

Abstract
STAT family members have been implicated in regulating the balance between B cell lymphoma (BCL)6 and B lymphocyte induced maturation protein (BLIMP)1 to control plasma cell differentiation. We previously showed that STAT5 induces BCL6 to block plasma cell differentiation and extend the life span of human B cells. The heterogeneity in STAT activation by cytokines and their effects on B cell differentiation prompted us to investigate the effect of STAT3 activation in plasma cell differentiation. First stimulation with IL-21, which promotes plasma cell differentiation, induced robust and prolonged STAT3 activation in primary human B cells. We then investigated effects of direct STAT3 activation on regulation of plasma cell genes, cellular phenotype, and Ig production. Activation of a tamoxifen-regulated STAT3-estrogen receptor fusion protein triggered BLIMP1 mRNA and protein up-regulation, plasma cell phenotypic features, and Ig secretion. When STAT3 was activated by IL-21 in B cells ectopically expressing BCL6, BLIMP1 was up-regulated, but only partial plasma cell differentiation was achieved. Lastly, through coexpression of BCL6 and STAT3-ER, we verified that STAT3 activation functionally mimicked IL-21 treatment and that STAT3-mediated BLIMP1 up-regulation occurred despite high BCL6 expression levels indicating that BCL6 is not the dominant repressor of BLIMP1. Thus, up-regulation of BLIMP1 alone is not sufficient for differentiation of primary human B cells into plasma cells; concomitant down-regulation of BCL6 is absolutely required for completion of the plasma cell differentiation program.
In the germinal center (GC),3 B and T cells communicate via cytokines and cell surface molecules. The GC reaction involves activation of CD4+ T cells leading to up-regulation of surface molecules and cytokine production, which help activated B cells to grow and differentiate. CD40L is expressed on the surface of activated T cells and binds to CD40 expressed on B cells to promote B cell activation (1). Following migration of GC B cells from the dark into the light zone, they differentiate either into memory or Ig-secreting plasma cells to establish humoral immunity (2). Several factors are thought to play a role in the development of memory or plasma cells from the GC. Interaction of B cells with cell surface molecules such as OX40 or ICOS on T cells have been shown to regulate memory B cell formation (3, 4). The strength of initial BCR signaling has also been implicated in differentiation of GC B cells into plasma cells (5) where high affinity BCR or high Ag load favor plasma cell differentiation (6).
It is appreciated that cytokines produced by activated T cells promote proliferation and/or differentiation of B cells into plasma cells. IL-2 and IL-4 promote B cell proliferation (7–9), while IL-10 and IL-21 drive proliferation and differentiation of B cells into plasma cells (10, 11). These cytokines signal through Jak-STAT pathways (12). There are four Jaks (Jak1, Jak2, Jak3, and Tyk2) and seven STATs (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6). Many groups have shown in various cell types that a given cytokine can activate multiple STATs. IL-2 activates STAT5 and STAT3 (13), IL-4 activates STAT6 and STAT5 (14), IL-10 activates STAT3 and STAT1 (15), and IL-21 has been shown to activate mainly STAT3, and to a lesser extent, STAT5 and STAT1 (16). We have recently shown that activation of STAT5 in human B cells blocks plasma cell differentiation and promotes proliferative self-renewal (17).
Transcriptional regulation plays an important role in plasma cell differentiation. B cell lymphoma (BCL)6 is a transcriptional repressor expressed in GC B cells (18) and has been shown to be required for GC formation in mice (19–21). It has been proposed that BCL6 blocks plasma cell differentiation due the fact that it negatively regulates expression of B lymphocyte induced maturation protein (BLIMP)1 (22). Gene targeting has shown that BLIMP1 is necessary for plasma cell differentiation in vivo (23) and is sufficient to drive plasma cell differentiation in B cell lines (24, 25). BLIMP1 expression has been correlated with plasma cell commitment in mice and humans (26–28). BLIMP1 initiates plasma cell differentiation by extinguishing MHC CIITA, Pax5, and c-myc expression (29–31), and by inducing increased X-box-binding protein (XBP)-1 expression (32). These genetic events result in decreased MHC class II expression, loss of B cell identity, cessation of proliferation, and an increase in the cellular machinery required for high-level protein production, respectively. Recent data have also shown that IFN regulatory factor (IRF)4 plays a crucial role in plasma cell differentiation (33, 34).
Although the molecules involved in regulating commitment to the plasma cell fate have been well studied (for review see Ref. 35), the initiating stimuli which affect these regulatory circuits are less clear. We showed previously that STAT5 signaling up-regulated BCL6 expression to inhibit plasma cell differentiation in primary human B cells (17). STAT3, in contrast, has been shown to up-regulate BLIMP1 gene expression and promote plasma cell differentiation of murine B cell lines (36). In this study, we show in primary human B cells that inducible activation of STAT3 triggered BLIMP1 expression, and promoted plasma cell differentiation and Ig production. With these findings, we expand the BCL6/BLIMP1 axis model of plasma cell differentiation to incorporate the influence of STAT3 activation on BLIMP1 regulation and initiation of plasma cell differentiation.
Materials and Methods
B cell isolation
B cells were obtained from buffy coats prepared from the peripheral blood of adults (Sanguin Bloodbank) by Ficoll-Paque separation and CD19 MACS microbeads (Miltenyi Biotec). Tonsils were obtained from routine tonsillectomies (AMC Department of Otolaryngology). Use of these tissues was approved by the medical ethical committee of the AMC and was contingent upon informed consent. Purity was ≥ 97% as determined by flow cytometry. For long-term culture (>2 wk) CD19-MACS-selected B cells were sorted to 99.9% purity for CD19+CD3− cells on a FACSAria (BD Biosciences). Purity was confirmed by reanalysis of sorted cells.
B cell phenotyping
The following mAbs against the human molecules CD3 (SK7), CD19 (4G7, SJ25C1, ID3, or HIB19), CD20 (2H7), CD25 (2A3), CD27 (L128), CD38 (HB7), CD86 (It2.2), HLA-DR (L243); Igκ (TB28 −2, JDC-12); and Igλ (G20 −193) were directly conjugated to FITC, PE, PerCP, allophycocyanin, PE-indotricarbocyanine, allophycocyanin-indotricarbocyanine, or to biotin and were purchased from BD Pharmingen. Streptavidin-PE-indotricarbocyanine conjugate (BD Pharmingen) was used to detect biotin staining. From DakoCytomation, we purchased PE-labeled anti-CD138 (MI15), FITC- and PE-conjugated anti-κ L chain (polyclonal rabbit Fab'2), and PE-conjugated anti-λ L chain (polyclonal rabbit Fab'2; DakoCytomation). Stained cells were analyzed on an LSR II (BD Biosciences) and flow cytometry data were processed using FlowJo software version 6 (TreeStar) on Apple computers. Histograms and dot plots use log10 fluorescence intervals.
Cell culture
B cells (5 × 105) were cocultured on γ-irradiated (100Gy) mouse L cell fibroblasts stably expressing CD40L (CD40L-L cells, 105 cells/ml). Cytokines used were IL-2 (40 U/ml), IL-4 (10 ng/ml R&D Systems), or rmIL-21 (50 ng/ml; R&D Systems). The 4-hydroxytamoxifen (4-HT, 1 μM) was from Sigma-Aldrich. Optimal cytokine concentrations were determined empirically by [3H]thymidine incorporation assay. Cells were routinely tested for mycoplasma and were found to be negative by PCR. EBV-mediated growth was also excluded by routine testing for LMP1, EBNA1, and EBNA2 gene expression, and culture without cytokines or CD40L-L cells.
Induction and determination of STAT phosphorylation
For acute cytokine stimulations (15 or 60 min) CD19+ B cells (freshly isolated or CD40L-activated) were rested 2 h at 37°C. Cells were then stimulated with medium alone or with rmIL-21 (200 ng/ml) for 15 or 60 min at 37°C. For long-term phospho (p)-STAT analysis (24−144 h) cells were cultured with CD40L stimulation in the presence of 50 ng/ml rmIL-21 and directly assayed. Cells were fixed with ice-cold methanol and permeabilized with the CytoPerm/Fix kit (BD Biosciences) and stained intracellularly with mAb against phospho-STAT1 (Y701) conjugated to Alexa-Fluor 488, phospho-STAT3 (Y705) conjugated to Alexa-Fluor 647, and phospho-STAT5 (Y694) conjugated to Alexa-Fluor 647 (all from BD Biosciences).
Retroviral constructs
We obtained wild-type (WT) human STAT3 from E. Caldenhoven (Erasmus University, Rotterdam, The Netherlands) and generated Lazarus (LZRS)-WT-STAT3-internal ribosomal entry site (IRES)-GFP. To fuse the WT STAT3 with C terminus of estrogen receptor (ER), we performed PCR to amplify the C terminus of STAT3 and modify the stop codon to introduce a BglII site using the primer set 5′-CGGCCGATGCTGGAG GAGAGAA-3′ and 5′-CGAGATCTATGGGGGAGGTAGCGCACTC-3′. This PCR product was digested with BamHI-BglII and ligated into BamHI-linearized pBlueScript ER-C-terminal (provided by H. Kurata, DNAX Institute). The C terminus STAT3ER fragment was released with BamHI/EcoRV and ligated into BamHI-SnaBI-digested LZRS-WTSTAT3-IRES-GFP to generate full length LZRS-STAT3ER-IRES-GFP. LZRS-BCL6-IRES-YFP (yellow fluorescent protein) and LZRS-BCL6-IRES-ΔNGFR (signaling-incompetent version of the nerve growth factor receptor) were made by isolating BCL6 from LZRS-BCL6-IRES-GFP (17). Generated vectors were verified by sequencing. LZRS-STAT5bER-IRES-GFP and LZRS-BCL6-IRES-GFP were previously described (17).
