

Abstract
The dissection of mechanisms that regulate glucose transport by insulin has revealed an intricate network of signaling molecules scattered from the insulin receptor to the intracellular glucose transporter GLUT4. It is also appreciated that some insulin receptor signals jaunt in different directions to regulate events essential for the efficient redistribution of GLUT4 to the plasma membrane. Moreover key assists in the process appear to be arranged by membrane lipids and cytoskeletal proteins. Following current considerations of insulin signals regulating GLUT4, this review will focus on in vitro and in vivo evidence that supports an essential role for phosphoinositides and actin filaments in the control of glucose transport. The discussion will visit recent cell culture, whole animal, and human data highlighting membrane and cytoskeletal aspects of insulin resistance.
Keywords: Diabetes, glucose transport, insulin resistance, signaling, trafficking
INTRODUCTION
Of the plethora of biological responses elicited by insulin, perhaps the most important is the removal of excess circulatory glucose by skeletal muscle and adipose tissue. A key aspect of this cellular response to insulin entails the redistribution of the glucose transporter GLUT4 from a specialized intracellular insulin-sensitive storage compartment to the plasma membrane. This regulated redistribution or translocation of GLUT4 involves a complicated network of cellular interactions, many of which appear critically coupled to the actin cytoskeleton. In addition, membrane phospholipids appear to play a pertinent role in insulin signaling and cytoskeletal control of GLUT4. Accumulating in vitro and in vivo evidence highlights that membrane and cytoskeletal abnormalities may render the control of GLUT4 ineffective. Interestingly, blood cells (neutrophils, mononuclear cells, erythrocytes) isolated from insulin-resistant individuals exhibit alterations in cortical actin structure and membrane lipid composition [1–4]. These phospholipid and cytoskeletal irregularities in human blood cells raise the question whether similar changes are present in human muscle and fat cells and explain, at least in part, their insulin-resistant state.
Insulin Signaling to GLUT4 Translocation
It is well established that activation of GLUT4 translocation by insulin requires a phosphatidylinositol 3-kinase (PI3K) signal involving the upstream insulin receptor (IR) and insulin receptor substrate (IRS) activators and the downstream protein kinase B (Akt) and C (PKC) target enzymes, however the identity of the downstream effector of this cascade remained elusive (Fig. 1). With the discovery of AS160 (Akt substrate of 160 kDa) in 2002 by Kane and colleagues, knowledge of the downstream signaling events to GLUT4 translocation after Akt activation became more complete [5]. Utilizing an antibody that recognizes the Akt phosphomotif RXRXXpS/T, this group initially identifed and characterized this protein, and since this initial report, AS160 protein expression has been confirmed in cultured adipocytes, rat and human skeletal muscle {Kane, 2002 #2006; Sano, 2003 #1731; Bruss, 2005 #2007; Karlsson, 2005 #2008}, and L6 myotubes [6]. Additionally a key property of AS160 is that it contains a GTPase-activating domain (GAP) for Rabs, small G proteins involved in vesicle trafficking [7, 8]. Intriguingly, Rabs 2A, 8A, 10, and 14 appear to be substrates of the AS160 GAP domain and are also associated with insulin-responsive GLUT4-containing vesicles [9, 10][11]. As Rab proteins have been shown to be necessary effectors in vesicle trafficking, docking and fusion, AS160 may represent a convergence between insulin signaling and vesicle trafficking. Two separate sets of studies utilizing AS160 knockdown and rescue have identified AS160 as a negative regulator of basal GLUT4 exocytosis [9, 12].. In combination, these studies have created the following model of AS160 function in GLUT4 translocation: Basally, AS160 associates with GLUT4 vesicles and intrinsic GAP activity maintains Rab proteins in their inactive form, complexed to GDP. Insulin-stimulated phosphorylation of AS160 inhibits GAP activity towards Rabs, causing a shift towards active Rab-GTP complexes and allowing for Rab-dependent GLUT4 translocation to occur [9, 12, 13].
Fig. 1.
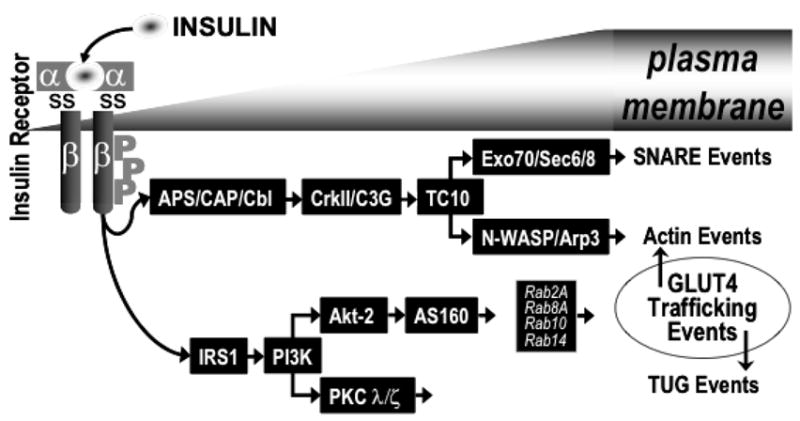
Model showing the various signaling events involved in GLUT4 translocation. TUG, tether containing a UBX domain for GLUT4; SNARE, soluble NSF-attachment factor receptor.
It has also been suggested that, a second pathway occurs as a consequence of Cbl tyrosine phosphorylation (Fig. 1) [14, 15]. Cbl and the adaptor protein CAP (c-Cbl-associated protein; [16]) are recruited to the insulin receptor by APS (adaptor molecules containing pleckstrin homology (PH) and Srchomology 2 (SH2) domains; [17]). Once tyrosine phosphorylated by the receptor, Cbl can recruit the adaptor protein CrkII to lipid rafts, along with the guanyl nucleotide exchange factor C3G [18]. C3G, in turn, can activate the GTP-binding protein TC10, which resides in lipid rafts [19]. The correct spatial compartmentalization of these signaling molecules in the lipid raft microdomain appears to be essential for insulin-stimulated GLUT4 translocation and glucose transport, as these insulin-mediated events are abolished by dominant-interfering mutants of CAP that prevent the localization of Cbl to lipid rafts [20]. Recent investigation suggests a role of TC10 in the regulation of actin dynamics [21–26] and phosphoinositides [27]. In particular, the actin-regulatory neural Wiskott-Aldrich syndrome protein (N-WASP) [23], the actin related protein-3 (Arp3) [23], and the exocyst protein complex [22] appear to be downstream targets of TC10. Whereas N-WASP/Arp3 apparently regulates actin polymerization [23], the exocyst protein complex is postulated to mediate the tethering/docking of GLUT4 vesicles at the plasma membrane [22]. This interaction is necessary for GLUT4 vesicle and plasma membrane fusion mediated by SNARE proteins (SNAP-23, syntaxin 4, Synip, Munc18c and VAMP2).
The burgeoning number of these GLUT4 effectors highlights a remarkably complex signaling architecture that includes key assists from membrane phospholipids and cytoskeletal proteins. Recent insights into the importance of membrane phospholipids such as phosphoinositol 4,5-bisphosphate (PIP2) and cytoskeletal filaments such as filamentous actin (F-actin) in GLUT4 regulation are the focus of this review. The discussion visits the importance of these components in insulin signaling and GLUT4 vesicular trafficking.
Phosphoinositides & GLUT4
Phosphoinositides are a large group of inositol containing phospholipids, of which the major precursor is phosphatidylinositol (PI). This glycerolipid is comprised of an inositol ring attached to a glycerol backbone by a phosphate at position 1. Other polyphosphoinositides are derived from PI by phosphorylation at various positions of the inositol ring by a variety of lipid kinases and phosphatases (Reviewed in [28–32]). PIs represent a very small portion of the total membrane lipid content in the plasma membrane [31]. These amounts vary from between 5 and 10% for phosphatidylinositol 4,5-bisphosphate (PIP2) and PI, respectively to as little as 0.25% for the 3′ phosphorylated phosphoinositides [32]. These lipids are known to play important roles as precursors to second messengers and as signaling and scaffold targeting molecules [33]. Several PIs are known to bind to and regulate the actions of proteins that bind to actin filaments to regulate actin dynamics. Early experimentation in this field utilizing permeabilized platelets demonstrated that PIs regulate the actin cytoskeleton [34], and recent experiments have resulted in additional evidence supporting this conclusion [35–51]. Actin dynamics, and their interaction with phosphoinositides, which are also factors in GLUT4 translocation, will be reviewed later in this paper. (Reviewed in [33].) Here we focus directly on the participation of PIs in the key steps of GLUT4 translocation.
