UC Berkeley mass spectrometry facility.
tert-Butyl 2,6-Dibromo-3,5-dihydroxybenzoate (1)
A solution containing 2,6-dibromo-3,5-dihydroxybenzoic acid (3.12 g, 10 mmol), di-tert-butyl dicarbonate (8.72 g, 40 mmol), and magnesium perchlorate (22 mg, 0.1 mmol) in 50 mL of nitromethane and 50 mL of acetone was heated at 40 °C for 16 h. The reaction mixture was then diluted with 200 mL of ethyl acetate and washed successively with saturated NH4Cl, saturated NaHCO3, water, and brine. The organic layer was dried over MgSO4 and concentrated. The residue was purified by column chromatography using an ethyl acetate/hexanes gradient of 1:2 to 2:1 (v/v). The product was obtained as a colorless solid (1.84 g, 50%). 1H NMR (acetone-d6) δ (ppm): 9.2 (br, 2H), 6.75 (s, 1H), 1.60 (s, 9H). 13C NMR (acetone-d6) δ (ppm): 164.4, 154.2, 139.8, 103.5, 96.7, 82.9, 27.3. FT-IR (cm−1): 3469, 3158, 2980, 2930, 2810, 2666, 2579, 2493, 1690, 1576, 1476, 1453, 1424, 1385, 1368, 1337, 1294, 1248, 1157, 1065, 1045, 971, 834, 807, 739, 690. LR-MS (FAB), Calcd for C11H12Br2O4: m/z 366. Found: m/z 367 ([M + H]+, 30), 312(30), 295(15), 154(100), 136(75). HR-MS (FAB), Calcd for C11H12Br2O4: m/z 365.9102. Found: m/z 365.9110. Anal. Calcd for C11H12Br2O4: C, 35.90; H, 3.29. Found: C, 36.20; H, 3.36.
tert-Butyl 2,6-Dibromo-3,5-di(benzyloxy)benzoate (2)
A portion of benzyl bromide (2.08 g, 12.2 mmol), 1 (2.00 g, 5.42 mmol), and 18-crown-6 (284 mg, 1.08 mmol) were dissolved in 40 mL of acetone. To this solution was added finely pulverized anhydrous K2CO3 (4.50 g, 32.6 mmol). After 16 h at gentle reflux, the reaction mixture was cooled to room temperature and diluted with ethyl acetate, filtered, and concentrated. The product was obtained as colorless needles (2.85 g, 96%) after recrystallization from a mixture of hexane and ethyl acetate (15:1, v/v). 1H NMR (CDCl3) δ (ppm): 7.38–7.32 (m, 10H), 6.50 (s, 1H), 5.06 (s, 4H), 1.64 (s, 9H). 13C NMR (CDCl3) δ (ppm): 164.7, 155.1, 140.0, 135.8, 128.8, 128.3, 127.0, 101.2, 101.1, 84.1, 71.5, 28.1. FT-IR (cm−1): 3087, 3065, 3032, 2978, 2932, 2875, 1730, 1572, 1497, 1454, 1437, 1392, 1369, 1337, 1246, 1226, 1198, 1157, 1089, 1047, 1027, 908, 843, 810, 791, 735, 697. LR-MS (EI), Calcd for C25H24Br2O4: m/z 546. Found: m/z 546 ([M]+, 20), 494(30), 492(15), 475(80), 473(40), 458(20), 456(10), 402(60), 400(30), 385(40), 383(20), 91(100). HR-MS (EI), Calcd. for C25H24Br2O4: m/z 546.0041. Found: m/z 546.0046. Anal. Calcd for C25H24Br2O4: C, 54.77; H, 4.41. Found: C, 54.40; H, 4.65.
tert-Butyl 3,5-Di(benzyloxy)-2,6-diphenylbenzoate (3)
A reaction mixture containing 2 (3.87 g, 7.05 mmol), phenylboronic acid (12.9 g, 106 mmol), Pd(PPh3)4 (2.03 g, 1.76 mmol), and K3PO4 (30.0 g, 141 mmol) in 150 mL of dry 1,2-dimethoxyethane was subjected to three freeze–pump–thaw cycles before heating at 95 °C for 72 h under an argon atmosphere. The crude mixture was diluted with diethyl ether and then extracted successively with saturated NH4Cl, saturated NaHCO3, water, and then brine. The organic layer was dried over MgSO4 and concentrated. The product was obtained as a colorless solid (2.75 g, 72%) after column chromatography using a gradient of 4:1 (v/v) chloroform/hexanes to chloroform. Recrystallization from hexanes yielded colorless needles. 1H NMR (CDCl3) δ (ppm): 7.40–7.25 (m, 16H), 7.16 (d, 2H, J 6.8 Hz), 6.66 (s, 1H), 4.96 (s, 4H), 0.95 (s, 9H). 13C NMR (CDCl3) δ (ppm): 166.8, 155.6, 138.1, 136.9, 135.9, 130.5, 128.4, 127.6, 127.6, 127.0, 126.7, 122.1, 101.0, 81.5, 70.8, 27.2. FT-IR (cm−1): 3087, 3061, 3030, 2974, 2931, 2875, 1723, 1602, 1585, 1497, 1454, 1368, 1325, 1303, 1255, 1222, 1169, 1083, 1051, 1029, 910, 849, 830, 818, 790, 762, 736, 698. LR-MS (FAB), Calcd for C37H34O4: m/z 542. Found: m/z 543 ([M + H]+, 100), 487(40), 469(25). HR-MS (FAB), Calcd for C37H34O4: m/z 542.2457. Found: m/z 542.2459. Anal. Calcd for C37H34O4: C, 81.89; H, 6.32. Found: C, 81.81; H, 6.51.
tert-Butyl 3,5-Dihydroxy-2,6-Diphenylbenzoate (4)
Hydrogenolysis of 3 (1.93 g, 3.55 mmol) was accomplished using a suspension of 10% Pd/C (200 mg) in 50 mL of a 1:1 (v/v) mixture of methanol and dichloromethane. The reaction vessel was evacuated and back-filled with hydrogen gas three times before stirring at ambient temperature for 16 h. The Pd/C was filtered though Celite, which was then washed several times with the same solvent mixture used in the reaction. After removal of the volatiles in vacuo, the product was obtained as a colorless solid (1.25 g, 97%) that required no further purification. 1H NMR (CDCl3) δ (ppm): 7.48−7.39 (m, 10H), 6.68 (s, 1H), 5.11 (s, 2H), 0.95 (s, 9H). 13C NMR (CDCl3) δ (ppm): 166.6, 153.3, 136.5, 133.7, 130.6, 129.1, 128.4, 118.3, 102.7, 81.6, 27.2. FT-IR (cm−1): 3505, 3485, 3058, 3023, 3004, 2977, 2931, 2249, 1721, 1696, 1608, 1593, 1496, 1472, 1460, 1392, 1368, 1320, 1253, 1153, 1073, 1040, 968, 910, 848, 790, 764, 733, 702. LR-MS (FAB), Calcd for C23H22O4: m/z 362. Found: m/z 362 ([M]+, 100), 307(90), 289(50). HR-MS (FAB), Calcd for C23H22O4: m/z 362.1518. Found: m/z 362.1521. Anal. Calcd for C23H22O4: C, 76.22; H, 6.12. Found: C, 76.12; H, 5.94.
[G-3]-Ester (6)
The second-generation bromide dendron 515 (1.62 g, 1.44 mmol) was dissolved in 10 mL of acetone along with 4 (250 mg, 0.691 mmol) and 18-crown-6 (18 mg, 0.068 mmol). To this solution was added finely pulverized anhydrous K2CO3 (571 mg, 4.14 mmol). After 16 h at gentle reflux, the reaction mixture was cooled and diluted with ethyl acetate, filtered, and concentrated. The product was obtained as a colorless glass (1.67 g, 99%) after column chromatography using a mixture of hexane and ethyl acetate (15:1, v/v) as eluent. 1H NMR (CDCl3) δ (ppm): 7.44 (d, 4H, J 7.2 Hz), 7.37 (t, 4H, J 7.3 Hz), 7.29 (t, 2H, J 7.2 Hz), 6.75 (s, 1H), 6.53 (s, 8H), 6.49 (s, 2H), 6.44 (s, 4H), 6.41 (s, 4H), 4.98 (s, 4H), 4.79 (s, 8H), 3.93 (t, 16H, J 6.6 Hz), 1.76 (m, 16H), 1.42 (m, 16H), 1.26 (m, 128H), 0.94 (s, 9H), 0.88 (t, 24H, J 6.8 Hz). 13C NMR (CDCl3) δ (ppm): 166.7, 160.5, 160.0, 155.8, 139.2, 138.9, 138.0, 136.0, 130.7, 127.6, 127.0, 122.0, 105.7, 104.9, 101.6, 100.7, 100.0, 81.5, 70.5, 70.0, 68.0, 31.9, 29.63, 29.60, 29.58, 29.56, 29.39, 29.31, 29.25, 26.0, 22.7, 14.1. FT-IR (cm−1): 2923, 2853, 1725, 1597, 1450, 1377, 1331, 1289, 1258, 1160, 1057, 830, 720, 698, 673. LR-MS (MALDI-TOF): Calcd. for C161H250O16: m/z 2439.9; Found: m/z 2462.4 ([M + Na]+, 100). Anal. Calcd for C161H250O16: C, 79.20; H, 10.32. Found: C, 79.53; H, 10.59.