Retroviral transduction
Transductions and virus production by Phoenix GalV packaging cells were performed essentially as described (17) except that B cells were activated on CD40L-L cells in the presence of rmIL-21 (50 ng/ml) for 36 h before transduction.
Confocal microscopy
STAT3ER-transduced B cells were cultured overnight with or without 1 μM 4-HT and were plated onto poly-L-lysine-coated cover slips, fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.1% Triton X-100 for 45 s at room temperature. Blocking and Ab incubations were conducted in 2% BSA. Anti-ERa (MC-20, Santa Cruz Biotechnology, 1/100) and Texas red-conjugated donkey anti-rabbit (Molecular Probes, 1/400) was used for detection. The 4′,6′-diamidino-2-phenylindole was used at 300 nM. Images were captured with a Leica TCSSP2 confocal microscope. Localization results were similar over multiple fields. Staining with secondary Ab alone did not display any signal in the Texas red channel.
RT-PCR
Total RNA was isolated with the RNeasy kit (Qiagen) and reverse transcribed using oligo (dT) (Promega) and SuperscriptIII reverse transcriptase (Invitrogen Life Technologies). Primer sequences for BCL6, BLIMP1, and ACTB (17) are published. Other primers used were: PAX-5: forward GGAGGAGTGAATCAGCTTGG, reverse GGCTTGATGCTTCCTGTCTC; IRF-4 forward ACCGAAGCTGGAGGGACTAC, reverse GTG GGGCACAAGCATAAAAG; XBP-1 forward TCACCCCTCCAGAACATCTC, reverse AAAGGGAGGCTGGTAAGGAA. Quantitative RT-PCR was conducted with a Bio-Rad iCycler and relative mRNA expression levels were normalized to HPRT and calculated using the 2 −ΔΔCT) method.
Immunoblotting
Whole cell extracts were isolated using Triton Lysis Buffer (20 mM Tris (pH 7.4), 137 mM NaCl, 25 mM β -glycerolphosphate, 2 mM EDTA (pH 7.4), 1% Triton X-100, 10% glycerol) supplemented with HALT protease inhibitor mixture (Roche) and 1 mM Na3VO4. In brief, 15−30 μg of lysate was separated on acrylamide gels and transferred to nitrocellulose (Schleicher-Schuell). Anti-ERα (MC-20), STAT3 (C-20), BCL6 (C-19), and IRF4 (M-17) Abs were purchased from Santa Cruz Biotechnology and rabbit anti-tubulin was purchased from Cell Signaling Technologies). Anti-BLIMP1 (37) was a gift of Dr. R. Tooze, (Leeds Institute for Molecular Medicine, Leeds, U.K.). Anti-PAX5 Ab (38) was a gift from Dr. Stephen Nutt (Walter and Eliza Hall Institute for Medical Research, Parkville, Victoria, Australia). HRP-conjugated anti-rabbit secondary Ab was from Pierce and anti-goat-, anti-mouse-, and anti-rat-HRP Abs were from DakoCytomation. Blots were developed by ECL (Pierce) and exposed to x-ray film (Amersham Hyperfilm).
ELISA
Plates were coated with capture Abs anti-human IgG, IgM, or IgA (Dako-Cytomation) at 5 μg/ml in 0.1 M NaHCO3 (pH 9.6) for 2 h at 37°C and washed in ELISA wash buffer (PBS, 0.5% Tween 20). Ten percent FCS in PBS was used as blocking agent and diluent for cell supernatants and for enzyme-conjugated detection Abs (Dilutions: 1/5000 for HRP-conjugated anti-IgG, 1/1000 for HRP-anti-IgM, and 1/1000 for alkaline phosphatase (AP) conjugated anti-IgA. TMB substrate/stop solution (Biosource) was used for development of IgG and IgM ELISAs, while AP substrate (Sigma-Aldrich) was used for anti-IgA ELISA.
ELISPOT for total IgG- and IgM-secreting cells
Immobilon-P membrane filter plates (Millipore) were coated overnight with either 5 μg/ml anti-human IgG mAb (Sanquin Reagents) or 5 μg/ml anti-human IgM mAb (Bethyl Laboratories) in PBS. Plates were blocked using IMDM containing 10% FCS. Cells were washed in IMDM with 10% FCS and incubated on the filter plates for 5 h at 37°C for the anti-IgG ELISPOT and 7.5 h for the anti-IgM ELISPOT. Bound IgG was detected with a mix of HRP-labeled IgG1 (1/1000), IgG2 (1/2000), IgG3 (1/2000) and IgG4 (1/1000) (Sanquin Reagents). Bound IgM was detected with HRP-labeled anti-human IgM polyclonal Ab (1/1000) (Bethyl Laboratories). Spots were developed using TMB-substrate for PELIspot (Sanquin Reagents) and analyzed using an AELvis reader (A.EL.VIS GmbH) and eli.analyze software version 4.0
Results
STAT3 activation is sufficient to promote plasma cell differentiation
To investigate the role of STAT3 in control of plasma cell differentiation we first chose to examine the STAT activation spectrum elicited by IL-21, a potent promoter of plasma cell differentiation. We exposed resting human peripheral blood CD19+ B cells to IL-21 and assessed activation of STAT3, STAT5, and STAT1 by flow cytometry with phospho-STAT (p-STAT)-specific Abs. In resting cells, IL-21 induced STAT3 activation, but failed to induce STAT1 or STAT5 activation (Fig. 1A). As positive controls IFN-γ induced p-STAT1, and IL-4 stimulation led to increased p-STAT5 (data not shown). Because B cells generally receive cytokine signals in the context of T cell help we examined STAT activation in CD40L- stimulated B cells. Acute (15 min) exposure to IL-21 induced STAT3 and STAT5 activation but not STAT1 in CD40L-stimulated B cells (Fig. 1B). After 60 min, p-STAT5 signal returned to baseline while p-STAT3 remained elevated (Fig. 1B). This prompted us to examine STAT activation during the period in which IL-21 induces plasma cell differentiation. B cells were cultured with CD40L and in the presence or absence of continuous IL-21 and p-STAT3, p-STAT5, and p-STAT1 were determined in time. STAT3 activation was apparent at 24 h, peaking at 36 h and still present at 96 h (Fig. 1C). Activation of STAT5 and STAT1 above baseline were not observed at any time during the culture period (Fig. 1C). Note that B cells activated for 36 h did not lose their capacity to activate STAT5 as they did activate STAT5 after acute exposure to IL-21 (Fig. 1B). These results showed that STAT3 signaling was more sustained than the brief STAT5 signal induced upon IL-21 triggering of primary human B cells. Given the ability of IL-21 to induce plasma cell differentiation, these results suggested that STAT3 may mediate this process.
FIGURE 1.
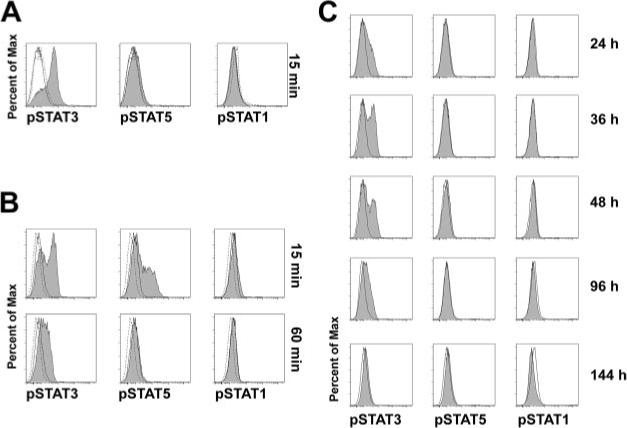
Sustained STAT3 phosphorylation after IL-21 activation in primary human B cells. A, Peripheral blood (PB) CD19+ B cells were freshly isolated and rested at 37°C for 2 h. Cells were then stimulated for 15 min with either medium alone (−, thin histogram), or rmIL-21 (200 ng/ml, shaded histogram). Levels of phospho-STAT3 (Y705), phospho-STAT1 (Y701), or phospho-STAT5 (Y694) were determined by FACS. Isotype control staining is depicted by a dashed line. B, CD19+ B cells were activated for 36 h on CD40L-transfected mouse L cell fibroblasts (CD40L-L cells). Cells were then rested followed by stimulation with medium alone (thin line) or IL-21 (shaded) for 15 or 60 min and levels of phospho-STAT3, STAT1, and STAT5 were determined by flow cytometry. C, CD19+ B cells were cultured with CD40L stimulation with medium alone (thin line) or with IL-21 (shaded) and STAT activation was measured at the indicated times.
To test the hypothesis that STAT3 activation could promote plasma cell differentiation, we generated an inducible system to specifically activate STAT3. We fused the mutated hormone-binding domain of the ERα (39) to the C terminus of full-length human STAT3. STAT3ER was cloned into the LZRS-IRES-GFP retroviral vector (40) for overexpression in human cells. Proper synthesis of STAT3ER fusion protein was verified by immunoblotting in 293T cells transfected with LZRS-IRES-STAT3ER. An anti-STAT3 Ab revealed two bands; the lower band representing endogenous STAT3 and the upper band representing STAT3ER (Fig. 2A), which was confirmed by blotting with an anti-ERα Ab, which showed only the upper band (Fig. 2A). Upon administration of 4-HT, STAT-ER proteins translocate from the cytosol to the nucleus to activate target genes (17, 41). 4-HT-dependent nuclear translocation of STAT3ER was verified in transduced primary B cells by confocal microscopy (Fig. 2B).
FIGURE 2.