Signaling
Involvement of PIs in signal transduction is well-characterized. With respect to signaling, PIs are most well-known for their ability to serve as substrates for phospholipase C (PLC) and PI kinases and phosphatases [37, 52, 53]. Of the nine characterized PIs, PIP2 is the most studied, most likely due to the fact that it is a versatile signaling molecule known to be a regulator of the actin cytoskeleton [38], membrane trafficking events, and plasma membrane ion channels and transporters (reviewed in [33]). It serves as a precursor to the second messengers IP3 and diacylglycerol (DAG) and may in and of itself be a second messenger. Specific to insulin signaling, PIP2 plays a major role as a substrate for PI3K in the synthesis of phosphatidylinositol 3,4,5-trisphospate (PIP3), which is a crucial second messenger propagating the insulin signal from the IR to GLUT4 [54]. In an effort to delineate the dynamics of 3-phosphoinositides (PI 3-P, PI 3,4-P2 and PIP3) in vivo, Dormann et al. analyzed the binding specificities for various PI species during Dictyostelium phagocytosis and chemotaxis [52]. These authors concluded that different PIP3 binding PH domains gave distinct spatial and temporal readouts of the same underlying PIP3 signal, demonstrating that distinct biological responses could be elicited from the same PIP3 signal. This lends support to the hypothesis that PIP3 might be able to participate in more than one of the cellular events involved in insulin-activated GLUT4 translocation [52].
As previously mentioned, insulin stimulated PI3K activity, generation of PIP3, and the subsequent activation of the downstream kinases Akt and PKC ζ/λ has been shown to be crucial for activation of glucose transport in both muscle and fat cells [55–61]. In addition, overexpression of various PI lipid phosphatases, such as the SH2-containing inositol phosphatase (SHIP) or skeletal muscle and kidney-enriched inositol phosphatase (SKIP), both 5′ phosphatases that act on PIP3, inhibits insulin-induced GLUT4 translocation [62–64]. In addition, similar data has been reported using overexpression of phosphatase and tensin homolog (PTEN), a 3′ phosphatase that produces PIP2 from PIP3 [65]. Although there is good evidence that PIP3 production is crucial for insulin-stimulated glucose transport, some controversy exists as to the nature of the specificity of insulin generated PIP3 for this process versus the PIP3 generated by other growth factors (EGF, IGF or PDGF). Data has suggested that this signal specificity of insulin lies in the threshold of duration and/or intensity of the PIP3 signal produced [66, 67]. Further, Sweeney et al. showed that intracellular delivery of PIP3 augmented plasma membrane GLUT4 content [68]. However, delivery of exogenous PIP3 was unable to stimulate the same increase in plasma membrane GLUT4 content like insulin, and it did not increase glucose uptake. Thus, while PIP3 may be necessary for stimulation of glucose transport, it does not completely recapitulate the signaling cascade for this process, indicating a further level of complexity.
Indeed, several studies have shown that in addition to the role PIP3 plays in GLUT4 regulation by insulin, direct roles also exist for PIP2 and various other PIs in insulin-regulated GLUT4 translocation. There is some evidence that insulin may activate phospholipase C (PLC) in a PI3K-dependent manner, which generates the second messengers diacylglycerol (DAG) and inositol trisphosphate (IP3) from PIP2. Several previous studies have suggested that PLC may play a role in insulin-stimulated GLUT4 translocation potentially via generation of phosphatidic acid from DAG and subsequent activation of PKCζ [69–71]. Additionally, PI 3-P also has also been experimentally demonstrated to play a role in insulin-induced GLUT4 translocation. Insulin increases the plasma membrane/lipid raft levels of PI 3-P in L6 cells and 3T3-L1 adipocytes, and this pool of PI 3-P is relatively resistant to PI3K inhibitors. Further, exogenously supplied PI 3-P induces the plasma membrane translocation of both over-expressed and endogenous GLUT4. It has also been shown that a dominant-negative mutant of TC10 inhibits the insulin-stimulated formation of PI 3-P, while a constitutively active mutant mimics the effect of insulin. Taken together, these data suggest that PI 3-P is produced downstream from insulin-mediated activation of TC10 to promote GLUT4 translocation [27].
Additional evidence that PI 5-P, is involved in GLUT4 translocation was suggested by Sbrissa et al. [72]. These investigators found that microinjected PI 5-P stimulated GLUT4 translocation in 3T3-L1 adipocytes, and that expression of protein-3xPHD, which selectively binds PI 5-P, blocked insulin-induced GLUT4 vesicle dynamics. These authors postulated that insulin could generate PI 5-P via recruitment of PIKfyve (an enzyme that produces PI 5-P from PI) to the plasma membrane, or via breakdown of PIP2 by an unidentified 4-phosphatase. Thus, PI 5-P may represent an additional PI signaling lipid involved in PI3K-independent insulin-signaling leading to GLUT4 translocation. The role of other PI lipids in insulin signal propagation remains an area for further exploration.
Trafficking, Docking, & Fusion
An extensive role of PIs in vesicle exocytosis is well documented in other vesicle trafficking systems, such as in nerve terminals, however new studies are needed aimed at dissecting the functionality of distinct PIs [73]. Recently, Ishiki et al. reported that PIP3 and PI 3-P directly regulate GLUT4 vesicle trafficking [27, 74]. These PIs seem to coordinate membrane arrival and fusion, and GLUT4 unmasking events critical in plasma membrane accumulation of GLUT4. Other cell systems also shed light on the importance of PI metabolism in membrane trafficking. For example, in chromaffin granules, PIP2 and other PIs participate in the priming stage of exocytosis [75, 76]. Specifically, PIP2 binds to the granule protein synaptotagmin, thought to function as a calcium sensor in neurotransmitter release. Also, PIP2, binds to rabphilin 3a, a peripheral membrane protein that may also bind calcium and the docking and fusion regulators Rab3A and Rab3C. Since PIs also regulate actin binding proteins and actin dynamics, it is believed that PIs on the membrane of secretory granules may serve to coordinate secretion of several secretory granule proteins, or they may modulate the necessary dynamic changes that occur in the actin cytoskeleton during exocytosis [77]. In agreement with this hypothesis, recent data from Bittner and Holz recently demonstrated that secretion in primary neuroendocrine cells relies upon the coordinate interactions of PIP2, actin, and other molecules involved in the secretory response [78]. As introduced above, similar to neuroendocrine cells, insulin responsive tissues express Rabs 2A, 8A, 10, and 14 that appear to be substrates of the AS160 GAP. In addition, it has also been suggested that Rab4 co-localizes with the GLUT4-enriched microsome fraction and may be involved in GLUT4 translocation [79–83].
Finally, evidence is emerging that PIs play roles in membrane docking, fusion and retrieval events. Adipocytes from mice null for the syntaxin 4 binding protein Munc18c have been reported to show increased sensitivity to insulin-stimulated GLUT4 vesicle fusion. Although in normal cells PI 3-P induces GLUT4 trafficking to the plasma membrane without causing vesicle fusion [74], in Munc18c-null mice, this lipid induced both GLUT4 trafficking to and fusion with the plasma membrane [84]. Thus, Munc18c may function as a negative regulator of PI-regulated GLUT4 vesicle docking and fusion, and disruption of the interaction between syntaxin 4 and Munc18c might result in enhancement of insulin-stimulated GLUT4 vesicle and plasma membrane fusion. In contrast, the internalization of GLUT4, which occurs through a dynamin-dependent mechanism [85–88], may entail PIP2 activation [89–93].