[G-3]-COOH (7)
Thermolysis of 6 (1.50 g, 0.614 mmol) was performed in 10 mL of quinoline at 220 °C for 16 h. The reaction mixture was diluted with 50 mL of ethyl acetate, washed with 2.0 M HCl (5 × 25 mL), dried, and concentrated. The title compound was obtained as a pale brown glass (1.40 g, 96%) after column chromatography using a mixture of hexane and ethyl acetate (9:1, v/v). 1H NMR (CDCl3) δ (ppm): 7.44 (d, 4H, J 7.2 Hz), 7.37 (t, 4H, J 7.2 Hz), 7.32 (t, 2H, J 7.2 Hz), 6.76 (s, 1H), 6.53 (s, 8H), 6.49 (s, 2H), 6.44 (s, 4H), 6.41 (s, 4H), 4.98 (s, 4H), 4.79 (s, 8H), 3.92 (t, 16H, J 6.6 Hz), 1.76 (m, 16H), 1.42 (m, 16H), 1.26 (m, 128H), 0.88 (t, 24H, J 6.8 Hz). 13C NMR (CDCl3) δ (ppm): 168.9, 160.5, 160.0, 155.9, 139.1, 138.9, 135.7, 135.4, 130.4, 127.9, 127.4, 122.3, 105.8, 105.0, 101.7, 100.8, 70.6, 70.1, 68.1, 32.0, 29.71, 29.67, 29.66, 29.63, 29.46, 29.39, 29.32, 26.1, 22.7, 14.2. FT-IR (cm−1): 2922, 2853, 1702, 1599, 1451, 1375, 1327, 1219, 1158, 1051, 920, 849, 830, 772, 704, 682. LR-MS (MALDI-TOF), Calcd for C157H242O16: m/z 2383.8. Found: m/z 2410.7 ([M + Na]+, 100), 2426.6 ([M + K]+, 20). Anal. Calcd for C157H242O16: C, 79.09; H, 10.22. Found: C, 79.21; H, 10.37.
[G-3]-COONa (8)
Compound 7 (130 mg, 54.5 μmol) was dissolved in 4 mL of THF, and to this solution was added 19 μL of 3 N aq NaOH, followed by 1 mL of MeOH to make the reaction mixture homogeneous. The solution was allowed to reflux for 5 h. The solvents were then removed under reduced pressure. The residue was dissolved in pentane and filtered through Celite. Pentane was evaporated to afford a light yellow solid, which was dried under vacuum at 80 °C overnight to yield 136 mg of product (100% yield). FT-IR (cm−1): 2923 (s), 2853 (m), 1599 (s), 1574 (s), 1456 (m), 1375 (m), 1326 (w), 1169 (s), 1055 (m), 830 (m), 761 (w), 722 (w), 700 (w), 682 (w). 1H NMR (CDCl3) δ (ppm): 7.47 m, (4H), 7.28 (m, 6H), 6.71 (s, 1H), 6.60 (d, 8H), 6.55 (s, 2H), 6.47 (s, 8H), 4.98 (s, 4H), 4.83 (s, 8H), 3.97 (t, 16H), 1.83 (m, 16H), 1.51 (m, 16H), 1.36 (m, 128H), 0.98 (t, 24H). LR-MS (ESI), Calcd for C157H241O16Na: m/z 2406. Found: m/z 2383 ([M – Na]–, 100)
[Fe2([G-3]COO)4(4-RPy)2] (9-R)
Compound 8 (96.2 mg, 40.0 μmol) and Fe(OTf)2·2MeCN (10.9 mg, 25.0 μmol) were mixed in THF and stirred at room temperature for 10 h. The volatile fraction was removed in vacuo, and the residue was dissolved in pentane. Insoluble materials were filtered off. Excess 4-R-pyridine (4-cyanopyridine, 4-CNPy; or 4-pyrrolidinopyridine, 4-PPy) was then added to the yellow filtrate, and the mixture was stirred vigorously for 2 h. Solvent was removed, pentane (2 mL) was added to dissolve the dendrimer product, and the solution was filtered. These procedures were repeated three times to afford the diiron(II) products as a red (for 4-CNPy) or yellow (for 4-PPy) sticky solid. 9-CN: Yield 95%. An 57Fe-enriched sample of 9-P was prepared from 57Fe-enriched (40%) Fe(OTf)2·2MeCN, which was synthesized by reaction of triflic acid with a mixture of 57Fe and natural Fe powder (2:3, w/w).13 FT-IR (cm−1): 2924 (s), 2853 (s), 2238 (w, νC≡N), 1598 (s), 1454 (m), 1378 (m), 1323 (m), 1167 (s), 1054 (m), 830 (m) 765 (w), 721 (w), 703 (w), 682 (w). Anal. Calcd for C640H992N4O74Fe2 (9-CN + 10H2O): C, 76.57; H, 9.96; N, 0.56. Found: C, 76.58; H, 9.86; N, 0.64. 9-P: Yield 90%. FT-IR (cm−1): 2923 (s), 2853 (s), 1599 (s), 1559 (w), 1527 (w), 1457 (m), 1378 (m), 1312 (w), 1296 (w), 1258 (w), 1226 (w), 1163 (s), 1055 (m), 1014 (w), 830 (m), 805 (w), 786 (w), 765 (w), 721 (w), 701 (w), 682 (w). Anal. Calcd for C646H1008N4O74Fe2 (9-P + 10H2O): C, 76.62; H, 10.03; N, 0.55. Found: C, 76.48; H, 9.80; N, 0.54.
Dioxygen Reactivity Studies
Measurements were carried out in a UV–vis cell equipped with a Dewar vessel for cooling. Diiron(II) complexes were dissolved to a concentration of 0.2−0.4 mM in a drybox and loaded into the cell, which was then sealed with a septum and cooled to −29 °C in a nitromethane/dry ice bath. After the initial spectrum was recorded, O2 gas was bubbled into the solution through a long needle for 30 s. Spectra were then recorded every 30 s for 15 min and with an increment of 20% in the cycle time thereafter. For substrate reactions, this procedure was continued for 1.5 h to achieve full conversion to the oxygenated intermediate 10. Ar was then bubbled through the solution for 15−20 min to remove excess O2. At this point, substrate was injected via an airtight syringe. The reaction temperature was maintained at −29 °C for 2 h, and then the solution was gradually allowed to warm up to room temperature overnight. During workup, Chelex was added to the reaction mixture, which was then stirred for two h to remove metal ions. The Chelex was filtered off and washed with a copious amount of CH2Cl2. The filtrates were combined, and the solvent was removed in vacuo. The products were analyzed by GC-MS with 1-bromodecane as an internal standard.
GC-MS Analyses
All gas chromatography was performed on an Agilent 6890 gas chromatograph connected to an Agilent 5973 Network mass selective detector. An HP-5ms (5%-phenyl)-methylpolysiloxane capillary column (30 m × 0.25 mm × 0.25 μm) was employed. Conditions for the 9,10-dihydroanthracene (DHA) reaction were as follows: initial temperature of 150 °C with initial time of 5 min, then the temperature was raised at 15 °C/min to 250 °C and held for a final time of 10 min. For the anthrone reaction: initial temperature = 150 °C, initial time = 5 min, ramp rate = 50 °C/min to 230 °C, final time = 10 min. Products were identified by their mass spectra and retention times compared with those of authentic samples. Quantification was made by comparison of the total ion count of the peaks interested with that of the internal standard. Calibration plots for the detector response were prepared from authentic samples of known concentration and with an internal standard.
Stopped-Flow Kinetic Studies
Kinetics experiments were carried out by using a Canterbury stopped-flow SF-40 and MG-6000 rapid diode array system (Hi-Tech Scientific) under ambient pressure. To avoid moisture and pre-oxidation, the instrument employs stainless-steel flow lines and an argon-purged anaerobic attachment. The mixing cell was maintained to ±0.1 °C. All lines of the instrument were extensively washed with dioxygen-free anhydrous solvent before experiment. Solutions of 9-P (75.2 μM in CH2Cl2 before mixing) were prepared in a drybox and stored in an airtight syringe prior to loading into the stopped-flow apparatus. A saturated solution of dioxygen was prepared by bubbling the gas through CH2Cl2 for 20 min in a septumsealed round-bottomed flask maintained at 20 °C, and the concentration was taken as 5.8 mM before mixing.16 A total of 300 scans were taken within 1050 s.