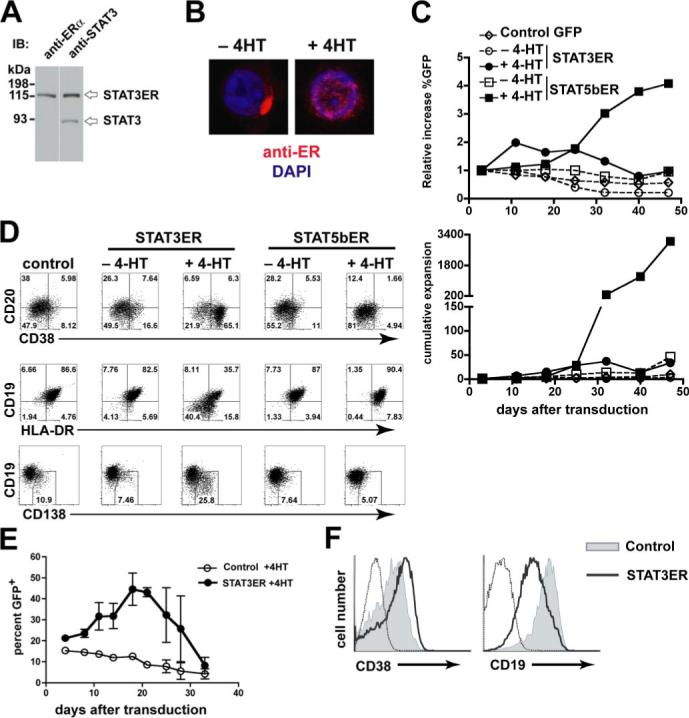
STAT3 activation leads to acquisition of plasma cell phenotype in vitro. A, Immunoblot analysis of 293T cells transfected with LZRS-IRES-STAT3ER. probed with anti-ERα or anti-STAT3 Abs. B, STAT3ER-transduced CD19+ B cells were cultured overnight with or without 1 μM 4-HT and then analyzed with anti-ERα Ab and 4′,6′-diamidino-2-phenylindole to show nuclear localization by confocal microscopy. C, Total PB CD19+ B cells were transduced with LZRS-control-IRES-GFP (Control GFP, ◇), LZRS-STAT3ER-IRES-GFP (STAT3ER, ○), or LZRS-STAT5b-ER-IRES-GFP (STAT5b-ER, □). Transduced cells were cultured on CD40L-L cells in the presence of IL-2 (40U/ml) and IL-4 (10 ng/ml) and in the presence (closed symbols) or absence (open symbols) of 1 μM 4-HT. Upper panel, Percentages of transduced cells were determined over time and the relative increase in GFP from the start of the culture is expressed. Lower panel, Absolute numbers of GFP+ cells were determined and the cumulative expansion is shown. D, At day 17 GFP+ cells were gated and analyzed for CD38, CD20, CD19, HLA-DR, and CD138 expression. Data are representative of three independent experiments using different donors. E, Tonsil CD19+ B cells were transduced with LZRS-control-IRES-GFP or LZRS-STAT3ER-IRES-GFP and cultured on CD40L-L cells with IL-2 plus IL-4 in the presence of 4-HT (1 μM). The percentage of GFP+ cells was determined continuously throughout the culture period. Data are means ± SD of two individual donors. F, At day 17 after transduction tonsil STAT3ER-B cells exhibited increased CD38 and decreased CD19 expression.
In mice, expression of STAT3 in B cells is important for T cell-dependent plasma cell differentiation (42). STAT3 activating cytokines such as IL-10, and IL-21 also promote this process in human B cells (10, 11, 43). In contrast STAT5 activation leads to long-term self-renewal of human B cells which are blocked in plasma cell differentiation (17). For this reason, we directly compared the effect of STAT3 and STAT5 activation on long-term proliferative capacity of B cells. Total CD19+ cells were transduced with LZRS-IRES-control GFP, LZRS-STAT3ER-IRES-GFP, or LZRS-STAT5bER-IRES-GFP and cultured on CD40L-L cells in the presence of IL-2, IL-4, and with or without 4-HT as described (17). The frequency and number (Fig. 2C) of STAT3ER-GFP+ cells peaked at week two and then progressively declined by the end of the culture period. In sharp contrast, STAT5bER-transduced cells exhibited an increase in frequency and in absolute number (Fig. 2C), apparent after approximately 3 wk in culture, consistent with previous findings (17).
As activation of STAT3ER did not lead to long-term self-renewal of B cells, we questioned whether these cells were differentiating into plasma cells. During plasma cell differentiation expression of CD38 and CD138 (Syndecan-1) increase, while expression of CD20, HLA-DR, and CD19 are decreased on plasma cells (2). Compared with GFP+ control cells and with STAT3ER-transduced B cells cultured without 4-HT, STAT3ER cells cultured with 4-HT exhibited an increase in cells that were CD38highCD20− CD19lowHLA-DRlow and CD138+ (Fig. 2D). This phenotype is consistent with plasma cell differentiation. STAT5bER cells cultured with 4-HT did not exhibit phenotypic changes associated with plasma cell differentiation (Fig. 2D) in accordance with previous observations (17). We observed the same effect of STAT3 activation in tonsil CD19+ B cells: lack of long-term growth, up-regulation of CD38, and down-regulation of CD19 (Fig. 2, E and F). Thus, STAT3 activation, in contrast to STAT5, did not confer a long-term growth advantage to primary human B cells, but rather, induced a phenotype consistent with plasma cell differentiation.
Prdm1, the gene encoding murine Blimp-1, is indispensable for plasma cell differentiation (23). Its homologue, BLIMP1, is highly expressed in human plasma cells (28). In murine B cell lines STAT3 has been shown to regulate prdm1 (36). Because the phenotypic changes brought on by STAT3 activation were consistent with plasma cell differentiation, we tested the effect of STAT3 activation on expression of genes involved in control of plasma cell differentiation. CD19+ B cells were transduced with STAT3ER or control virus. GFP+ cells were then sorted and cultured in the presence or absence of 4-HT and expression of PRDM1, IRF4, and XBP1 (44) were determined by quantitative (q)RT-PCR. For comparison, we also examined gene expression levels in in vitro-derived plasma cells (ivPC), which were generated by culture of peripheral blood B cells for 3 days on CD40L-L cells with IL-2 and IL-21, followed by 4 days of culture in the absence of CD40L-L cells, but with IL-2 and IL-21. Similar culture conditions have been shown to drive proliferation and differentiation of human B cells into Ab-secreting PCs (11, 45). As expected, BLIMP1, XBP1, and IRF4 mRNA levels were high and BCL6 and PAX5 levels were low in ivPC (Fig. 3A). BLIMP1 levels were strongly induced by IL-21 while IRF4 and XBP1 expression were also induced, but to a lesser extent (Fig. 3A). Consistent with the principal activation of STAT3 by IL-21 (Fig. 1C), we found that specific STAT3 activation induced BLIMP1 to a similar extent as IL-21 (Fig. 3A). Moreover, IRF4 and XBP1 mRNA expression were increased by STAT3ER activation (Fig. 3A). IL-21 induced BCL6 gene expression (Fig. 3A), but specific STAT3 activation did not significantly increase BCL6 mRNA levels (Fig. 3A). IL-21-mediated up-regulation of BCL6 has been also seen by others (11, 46), but the significance of this effect is not clear. Except for the IL-21-stimulated cells, BCL6 mRNA levels were uniformly low in agreement with down-modulation of BCL6 gene expression by CD40L (47) and were not further decreased by STAT3ER activation. PAX5 also inhibits plasma cell differentiation (48, 49), but neither IL-21 nor activation of STAT3ER affected PAX5 mRNA levels (Fig. 3A).
FIGURE 3.