Actin & GLUT4
An abundance of cellular functions have been ascribed to cytoskeletal actin including maintenance of cell polarity [94], activation of osteoclasts during bone remodeling [95], maturation of granulocytes [96], platelet function [34, 97, 98], and sperm capacitation and acrosomal reaction [99]. With regard to insulin action and glucose transport, an increasing body of evidence supports a critical role for actin in GLUT4 translocation (Fig. 2) [100–105]. One of the best documented actin events engaged by insulin is “membrane ruffling”, which is an energy-dependent cytoskeleton-driven process controlled by PI3K. Further, the PI3K products PI 3,4-P2 and PIP3—as well as PIP2—can bind to the cytoskeletal protein profilin, which serves as an organizational site for actin remodeling [106]. It has been reported that insulin elicits the formation of F-actin, and that pharmaceutical disruption of cortical actin filament formation with latrunculin B inhibits insulin-stimulated GLUT4 translocation [25, 100, 107]. Just as the number of signaling proteins coupling the IR to GLUT4 action has escalated, several actin binding proteins seem to be decorating the GLUT4-regulatory network. For example, as presented above under the Insulin Signaling & GLUT4 section, in vitro study suggests a role for the actin regulatory protein N-WASP in mediating the effects of insulin on the actin filament network. The subsections below detail findings suggestive of a role of actin in insulin-regulated GLUT4 function.
Fig. 2.
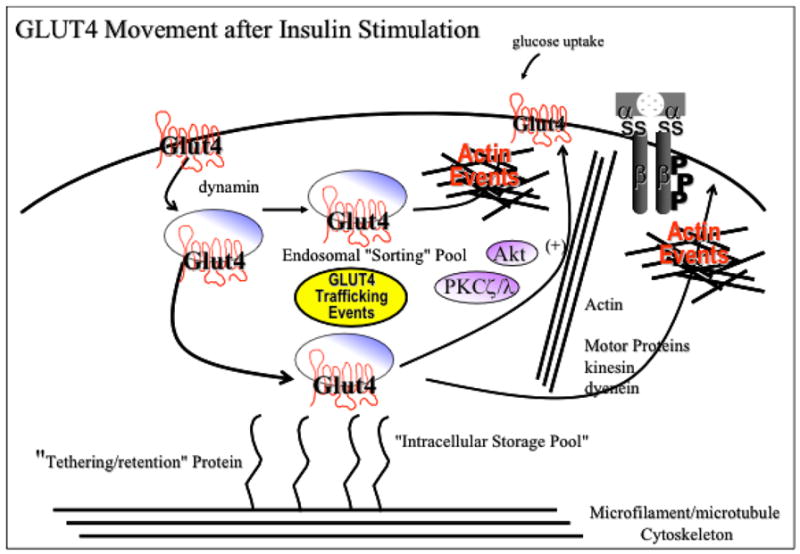
Proposed model of GLUT4 translocation incorporating signaling through phosphoinositides and integration with actin/cytoskeletal remodeling.
Signaling
As previously mentioned, data exists indicating that the actin cytoskeleton forms a scaffold for signaling molecules in a variety of cellular processes [34], [50], [96], [108],[109], [110], [111]. Analogously, it is hypothesized that actin may play a similar role in the insulin signaling pathway [104]. Additionally, evidence exists that there are interactions between the actin cytoskeleton and PI metabolism [39] discussed below under Phosphoinositides & Actin.
With regards to insulin, this hormone elicits a rapid, dynamic remodeling of actin filaments into a cortical mesh, and in a variety of insulin-sensitive cell types, such as differentiated L6 muscle cells, 3T3-L1 adipocytes, primary fat cells, and primary muscle cells, this mesh is a necessity for GLUT4 translocation [23, 24, 112–115]. Since insulin signaling involves activation of PI3K, it is believed that the insulin-induced formation of a cortical F-actin mesh serves to spatially localize the generated PIP3 and/or downstream signaling components in order to elicit the appropriate downstream effects. Thus, the actin cytoskeleton may comprise a matrix in which the enzymes involved in PI metabolism can associate, and segregate the responses in the insulin signaling pathway (Fig. 2). In agreement with this idea, Patel et al. demonstrated that when insulin-stimulated actin remodeling is disrupted using excess PIP3, this resulted in the inhibition of GLUT4 translocation [112].
It has recently been reported by Martin et al. that activated PI3K was itself sufficient to mediate actin rearrangement and GLUT4 translocation in 3T3-L1 adipocytes, suggesting that insulin’s ability to produce these two effects must depend upon its ability to trigger a PI3K-induced signaling cascade [116]. However, at least one other PI3K independent pathway has been hypothesized to be involved in actin remodeling and GLUT4 translocation, which involves the insulin stimulated activation of TC10, as described under Insulin Signaling & GLUT4. Although the significance of this pathway remains controversial [18, 19, 21, 23] its connection to the actin network via N-WASP is intriguing. Moreover, insulin mimetic stimulation of GLUT4 translocation by osmotic shock, which does not require PI3K activation, does require TC10 and presumably N-WASP [117]. Interestingly, expression of a dominant negative N-WASP lacking the Arp and actin binding domains attenuated cortical F-actin rearrangements by insulin and inhibited insulin-induced GLUT4 translocation in cultured adipocytes [23]. Additionally, expression of an inhibitory TC10 mutant inhibited cortical localization of N-WASP and F-actin formation in response to insulin. Taken together, these data reveal a pathway in which N-WASP likely functions downstream of both PI3K and TC10 to mobilize cortical F-actin, thereby promoting GLUT4 translocation to the plasma membrane. Nonetheless, while these studies demonstrate a necessary role for TC10 activation in actin remodeling events required for GLUT4 translocation in adipocytes, there is evidence that this TC10 pathway may not function in muscle cells [118, 119].
Trafficking, Docking, & Fusion
Although it has been shown that the cell cytoskeleton plays important roles in membrane trafficking events, it is still unclear as to whether GLUT4 vesicle trafficking involves both the microtubule and the actin cytoskeleton [100, 101, 120–127]. Since the majority of the evidence in this area concerns the actin cytoskeleton, this review will focus on the involvement of this structure. As previously indicated, there is considerable evidence implicating the cortical actin cytoskeleton in formation of GLUT4 vesicle actin comet tail formation and translocation in both adipocytes and muscle cells [25, 105, 128]. One of the major components of GLUT4-containing vesicles is the zinc-dependent protease designated insulin-responsive aminopeptidase (IRAP) that traffics in a similar or identical fashion to GLUT4 [129–134]. There is some evidence that the N terminus of IRAP functions in the regulation of the intracellular retention and trafficking of GLUT4 vesicles via interaction with the cytoskeleton in an insulin dependent manner [135]. Indeed, adenoviral overexpression of the IRAP interacting protein FHOS (formin homolog overexpressed in spleen) enhances glucose uptake in L6 muscle cells. Since FHOS has been shown to have a skeletal muscle isoform with a binding affinity for profiling, this may suggest that FHOS acts as a linker protein between GLUT4-containing vesicles and the actin cytoskeleton [136].
It has recently been reported that the calpain family of cysteine proteases have a potential role in insulin-induced glucose uptake. Intriguingly, a SNP in this protease has been shown to be associated with development of diabetes. Paul et al. reported that inhibition of calpain activity in 3T3-L1 adipocytes reduced insulin-induced GLUT4 translocation [137]. Significant to the point of this review, the inhibition of calpain activity was associated with an inhibition of insulin-stimulated actin reorganization, with no effect on the immediate steps of insulin signaling. Therefore, these data would lead to the hypothesis that calpain-10 may facilitate GLUT4 translocation via reorganization of the cortical actin cytoskeleton [138]. Hence, in addition to identifying calpain as a potential novel factor involved in the trafficking of GLUT4 vesicles, this study also provided more evidence of the necessary role of actin dynamics in the overall process of GLUT4 translocation.
In addition to the exocytosis of GLUT4 in adipocytes, PIP2-regulated cortical F-actin structure appears essential for the release of adiponectin and may explain why the levels of this adipokine are reduced in insulin resistant states [139]. In other secretory cells, the actin cytoskeleton has also been observed to have profound influence on exocytosis. In a study of pancreatic acinar cells undergoing regulated exocytosis, it was observed that in response to the secretagogue carbamylcholine, zymogen granules participating in exocytosis became coated with F-actin and released rab3d. These observations suggest that rab3d is involved in the regulation of actin polymerization around secretory granules and that the subsequent coating of the vesicles with actin facilitates the movement of granules across the subapical actin network and toward their fusion site [140]. Other studies of secretory cells have suggested that cortical F-actin instead functions as a physical barrier to vesicle docking based upon its transient depolymerization during exocytosis, and that secretion preferentially occurs at sites where the actin cortex is relatively thin. Moreover, to add to the complexity of this area in some cell systems, disruption of the actin cytoskeleton markedly potentiates agonist-stimulated secretion, while in others, depletion of F-actin structures either by sequestering actin monomers or by stimulation of actin severing results in inhibition of agonist-induced secretion [141–147].