Mössbauer Spectroscopy
Spectra were recorded on an MSI spectrometer (WEB Research Co.) using a 57Co source in a Rh matrix. All samples were prepared with 57Fe-enriched (40%) compounds. The solid sample of 9-P was prepared by mixing ∼25 mg of the compound in Apiezon N grease, coating the mixture on the lid of a nylon holder, and freezing in liquid nitrogen. The frozen toluene solution sample of 10 was prepared by the following procedure. Dioxygen was introduced into a toluene solution (400 μL) of 9-P (∼25 mg) at −29 °C. After 1.5 h, the reaction mixture was cooled in an acetone/CO2 bath and transferred via pipet to a sample holder where the lid was permanently assembled using epoxy. The sample was then immersed in liquid nitrogen to freeze the liquid. After the measurement, the sample was warmed in the holder to room temperature for 1 h to obtain the fully oxidized product (11) and then frozen again for measurement. Data were collected at 4.2 K. Calibration was done with natural iron foil at room temperature for isomer shift values. The WMOSS plot and fit program17 was used to fit the spectra to Gaussian lines.
Electron Paramagnetic Resonance (EPR) Spectroscopy
X-band EPR spectra were recorded on a Bruker Model 300 ESP spectrometer. The temperature was controlled with a liquid-helium EPR 900 cryostat (Oxford Instruments). Samples of 9-P were prepared as a 1 − 1.5 mM solution in toluene or CH2Cl2 in the drybox. One sample was frozen to 77 K immediately when it was brought outside of the drybox. Another was cooled at −29 °C. O2 was bubbled for 30 s through the solution, which was frozen to 77 K after 1.5 h. Spin quantification was done with copper triflate as a concentration standard. Values obtained by double integration of the signals were divided by a correcting factor that is a function of the principal g values.18
X-ray Absorption Spectroscopy (XAS)
Samples of 9-P, 10, and 11 were prepared as 2.56 mM solutions in toluene and kept in liquid N2 until used. Procedures similar to those used in the Mössbauer sample preparation were adopted to generate 10 and 11, which were then transferred to Lucite sample cells with a Kapton tape window via pipet. Samples A–C were ground with BN and pressed into a 1 mm thick Al sample holder under an inert atmosphere. The X-ray absorption spectra were recorded at the Stanford Synchrotron Radiation Laboratory (SSRL) on focused 20-pole wiggler beam line 7−3 for 9-P, 10, 11, and C and the 2−3 beam line for A and B, with the ring operating at 3 GeV, 50−100 mA. A Si(220) double-crystal monochromator was utilized for energy selection at the Fe K edge, detuned 50% at 8000 eV to minimize contamination of the radiation by higher harmonics on beam line 2−3. On beam line 7−3, a Rh-coated mirror upstream of the monochromator was used for harmonic rejection. The samples were maintained at 10 K during data collection by using an Oxford Instruments CF1208 continuous-flow liquid-helium cryostat. Data were measured in fluorescence mode for 9-P, 10, and 11 with a Canberra Ge 30-element array detector and in transmission mode for A-C, using ionization chambers filled with N2. Internal energy calibration was performed by simultaneous measurement of the absorption of Fe foil placed between two ionization chambers located after the sample. The first inflection point of the foil was assigned to 7111.20 eV. No photoreduction effects were observed for any of the data sets. The averaged data included 33 scans for 9-P, 21 scans for 10, 15 scans for 11, 11 scans for A, and 9 scans for B and C.
The averaged data were normalized with the program XFIT19 by first subtracting a polynomial background absorbance that was fit to the preedge region and extended over the postedge with control points, followed by fitting a three-region polynomial spline of orders 2, 3, and 3 over the postedge region. The data were normalized to an edge jump of 1.0 between the background and spline curves at 7130 eV. Theoretical extended X-ray absorption fine structure (EXAFS) signals χ(k) were calculated using FEFF (version 7.02)20 and fit to the data by EXAFSPAK (G. N. George, SSRL). The experimental energy threshold, E0, the point at which the photoelectron wavevector k = 0, was chosen as 7130 eV and was varied in each fit as a common value for every component. The scale factor, S02, was set to 1.0. The structural parameters that were varied during the refinements included the bond distance (R) and the bond variance (σ2). Atom types and coordination numbers were systematically varied during the course of the analysis, but were not allowed to vary within a given fit. Data were fit over the region of k = 2−15 Å−1.
The intensities and energies of the preedge features for all samples were quantified by using the fitting program EDG_FIT (G. N. George, SSRL) and established methodology.21 All spectra were fit over several energy ranges (7108−7116, 7108−7117, 7108−7118, and 7108−7119 eV), with varied backgrounds to give nine fits per sample. Each preedge feature was modeled with pseudo-Voigt line shapes of a fixed 1:1 ratio of Lorentzian to Gaussian contributions. The backgrounds were chosen to give a best fit to the preedge area while reproducing edge features. Each fit was considered successful if it simultaneously reproduced the data and the second derivative of the data over the entire energy range. The total area is the sum of the areas of all preedge features, where the area is approximated by peak height multiplied by the full width at half-maximum, scaled by 100. Error was calculated by determining the standard deviation for peak heights and half widths for each preedge feature from all successful fits.
Results and Discussion
Design, Synthesis, and Characterization of Diiron Dendrimer Complexes
The molecular design of dendritic ligands capable of assembling uniquely only two iron moieties at a central carboxylate-rich core was inspired by the family of terphenyl carboxylates.5–7,9,10,22 The basic structure of this ligand was modified so as to incorporate convenient points for dendronization with C12-terminated Fréchet-type poly(benzyl ether)dendritic wedges using a convergent approach.23 A short synthetic route to the central core unit 4 was devised from 2,6-dibromo-3,5-dihydroxybenzoic acid as a starting material (Scheme 1).14 Magnesium perchlorate was employed as a Lewis acid catalyst in the selective decarboxylative esterification of the benzoate functionality (1).24 The phenolic moieties were then alkylated with benzyl bromide in a Williamson-type etherification facilitated by 18-crown-6 as a phase transfer catalyst. The arylation of 2 via palladium-catalyzed cross-coupling was nontrivial given the hindered nature of the reactant. After several attempts, Suzuki coupling with Pd(PPh3)4 as the catalyst demonstrated superior yields. The role of the base in this step was also noteworthy because neither Cs2CO3 nor K2CO3 gave the desired product, whereas K3PO4 was able to furnish 3 readily in 72% yield. The benzyl groups of 3 were removed after hydrogenolysis using Pd/C to give the central core unit poised for further dendronization.
Scheme 1.

Synthetic Route to tert-Butyl-3,5-dihydroxy-2,6-diphenylbenzoate, 4
C12-terminated Fréchet-type poly(benzyl ether)dendritic wedges were chosen for the dendronization of 4 so that the fully assembled dendritic complex would be both highly globular and soluble in a range of organic solvents (Scheme 2). Our previous experience with these wedges and dendrimers thereof suggested that they would be ideal candidates for the encapsulation and protection of the diiron active site, for their performance in other catalyst constructs has been similarly successful.15 The second-generation dendritic wedges 5 were coupled to 4 by standard methodologies,23 giving the tert-butyl-protected ligands 6 in high yields. The tert-butyl group of 6 was then removed via thermolysis in quinoline at 220 °C to give the carboxylic acid derivative 7. We report here diiron complexes derived from 7.
Scheme 2.

Synthesis of the Third-Generation Dendrimer-Appended Carboxylate Ligand
A full comparison of compounds with both the third- and fourth-generation ligands will be made in due course.
The standard synthetic protocol for the preparation of carboxylate-rich diiron complexes involves deprotonation of carboxylic acid to ensure efficient complexation.9 Compound 7 could not be deprotonated by NaOH at room temperature, however. A modified procedure25 was therefore adopted, utilizing a mixed-solvent system comprising THF, water, and MeOH to make a homogeneous reaction mixture. The solution was then brought to reflux for 5 h, during which time the acid was fully deprotonated by NaOH to afford 8. Its formation was demonstrated by loss of the C=O stretch of the free acid (1702 cm−1) and the appearance of two C=O stretches arising from the carboxylate anion (1599 and 1574 cm−1) in the IR spectrum. Compound 8 was then mixed with Fe(OTf)2·2MeCN in THF for over 10 h (Scheme 3), resulting in a clear yellow solution. This behavior is the same as that observed for the simple terphenyl carboxylate ArTolCO2−,9 indicating the formation of the corresponding diiron complex with THF ligands. After removing the THF solvent, pentane was added and the insoluble triflate salts were filtered off. Without further purification, the filtrate was further exposed to solid 4-RPy suspended in pentanes. Unreacted 4-RPy was removed by copious pentane dissolution/filtration from the heterogeneous mixture.