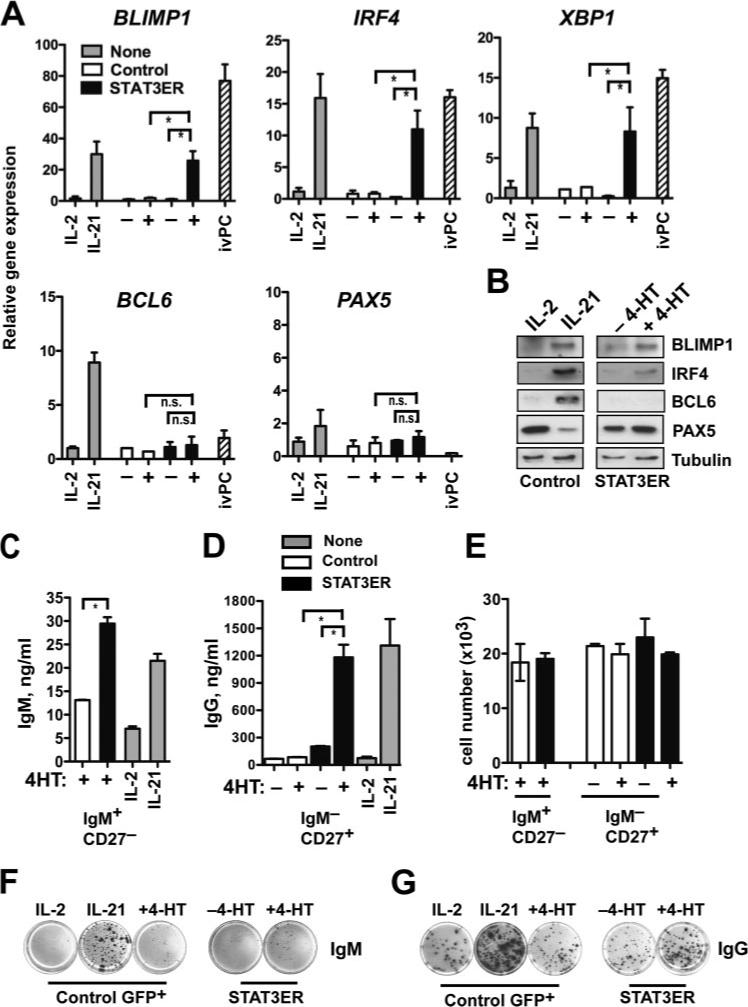
STAT3 activation leads to BLIMP1 up-regulation and Ig secretion. A, PB CD19+ B cells were transduced with either LZRS-control-IRES-GFP (Control, □) or LZRS-STAT3ER-IRES-GFP (STAT3ER, ■). GFP+ cells were sorted and cultured on CD40L-L cells in the presence of IL-2 (40U/ml) and in the presence (+) or absence (−) of 1 μM 4-HT for 5 days. ivPC made from total PB CD19+ B cells (see Results) and nontransduced CD19+ cells (None,  ) cultured in the presence of IL-2 or rmIL-21 (50 ng/ml) for 5 days were included for comparison. mRNA expression of BLIMP1, IRF4, XBP1, BCL6, and PAX5 were determined by quantitative (q)RT-PCR. Results represent means ± SD. *, p < 0.05 vs STAT3ER −4-HT or vs control +4-HT by Student's t test (n = 4). n.s. denotes not significant. B, GFP+ sorted control– or STAT3ER-transduced CD19+ B cells were cultured on CD40L and with IL-2 or IL-21, or with IL-2 in the presence (+) or absence (−) of 4-HT for 5 days. Whole cell extracts were analyzed for BLIMP1, IRF4, BCL6, PAX5, and tubulin expression by immunoblotting. C, Nonswitched CD19+IgM+CD27– and (D) switched CD19+IgM–CD27+ B cells were isolated by FACS from PB and transduced with LZRS-control-IRES-GFP (□) or LZRS-STAT3ER-IRES-GFP (■) virus. Sorted GFP+ cells were cultured with CD40L stimulation in the presence of IL-2 and with (+) or without (−) of 1 μM 4-HT for 5 days. Nontransduced cells of the corresponding phenotype (
) cultured in the presence of IL-2 or rmIL-21 (50 ng/ml) for 5 days were included for comparison. mRNA expression of BLIMP1, IRF4, XBP1, BCL6, and PAX5 were determined by quantitative (q)RT-PCR. Results represent means ± SD. *, p < 0.05 vs STAT3ER −4-HT or vs control +4-HT by Student's t test (n = 4). n.s. denotes not significant. B, GFP+ sorted control– or STAT3ER-transduced CD19+ B cells were cultured on CD40L and with IL-2 or IL-21, or with IL-2 in the presence (+) or absence (−) of 4-HT for 5 days. Whole cell extracts were analyzed for BLIMP1, IRF4, BCL6, PAX5, and tubulin expression by immunoblotting. C, Nonswitched CD19+IgM+CD27– and (D) switched CD19+IgM–CD27+ B cells were isolated by FACS from PB and transduced with LZRS-control-IRES-GFP (□) or LZRS-STAT3ER-IRES-GFP (■) virus. Sorted GFP+ cells were cultured with CD40L stimulation in the presence of IL-2 and with (+) or without (−) of 1 μM 4-HT for 5 days. Nontransduced cells of the corresponding phenotype ( ) were cultured in the presence of IL-2 or IL-21. IgM (left panel) and IgG (right panel) were then measured in culture supernatants by ELISA. *, p < 0.05 vs STAT3ER −4-HT or control +4-HT. Results represent means ± SD. *, p < 0.05 vs STAT3ER −4-HT or vs control + 4-HT by Student's t test (n = 4). E, Absolute cell numbers of cells from B and C. F, Nonswitched PB CD19+IgM+CD27– and (G) switched CD19+IgM–CD27+ were transduced with control- or STAT3ER virus, GFP-sorted, and cultured with IL-2, IL-21, or IL-2 in the presence or absence of 4HT. After 7 days, equal numbers of cells were plated onto membranes in serial dilutions and in triplicate and IgM (F) and IgG (G) secretion were determined by ELISPOT. Representative individual wells are shown.
) were cultured in the presence of IL-2 or IL-21. IgM (left panel) and IgG (right panel) were then measured in culture supernatants by ELISA. *, p < 0.05 vs STAT3ER −4-HT or control +4-HT. Results represent means ± SD. *, p < 0.05 vs STAT3ER −4-HT or vs control + 4-HT by Student's t test (n = 4). E, Absolute cell numbers of cells from B and C. F, Nonswitched PB CD19+IgM+CD27– and (G) switched CD19+IgM–CD27+ were transduced with control- or STAT3ER virus, GFP-sorted, and cultured with IL-2, IL-21, or IL-2 in the presence or absence of 4HT. After 7 days, equal numbers of cells were plated onto membranes in serial dilutions and in triplicate and IgM (F) and IgG (G) secretion were determined by ELISPOT. Representative individual wells are shown.
We also determined protein expression of BLIMP1, IRF4, BCL6, and PAX5 in response to specific STAT3 activation. In agreement with the mRNA profile, IL-21 induced expression of BLIMP1, IRF4, and BCL6 protein, while STAT3ER activation induced BLIMP1 and IRF4, but not BCL6 (Fig. 3B). PAX5 protein was decreased by IL-21 stimulation, but not appreciably affected by STAT3ER activation. Our results suggest that specific STAT3 activation was sufficient to initiate the plasma cell gene expression program by inducing BLIMP1.
Plasma cell differentiation leads to secretion of Ig. To determine the effect of STAT3ER activation on Ig production, naive (CD19+IgM+CD27−) and switched memory B cells (CD19+IgM−CD27+) cells were transduced with control-IRES-GFP or STAT3ER-IRES-GFP. GFP+ cells were sorted and cultured in the presence or absence of 4-HT. For comparison nontransduced cells were also cultured in the presence or absence of IL-21. Five days later IgM and IgG secretion were measured by ELISA. We found that activation of STAT3ER with 4-HT in naive B cells resulted in enhanced IgM secretion (Fig. 3C). Low amounts of IgG were detected in IL-21-treated naive cells (data not shown), consistent with a role for IL-21 in isotype switching (50), but no IgG was detected in supernatants of control- or STAT3ER-transduced cultures cultured with 4-HT (data not shown), suggesting that specific STAT3 activation did not induce isotype switching. In switched memory cells, IgG secretion was strongly induced upon STAT3ER activation as well as by as IL-21 treatment (Fig. 3D). Increased Ig secretion in response to STAT3ER activation was not due to increased cell number (Fig. 3E). To confirm these results we performed ELISPOT analysis for total IgM and IgG in IL-21-treated and STAT3ER-activated B cells. Naive (CD19+IgM+CD27−) and switched memory B cells (CD19+IgM−CD27+) expressing control-IRES-GFP or STAT3ER-IRES-GFP were stimulated for 7 days and IgM and IgG production were determined. IL-21 induced strong increases in the frequency (8 ± 1% for IL-2 vs 32 ± 8% for IL-21) and intensity (100 ± 10 arbitrary units (AU) for IL-2 vs 715 ± 23 AU for IL-21) of IgM-secretion in naive B cells, while STAT3ER activation more mildly increased the frequency (3 ± 1% for −4-HT vs 7 ± 1% for + 4-HT) and spot intensity (48 ± 5 AU for −4-HT vs 103 ± 25 AU for + 4-HT) of IgM-producing cells (Fig. 3F). Of note specific STAT3 activation, unlike IL-21 stimulation did not induce IgG secretion in naive B cells (data not shown), in support of results obtained by ELISA. In switched memory B cells, IL-21 increased the frequency (7 ± 1% for IL-2 vs 18 ± 2% for IL-21) and intensity (372 ± 50 AU for IL-2 vs 824 ± 54 AU for IL-21) of IgG-secretion in switched memory B cells, while STAT3ER activation more mildly increased the frequency (9 ± 1% for −4-HT vs 15 ± 2% for + 4-HT) and spot intensity (288 ± 21 AU for −4-HT vs 740 ± 120 AU for + 4-HT) of IgG-producing cells (Fig. 3G). Taken together these results demonstrate that specific activation of STAT3 in primary human B cells resulted in BLIMP1 up-regulation and enhanced Ig secretion but not to isotype switching.
BCL6 expression prevents IL-21-induced plasma cell differentiation
BCL6 is expressed in GC B cells and functions to repress BLIMP1 and plasma cell differentiation. It has been suggested that BCL6 and STAT3 compete for binding at the murine prdm1 promoter (36), which may imply that BLIMP1 expression could not be induced by STAT3 signaling in GC B cells expressing high BCL6. We then asked whether BLIMP1 could be induced by STAT3 signaling if BCL6 expression was already established and, if so, what are the consequences in terms of cellular differentiation, function, and growth? To address this we made use of the known induction of BLIMP1 expression by treatment with IL-21 in combination with BCL6 overexpression. CD19+ B cells were transduced with an LZRS-BCL6-IRES-YFP (yellow fluorescent protein) vector, which was verified by culturing all cells with CD40L stimulation in the presence of IL-2 and IL-4 until the transduced cells reached 100% of the culture (data not shown) as previously shown for BCL6-IRES-GFP (17, 51). These BCL6+ cells were then cultured in the presence of IL-2 and IL-4 or IL-21 alone and BLIMP1 levels were measured by qRT-PCR. We observed that BLIMP1 mRNA levels were rapidly increased and maintained by IL-21 in BCL6+ cells over a 3-day period (Fig. 4A) and for more prolonged periods (up to 21 days, data not shown). Identical results were obtained with cells transduced with other BCL6-IRES overexpression vectors with either GFP (51), or signaling incompetent nerve growth factor receptor (ΔNGFR) as transduction markers. BCL6 can undergo posttranslational modifications that result in degradation (52), so it was possible that BLIMP1 induction by IL-21 in BCL6-transduced cells was due to decreased functional BCL6 protein. We assessed BCL6 and BLIMP1 protein expression in these cells and found that IL-21 strongly induced BLIMP1 protein, and even a slight increase in BCL6 protein after 7 days in culture (Fig. 4B).