It is still unclear as to whether GLUT4 vesicles have properties more consistent with secretory granules or synaptic vesicles, although recent evidence points to them having properties unique from both. In muscle cells the insulin-dependent gain in surface GLUT4 functionally requires the fusogenic v-SNARE VAMP2 [148]. Additionally, in response to insulin, VAMP2-containing vesicles partially localize with the actin structures induced by the hormone. Thus, insulin-induced actin remodeling may be required to specifically segregate VAMP2-containing GLUT4 vesicles and allow their recruitment to the plasma membrane [112, 119, 148, 149]. VAMP2 would then act as a fusogen either at the plasma membrane, or through additional intermediate fusion (and fission) steps [150]. An interesting report suggests that insulin-stimulated activation of PKCζ, in a PI3K-dependent manner, induces serine phosphorylation of VAMP2 in the GLUT4 compartment, which was hypothesized to lead to GLUT4 translocation. It is unclear, however, how phosphorylation of this protein could initiate this process or trigger the association of VAMP2 containing vesicles with the actin cytoskeleton, suggesting there are yet unknown interactions between GLUT4 vesicles and the actin cytoskeleton that modulate exocytosis and fusion [151, 152].
Phosphoinositides & Actin
The coordination of rearrangements of the actin cytoskeleton depends on its tight connection to the plasma membrane, and is thought to occur through signals transmitted to the underlying actin cytoskeleton via PIP2. This lipid messenger binds to, and influences the activity of, several actin-associated proteins in vitro that regulate the architecture of the actin cytoskeleton, such as the Rho GTPase Cdc42 [33, 153, 154]. Conflicting data exists, however, that suggests Cdc42 in fact does not bind to PIP3, which implies that it may not be a key player in the PI3K signaling pathway involved in insulin-induced GLUT4 translocation. Moreover, other data exists showing that the Rho GTPases Rac1 and RhoA are capable of binding to PIP3, which lends support to the possibility that these molecules may have a role in this process [155]. Complicating this issue is the fact that ARF6, another well-known small GTP binding protein, appears to be involved in insulin-induced actin cytoskeletal rearrangement, but does not seem to affect GLUT4 translocation [156, 157]. This may point to the importance of cellular compartmentalization in regulating intracellular trafficking.
Recent work by Kanzaki et al. supports this hypothesis and illustrates the potential role of membrane cholesterol content in this process. These authors produced elevated levels of PIP2 in both 3T3-L1 fibroblasts and adipocytes via overexpression of PIP5K. In fibroblasts PIP5K expression yielded motile actin comet tails, whereas in differentiated adipocytes the formation of vacuoles coated with F-actin, cortactin, dynamin, and N-WASP, and an inhibition GLUT4 endocytosis was noted. Further, these authors demonstrated that cholesterol loading of fibroblasts expressing PIP5K promoted formation of similar vacuoles, leading these authors to hypothesize that the effect of cholesterol on membrane thickness and curvature or its influence on PIP2 levels and distribution—both of which affect actin cytoskeletal organization and membrane trafficking events such as GLUT4 endocytosis—is involved [128].
Concordant with the insulin-sensitizing and cholesterol-lowering effects of CrPic [158–160], this dietary supplement improves insulin sensitivity by lowering membrane cholesterol from insulin-resistant, cholesterol-laden PMs [161]. These beneficial actions of CrPic are consistent with newly recognized effects of statins on improving insulin resistance in dyslipidemic patients [162–164]. The mechanism by which CrPic decreases cholesterol is speculative but may occur by means similar to that used by the anti-diabetic drug metformin. For example, CrPic [161, 165] and metformin [166, 167] have recently been reported to activate 5′-AMP-activated kinase AMPK. Metformin stimulated AMPK activity has been shown to suppress expression of a sterol regulatory element binding protein, SREBP-1 [167] SREBP-1 belongs to a family of key lipogenic transcription factors directly involved in the expression of more than 30 genes dedicated to the synthesis and uptake of cholesterol, fatty acids, triglycerides, and phospholipids, as well as the NADPH cofactor required to synthesize these molecules [168] More recent work also shows that acute contraction restores insulin-stimulated glucose uptake in muscle of obese insulin-resistant Zucker rats to levels seen in lean controls [169]. As predicted, contraction decreased intramuscular triacylglycerol content; however, diacylglycerol and long chain acyl-CoAs, lipid intermediates presumed to trigger insulin resistance, were either unchanged or increased [169]. Also, it was observed that only a partial improvement in insulin signaling to AS160 occurred, thus implicating another target of the beneficial effects of exercise. Together, these findings raise the question whether metformin and/or acute exercise decreases PM cholesterol.
In further support of an association between PIP2 and zones of actin re-organization, Huang et al. showed that PIP2-rich plasma membrane patches (PRMPs) organize active zones of endocytosis and membrane ruffling in cultured adipocytes [54, 170]. Although their data provided a correlation between the location of PRMPs and zones of concentrated F-actin where endocytic activity was occurring, it did not provide evidence of a causative link between the two. To explore this relationship, these authors conducted additional experiments using the molecular motor protein Myo1c, a mediator of membrane ruffling which requires F-actin for proper function. Data from these experiments suggested that PRMPs may function to locally concentrate F-actin and Myo1c to enhance their ability to promote membrane ruffling [54, 170]. Previous evidence has also indicated that Myo1c may play some role in GLUT4 translocation and insulin-stimulated GLUT4 recycling [102]; thus it is also possible that PRMPs may promote this process as well [54]. (For a review of spatial control of actin-based motility through plasmalemmal PIP2-rich raft assemblies, see [171])
Insulin Resistance: A Disease of the Actin Cytoskeleton?
It is clear that the molecular etiology of insulin resistance involves multiple genetic and non-genetic mechanisms contributing to the final phenotype [172]. Furthermore, the resultant hyperglycemia of the syndrome and the compensatory rise in insulin in turn exacerbate insulin resistance, contributing significantly to the pathogenesis of the disease [173]. Superimposed on that backdrop is the negative contribution of the obese state [174]. Although pinpointing the site of the abnormality induced by these conditions is still an intensive area of current research, evidence strongly suggests that t[175]he defects induced by each occur at different loci in the insulin-signaling network. Previous studies show that prolonged insulin treatment of primary rat adipocytes and 3T3-L1 adipocytes cause a state of insulin resistance as manifested by decreased insulin-stimulated glucose transport [176, 177]. This defect is associated with an impaired net GLUT4 translocation to the cell surface of adipocytes [178, 179]. Examination of the protein and activity levels of the IR, PI3K and Akt demonstrate a clear progressive disturbance in the insulin stimulated sequential communication of these signaling molecules [180, 181]. Interestingly, in support of this signaling defect being corrected by changing membrane fluidity, the decreased IR, PI3K and Akt activities were reversed when metformin was included in the chronic insulin culture [181].
In contrast to an insulin receptor abnormality as a basis of signal interference, multiple studies suggest that lipid oversupply contributes to the development of insulin resistance, perhaps by promoting the accumulation of ceramide or a ceramide metabolite that in turn inhibits insulin stimulation of Akt without a decrease in the tyrosine phosphorylation of the IR or IRS1 or a decrease in the interaction of IRS1 with PI3K [182–189]. In addition, work suggests that defects in the IR, IRS1, PI3K, Akt, and Cbl do not readily account for decreased insulin responsiveness induced by increasing the flux of glucose through the hexosamine biosynthesis pathway [179, 188, 190–194]. Buse et al. [195] recently demonstrated that chronically increased glucose flux elevated muscle UDP-N-acetylglucosamine levels and this in turn induced O-linked glycosylation of membrane proteins in vivo.