Scheme 3.

Synthesis of Dendrimer-Appended Diiron(II) Complexes 9-R
The flexibility of the dendrimer unit makes it impossible to obtain crystal structures to characterize the resulting products, 9-CN and 9-P, but we have a sufficiently good understanding of related complexes through spectroscopic and other characterization methods that we can assign the chemical composition and probable structure with confidence. First, the elemental analyses establish the presence of one N donor and two dendrimer ligands per iron atom. Although from this information alone we cannot rule out the possibility of a mononuclear iron complex with two carboxylates and one N-donor ligand, previous work on related mononuclear Fe(II) compounds revealed that two carboxylate and two N-donor ligands are typically associated with the metal center unless the N donor is bidentate.26 In addition, no mononuclear iron(II) complex formed when [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] was allowed to react with excess 4-cyanopyridine (4-CNPy). Second, the product of the reaction of 4-CNPy with the dendrimer precursor has an optical absorbance at 463 nm (ε = 2600 cm−1 M−1) in anhydrous CH2Cl2. The value is quite similar to that (λmax 460 nm, ε = 2200 cm−1 M−1) of [Fe2(O2CArTol)2(μ-O2CArTol)2(4-CNPy)2(H2O)2], a windmill-structured dinuclear species.11,27 A more direct, third piece of evidence is provided by both X- and Q-band EPR spectra, in which a broad g = 16 signal characteristic of a coupled diiron(II) center clearly indicates the presence of a dinuclear complex (Figure S1, Supporting Information). This assignment is also consistent with the measurements described in the XAS section (vide infra). Thus, taken together, the data indicate that dinuclear windmill complexes form with the dendrimer-appended ligand. The use of water in the synthesis of 8 and the elemental analysis are consistent with the formula [Fe2([G-3]COO)4(4-RPy)2]·10H2O.
Dioxygen Reactivity
Reaction of a toluene solution of 9-CN with dioxygen at −29 °C led to a slow reduction in intensity of the 475 nm absorption band with no indication of the formation of an intermediate (Figure S2, Supporting Information). The kinetics of this reaction were fit to a pseudo-first-order equation with kobs = 6.6(9) × 10−4 s−1. The reaction of the analogous paddlewheel compound [Fe2(μ-O2CArTol)4(4-CNPy)2] with dioxygen occurs with a rate constant almost 300-fold larger at this temperature.27 We attribute this difference to the dendrimer framework, which limits diffusion of dioxygen into the diiron core and provides an increased energy barrier for structural rearrangements that may be required for the chemistry associated with dioxygen binding.
When O2 was bubbled into a CH2Cl2 solution of 9-P at −29 °C, an optical band at 442 nm (ε = 6000 cm−1 M−1) grew in, reaching a maximum at about 1.5 h (Figure 1a). This oxygenated intermediate, 10, was stable as long as the temperature was maintained below −5 °C, judging by the intensity of this absorbance. The same behavior was also observed in other solvents, specifically chloroform and toluene. When Ar was used to remove excess dioxygen, the intensity of the 442 nm peak remained the same, indicating irreversible binding. Upon warming to room temperature, the absorption maximum shifted to 429 nm (ε = 5000 cm−1 M−1), which we attribute to formation of the diiron(III) species 11, discussed below.
Figure 1.
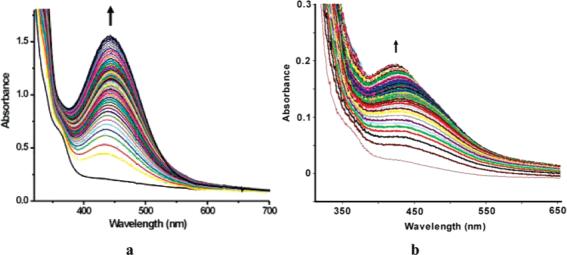
Optical changes observed when 9-P reacts with O2 in CH2Cl2. (a) At −29 °C (0.252 mM). Spectra were recorded every 30 s up to 15 min and with an increment of 20% in the sampling thereafter. (b) At 20 °C (0.0376 mM). A total of 300 scans were taken in 1050 s.
Reaction of 9-P with O2 at 20 °C leads to the formation of 10, which dominates the spectrum for about 100 s as indicated by its absorbance at 442 nm (Figure 1b). Compound 10 subsequently decays to 11, overwhelming the spectral window. The intensity change at 442 nm was fit to an A → B → C process as indicated by eq 1, where k1′ = k1[O2], since excess dioxygen ensures pseudo-first-order conditions.
| (1) |
When ε10 and ε9-P were fixed at 6000 and 1000 cm−1 M−1, respectively, and the initial concentration of 9-P was 0.0376 mM, the fit (Figure S3, Supporting Information) yielded k1′ = 0.0132(8) s−1, k2 = 0.04 (2) s−1, and ε11 = 4523(5) cm−1 M−1. The rate constants k1′ and k2 are of the same order of magnitude, consistent with the fact that both 10 and 11 appeared within a short period of time at this temperature. This chemistry is attributed to eq 2, and the nature of 10 and 11 is further addressed below.
| (2) |
Mössbauer and EPR Studies
The Mössbauer spectrum of 9-P (Figure 2a) displays a single sharp quadrupole doublet, indicating the two iron sites to be indistinguishable. The isomer shift (δ = 1.15 mm/s) and quadrupole splitting (ΔEQ = 2.76 mm/s) are consistent with those of high-spin diiron(II) compounds in an oxygen-rich coordination environment.22,28
Figure 2.
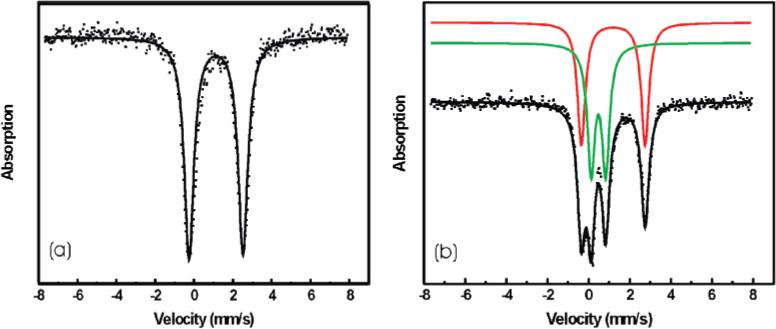
Mössbauer spectra of (a) 9-P and (b) oxygenated intermediate 10. The experimental data are displayed as dots, and the fits are displayed as solid lines.
The Mössbauer spectrum of 10 (Figure 2b) displays two quadrupole doublets, indicating two different iron sites with the isomer shift and quadrupole splitting values of δ = 1.20 mm/s, ΔEQ = 3.10 mm/s and δ = 0.48 mm/s, and ΔEQ = 0.71 mm/s, respectively. The fit gave a 1:1 ratio for the two sites. By comparison with the parameters reported previously, we could assign the former as a high-spin iron(II) center and the latter as a high-spin iron(III) center.22,28 These results strongly suggest that the oxygenated intermediate 10 is a mixed-valent FeIIFeIII species, lending further support to the assigned dinuclear structure of 9-R. The EPR spectrum of 10 displays a weak broad absorbance at g = 2 and relatively strong signals at g = 4.24 and 4.04 (Figure S4, Supporting Information), which is typical for mononuclear Fe(III) in a rhombic environment.29 Quantification indicated that the signals comprise less than 3.5% of total iron, the remainder being EPR-silent. This intermediate is capable of oxidizing substrates 9,10-dihydroanthracene (DHA) to anthracene and anthraquinone and also anthrone to anthraquinone, as will be discussed later. An FeIIFeIII cation, previously generated by oxygenation of a diferrous compound, could not activate substrates because there was no oxygen-containing species bound to Fe(III).6 We therefore believe that the FeIIFeIII intermediate obtained here contains a reduced form of dioxygen, presumably superoxide (O2−), since it can perform multi-electron oxidations. In addition, the unpaired electron of a bound superoxo ion would be strongly coupled to the Fe(III) center, accounting for the absence of an EPR signal in the X-band spectrum. Intensive efforts were undertaken to detect an isotope-sensitive O–O stretching band by resonance Raman spectroscopy, but no mode could be identified. This result may reflect either a very efficient and possibly reversible photodissociation process or the lack of an optical band involving the superoxide ion for resonance enhancement. The fact that the optical spectrum of a mixture of Et4N[FeCl4] and 4-PPy is similar to that of 10 is consistent with the presence of an Fe–Npyridine bond in the latter. Recently, an oxygenated diiron(III) intermediate with no obvious optical absorbance was discovered for a transient species formed in the reaction with dioxygen of both a mutant and wild-type forms of reduced toluene/o-xylene monooxygenase.30 Taken together, these results suggest that the strong absorption band observed for intermediate 10 originates from a ligand-to-metal charge transfer involving 4-PPy and Fe(III) and that no optical band derives from the superoxide ligand. Therefore, the 442 nm absorption would not provide significant enhancement to vibrations implicating the Fe–O–O fragment in a resonance Raman experiment.