FIGURE 4.
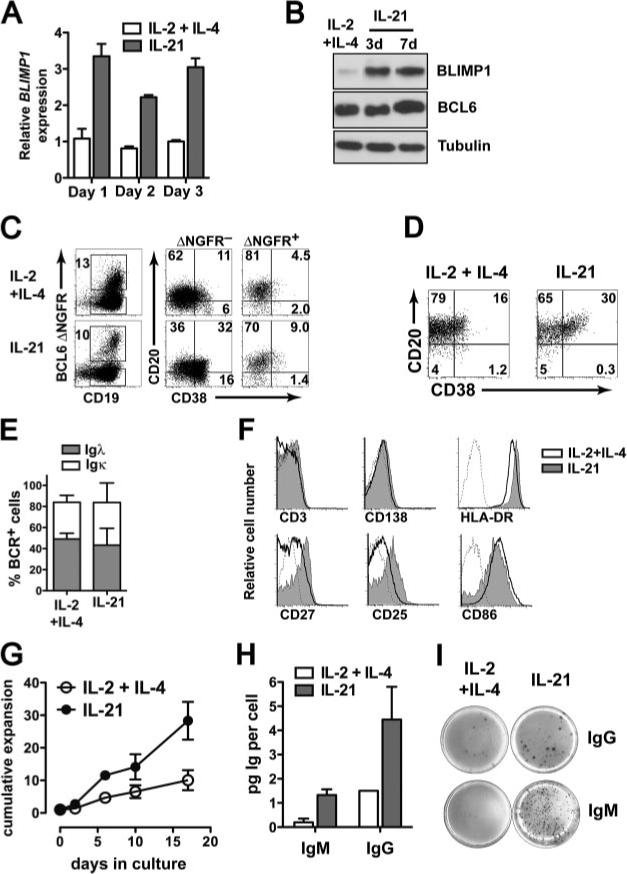
IL-21 drives partial plasma cell differentiation in BCL6-transduced B cells. A, PB CD19+ B cells expressing BCL6-IRES-YFP were cultured in the presence of IL-2 and IL-4 or with rmIL-21 alone. At the indicated time points expression of BLIMP1 and ACTB were measured by qRT-PCR. BLIMP1 expression was normalized to ACTB expression within each sample and the means ± SD of triplicate measurements in two different donors is plotted. B, BCL6-YFP expressing B cells were cultured with CD40L in the presence of IL-2 and IL-4 or with IL-21 and BLIMP1, BCL6, and tubulin protein expression were determined by immunoblotting 3 or 7 days after IL-21 treatment. C, CD19+ B cells were transduced with LZRS-BCL6-IRES-ΔNGFR and cultured for 10 days on CD40L in the presence of IL-2 and IL-4 or with IL-21 alone. Cells were stained with anti-CD19, anti-NGFR, anti-CD38, and anti-CD20 and analyzed by flow cytometry. The rightmost four plots show CD38/CD20 expression on CD19+BCL6–ΔNGFR– and CD19+BCL6–ΔNGFR+ cells. D, CD38/CD20 expression on BCL6-IRES-YFP-transduced B cells after 21 days of culture with IL-2 and IL-4 or with IL-21 alone. E, Surface Igλ and Igκ BCR expression on cells cultured as in C. F, Expression of CD138, HLA-DR, CD86, CD27, CD25, and CD3 on BCL6+ B cells cultured as in C. Dotted histogram is iso-type control. G, CD19+ BCL6-overexpressing B cells maintained on CD40L with either IL-2 and IL-4 (○)or with IL-21 (•). Cumulative expansion was calculated based on absolute cell numbers over a 17-day period. Data shown are means ± SD of three independent experiments involving different donors and constructs (BCL6-IRES-ΔNGFR or BCL6-IRES-GFP). H, BCL6-IRES-ΔNGFR+ cells were cultured for 6 days on CD40L-L cells in the presence of IL-2 and IL-4 or with IL-21. Cell numbers were counted and IgM and IgG in culture supernatants were measured by ELISA to obtain per cell values of IgM and IgG. Data are representative of three independent experiments. I, Total PB CD19+ BCL6-GFP+ cells were cultured with CD40L in the presence of IL-2 and IL-4 or with IL-21. After 7 days, equal numbers of cells were plated in serial dilutions and in triplicate and IgG and IgM production were determined by ELISPOT.
We next determined whether the IL-21-mediated BLIMP1 increase in BCL6-transduced cells had an effect on their cell surface phenotype. CD19+ B cells were transduced with BCL6-IRES-ΔNGFR and cultured in the presence of IL-2 and IL-4 or with IL-21 alone. In the presence of IL-21 a proportion of the ΔNGFR− cells were CD38highCD20+ and CD38highCD20− (Fig. 4C), which correspond to in vitro-derived plasmablasts and plasma cells, respectively (45, 53). Compared with ΔNGFR− cells in either IL-2 and IL-4 or IL-21 cultures, the frequency of CD38high cells among BCL6-ΔNGFR+ cells was reduced (Fig. 4C), Despite the presence of high BCL6 expression, IL-21 elicited an increase in the proportion of CD38highCD20+ cells among BCL6-ΔNFGR+ cells (Fig. 4C). However, these BCL6+ cells cultured with IL-21 did not differentiate to the CD38highCD20− stage (Fig. 4C). Similar results were obtained when using control ΔNGFR-transduced cells instead of BCL6–ΔNGFR– cells. The percentage of CD38highCD20+ cells reached a plateau of around 30% in the prolonged presence of IL-21 (Fig. 4D). Surface BCR expression was equivalent with similar distribution of Igκ and Igλ on IL-21-cultured cells compared with those cultured with IL-2 and IL-4 (Fig. 4E). IL-21 culture led to higher expression of CD27 and CD25, slightly higher CD138 and HLA-DR expression, and slightly decreased CD86 expression (Fig. 4F). These cells, therefore, have an activated (elevated CD25 expression), but partially differentiated phenotype (elevated CD38, CD27 expression with maintained expression of CD20, HLA-DR, BCR, and costimulatory molecules). BCL6+ B cells cultured with IL-21 increased 30 ± 5-fold in number after 18 days while IL-2 and IL-4-cultured cells increased 9 ± 3-fold in number over the same period (Fig. 4G), further supporting the notion that functional BCL6 protein persisted in IL-21-treated BCL6-transduced cells. Given the BLIMP1 up-regulation and phenotypic changes induced by IL-21, we expected that IL-21 treatment would lead to increased Ig production in BCL6+ B cell cultures. Indeed in CD19+ B cells expressing BCL6 IgM and IgG production were increased on a per cell basis after culture with IL-21 as measured by ELISA (Fig. 4H) and in frequency as measured by ELISPOT (Fig. 4I). In total, CD19+ cells transduced with BCL6, IL-21 increased the frequency of IgG and IgM-producing cells (IgG: 4 ± 1% for IL-2 and IL-4 vs 10 ± 1% for IL-21; IgM: 17 ± 4% for IL-2+IL-4 vs 30 ± 4% for IL-21). Also, IL-21 increased the intensity of both IgG (157 ± 17 A.U. for IL-2 and IL-4 vs 364 ± 90 A.U. for IL-21) and IgM (28 ± 5 A.U. for IL-2 and IL-4 vs 540 ± 60 A.U. for IL-21) secretion in BCL6-transduced B cells. These results show that IL-21 promotes BLIMP1 expression, an intermediate plasma cell phenotype, and increased Ig production even in cells expressing high levels of the differentiation inhibitor BCL6.
Functional mimicry of IL-21 by STAT3 activation
To further establish the link between the effects of IL-21 and STAT3 activation, we tested whether STAT3 activation could functionally substitute for the effects of IL-21 in BCL6-overex-pressing cells. To address this, BCL6-YFP+ cells were transduced with LZRS-STAT3ER-IRES-GFP or LZRS-control-IRES-GFP. Double transduced cells were then sorted and cultured in the presence of either medium alone, IL-2 and IL-4, or only 4-HT. First, we observed that activation of STAT3ER in BCL6-transduced resulted in a cumulative increase in cell number overa3wk period (Fig. 5A). This is in agreement with the proliferative effect of IL-21 in BCL6-transduced B cells (Fig. 4G). Next, we found a robust increase in IgG (Fig. 5B, left panel) and IgA secretion (Fig. 5B, right panel) in BCL6/STAT3ER+ cells treated with 4-HT for 4 days compared with those cultured in the absence of 4-HT. Although the STAT3ER-mediated proliferative increase was modest after 4 days, we also verified an increase in Ig production at the per cell level. Finally, we examined whether STAT3ER activation itself was sufficient to induce BLIMP1 in GC-like BCL6- expressing cells. This was indeed the case, as addition of 4-HT to BCL6/ STAT3ER+ cells cultured on CD40L-L cells alone induced a clear increase in BLIMP1 mRNA levels while, without 4-HT, BLIMP1 expression was undetectable (Fig. 5C). At the protein level, 4-HT induced BLIMP1 expression, while not substantially affecting BCL6 protein expression in BCL6/STAT3ER-double transduced cells (Fig. 5D). We confirmed these findings in Raji B cells that were stably transduced with STAT3ER, and induced with 4-HT. Also in these lymphoma cells which express high levels of endogenous BCL6, activation of STAT3ER was sufficient to up-regulate BLIMP1 (Fig. 5E), but did not significantly affect BCL6 levels (Fig. 5F). Taken together these results demonstrate that STAT3 activation alone is sufficient to induce BLIMP1 expression in both primary and transformed human B cells, even in the presence of high BCL6 expression.