In the same way that increased hexosamine biosynthesis contributes, at least in part, to the insulin resistant state without insulin signaling defects, new perspective shows a membrane/cytoskeletal basis of hyperinsulinemia-induced insulin resistance. This finding has significant merit as a preliminary component of insulin resistance is hyperinsulinemia [196]. This compensatory increase in insulin during the insulin resistant state initially offsets insulin’s reduced ability to stimulate GLUT4 translocation and glucose uptake, which is essential for the maintenance of blood glucose homeostasis. However, in vivo [197, 198] and in vitro [180, 199] studies demonstrate that the hyperinsulinemic state has a negative effect on insulin action. To date, extensive study has built a good understanding of how insulin itself can be involved in the staging of diabetes. For example, chronic exposure to pharmacological doses of insulin (≥100 nM) have been shown by several laboratories to markedly attenuate expression levels and/or activity states of the IR, IRS-1, PI3K, Akt and GLUT4 proteins in 3T3-L1 adipocytes and thus, produce a defect in the cell’s ability to respond to subsequent acute insulin stimulation with an increase in GLUT4 translocation and glucose transport [181, 200, 201]. On the other hand, similar defects in the ability of insulin to acutely regulate the glucose transport system can be induced by lower, more pathophysiological concentrations of insulin (≤5 nM) that are not associated with disturbances in early insulin signaling [6, 103, 193, 202]. Therefore, defects positioned more distal in the insulin signaling pathway may contribute to the insulin-mediated cellular insulin resistance.
Recent study has found that plasma membrane phosphoinositide-regulated cortical F-actin structure is perturbed in muscle and fat cells made insulin resistant by hyperinsulinemia [6, 103]. Consistent with previous studies that induce insulin resistance via overnight treatment of cells with a more pathophysiological dose of insulin [193], the impaired ability of insulin to acutely regulate the glucose transport process did not correlate with reduced protein levels or activation of several known insulin signaling intermediates. In contrast, there was an associated loss of PIP2 and cortical F-actin during the insulin-induced state of insulin resistance. Particularly striking was that correction of that via PIP2 replenishment restored insulin sensitivity. Thus, these recent data define a novel diabetic-state alteration and reveal a counteractive tactic to normalize insulin sensitivity. Data from another set of studies demonstrates a strikingly similar importance of plasma membrane PIP2 and cortical F-actin in the development of insulin resistance induced by endothelin-1 [203, 204]. This vasoactive peptide is well-documented to diminish insulin sensitivity by a direct effect on one or more mechanisms involved in insulin-stimulated glucose transport and not from a vasoconstrictive decrease in skeletal muscle blood flow [205–208]. As this vasoactive peptide also displays insulin mimetic activity [207, 209–211], it is tempting to speculate that both endothelin-1- and insulin-induced insulin resistance result from stimulated glucose transport and increased hexosamine biosynthesis, which as indicated above may have ties to cell surface abnormalities.
The cortical actin network also interacts with the extracellular cytoskeletal matrix proteins (dystrophin, α/β-dystroglycan, spectrin, etc.), which serves to stabilize the structure of the plasma membrane. Thus, it would be expected that disruption of either of these structures could influence insulin signaling and GLUT4 translocation. In agreement with this idea, it has been reported that mdx mice, an animal model of Duchene’s Muscular Dystrophy which possess a mutation of the dystrophin gene, exhibit impaired regulation of carbohydrate metabolism [212]. Conversely, and more pertinant to the topic of this review, it has recently been reported that the diabetic Goto-Kakizaki rat exhibits diminished levels of the dystrophin and α/β dystroglycan isoforms that form a link between the extracellular cytoskeletal matrix and the actin membrane cytoskeleton [213]. Thus, impaired anchoring of the actin cytoskeleton to the plasma membrane may contribute to the reduced insulin stimulated GLUT4 translocation observed in this model. This may be of relevance to humans, as muscular weakness has long been recognized as a phenomenon observed in human diabetics [214]. In light of this observation, it has recently been reported that endurance exercise, which has insulin sensitizing effects, is associated with an increase in the phosphorylation of filamin A [175], an additional protein that along with dystrophin, is involved in the linkage of the cortical actin cytoskeleton to the extracellular matrix. Although the importance of this event remains to be determined, it is tempting to speculate that muscular contraction may be positively modulating the interaction between the extracellular matrix and the actin cytoskeleton to facilitate improved insulin signaling and GLUT4 translocation. However, the true significance of this finding remains to be determined through future investigation.
CONCLUSIONS
Among the many facets of insulin signaling currently being studied, intense effort is being focused on insulin’s ability to effect GLUT4 translocation. Although there is still much to learn about the complex regulation of GLUT4 translocation by insulin; as highlighted in this review, it is clear that regulatory events involving actin and phosphoinositides and are major players in this process. What remains unclear, however, is how elevations in insulin, perhaps in combination with elevated plasma lipids, alter the plasma membrane and actin cytoskeleton to induce insulin resistance. This, for certain will be an area of concerted study in the years to come.
Acknowledgments
This work was supported in part by a National Center for Complementary and Alternative Medicine Grant R01-AT001846 (JSE) and an American Diabetes Association Research Award 07-05-RA-37 (JSE).
References
- 1.Advani A, Marshall SM, Thomas TH. Impaired neutrophil actin assembly causes persistent CD11b expression and reduced primary granule exocytosis in Type II diabetes. Diabetologia. 2002;45(5):719–27. doi: 10.1007/s00125-002-0802-0. [DOI] [PubMed] [Google Scholar]
- 2.Advani A, Marshall SM, Thomas TH. Increasing neutrophil F-actin corrects CD11b exposure in Type 2 diabetes. Eur J Clin Invest. 2004;34(5):358–64. doi: 10.1111/j.1365-2362.2004.01346.x. [DOI] [PubMed] [Google Scholar]
- 3.Candiloros H, Zeghari N, Ziegler O, Donner M, Drouin P. Hyper-insulinemia is related to erythrocyte phospholipid composition and membrane fluidity changes in obese nondiabetic women. J Clin Endocrinol Metab. 1996;81(8):2912–8. doi: 10.1210/jcem.81.8.8768851. [DOI] [PubMed] [Google Scholar]
- 4.Younsi M, Quilliot D, Al-Makdissy N, et al. Erythrocyte membrane phospholipid composition is related to hyperinsulinemia in obese nondiabetic women: effects of weight loss. Metabolism. 2002;51(10):1261–8. doi: 10.1053/meta.2002.35184. [DOI] [PubMed] [Google Scholar]
- 5.Kane S, Sano H, Liu SC, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277(25):22115–8. doi: 10.1074/jbc.C200198200. Epub 2002 May 6. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy AM, Elmendorf JS. Phosphatidylinostol 4,5-bisphosphate and cortical F-actin abnormalities in insulin resistant skeletal muscle. Diabetes. 2005;54(Suppl 1):A319. [Google Scholar]
- 7.Sano H, Kane S, Sano E, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278(17):14599–602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 8.Jordens I, Marsman M, Kuijl C, Neefjes J. Rab proteins, connecting transport and vesicle fusion. Traffic. 2005;6(12):1070–7. doi: 10.1111/j.1600-0854.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 9.Larance M, Ramm G, Stockli J, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280(45):37803–13. doi: 10.1074/jbc.M503897200. Epub 2005 Sep 8. [DOI] [PubMed] [Google Scholar]