When allowed to warm up, a solution of 10 decayed to the high-spin diiron(III) species 11, for which the quadrupole splitting ΔEQ is 0.66 mm/s and the isomer shift δ is 0.48 mm/ s. From the difference in the ΔEQ parameters, the Fe(III) atoms in 10 and the final product must have different coordination environments.
X-ray Absorption Spectroscopy (XAS)
The Fe K-edge X-ray absorption edge shifts to higher energy from 9-P to 10 to 11, indicating an increase in the average iron oxidation state across the series (Figure 3). The main edge transition for 9-P overlays well with those for the diiron(II) complexes A and B (Figure S5, Supporting Information), whereas the spectrum of 11 overlays with that for the diiron(III) complex C (Figure S6, Supporting Information), supporting the iron(II) and iron(III) oxidation level for 9-P and 11, respectively. The midway edge energy position for 10 between those for 9-P and 11 suggests that 10 contains equal amounts of iron(II) and iron(III). A linear combination of 50% of 9-P and 50% of 11, however, does not reproduce the data for 10 completely (Figure S7, Supporting Information), especially in the preedge region, indicating that the iron centers in 10 are of different nature than those in the precursor and decay species.
Figure 3.
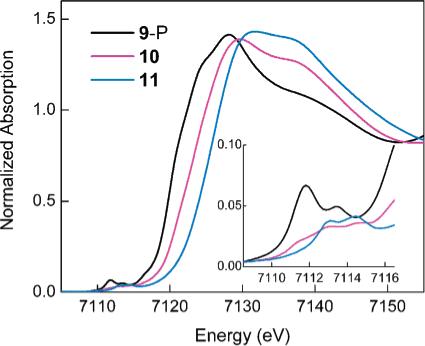
Fe K-edge spectra for 9-P, 10, and 11. The inset shows the magnified preedge region.
The preedge region of the X-ray absorption spectrum is extremely sensitive to the oxidation state and geometry at an average iron site in a molecule.21 The preedge spectrum of 9-P consists of two peaks centered at 7111.8 and 7113.5 eV (Figure 3, inset; Table 1). This preedge energy range is characteristic of ferrous complexes, and the total preedge area of 12.3 units and the area distribution between the two peaks indicate high-spin five-coordinate iron centers in 9-P.21 The preedge fit for 11 reveals two peaks of comparable intensity at 7113.0 and 7114.5 eV with the total area of 7.1 units (Table 1). This energy range and area distribution are typical for high-spin ferric complexes with distorted-octahedral geometry.21 In addition, this pattern is distinct from that observed for (μ-oxo)diiron(III) complexes,21 and thus an oxo bridge can be ruled out for 11. Moreover, this assignment is also consistent with the small quadrupole splitting (0.66 mm/sec) of 11 determined by Mössbauer spectroscopy; oxo bridges typically give rise to significantly larger values.31 The preedge of 10 comprises three peaks at 7111.5, 7112.9, and 7114.3 eV, encompassing the energy regions of both iron(II) and iron(III), and thus further supporting the mixed-valent description for 10. The relatively low total preedge intensity of 6.2 units indicates that both iron centers in 10 are in distorted-octahedral environments and contain no bridging oxo group. Finally, the highest energy peak for 10 at 7114.3 eV, which can be assigned to the iron(III) center, is shifted by 0.2 eV from the analogous peak for 11, suggesting that the iron(III) environments are different in 10 and 11. Hence, the Fe K preedge data indicate that both Fe(II) and Fe(III) centers in 10 are different in nature than Fe(II) in 9-P and Fe(III) in 11, respectively.
Table 1.
XAS Preedge Fit Results for 9-P, 10, and 11
| sample | preedge peak energy | preedge peak intensitya | total preedge peak intensitya |
|---|---|---|---|
| 9-P | 7111.8 (0.01) | 9.0 (0.1) | 12.3 (0.2) |
| 7113.5 (0.01) | 3.3 (0.2) | ||
| 10 | 7111.5 (0.01) | 1.8 (0.1) | 6.2 (0.1) |
| 7112.9 (0.01) | 2.7 (0.1) | ||
| 7114.3 (0.01) | 1.7 (0.1) | ||
| 11 | 7113.0 (0.03) | 4.1 (0.1) | 7.1 (0.5) |
| 7114.5 (0.01) | 3.1 (0.6) |
Values reported for the preedge intensity are multiplied by 100 for convenience.
The Fourier transforms of the EXAFS data shown in Figure 4 illustrate major structural differences between the samples. In particular, the first-shell scattering is significantly less intense for 9-P than that for 10 and 11, implying a broad distribution of the first-shell bond lengths in 9-P. In addition, the Fourier transforms for 10 and 11 display an intense second-shell peak between 2 and 3 Å, similar to the one observed for diiron complexes and proteins with a short Fe–Fe separation32,33 and absent in the data for the precursor 9-P.
Figure 4.
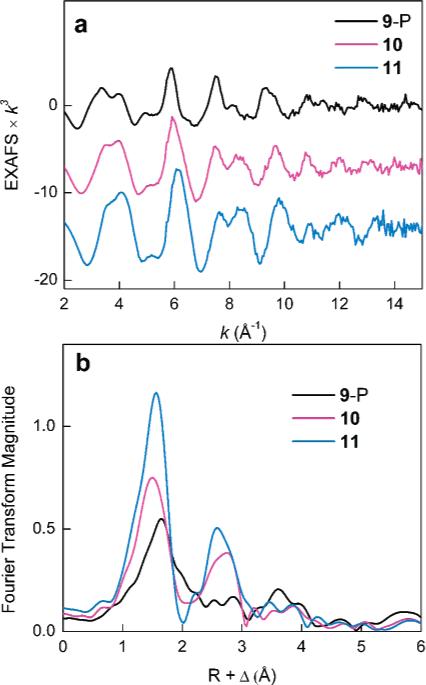
(a) Fe K-edge EXAFS data for 9-P, 10, and 11. (b) Fourier transforms of the EXAFS data.
The EXAFS fits for 9-P yield an average first-shell Fe–ligand distance of 2.10–2.12 Å and are significantly improved upon inclusion of a scattering component at 2.50 Å (Table 2, fits 1 and 2; Figure S8, Supporting Information). A similar component at 2.44 Å is required in the data for the dibridged [FeII2(μ-O2−CArTol)2(O2CArTol)2] complex A, where it can be attributed to an average of the Fe–O and Fe–C distances of the terminal bidentate carboxylate ligand in correspondence with the crystal structure (Table S1 and Figure S9, Supporting Information). This result suggests that one terminal bidentate carboxylate per iron atom is present in 9-P. The data for 9-P do not accommodate a short Fe–Fe vector (fit 3), which would be representative of a tetrabridged structure similar to that of the [FeII2(μ-O2CArTol)4] complex B with an Fe–Fe separation of 2.82 Å. Particularly, the addition of a short Fe–Fe vector in fit 3 results in a very low coordination number of 0.2 Fe and improves the fit error only marginally in comparison to that of fit 2. Since this short metal–metal distance is readily detected in the EXAFS analysis of B (Table S2 and Figure S10, Supporting Information), we conclude that a tetrabridged structure with a short Fe–Fe separation is incompatible with the data for 9-P and may only be valid for 20% or less of all species in the solution. Therefore, the local structure around the iron centers of 9-P is more similar to that of A than of B. The average Fe–ligand distances are longer for 9-P (2.12 Å) than those for A (2.00 Å), reflecting steric demands imposed by the dendrimer ligands.
Table 2.