FIGURE 5.
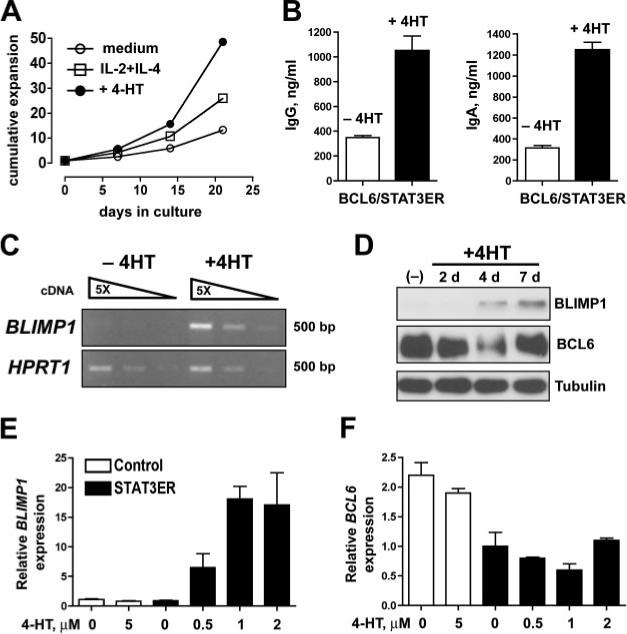
Specific activation of STAT3 in BCL6-transduced B cells. 100% pure PB CD19+ BCL6-IRES-YFP+ B cells were transduced with LZRS-STAT3ER-IRES-GFP and expanded on CD40L-L cells with IL-2 and IL-4. Double positive GFP+YFP+ cells were sorted by FACS. A, Cumulative expansion of BCL6/ STAT3ER cells cultured with medium only, with IL-2 and IL-4, or with only 1 μM 4-HT for 22 days. B, IgG and IgA production in BCL6-YFP+/STAT3ER-GFP+ cells treated for 4 days in the presence or absence of 4-HT. IgM was not detected in these cultures. C, Semi-quantitative RT-PCR for BLIMP1 and HPRT1 mRNA expression in BCL6-YFP+/STAT3ER-GFP+ cells cultured for 4 days in the presence or absence of 4-HT. Triangles represent 5-fold dilutions of cDNA in the PCR reaction. Data are representative of two independent experiments. D, PB CD19+ B cells coexpressing BCL6 and STAT3ER were cultured in the presence or absence of 4-HT and at different time points after 4-HT addition, BLIMP1, BCL6, and tubulin protein levels were determined by immunoblot analysis. E and F, Raji cells expressing control-IRES-GFP or STAT3ER-IRES-GFP were cultured for 4 days in the presence of 1 μM 4-HT. E, BLIMP1 and BCL6 (F) mRNA levels were determined by qRT-PCR and normalized to ACTB expression.
Discussion
In this study, we report two principal findings: 1) activation of STAT3 in human B cells induced BLIMP1 up-regulation and drove plasma cell differentiation and 2) STAT3 activation was sufficient to induce BLIMP1 even in the presence of BCL6. STAT3 activation in these cells led to increased Ig production but not full plasma cell differentiation. Thus, although STAT3 induction by cytokines such as IL-21 may be an initiating step in plasma cell differentiation in GC B cells, BCL6 down-regulation is also required for completion of this process.
Cytokines that have been shown to activate STAT3 in various cell types (such as IL-6, IL-10, and IL-21) have all been implicated in plasma cell differentiation (10, 11, 43, 54). IL-21, which is exclusively T cell-derived, is particularly potent in driving this process (11, 46, 50, 55). IL-21 activates STAT3 in human splenic B cells (56), both STAT1 and STAT3 in EBV+ B cell lines (57) and in multiple myeloma cells (58), and STAT3, STAT1, and STAT5 in activated chronic lymphocytic leukemia B cells (59). In addition to JAK-STAT signaling, IL-21 also utilizes MAPK and PI3K pathways in murine T and B cells (60). Our present findings show that IL-21 predominantly activated STAT3, although brief activation of STAT5 was observed in CD40L-stimulated B cells. This finding was not expected given the potency of IL-21 in promoting plasma cell differentiation and our previous findings that STAT5 activation blocked differentiation (17). However, the IL-21-induced STAT5 response waned after just 15 min after cytokine exposure, while STAT3 activation remained even after several days in culture. In agreement with this, STAT3 phosphorylation persisted longer than STAT5 or STAT1 phosphorylation after IL-21 treatment of murine CD8+ T cells (60). Recent work from the Th17 field has also addressed the differential effects of STAT3 and STAT5 activation. IL-21 activates both STAT3 and STAT5 in T cells (60) and has also been shown to play a role in Th17 development (61). However, while STAT3 is essential for Th17 cell development (62), STAT5 activation itself has been shown to be inhibitory for Th17 development (63). Thus, although IL-21 activates both STAT3 and STAT5 in both mouse and human lymphocytes the STAT3-mediated effects of IL-21 downstream signaling are more prominently manifested. Additionally because we showed that BCL6 is positively regulated by STAT5 (17), this result may also explain why BCL6 levels increased after IL-21 treatment, but not by STAT3 activation (11, 46).
To further explore this dichotomy in STAT3/STAT5 signaling, we directly compared the effects of their individual activation on self-renewal capacity and differentiation in vitro. As we did for STAT5 (17) we generated a tamoxifen-inducible STAT3ER construct. Although STAT5ER activation led to long-term proliferation, STAT3ER activation did not. STAT3 activation alone was sufficient to promote plasma cell differentiation as determined by phenotype, plasma cell gene up-regulation, and Ig secretion. In line with our results, inhibition of STAT3 using a dominant negative mutant in murine B cell lines resulted in loss of cytokine-induced terminal differentiation (36). Mice harboring a B cell-specific STAT3 deficiency exhibit a selective defect in development of IgG-secreting plasma cells (42). STAT3 activation is also essential for survival of multiple myeloma cells, a tumor derived from plasma cells (64). Wang et al. (65) found that strong BCR activation led to activation of STAT3, a finding which correlates with recent studies showing that high affinity BCR signaling promotes plasma cell differentiation (6). Our data directly show a positive role for STAT3 in human plasma cell differentiation.
BCL6 and BLIMP1 form a regulatory circuit due to their repression of each other at the level of gene expression to control plasma cell differentiation (22, 31). Mice lacking Bcl6 contain elevated numbers of Blimp-1+ plasma cells (66). This also applies to human B cells as reducing BCL6 expression by RNA interference results in increased BLIMP1 (S.A. Diehl, unpublished data), and BLIMP1 levels are very low in B cells overexpressing BCL6 (17). Although important for controlling plasma cell differentiation, this model does not take in to account other inputs such as cytokine signaling that can affect this balance. From this and our previous work (17), we hypothesize that the Jak/STAT pathway may play a role in establishing the BCL6/BLIMP1 balance in B cells.
STAT3 activation has been implicated in positive regulation of pdrm1 in the mouse (36) and we show in this study that this holds true in human. BCL6 inhibits BLIMP1 expression (17, 22, 67), but not absolutely because we show elevated BLIMP1 in BCL6+ cells exposed to STAT3 activation. Cumulatively, these findings suggested that BCL6 and STAT3 exert opposing functions at the BLIMP1 locus to control plasma cell differentiation. The DNA consensus binding sites for BCL6 (TTC[C/T][T/A][G/A]GAA) and for STATs are similar (TTCTC[A/T]GAA). For this reason, Reljic and coworkers (36) proposed that BCL6 represses limp-1 expression by competing with STAT3 binding at the promoter of PRDM1, the gene encoding limp-1. However, it was subsequently shown that BCL6 and STAT3 do not compete for binding at key intronic regulatory sites of the PRDM1 locus (66). To discern between these two possibilities, we forced expression of BCL6 and asked whether STAT3 activation could turn on BLIMP1 expression. We demonstrate in this study that even in the presence of high BCL6 expression, BLIMP1 expression could be increased by STAT3, either directly or via IL-21 stimulation. These results, therefore, show that BCL6 and STAT3 are not mutually exclusive in their activities, but rather they can be integrated to generate long-lived B cells that maintain BLIMP1 expression and Ig-producing capacity. Our results do not formally exclude the possibility that BCL6 and STAT3 compete for binding at the PRDM1 promoter, but our results strongly indicate that this is not the dominant mechanism for control of this locus because intronic binding of BCL6 in the PRDM1 gene also plays a crucial role in the repression of this locus (66, 67). We also observed an increase in IRF4 and XBP-1 after STAT3 activation. XBP-1 has been shown to be downstream of BLIMP1 (32), but can also be induced in the absence of BLIMP1 (38). IRF4 plays an important role in maintenance of the plasma cell phenotype (33, 34), but whether IRF4 is up- or downstream of BLIMP1 is not entirely clear. Due to the unresolved relationships between BLIMP1 and IRF4 or XBP-1, we cannot say whether STAT3-mediated up-regulation of XBP-1 and IRF4 are dependent upon BLIMP1. We do propose, however, that STAT3 strongly promoted a genetic program consistent with plasma cell differentiation.