- 10.Miinea CP, Sano H, Kane S, et al. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391(Pt 1):87–93. doi: 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elmendorf JS, Pessin JE. Insulin signaling regulating the trafficking and plasma membrane fusion of GLUT4-containing intracellular vesicles. Exp Cell Res. 1999;253(1):55–62. doi: 10.1006/excr.1999.4675. [DOI] [PubMed] [Google Scholar]
- 12.Eguez L, Lee A, Chavez JA, et al. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2(4):263–72. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Zeigerer A, McBrayer MK, McGraw TE. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell. 2004;15(10):4406–15. doi: 10.1091/mbc.E04-04-0333. Epub 2004 Jul 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ribon V, Saltiel AR. Insulin stimulates tyrosine phosphorylation of the protooncogene product of c-Cbl in 3T3-L1 adipocytes. Biochem J. 1997;324:839–846. doi: 10.1042/bj3240839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, DeYoung SM, Hwang JB, O’Leary EE, Saltiel AR. The roles of Cbl-b and c-Cbl in insulin-stimulated glucose transport. J Biol Chem. 2003;278(38):36754–62. doi: 10.1074/jbc.M300664200. [DOI] [PubMed] [Google Scholar]
- 16.Ribon V, Printen JA, Hoffman NG, Kay BK, Saltiel AR. A novel, multifuntional c-Cbl binding protein in insulin receptor signaling in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18(2):872–9. doi: 10.1128/mcb.18.2.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J, Kimura A, Baumann CA, Saltiel AR. APS facilitates c-Cbl tyrosine phosphorylation and GLUT4 translocation in response to insulin in 3T3-L1 adipocytes. Mol Cell Biol. 2002;22(11):3599–609. doi: 10.1128/MCB.22.11.3599-3609.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiang SH, Baumann CA, Kanzaki M, et al. Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature. 2001;410(6831):944–8. doi: 10.1038/35073608. [DOI] [PubMed] [Google Scholar]
- 19.Watson RT, Shigematsu S, Chiang SH, et al. Lipid raft microdomain compartmentalization of TC10 is required for insulin signaling and GLUT4 translocation. J Cell Biol. 2001;154(4):829–40. doi: 10.1083/jcb.200102078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumann CA, Ribon V, Kanzaki M, et al. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407(6801):202–7. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- 21.Chunqiu Hou J, Pessin JE. Lipid Raft Targeting of the TC10 Amino Terminal Domain Is Responsible for Disruption of Adipocyte Cortical Actin. Mol Biol Cell. 2003;14(9):3578–91. doi: 10.1091/mbc.E03-01-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422(6932):629–33. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- 23.Jiang ZY, Chawla A, Bose A, Way M, Czech MP. A phosphatidylinositol 3-kinase-independent insulin signaling pathway to N-WASP/Arp2/3/F-actin required for GLUT4 glucose transporter recycling. J Biol Chem. 2002;277(1):509–15. doi: 10.1074/jbc.M108280200. [DOI] [PubMed] [Google Scholar]
- 24.Kanzaki M, Pessin JE. Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J Biol Chem. 2001;276(45):42436–44. doi: 10.1074/jbc.M108297200. [DOI] [PubMed] [Google Scholar]
- 25.Kanzaki M, Watson RT, Hou JC, Stamnes M, Saltiel AR, Pessin JE. Small GTP-binding protein TC10 differentially regulates two distinct populations of filamentous actin in 3T3L1 adipocytes. Mol Biol Cell. 2002;13(7):2334–46. doi: 10.1091/mbc.01-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanzaki M, Pessin JE. Caveolin-associated filamentous actin (Cavactin) defines a novel F-actin structure in adipocytes. J Biol Chem. 2002;277(29):25867–9. doi: 10.1074/jbc.C200292200. [DOI] [PubMed] [Google Scholar]
- 27.Maffucci T, Brancaccio A, Piccolo E, Stein RC, Falasca M. Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. Embo J. 2003;22(16):4178–89. doi: 10.1093/emboj/cdg402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Downes CP, Gray A, Lucocq JM. Probing phosphoinositide functions in signaling and membrane trafficking. Trends Cell Biol. 2005;15(5):259–68. doi: 10.1016/j.tcb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Parker PJ. The ubiquitous phosphoinositides. Biochem Soc Trans. 2004;32(Pt 6):893–8. doi: 10.1042/BST0320893. [DOI] [PubMed] [Google Scholar]
- 30.Roth MG. Phosphoinositides in constitutive membrane traffic. Physiol Rev. 2004;84(3):699–730. doi: 10.1152/physrev.00033.2003. [DOI] [PubMed] [Google Scholar]
- 31.Michell RH, Hawthorne JN, Coleman R, Karnovsky ML. Extraction of polyphosphoinositides with neutral and acidified solvents. A comparison of guinea-pig brain and liver, and measurements of rat liver inositol compounds which are resistant to extraction. Biochim Biophys Acta. 1970;210(1):86–91. doi: 10.1016/0005-2760(70)90064-0. [DOI] [PubMed] [Google Scholar]
- 32.Vanhaesebroeck B, Leevers SJ, Ahmadi K, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 33.Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol. 2003;65:761–89. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 34.Hartwig JH, Bokoch GM, Carpenter CL, et al. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82(4):643–53. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 35.Kwik J, Boyle S, Fooksman D, Margolis L, Sheetz MP, Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc Natl Acad Sci U S A. 2003;100(24):13964–9. doi: 10.1073/pnas.2336102100. Epub 2003 Nov 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Rheenen J, Jalink K. Agonist-induced PIP(2) hydrolysis inhibits cortical actin dynamics: regulation at a global but not at a micrometer scale. Mol Biol Cell. 2002;13(9):3257–67. doi: 10.1091/mbc.E02-04-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foti M, Audhya A, Emr SD. Sac1 lipid phosphatase and Stt4 phosphatidylinositol 4-kinase regulate a pool of phosphatidylinositol 4-phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol Biol Cell. 2001;12(8):2396–411. doi: 10.1091/mbc.12.8.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DiNubile MJ, Huang S. High concentrations of phosphatidylinositol-4,5-bisphosphate may promote actin filament growth by three potential mechanisms: inhibiting capping by neutrophil lysates, severing actin filaments and removing capping protein-beta2 from barbed ends. Biochim Biophys Acta. 1997;1358(3):261–78. doi: 10.1016/s0167-4889(97)00076-1. [DOI] [PubMed] [Google Scholar]
- 39.Payrastre B, Missy K, Giuriato S, Bodin S, Plantavid M, Gratacap M. Phosphoinositides: key players in cell signalling, in time and space. Cell Signal. 2001;13(6):377–87. doi: 10.1016/s0898-6568(01)00158-9. [DOI] [PubMed] [Google Scholar]
- 40.Corgan AM, Singleton C, Santoso CB, Greenwood JA. Phosphoinositides differentially regulate alpha-actinin flexibility and function. Biochem J. 2004;378(Pt 3):1067–72. doi: 10.1042/BJ20031124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fievet BT, Gautreau A, Roy C, et al. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol. 2004;164(5):653–9. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraley TS, Pereira CB, Tran TC, Singleton C, Greenwood JA. Phosphoinositide binding regulates alpha-actinin dynamics: mechanism for modulating cytoskeletal remodeling. J Biol Chem. 2005;280(15):15479–82. doi: 10.1074/jbc.M500631200. Epub 2005 Feb 13. [DOI] [PubMed] [Google Scholar]
- 43.Hamada K, Shimizu T, Matsui T, Tsukita S, Hakoshima T. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. Embo J. 2000;19(17):4449–62. doi: 10.1093/emboj/19.17.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heiska L, Alfthan K, Gronholm M, Vilja P, Vaheri A, Carpen O. Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4, 5-bisphosphate. J Biol Chem. 1998;273(34):21893–900. doi: 10.1074/jbc.273.34.21893. [DOI] [PubMed] [Google Scholar]