EXAFS Curve-Fitting Results for 9-P, 10, and 11a
| sample | fit no. | R (Å) | σ2 × 106 (Å2) | ΔE0 (eV) | Fb | |
|---|---|---|---|---|---|---|
| 9-P | 1 | 5 O/Nc | 2.10 | 1200 | −6.2 | 0.497 |
| 4 C | 3.06 | 660 | ||||
| 2 | 4 O/N | 2.12 | 900 | −3.9 | 0.400 | |
| 2 C/O | 2.50 | 830 | ||||
| 4 C | 3.07 | 600 | ||||
| 3 | 4 O/N | 2.12 | 900 | −3.8 | 0.393 | |
| 2 C/O | 2.49 | 860 | ||||
| 0.2 Fe | 2.92 | 650 | ||||
| 4 C | 3.09 | 510 | ||||
| 10 | 4 | 6 O/N | 2.05 | 1300 | 0.7 | 0.331 |
| 5 C | 3.04 | 340 | ||||
| 5 | 6 O/N | 2.05 | 1280 | 0.1 | 0.280 | |
| 1 Fed | 3.03 | 400 | ||||
| 5 Cd | 3.00 | 1520 | ||||
| 11 | 6 | 6 O/N | 2.02 | 840 | 0.7 | 0.298 |
| 5 C | 3.00 | 270 | ||||
| 7 | 6 O/N | 2.02 | 820 | 0.0 | 0.241 | |
| 1 Fed | 3.00 | 400 | ||||
| 5 Cd | 2.94 | 1160 |
Errors are estimated to be 25% for coordination numbers and 0.01 − 0.03 Å for distances.
Error (F) is defined as F = [Σk6(χexptl — χcalcd)2]/Σk6| χexptl2].
Scatterers differing by Z = ±1 are not distinguishable by EXAFS. The first element in a pair indicates the type of atom used to model the backscattered wave in the theoretical fit.
Due to a high degree of correlation, the errors for the Fe—Fe and Fe—C distances at ∼3.0 Å are estimated to be around 0.05 Å.
The first shell for the decay compound 11 can be modeled with 6 O/N ligands at 2.02 Å (Table 2, fit 6), consistent with expected bond lengths for a high-spin ferric complex. However, the shell of five Fe–C(pyridine, carboxylate) interactions at 3.00 Å has a very low Debye–Waller parameter σ2, even when additional shells at longer distances are modeled in the fit (Table S3 and Figure S11, Supporting Information). Inclusion of an Fe–Fe vector at 3.00 ± 0.05 Å improves the σ2 value and results in reasonable parameters for the Fe–Fe component (fit 7). This Fe–Fe distance is 0.15 Å longer than that in the tetrabridged [FeIII2(μ-OH)2(μ-O2CArTol)2] complex C, suggesting that the number, nature, and/or geometry of the bridging ligands are different in 11 and C. The Fe–Fe separation of 3.00 Å in 11 matches well the Fe–Fe distance of 2.99 Å in the oxidized form of MMOH, which features a μ-1,3 carboxylate and two hydroxo bridging ligands.33
The EXAFS data for the intermediate complex 10 are best modeled with 6 O/N scatterers at 2.05 Å, consistent with the intermediate valence state (Table 2, fit 4). As in the case of 11, an Fe–Fe component at 3.05 ± 0.05 Å can be included in the second shell of the data (fit 5), improving σ2 for the carbon shell. Although more detailed fits to the data of 10 have also been performed (Table S4 and Figure S12, Supporting Information), it is not possible to assign any of the EXAFS-derived interactions specifically to a bound superoxo ligand because of the broad range of Fe–O(carboxylate) and Fe–N(pyridine) bond lengths when Fe(II) and Fe(III) centers are simultaneously present.
Although no FeIIFeIII superoxo intermediate has yet been detected in experimental studies of the oxygenation of nonheme diiron enzymes,30,34 theoretical work on dioxygen activation of soluble MMOH has suggested the existence of an FeIIFeIII superoxo species, the structure of which is portrayed in Scheme 4.35,36 An Fe–Fe distance of 3.44 Å was computed recently by QM/MM, reduced by about 0.3 Å compared with that previously reported by a pure QM calculation.36 Although the Fe–Fe distance from theoretical calculation for MMOHred agrees reasonably well with that from crystallographic studies, the value calculated for Q is significantly larger than that (2.46 Å) obtained from XAS.36,37 It is possible to obtain shorter values if other models are used. Therefore, intermediate 10 may bear a core structure similar to that shown in Scheme 4. We stress, however, that we do not rule out other possible binding modes for the superoxide ion. We also note the absence of an optical absorbance band at 442 nm in a published report of an η1-superoxo iron(III) complex.38
Scheme 4.
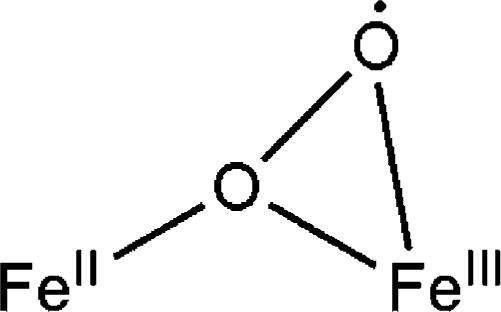
Possible Structure of the FeIIFeIII Superoxo Intermediate 10
Reaction of the Oxygenated Intermediate 10 with External Reagents
a. Cp*2Fe
Cp*2Fe did not react with 10 at −29 °C, judging by the failure to generate the characteristic spectrum of Cp*2Fe+. Upon warming, however, a broad absorbance around 780 nm grew in, which is diagnostic of the decamethylferrocenium cation.39 The relative stability of 10 toward reduction at the lower temperature may reflect the inability of Cp*2Fe to penetrate the dendrimer sheath at low temperatures.
b. Oxidation of 9,10-Dihydroanthracene (DHA)
DHA is a commonly used substrate for oxygenation because its weak C–H bond (BDE = 78 kcal/mol) is easy to activate.40 After the FeIIFeIII intermediate 10 was generated and reached its maximum concentration, Ar was bubbled through the reaction mixture for 20 min to remove unreacted O2. DHA was then injected through an airtight syringe. The temperature was maintained at −29 °C for 2 h, and no change was observed in the UV–vis spectrum (Figure 5a). Upon warming, however, the absorption maximum began to decrease (Figure 5b), indicating reaction of the substrate with 10. As was the case for decamethylferrocene, the diiron(II) center appears to be protected by the dendrimer shell, reacting with dioxygen only above −30 °C. The oxygenated intermediate is also very stable and does not react with substrate without warming.
Figure 5.
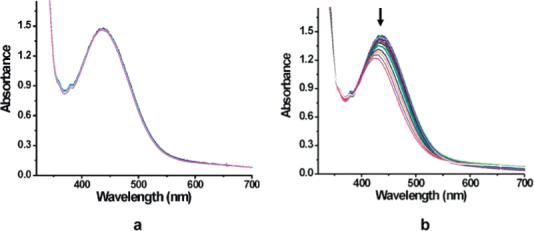
Optical changes observed when 10 reacted with DHA (a) at −29 °C for two h and (b) upon warming to room temperature.
After workup of the reaction mixture, the products (Scheme 5) were analyzed by GC-MS. As listed in Table 3, 13% of the substrate converts to anthracene and 25% to anthraquinone. No anthrone was detected in this experiment. Products were identified by comparing their retention times and mass spectral patterns with those of authentic standards. There were two additional small peaks in the GC trace (Figure S13, Supporting Information) that we could not identify. The mass spectrum of one showed its most intense signal at 178+, identical to that of anthracene, and very weak signals at 194+ and 372+, respectively, which could arise from coupling of DHA and anthrone radicals. However, the retention time is less than that of anthrone itself. Considering the larger molecular mass expected for a coupled compound, this assignment therefore seems unlikely. The mass spectrum of another peak displayed numerous signals, and no conclusion could be drawn concerning its identity. Excluding these contributions, the mass balance was calculated to be 62%. A control experiment using the exact procedure but performed in the absence of 9-P showed no DHA oxidation and 93% recovery of the substrate.
Scheme 5.

Oxidation of DHA in the Presence of FeIIFeIII Species 10 and O2
Table 3.
Product Distribution in the Reaction of DHA with 10, Which Was Generated at −29 °C, Followed by Substrate Addition at The Same Temperature
| reaction conditions | anthracene (%) | anthraquinonea (%) |
|---|---|---|
| Ar, warm up from −29 °C to RT over 12 h | 13 | 25 |
| O2, warm up from −29 °C to RT over 12 h | 13 | 9 |
| Ar, warm up from −29 °C to RT over 15 min | 11 | 5 |
Product yield was calculated based on 10.