In this study, and in our earlier work (17), we show that STAT5 activation promotes self-renewal of proliferating B cells which are blocked in their terminal differentiation through up-regulation of the repressor BCL6. In contrast to our results, a recent study has shown a negative role for STAT5 in regulating BCL6 expression in various hematopoeitic tumor cell lines including BCL1, NKL, 32D, and Ba/f3 cells (68). BCL6 is a repressor of differentiation in B cells and our results clearly demonstrated that direct activation of STAT5 activation blocks plasma cell differentiation, supporting a positive relationship between STAT5 and BCL6. In fact, withdrawal of 4-HT from STAT5bER B cells resulted in spontaneous appearance of plasma cells. Thus, it is possible that the regulatory relationship between STAT5 and BCL6 in these transformed cell lines is different from that of primary human B cells. Another study proposed that, via AP-1 and STAT3 activation, IL-21 positively regulated BCL6 expression in mouse B cells (69). Although we and others have also found that IL-21 increased BCL6 expression (11, 46), we did not find that specific STAT3 activation affected BCL6 mRNA or protein levels. STAT3 activation, however, did increase BLIMP1 expression and might therefore be an initiating stimulus to push the BCL6/BLIMP balance in favor of BLIMP1, leading to human plasma cell differentiation.
The current model of transcriptional regulation of GC-based plasma cell differentiation states that BCL6 expression is high in GC centroblasts and centrocytes. The mechanism by which this occurs is still unknown. It is well known, however, that BCL6 expression is absent in plasma cells (28) due to a repressive regulatory circuit with BLIMP1 (31), which is high in plasma cells (28). Thus the question arises, how can BLIMP1-dependent plasma cell differentiation be initiated in GC B cells expressing BCL6? Two possibilities are as follows: 1) that either BCL6 needs to first decrease to allow BLIMP1 to increase or 2) that BLIMP1 can be increased in BCL6high GC B cells until BLIMP1 levels reach a critical threshold to tip the balance toward plasma cell differentiation. Our results do not exclude the possibility that first mechanism may occur, for example, through CD40L ligation (47) with GC T cells. In contrast, we provide evidence that the latter mechanism is also feasible. We show that STAT3 activation initiated plasma cell differentiation by triggering expression of BLIMP1, and that this occurred in cells expressing high levels of BCL6, suggesting that this factor is not the dominant repressor of BLIMP1 expression. Using a BCL6 transgenic mouse model, Cattoretti et al. (70) showed that high BCL6 expression occurred in a subset (15%) of Ig-secreting, CD138+ plasma cells, suggesting that forced BCL6 expression could not completely block plasma cell formation in vivo. It is possible that in this system a selection of plasma cells exhibiting weak BCL6 expression occurred. Our data suggest that BCL6 expression nearly completely blocks in vitro plasma cell differentiation. In light of the data of Cattoretti et al., it is possible that in our hands forced BCL6 expression negatively affects the survival of human plasma cells in vitro, while promoting survival of nondifferentiated cells. In addition to BCL6, down-regulation of the repressive activity of other factors such as PAX5 can lead to plasma cell differentiation (48, 49). Indeed, such a mechanism of “alleviated repression” was recently identified as a mechanism underlying PAX5-mediated blockade of plasma cell differentiation (38). However, we were unable to detect significant changes in PAX5 expression through manipulation of STAT3 activity.
Thus, by induction of STAT3 signaling, either directly or with IL-21, we demonstrate BLIMP1 up-regulation under conditions that may resemble the GC B cell environment-high BCL6 expression by GC B cells, CD40 triggering, and exposure to cytokines. Furthermore, we show that while BCL6 did not inhibit initiation of plasma cell differentiation, its down-regulation was required for completion of this process. Considering our previous data showing that STAT5 positively controls BCL6 expression (17) together with the results presented in this study, finding the source of STAT5 or STAT3- activating cytokines may be of interest. CXCR5+ CD4+ Follicular helper T cells are a possible candidate as subsets of these cells have been shown an array of cytokines such as IL-21, IL-4, and IL-2 (71, 72). We propose that the balance of activated STAT3 and STAT5 is an important factor in determining whether GC B cells differentiate into self-renewing B cells (STAT5 activation) or into plasma cells (STAT3 activation) in the GC.
Acknowledgments
We thank Berend Hooibrink (Academic Medical Center) for expert cell sorting and maintenance of the FACS facility and Bianca Blom (Academic Medical Center) for critical reading of the manuscript. We thank E. Caldenhoven (Erasmus University) for the gift of STAT3 cDNA, R. Tooze (Leeds Institute of Molecular Medicine, Leeds, U.K.) for the gift of anti-BLIMP1 Ab, and Stephen Nutt (Walter and Eliza Hall Institute of Medical Research, Australia) for anti-PAX-5 Ab.
Footnotes
This work is supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases Grant F32AI063846 (to S.A.D).
Abbreviations used in this paper: GC, germinal center; BCL, B cell lymphoma; BLIMP, B lymphocyte induced maturation protein; XBP, X-box-binding protein; CD40L-L, L cell fibroblasts stably expressing CD40L; 4-HT, 4-hydroxytamoxifen; p, phospho; WT, wild-type; LZRS, Lazarus; ER, estrogen receptor; IRES, internal ribosomal entry site; ivPC, in vitro-derived plasma cell; NGFR, nerve growth factor receptor.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Clark LB, Foy TM, Noelle RJ. CD40 and its ligand. Adv. Immunol. 1996;63:43–78. doi: 10.1016/s0065-2776(08)60854-8. [DOI] [PubMed] [Google Scholar]
- 2.Liu YJ, Arpin C. Germinal center development. Immunol. Rev. 1997;156:111–126. doi: 10.1111/j.1600-065x.1997.tb00963.x. [DOI] [PubMed] [Google Scholar]
- 3.Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, Eibel H, Fischer B, Schaffer AA, Mages HW, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat. Immunol. 2003;4:261–268. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 4.Stuber E, Strober W. The T cell-B cell interaction via OX40-OX40L is necessary for the T cell-dependent humoral immune response. J. Exp. Med. 1996;183:979–989. doi: 10.1084/jem.183.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarlinton DM, Smith KG. Dissecting affinity maturation: a model explaining selection of antibody-forming cells and memory B cells in the germinal centre. Immunol. Today. 2000;21:436–441. doi: 10.1016/s0167-5699(00)01687-x. [DOI] [PubMed] [Google Scholar]
- 6.Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 2006;203:1081–1091. doi: 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leibson HJ, Marrack P, Kappler JW. B cell helper factors: I. Requirement for both interleukin 2 and another 40,000 mol wt factor. J. Exp. Med. 1981;154:1681–1693. doi: 10.1084/jem.154.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howard M, Farrar J, Hilfiker M, Johnson B, Takatsu K, Hamaoka T, Paul WE. Identification of a T cell-derived b cell growth factor distinct from interleukin 2. J. Exp. Med. 1982;155:914–923. doi: 10.1084/jem.155.3.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banchereau J, de Paoli P, Valle A, Garcia E, Rousset F. Long-term human B cell lines dependent on interleukin-4 and antibody to CD40. Science. 1991;251:70–72. doi: 10.1126/science.1702555. [DOI] [PubMed] [Google Scholar]
- 10.Rousset F, Garcia E, Defrance T, Peronne C, Vezzio N, Hsu DH, Kastelein R, Moore KW, Banchereau J. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc. Natl. Acad. Sci. USA. 1992;89:1890–1893. doi: 10.1073/pnas.89.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, Leonard WJ, Lipsky PE. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J. Immunol. 2005;175:7867–7879. doi: 10.4049/jimmunol.175.12.7867. [DOI] [PubMed] [Google Scholar]
- 12.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 13.Frank DA, Robertson MJ, Bonni A, Ritz J, Greenberg ME. Interleukin 2 signaling involves the phosphorylation of Stat proteins. Proc. Natl. Acad. Sci. USA. 1995;92:7779–7783. doi: 10.1073/pnas.92.17.7779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rolling C, Treton D, Pellegrini S, Galanaud P, Richard Y. IL4 and IL13 receptors share the c chain and activate STAT6, STAT3, and STAT5 proteins in normal human B cells. FEBS Lett. 1996;393:53–56. doi: 10.1016/0014-5793(96)00835-6. [DOI] [PubMed] [Google Scholar]
- 15.Riley JK, Takeda K, Akira S, Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway: requirement for two distinct receptor-derived signals for anti-inflammatory action. J. Biol. Chem. 1999;274:16513–16521. doi: 10.1074/jbc.274.23.16513. [DOI] [PubMed] [Google Scholar]
- 16.Leonard WJ, Spolski R. Interleukin-21: a modulator of lymphoid proliferation, apoptosis, and differentiation. Nat. Rev. Immunol. 2005;5:688–698. doi: 10.1038/nri1688. [DOI] [PubMed] [Google Scholar]
- 17.Scheeren FA, Naspetti M, Diehl S, Schotte R, Nagasawa M, Wijnands E, Gimeno R, Vyth-Dreese FA, Blom B, Spits H. STAT5 regulates the self-renewal capacity and differentiation of human memory B cells and controls Bcl-6 expression. Nat. Immunol. 2005;6:303–313. doi: 10.1038/ni1172. [DOI] [PubMed] [Google Scholar]
- 18.Cattoretti G, Chang CC, Cechova K, Zhang J, Ye BH, Falini B, Louie DC, Offit K, Chaganti RS, Dalla-Favera R. BCL-6 protein is expressed in germinal-center B cells. Blood. 1995;86:45–53. [PubMed] [Google Scholar]
- 19.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 20.Fukuda T, Yoshida T, Okada S, Hatano M, Miki T, Ishibashi K, Okabe S, Koseki H, Hirosawa S, Taniguchi M, et al. Disruption of the Bcl6 gene results in an impaired germinal center formation. J. Exp. Med. 1997;186:439–448. doi: 10.1084/jem.186.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti RS, et al. The BCL-6 proto-onco-gene controls germinal-centre formation and Th2-type inflammation. Nat. Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 22.Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000;13:199–212. doi: 10.1016/s1074-7613(00)00020-0. [DOI] [PubMed] [Google Scholar]