- 45.Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109(5):599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- 46.Imai K, Nonoyama S, Miki H, et al. The pleckstrin homology domain of the Wiskott-Aldrich syndrome protein is involved in the organization of actin cytoskeleton. Clin Immunol. 1999;92(2):128–37. doi: 10.1006/clim.1999.4746. [DOI] [PubMed] [Google Scholar]
- 47.Kam JL, Miura K, Jackson TR, et al. Phosphoinositide-dependent activation of the ADP-ribosylation factor GTPase-activating protein ASAP1. Evidence for the pleckstrin homology domain functioning as an allosteric site. J Biol Chem. 2000;275(13):9653–63. doi: 10.1074/jbc.275.13.9653. [DOI] [PubMed] [Google Scholar]
- 48.Xian W, Janmey PA. Dissecting the gelsolin-polyphosphoinositide interaction and engineering of a polyphosphoinositide-sensitive gelsolin C-terminal half protein. J Mol Biol. 2002;322(4):755–71. doi: 10.1016/s0022-2836(02)00841-0. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto M, Hilgemann DH, Feng S, et al. Phosphatidylinositol 4,5-bisphosphate induces actin stress-fiber formation and inhibits membrane ruffling in CV1 cells. J Cell Biol. 2001;152(5):867–76. doi: 10.1083/jcb.152.5.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang SA, Carpenter CL, Abrams CS. Rho and Rho-kinase mediate thrombin-induced phosphatidylinositol 4-phosphate 5-kinase trafficking in platelets. J Biol Chem. 2004;279(40):42331–6. doi: 10.1074/jbc.M404335200. Epub 2004 Jul 23. [DOI] [PubMed] [Google Scholar]
- 51.Yonemura S, Matsui T, Tsukita S. Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: an essential role for polyphosphoinositides in vivo. J Cell Sci. 2002;115(Pt 12):2569–80. doi: 10.1242/jcs.115.12.2569. [DOI] [PubMed] [Google Scholar]
- 52.Dormann D, Weijer G, Dowler S, Weijer CJ. In vivo analysis of 3-phosphoinositide dynamics during Dictyostelium phagocytosis and chemotaxis. J Cell Sci. 2004;117(Pt 26):6497–509. doi: 10.1242/jcs.01579. Epub 2004 Nov 30. [DOI] [PubMed] [Google Scholar]
- 53.Shisheva A. Regulating Glut4 vesicle dynamics by phosphoinositide kinases and phosphoinositide phosphatases. Front Biosci. 2003;8:s945–6. doi: 10.2741/1101. [DOI] [PubMed] [Google Scholar]
- 54.Huang S, Lifshitz L, Patki-Kamath V, Tuft R, Fogarty K, Czech MP. Phosphatidylinositol-4,5-bisphosphate-rich plasma membrane patches organize active zones of endocytosis and ruffling in cultured adipocytes. Mol Cell Biol. 2004;24(20):9102–23. doi: 10.1128/MCB.24.20.9102-9123.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Standaert ML, Galloway L, Karnam P, Bandyopadhyay G, Moscat J, Farese RV. Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J Biol Chem. 1997;272(48):30075–82. doi: 10.1074/jbc.272.48.30075. [DOI] [PubMed] [Google Scholar]
- 56.Standaert ML, Bandyopadhyay G, Perez L, et al. Insulin activates protein kinases C-zeta and C-lambda by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem. 1999;274(36):25308–16. doi: 10.1074/jbc.274.36.25308. [DOI] [PubMed] [Google Scholar]
- 57.Bandyopadhyay G, Standaert ML, Galloway L, Moscat J, Farese RV. Evidence for involvement of protein kinase C (PKC)-zeta and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubes. Endocrinology. 1997;138(11):4721–31. doi: 10.1210/endo.138.11.5473. [DOI] [PubMed] [Google Scholar]
- 58.Kotani K, Ogawa W, Matsumoto M, et al. Requirement of atypical protein kinase clambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18(12):6971–82. doi: 10.1128/mcb.18.12.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mendez R, Kollmorgen G, White MF, Rhoads RE. Requirement of protein kinase C zeta for stimulation of protein synthesis by insulin. Mol Cell Biol. 1997;17(9):5184–92. doi: 10.1128/mcb.17.9.5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang C, Watson RT, Elmendorf JS, Sacks DB, Pessin JE. Calmodulin antagonists inhibit insulin-stimulated GLUT4 (glucose transporter 4) translocation by preventing the formation of phosphatidylinositol 3,4,5-trisphosphate in 3T3L1 adipocytes. Mol Endocrinol. 2000;14(2):317–26. doi: 10.1210/mend.14.2.0425. [DOI] [PubMed] [Google Scholar]
- 61.Tengholm A, Teruel MN, Meyer T. Single cell imaging of PI3K activity and glucose transporter insertion into the plasma membrane by dual color evanescent wave microscopy. Sci STKE. 2003;2003(169):L4. doi: 10.1126/stke.2003.169.pl4. [DOI] [PubMed] [Google Scholar]
- 62.Vollenweider P, Clodi M, Martin SS, Imamura T, Kavanaugh WM, Olefsky JM. An SH2 domain-containing 5′ inositolphosphatase inhibits insulin-induced GLUT4 translocation and growth factor-induced actin filament rearrangement. Mol Cell Biol. 1999;19(2):1081–91. doi: 10.1128/mcb.19.2.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wada T, Sasaoka T, Funaki M, et al. Overexpression of SH2-containing inositol phosphatase 2 results in negative regulation of insulin-induced metabolic actions in 3T3-L1 adipocytes via its 5′-phosphatase catalytic activity. Mol Cell Biol. 2001;21(5):1633–46. doi: 10.1128/MCB.21.5.1633-1646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ijuin T, Takenawa T. SKIP negatively regulates insulin-induced GLUT4 translocation and membrane ruffle formation. Mol Cell Biol. 2003;23(4):1209–20. doi: 10.1128/MCB.23.4.1209-1220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakashima N, Sharma PM, Imamura T, Bookstein R, Olefsky JM. The tumor suppressor PTEN negatively regulates insulin signaling in 3T3-L1 adipocytes. J Biol Chem. 2000;275(17):12889–95. doi: 10.1074/jbc.275.17.12889. [DOI] [PubMed] [Google Scholar]
- 66.Tengholm A, Meyer T. A PI3-kinase signaling code for insulin-triggered insertion of glucose transporters into the plasma membrane. Curr Biol. 2002;12(21):1871–6. doi: 10.1016/s0960-9822(02)01223-x. [DOI] [PubMed] [Google Scholar]
- 67.Oatey PB, Venkateswarlu K, Williams AG, et al. Confocal imaging of the subcellular distribution of phosphatidylinositol 3,4,5-trisphosphate in insulin- and PDGF-stimulated 3T3-L1 adipocytes. Biochem J. 1999;344(Pt 2):511–8. [PMC free article] [PubMed] [Google Scholar]
- 68.Sweeney G, Garg RR, Ceddia RB, et al. Intracellular delivery of phosphatidylinositol (3,4,5)-trisphosphate causes incorporation of glucose transporter 4 into the plasma membrane of muscle and fat cells without increasing glucose uptake. J Biol Chem. 2004;279(31):32233–42. doi: 10.1074/jbc.M402897200. Epub 2004 May 27. [DOI] [PubMed] [Google Scholar]
- 69.Eichhorn J, Kayali AG, Austin DA, Webster NJ. Insulin activates phospholipase C-gamma1 via a PI-3 kinase dependent mechanism in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2001;282(2):615–20. doi: 10.1006/bbrc.2001.4616. [DOI] [PubMed] [Google Scholar]
- 70.Kayali AG, Eichhorn J, Haruta T, et al. Association of the insulin receptor with phospholipase C-gamma (PLCgamma) in 3T3-L1 adipocytes suggests a role for PLCgamma in metabolic signaling by insulin. J Biol Chem. 1998;273(22):13808–18. doi: 10.1074/jbc.273.22.13808. [DOI] [PubMed] [Google Scholar]
- 71.Van Epps-Fung M, Gupta K, Hardy RW, Wells A. A role for phospholipase C activity in GLUT4-mediated glucose transport. Endocrinology. 1997;138(12):5170–5. doi: 10.1210/endo.138.12.5596. [DOI] [PubMed] [Google Scholar]
- 72.Sbrissa D, Ikonomov OC, Strakova J, Shisheva A. Role for a novel signaling intermediate, phosphatidylinositol 5-phosphate, in insulin-regulated F-actin stress fiber breakdown and GLUT4 translocation. Endocrinology. 2004;145(11):4853–65. doi: 10.1210/en.2004-0489. Epub 2004 Jul 29. [DOI] [PubMed] [Google Scholar]
- 73.Wenk MR, De Camilli P. Protein-lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc Natl Acad Sci U S A. 2004;101(22):8262–9. doi: 10.1073/pnas.0401874101. Epub 2004 May 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishiki M, Randhawa VK, Poon V, Jebailey L, Klip A. Insulin regulates the membrane arrival, fusion and C-terminal unmasking of Glut4 via distinct phosphoinositides. J Biol Chem. 2005;13:13. doi: 10.1074/jbc.M500501200. [DOI] [PubMed] [Google Scholar]