The generation of anthracene and anthraquinone most likely occurs via separate reaction pathways because no anthracene was oxidized to anthraquinone in a control experiment. To supply more information about the mechanism, two similar reactions were performed. In one case, Ar was not used to provide an O2-free environment prior to substrate addition. Although the same amount of anthracene formed, production of anthraquinone was significantly decreased (Table 3). In another experiment, the reaction was brought to room temperature under Ar over a shorter period of time (15 min) after substrate addition, and yields of 11% for anthracene and 5% for anthraquinone were recorded. In an experiment where anthraquinone was used as substrate, no reaction occurred. These observations suggest that the formation of anthracene is not affected by the presence of free dioxygen or the temperature, but that the intermediate steps to form anthraquinone are greatly influenced by these factors. The decrease in oxygenated product in the presence of O2 also suggests that radical chain chemistry, which was reported previously with a ferric bi-imidazoline complex,41 is unlikely to be occurring here.
c. Oxidation of Anthrone Substrate
To eliminate the pathway to form anthracene, anthrone was introduced as the substrate. Similar optical changes were observed upon the introduction of dioxygen to 9-P, generating 10, which was stable and unreactive at low temperature for 2 h (Figure 6a). Reaction with substrate ensued when the solution was allowed to warm up (Figure 6b). Approximately 75% conversion to anthraquinone, and only this one product, was observed when Ar was used to flush free O2 from the system prior to warm up. The higher yield strongly implies that anthrone is more reactive than DHA, perhaps because it can tautomerize to 9-hydroxyanthracene, the O–H bond in which is more readily oxidized than the C–H bond.42 Such behavior would account for the failure to detect anthrone in the DHA reaction.
Figure 6.
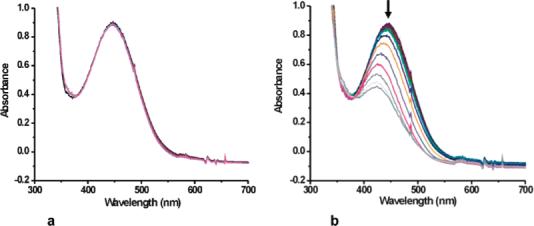
Optical changes observed when 10 reacted with anthrone (a) at −29 °C for 2 h and (b) upon warming to room temperature.
When anthrone was allowed to react with 10 in the presence of excess O2 and with gradual warm up, however, catalytic oxidation of anthrone to anthraquinone was observed (Scheme 6), although we have no evidence that 10 is the catalyst. Table 4 lists the conditions and the product distribution for reactions occurring in CH2Cl2. At a higher ratio of substrate to 10, there is significantly higher turnover. The highest turnover was obtained when the reaction was conducted at room temperature. A control experiment in the absence of 10 and in the presence of Fe(OTf)2·2MeCN showed no conversion to product, although when 4-PPy was present there was quantitative formation of anthraquinone from anthrone. In experiments where toluene was used as the solvent, catalytic reactivity was still observed (Table 5), so it is unlikely that a solvent-derived radical is involved in the chemistry.
Scheme 6.

Catalytic Oxidation of Anthrone to Anthraquinone in the Presence of 10 and O2
Table 4.
Product Distribution When Anthrone Was Allowed to Reacted with 10 in CH2Cl2. The Yields Were Calculated Based on Substrate
| substrate/10 | anthraquinone (%) | turnover |
|---|---|---|
| 11:1 | 53 | 5.6 |
| 18:1 | 53 | 9.5 |
| 129:1 | 11 | 14 |
| 103:1 (RT) | 43 | 44 |
Table 5.
Product Distribution When Anthrone Was Allowed to React with 10 in Toluene. The Yields Were Calculated Based on Substrate
| reaction condition | substrate/10 | anthraquinone (%) | turnover |
|---|---|---|---|
| −29 °C, slowly warm | 10.5:1 | 30 | 3.2 |
| RT | 103:1 | 24 | 25 |
| 71 °C | 124:1 | 13 | 16 |
In order to test whether the mixed-valent intermediate 10 may promote substrate oxidation, we generated 10 with 18O2 and conducted similar experiments. When excess isotopically labeled dioxygen was present, there was 100% incorporation of 18O into the anthroquinone product. When the reaction was allowed to continue under inert gas, surprisingly, we observed 16O incorporation, the source of which is unclear at this time. It is difficult to detect the product of a single turnover reaction, however, and there is usually some anthraquinone impurity in anthrone. A control experiment in which 18OH2 was used in the preparation of 9-P did not affect the outcome, indicating no exchange with water. Taken together, these results prevent us from assigning 10 as being responsible for the catalysis, although in a preliminary double-mixing stopped-flow experiment, where 10 was generated at 20 °C in the first chamber and mixed with anthrone after 100 s, the substrate significantly decreased the lifetime of the intermediate compared with its decay in the absence of the hydrocarbon. Further studies are planned to establish the mechanism of catalysis.
Conclusion
Dinuclear iron(II) complexes of general formula [Fe2([G-3]-COO)4(4-RPy)2] were successfully prepared from dendrimer-appended carboxylate ligands. The compounds were characterized by UV–vis, EPR, Mössbauer, and XAS spectroscopy in addition to elemental analysis. The oxygenation of such complexes is retarded by about 300-fold compared with that of related compounds derived from the sterically less demanding simple terphenyl carboxylate ([G-1]COO−). A metastable FeIIFeIII intermediate was detected in the reaction of [Fe2([G-3]-COO)4(4-PPy)2] with dioxygen. This intermediate was characterized by Mössbauer and XAS spectroscopy. Although no oxygen-sensitive band could be observed by resonance Raman spectroscopy, we formulate this intermediate as an FeIIFeIII superoxo species because it is EPR-silent and can react with external substrates, such as DHA and anthrone, to form oxygenated products. Catalytic oxidation was discovered for anthrone reacting in the presence of the intermediate and excess O2.
The present results provide one of few examples in non-heme diiron chemistry where external substrate oxidation can be achieved with dioxygen43 as the terminal oxidant instead of a peroxide.44 In addition, the chemistry provides an FeIIFeIII superoxo intermediate, which has been proposed in theoretical work but not yet observed for protein systems. Apparently dendritic encapsulation suppresses simple dioxygen-initiated outer-sphere one-electron oxidation to yield FeIIFeIII mixed-valent compounds with no bound O2 derivative, which has been reported for “smaller” diiron(II) models.6,45 The spectroscopic characterization of this intermediate leads us to propose a bridging superoxo structure, analogous to that predicted by theory.36 This structure differs from others by its unique μ-σ2,σ1 binding mode.
Supplemental Materials
Optical changes during oxygenation (Figures S2 and S3); physical characterizations of reaction intermediate and products (Figures S1, S4, and S13); and XAS analysis for 9-P, 10, 11, and A-C including Figures S5-S12 and Tables S1-S4. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
This work was supported by grants from the National Institute of General Medical Sciences (GM32134 to S.J.L.), the National Science Foundation (DMR-0317514 to J.M.J.F.), and the Department of Energy, Office of Basic Energy Sciences through the Biomolecular Science Program at LBNL (DEAC02-05CH11231 to J.M.J.F.), and the National Institutes of Health (P41 RR001209 to K.O.H.) The Structural Molecular Biology program at SSRL is funded by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the Department of Energy, Office of Biological and Environmental Research. We thank Dr. Rayane Moreira for some initial work and Dr. Shawn Burdette for initiating the collaboration between the laboratories of S.J.L. and J.M.J.F. We thank Dr. Roman Davydov and Professor Brian M. Hoffman for measuring the X- and Q-band EPR spectra of the diiron(II) compound 9-P. This project was in part made possible by Grant Number 5 P41 RR001209 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