- 23.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–620. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- 24.Turner CA, Jr., Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell. 1994;77:297–306. doi: 10.1016/0092-8674(94)90321-2. [DOI] [PubMed] [Google Scholar]
- 25.Messika EJ, Lu PS, Sung YJ, Yao T, Chi JT, Chien YH, Davis MM. Differential effect of B lymphocyte-induced maturation protein (Blimp-1) expression on cell fate during B cell development. J. Exp. Med. 1998;188:515–525. doi: 10.1084/jem.188.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J. Immunol. 2000;165:5462–5471. doi: 10.4049/jimmunol.165.10.5462. [DOI] [PubMed] [Google Scholar]
- 27.Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, Nutt SL. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J. Exp. Med. 2004;200:967–977. doi: 10.1084/jem.20040973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cattoretti G, Angelin-Duclos C, Shaknovich R, Zhou H, Wang D, Alobeid B. PRDM1/Blimp-1 is expressed in human B-lymphocytes committed to the plasma cell lineage. J. Pathol. 2005;206:76–86. doi: 10.1002/path.1752. [DOI] [PubMed] [Google Scholar]
- 29.Lin Y, Wong K, Calame K. Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science. 1997;276:596–599. doi: 10.1126/science.276.5312.596. [DOI] [PubMed] [Google Scholar]
- 30.Piskurich JF, Lin KI, Lin Y, Wang Y, Ting JP, Calame K. BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat. Immunol. 2000;1:526–532. doi: 10.1038/82788. [DOI] [PubMed] [Google Scholar]
- 31.Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 32.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee A-N, Qian S-B, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, et al. XBP1, downstream of Blimp-1, Expands the secretory apparatus and other organelles, and increases protein sysnthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 33.Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, Ludwig T, Rajewsky K, Dalla-Favera R. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat. Immunol. 2006;7:773–782. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 34.Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25:225–236. doi: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 36.Reljic R, Wagner SD, Peakman LJ, Fearon DT. Suppression of signal transducer and activator of transcription 3-dependent B lymphocyte terminal differentiation by BCL-6. J. Exp. Med. 2000;192:1841–1848. doi: 10.1084/jem.192.12.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doody GM, Stephenson S, Tooze RM. BLIMP-1 is a target of cellular stress and downstream of the unfolded protein response. Eur. J. Immunol. 2006;36:1572–1582. doi: 10.1002/eji.200535646. [DOI] [PubMed] [Google Scholar]
- 38.Kallies A, Hasbold J, Fairfax K, Pridans C, Emslie D, McKenzie BS, Lew AM, Corcoran LM, Hodgkin PD, Tarlinton DM, Nutt SL. Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity. 2007;26:555–566. doi: 10.1016/j.immuni.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 39.Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heemskerk MH, Blom B, Nolan G, Stegmann AP, Bakker AQ, Weijer K, Res PC, Spits H. Inhibition of T cell and promotion of natural killer cell development by the dominant negative helix loop helix factor Id3. J. Exp. Med. 1997;186:1597–1602. doi: 10.1084/jem.186.9.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurata H, Lee HJ, O'Garra A, Arai N. Ectopic expression of activated Stat6 induces the expression of Th2-specific cytokines and transcription factors in developing Th1 cells. Immunity. 1999;11:677–688. doi: 10.1016/s1074-7613(00)80142-9. [DOI] [PubMed] [Google Scholar]
- 42.Fornek JL, Tygrett LT, Waldschmidt TJ, Poli V, Rickert RC, Kansas GS. Critical role for Stat3 in T-dependent terminal differentiation of IgG B cells. Blood. 2006;107:1085–1091. doi: 10.1182/blood-2005-07-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for nonmalignant human plasmablasts. Blood. 2001;97:1817–1822. doi: 10.1182/blood.v97.6.1817. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 45.Arpin C, Dechanet J, Van Kooten C, Merville P, Grouard G, Briere F, Banchereau J, Liu YJ. Generation of memory B cells and plasma cells in vitro. Science. 1995;268:720–722. doi: 10.1126/science.7537388. [DOI] [PubMed] [Google Scholar]
- 46.Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF, Hwu P, Shaffer DJ, Akilesh S, Roopenian DC, et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J. Immunol. 2004;173:5361–5371. doi: 10.4049/jimmunol.173.9.5361. [DOI] [PubMed] [Google Scholar]
- 47.Allman D, Jain A, Dent A, Maile RR, Selvaggi T, Kehry MR, Staudt LM. BCL-6 expression during B-cell activation. Blood. 1996;87:5257–5268. [PubMed] [Google Scholar]
- 48.Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity. 2006;24:269–281. doi: 10.1016/j.immuni.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 49.Nera KP, Kohonen P, Narvi E, Peippo A, Mustonen L, Terho P, Koskela K, Buerstedde JM, Lassila O. Loss of Pax5 promotes plasma cell differentiation. Immunity. 2006;24:283–293. doi: 10.1016/j.immuni.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Pene J, Gauchat JF, Lecart S, Drouet E, Guglielmi P, Boulay V, Delwail A, Foster D, Lecron JC, Yssel H. Cutting edge: IL-21 is a switch factor for the production of IgG1 and IgG3 by human B cells. J. Immunol. 2004;172:5154–5157. doi: 10.4049/jimmunol.172.9.5154. [DOI] [PubMed] [Google Scholar]
- 51.Shvarts A, Brummelkamp TR, Scheeren F, Koh E, Daley GQ, Spits H, Bernards R. A senescence rescue screen identifies BCL6 as an inhibitor of anti-proliferative p19(ARF)-p53 signaling. Genes Dev. 2002;16:681–686. doi: 10.1101/gad.929302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niu H, Ye BH, Dalla-Favera R. Antigen receptor signaling induces MAP kinase-mediated phosphorylation and degradation of the BCL-6 transcription factor. Genes Dev. 1998;12:1953–1961. doi: 10.1101/gad.12.13.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Avery DT, Ellyard JI, Mackay F, Corcoran LM, Hodgkin PD, Tangye SG. Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. J. Immunol. 2005;174:4034–4042. doi: 10.4049/jimmunol.174.7.4034. [DOI] [PubMed] [Google Scholar]
- 54.Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC, III, Liu C, Schwartzberg PL, Leonard WJ. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 55.Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, Johnston J, Madden K, Xu W, West J, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408:57–63. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- 56.Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J. Immunol. 2006;177:5236–5247. doi: 10.4049/jimmunol.177.8.5236. [DOI] [PubMed] [Google Scholar]
- 57.Konforte D, Paige CJ. Identification of cellular intermediates and molecular pathways induced by IL-21 in human B cells. J. Immunol. 2006;177:8381–8392. doi: 10.4049/jimmunol.177.12.8381. [DOI] [PubMed] [Google Scholar]
- 58.Brenne AT, Ro TB, Waage A, Sundan A, Borset M, Hjorth-Hansen H. Interleukin-21 is a growth and survival factor for human myeloma cells. Blood. 2002;99:3756–3762. doi: 10.1182/blood.v99.10.3756. [DOI] [PubMed] [Google Scholar]
- 59.de Totero D, Meazza R, Zupo S, Cutrona G, Matis S, Colombo M, Balleari E, Pierri I, Fabbi M, Capaia M, et al. Interleukin-21 receptor (IL-21R) is up-regulated by CD40 triggering and mediates pro-apoptotic signals in chronic lymphocytic leukemia B cells. Blood. 2006;107:3708–3715. doi: 10.1182/blood-2005-09-3535. [DOI] [PubMed] [Google Scholar]
- 60.Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The molecular basis of IL-21-mediated proliferation. Blood. 2007;109:4135–4142. doi: 10.1182/blood-2006-10-054973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 62.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 63.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 64.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 65.Wang L, Kurosaki T, Corey SJ. Engagement of the B-cell antigen receptor activates STAT through Lyn in a Jak-independent pathway. Oncogene. 2007;26:2851–2859. doi: 10.1038/sj.onc.1210092. [DOI] [PubMed] [Google Scholar]
- 66.Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J. Immunol. 2004;173:1158–1165. doi: 10.4049/jimmunol.173.2.1158. [DOI] [PubMed] [Google Scholar]
- 67.Parekh S, Polo JM, Shaknovich R, Juszczynski P, Lev P, Ranuncolo SM, Yin Y, Klein U, Cattoretti G, Dalla Favera R, et al. BCL6 programs lymphoma cells for survival and differentiation through distinct biochemical mechanisms. Blood. 2007;110:2067–2074. doi: 10.1182/blood-2007-01-069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walker SR, Nelson EA, Frank DA. STAT5 represses BCL6 expression by binding to a regulatory region frequently mutated in lymphomas. Oncogene. 2007;26:224–233. doi: 10.1038/sj.onc.1209775. [DOI] [PubMed] [Google Scholar]
- 69.Arguni E, Arima M, Tsuruoka N, Sakamoto A, Hatano M, Tokuhisa T. JunD/AP-1 and STAT3 are the major enhancer molecules for high Bcl6 expression in germinal center B cells. Int. Immunol. 2006;18:1079–1089. doi: 10.1093/intimm/dxl041. [DOI] [PubMed] [Google Scholar]
- 70.Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q, Mo T, Murty VV, Dalla-Favera R. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell. 2005;7:445–455. doi: 10.1016/j.ccr.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 71.Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat. Rev. Immunol. 2005;5:853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- 72.Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, de Waal Malefyt R, Tangye SG. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J. Immunol. 2007;179:8180–8190. doi: 10.4049/jimmunol.179.12.8180. [DOI] [PubMed] [Google Scholar]