References
- 1.Hecht S, Fréchet JMJ. Angew. Chem., Int. Ed. 2001;40:74–91. doi: 10.1002/1521-3773(20010105)40:1<74::aid-anie74>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 2.Collman JP, Fu L, Zingg A, Diederich F. Chem. Commun. 1997:193–194. [Google Scholar]; Jiang D-L, Aida T. Chem. Commun. 1996:1523–1524. [Google Scholar]; Jiang D-L, Aida T. J. Macromol. Sci. Pure Appl. Chem. 1997;A34:2047–2055. [Google Scholar]; Zingg A, Felber B, Gramlich V, Fu L, Collman JP, Diederich F. Helv. Chim. Acta. 2002;85:333–351. [Google Scholar]
- 3.Bhyrappa P, Young JK, Moore JS, Suslick KS. J. Am. Chem. Soc. 1996;118:5708–5711. [Google Scholar]
- 4.Enomoto M, Aida T. J. Am. Chem. Soc. 2002;124:6099–6108. doi: 10.1021/ja012589n. [DOI] [PubMed] [Google Scholar]
- 5.Lee D, Lippard SJ. J. Am. Chem. Soc. 1998;120:12153–12154. [Google Scholar]
- 6.Lee D, Pierce B, Krebs C, Hendrich MP, Huynh BH, Lippard SJ. J. Am. Chem. Soc. 2002;124:3993–4007. doi: 10.1021/ja012251t. [DOI] [PubMed] [Google Scholar]
- 7.Tolman WB, Que L., Jr. J. Chem. Soc., Dalton Trans. 2002:653–660. [Google Scholar]
- 8.Lee D, Du Bois J, Petasis D, Hendrich MP, Krebs C, Huynh BH, Lippard SJ. J. Am. Chem. Soc. 1999;121:9893–9894. [Google Scholar]
- 9.Lee D, Lippard SJ. Inorg. Chem. 2002;41:2704–2719. doi: 10.1021/ic020186y. [DOI] [PubMed] [Google Scholar]
- 10.Yoon S, Lippard SJ. J. Am. Chem. Soc. 2004;126:2666–2667. doi: 10.1021/ja031603o. [DOI] [PubMed] [Google Scholar]
- 11.Yoon S, Lippard SJ. J. Am. Chem. Soc. 2005;127:8386–8397. doi: 10.1021/ja0512531. [DOI] [PubMed] [Google Scholar]
- 12.Lee D, Lippard SJ. Inorg. Chem. 2002;41:827–837. doi: 10.1021/ic011008s. [DOI] [PubMed] [Google Scholar]
- 13.Hagen KS. Inorg. Chem. 2000;39:5867–5869. doi: 10.1021/ic000444w. [DOI] [PubMed] [Google Scholar]
- 14.Borchardt RT, Sinhababu AK. J. Org. Chem. 1981;46:5021–5022. [Google Scholar]
- 15.Helms B, Liang CO, Hawker CJ, Fréchet JMJ. Macromolecules. 2005;38:5411–5415. [Google Scholar]
- 16.Battino R. Oxygen and Ozone. 1st ed. Pergamon Press; Oxford and New York: p. 1981. [Google Scholar]
- 17.Kent TA. WMOSS, version 2.5. WEB Research Co.; Minneapolis, MN: 1998. [Google Scholar]
- 18.Aasa R, Vaänngård T. J. Magn. Reson. 1975;19:308–315. [Google Scholar]
- 19.Ellis PJ, Freeman HC. J. Synchroton Radiat. 1995;2:190–195. doi: 10.1107/S0909049595006789. [DOI] [PubMed] [Google Scholar]
- 20.Ankudinov AL, Rehr JJ. Phys. Rev. B: Condens. Matter Mater. Phys. 1997;56:R1712–R1715. [Google Scholar]
- 21.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 1997;119:6297–6314. [Google Scholar]
- 22.Tshuva EY, Lippard SJ. Chem. Rev. 2004;104:987–1011. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- 23.Hawker CJ, Fréchet JMJ. J. Am. Chem. Soc. 1990;112:7638–7647. [Google Scholar]
- 24.Goossen L, Döhring A. Adv. Synth. Catal. 2003;345:943–947. [Google Scholar]
- 25.Kawa M, Fréchet JMJ. Chem. Mater. 1998;10:286–296. [Google Scholar]
- 26.Lee D, Lippard SJ. Inorg. Chim. Acta. 2002;341:1–11. [Google Scholar]; Hagadorn JR, Que L, Jr., Tolman WB. Inorg. Chem. 2000;39:6086–6090. doi: 10.1021/ic000531o. [DOI] [PubMed] [Google Scholar]
- 27.Zhao M, Song D, Lippard SJ. Inorg. Chem. 2006;45:6323–6330. doi: 10.1021/ic0602906. [DOI] [PubMed] [Google Scholar]
- 28.Münck E. In: Physical Methods in Bioinorganic Chemistry: Spectroscopy and Magnetism. Que L Jr., editor. University Science Books; Sausalito, CA: 2000. pp. 287–319. [Google Scholar]
- 29.Palmer G. In: Physical Methods in Bioinorganic Chemistry: Spectroscopy and Magnetism. Que L Jr., editor. University Science Books; Sausalito, CA: 2000. pp. 121–185. [Google Scholar]
- 30.Murray LJ, García-Serres R, Naik S, Huynh BH, Lippard SJ. J. Am. Chem. Soc. 2006;128:7458–7459. doi: 10.1021/ja062762l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizoguchi TJ, Davydov RM, Lippard SJ. Inorg. Chem. 1999;38:4098–4103. [Google Scholar]
- 32.Hsu H-F, Dong Y, Shu L, Young VG, Jr., Que L., Jr. J. Am. Chem. Soc. 1999;121:5230–5237. [Google Scholar]; Hedman B, Co MS, Armstrong WH, Hodgson KO, Lippard SJ. Inorg. Chem. 1986;25:3708–3711. [Google Scholar]
- 33.Rudd DJ, Sazinsky MH, Merkx M, Lippard SJ, Hedman B, Hodgson KO. Inorg. Chem. 2004;43:4579–4589. doi: 10.1021/ic049716b. [DOI] [PubMed] [Google Scholar]
- 34.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew. Chem., Int. Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; Kopp DA, Lippard SJ. Curr. Opin. Chem. Biol. 2002;6:568–576. doi: 10.1016/s1367-5931(02)00366-6. [DOI] [PubMed] [Google Scholar]; Stubbe J. Curr. Opin. Chem. Biol. 2003;7:183–188. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]; Moënne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM., Jr. Biochemistry. 1998;37:14659–14663. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]; Riggs-Gelasco PJ, Shu L, Chen S, Burdi D, Huynh BH, Que L, Jr., Stubbe J. J. Am. Chem. Soc. 1998;120:849–860. [Google Scholar]; Kryatov SV, Chavez FA, Reynolds AM, Rybak-Akimova EV, Que L, Jr., Tolman WB. Inorg. Chem. 2004;43:2141–2150. doi: 10.1021/ic049976t. [DOI] [PubMed] [Google Scholar]
- 35.Gherman BF, Baik M-H, Lippard SJ, Friesner RA. J. Am. Chem. Soc. 2004;126:2978–2990. doi: 10.1021/ja036506+. [DOI] [PubMed] [Google Scholar]
- 36.Rinaldo D, Philipp DM, Lippard SJ, Friesner RA. J. Am. Chem. Soc. 2007;129:3135–3147. doi: 10.1021/ja0654074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 38.Shan X, Que L., Jr. Proc. Natl. Acad. Sci. U.S.A. 2005;102:5340–5345. doi: 10.1073/pnas.0409640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaucher S, Charmant JPH, Sorace L, Gatteschi D, Mann S. Polyhedron. 2001;20:2467–2472. [Google Scholar]
- 40.Bordwell FG, Cheng J, Ji GZ, Satish AV, Zhang X. J. Am. Chem. Soc. 1991;113:9790–9795. [Google Scholar]
- 41.Roth JP, Mayer JM. Inorg. Chem. 1999;38:2760–2761. doi: 10.1021/ic990251c. [DOI] [PubMed] [Google Scholar]
- 42.Roth JP, Yoder JC, Won T-J, Mayer JM. Science. 2001;294:2524–2526. doi: 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- 43.Dong Y, Ménage S, Brennan BA, Elgren TE, Jang HG, Pearce LL, Que L., Jr. J. Am. Chem. Soc. 1993;115:1851–1859. [Google Scholar]; Carson EC, Lippard SJ. Inorg. Chem. 2006;45:837–848. doi: 10.1021/ic051476s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryu JY, Kim J, Costas M, Chen K, Nam W, Que L., Jr. Chem. Commun. 2002:1288–1289. doi: 10.1039/b203154j. [DOI] [PubMed] [Google Scholar]; Jensen MP, Lange SJ, Mehn MP, Que EL, Que L., Jr. J. Am. Chem. Soc. 2003;125:2113–2128. doi: 10.1021/ja028478l. [DOI] [PubMed] [Google Scholar]; Costas M, Tipton AK, Chen K, Jo D-H, Que L., Jr. J. Am. Chem. Soc. 2001;123:6722–6723. doi: 10.1021/ja015601k. [DOI] [PubMed] [Google Scholar]; Oldenburg PD, Shteinman AA, Que L., Jr. J. Am. Chem. Soc. 2005;127:15672–15673. doi: 10.1021/ja054947i. [DOI] [PubMed] [Google Scholar]; Kim J, Larka E, Wilkinson EC, Que L., Jr. Angew. Chem., Int. Ed. 1995;34:2048–2051. [Google Scholar]
- 45.Lee D, DuBois JL, Pierce B, Hedman B, Hodgson KO, Hendrich MP, Lippard SJ. Inorg. Chem. 2002;41:3172–3182. doi: 10.1021/ic011050n. [DOI] [PubMed] [Google Scholar]; Hagadorn JR, Que L, Jr., Tolman WB, Prisecaru I, Münck E. J. Am. Chem. Soc. 1999;121:9760–9761. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Optical changes during oxygenation (Figures S2 and S3); physical characterizations of reaction intermediate and products (Figures S1, S4, and S13); and XAS analysis for 9-P, 10, 11, and A-C including Figures S5-S12 and Tables S1-S4. This material is available free of charge via the Internet at http://pubs.acs.org.