- 75.Hay JC, Fisette PL, Jenkins GH, et al. ATP-dependent inositide phosphorylation required for Ca(2+)-activated secretion. Nature. 1995;374(6518):173–7. doi: 10.1038/374173a0. [DOI] [PubMed] [Google Scholar]
- 76.Hay JC, Martin TF. Phosphatidylinositol transfer protein required for ATP-dependent priming of Ca(2+)-activated secretion. Nature. 1993;366(6455):572–5. doi: 10.1038/366572a0. [DOI] [PubMed] [Google Scholar]
- 77.Eberhard DA, Cooper CL, Low MG, Holz RW. Evidence that the inositol phospholipids are necessary for exocytosis. Loss of inositol phospholipids and inhibition of secretion in permeabilized cells caused by a bacterial phospholipase C and removal of ATP. Biochem J. 1990;268(1):15–25. doi: 10.1042/bj2680015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bittner MA, Holz RW. Phosphatidylinositol-4,5-bisphosphate: actin dynamics and the regulation of ATP-dependent and -independent secretion. Mol Pharmacol. 2005;67(4):1089–98. doi: 10.1124/mol.104.008474. Epub 2005 Jan 5. [DOI] [PubMed] [Google Scholar]
- 79.Cormont M, Tanti JF, Zahraoui A, Van Obberghen E, Tavitian A, Le Marchand-Brustel Y. Insulin and okadaic acid induce Rab4 redistribution in adipocytes. J Biol Chem. 1993;268(26):19491–7. [PubMed] [Google Scholar]
- 80.Cormont M, Van Obberghen E, Zerial M, Le Marchand-Brustel Y. Insulin induces a change in Rab5 subcellular localization in adipocytes independently of phosphatidylinositol 3-kinase activation. Endocrinology. 1996;137(8):3408–15. doi: 10.1210/endo.137.8.8754768. [DOI] [PubMed] [Google Scholar]
- 81.Shibata H, Omata W, Suzuki Y, Tanaka S, Kojima I. A synthetic peptide corresponding to the Rab4 hypervariable carboxyl-terminal domain inhibits insulin action on glucose transport in rat adipocytes. J Biol Chem. 1996;271(16):9704–9. doi: 10.1074/jbc.271.16.9704. [DOI] [PubMed] [Google Scholar]
- 82.Mora S, Monden I, Zorzano A, Keller K. Heterologous expression of rab4 reduces glucose transport and GLUT4 abundance at the cell surface in oocytes. Biochem J. 1997;324(Pt 2):455–9. doi: 10.1042/bj3240455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li L, Omata W, Kojima I, Shibata H. Direct interaction of Rab4 with syntaxin 4. J Biol Chem. 2001;276(7):5265–73. doi: 10.1074/jbc.M003883200. [DOI] [PubMed] [Google Scholar]
- 84.Kanda H, Tamori Y, Shinoda H, et al. Adipocytes from Munc18c-null mice show increased sensitivity to insulin-stimulated GLUT4 externalization. J Clin Invest. 2005;115(2):291–301. doi: 10.1172/JCI22681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kao AW, Ceresa BP, Santeler SR, Pessin JE. Expression of a dominant interfering dynamin mutant in 3T3L1 adipocytes inhibits GLUT4 endocytosis without affecting insulin signaling. J Biol Chem. 1998;273(39):25450–7. doi: 10.1074/jbc.273.39.25450. [DOI] [PubMed] [Google Scholar]
- 86.Ceresa BP, Kao AW, Santeler SR, Pessin JE. Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol Cell Biol. 1998;18(7):3862–70. doi: 10.1128/mcb.18.7.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Volchuk A, Narine S, Foster LJ, Grabs D, De Camilli P, Klip A. Perturbation of dynamin II with an amphiphysin SH3 domain increases GLUT4 glucose transporters at the plasma membrane in 3T3-L1 adipocytes. Dynamin II participates in GLUT4 endocytosis. J Biol Chem. 1998;273(14):8169–76. doi: 10.1074/jbc.273.14.8169. [DOI] [PubMed] [Google Scholar]
- 88.Al-Hasani H, Hinck CS, Cushman SW. Endocytosis of the glucose transporter GLUT4 is mediated by the GTPase dynamin. J Biol Chem. 1998;273(28):17504–10. doi: 10.1074/jbc.273.28.17504. [DOI] [PubMed] [Google Scholar]
- 89.Hoy M, Efanov AM, Bertorello AM, et al. Inositol hexakisphosphate promotes dynamin I- mediated endocytosis. Proc Natl Acad Sci U S A. 2002;99(10):6773–7. doi: 10.1073/pnas.102157499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barylko B, Binns DD, Albanesi JP. Activation of dynamin GTPase activity by phosphoinositides and SH3 domain-containing proteins. Methods Enzymol. 2001;329:486–96. doi: 10.1016/s0076-6879(01)29110-1. [DOI] [PubMed] [Google Scholar]
- 91.Lin HC, Barylko B, Achiriloaie M, Albanesi JP. Phosphatidylinositol (4,5)-bisphosphate-dependent activation of dynamins I and II lacking the proline/arginine-rich domains. J Biol Chem. 1997;272(41):25999–6004. doi: 10.1074/jbc.272.41.25999. [DOI] [PubMed] [Google Scholar]
- 92.Salim K, Bottomley MJ, Querfurth E, et al. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. Embo J. 1996;15(22):6241–50. [PMC free article] [PubMed] [Google Scholar]
- 93.Lin HC, Gilman AG. Regulation of dynamin I GTPase activity by G protein betagamma subunits and phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 1996;271(45):27979–82. doi: 10.1074/jbc.271.45.27979. [DOI] [PubMed] [Google Scholar]
- 94.von Stein W, Ramrath A, Grimm A, Muller-Borg M, Wodarz A. Direct association of Bazooka/PAR-3 with the lipid phosphatase PTEN reveals a link between the PAR/aPKC complex and phosphoinositide signaling. Development. 2005;132(7):1675–86. doi: 10.1242/dev.01720. Epub 2005 Mar 2. [DOI] [PubMed] [Google Scholar]
- 95.Wang Q, Xie Y, Du QS, et al. Regulation of the formation of osteoclastic actin rings by proline-rich tyrosine kinase 2 interacting with gelsolin. J Cell Biol. 2003;160(4):565–75. doi: 10.1083/jcb.200207036. Epub 2003 Feb 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bertagnolo V, Brugnoli F, Marchisio M, Capitani S. Inositide-modifying enzymes: a cooperative role in regulating nuclear morphology during differentiation of myeloid cells. J Biol Regul Homeost Agents. 2004;18(3–4):381–6. [PubMed] [Google Scholar]
- 97.Bodin S, Soulet C, Tronchere H, et al. Integrin-dependent interaction of lipid rafts with the actin cytoskeleton in activated human platelets. J Cell Sci. 2005;118(Pt 4):759–69. doi: 10.1242/jcs.01648. Epub 2005 Jan 25. [DOI] [PubMed] [Google Scholar]
- 98.Hartwig JH, Barkalow K, Azim A, Italiano J. The elegant platelet: signals controlling actin assembly. Thromb Haemost. 1999;82(2):392–8. [PubMed] [Google Scholar]
- 99.Cabello-Agueros JF, Hernandez-Gonzalez EO, Mujica A. The role of F-actin cytoskeleton-associated gelsolin in the guinea pig capacitation and acrosome reaction. Cell Motil Cytoskeleton. 2003;56(2):94–108. doi: 10.1002/cm.10135. [DOI] [PubMed] [Google Scholar]
- 100.Omata W, Shibata H, Li L, Takata K, Kojima I. Actin filaments play a critical role in insulin-induced exocytotic recruitment but not in endocytosis of GLUT4 in isolated rat adipocytes. Biochem J. 2000;346(Pt 2):321–8. [PMC free article] [PubMed] [Google Scholar]
- 101.Tsakiridis T, Bergman A, Somwar R, et al. Actin filaments facilitate insulin activation of the src and collagen homologous/mitogen-activated protein kinase pathway leading to DNA synthesis and c-fos expression. J Biol Chem. 1998;273(43):28322–31. doi: 10.1074/jbc.273.43.28322. [DOI] [PubMed] [Google Scholar]
- 102.Bose A, Guilherme A, Robida SI, et al. Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature. 2002;420(6917):821–4. doi: 10.1038/nature01246. [DOI] [PubMed] [Google Scholar]
- 103.Chen G, Raman P, Bhonagiri P, Strawbridge AB, Pattar GR, Elmendorf JS. Protective effect of phosphatidylinositol 4,5-bisphosphate against cortical filamentous actin loss and insulin resistance induced by sustained exposure of 3T3-L1 adipocytes to insulin. J Biol Chem. 2004;279(38):39705–9. doi: 10.1074/jbc.C400171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Eyster CA, Duggins QS, Olson AL. Expression of constitutively active Akt/protein kinase B signals GLUT4 translocation in the absence of an intact actin cytoskeleton. J Biol Chem. 2005;280(18):17978–85. doi: 10.1074/jbc.M409806200. Epub 2005 Feb 28. [DOI] [PubMed] [Google Scholar]
- 105.Kanzaki M, Watson RT, Khan AH, Pessin JE. Insulin stimulates actin comet tails on intracellular GLUT4-containing compartments in differentiated 3T3L1 adipocytes. J Biol Chem. 2001;276(52):49331–6. doi: 10.1074/jbc.M109657200. [DOI] [PubMed] [Google Scholar]
- 106.Lu PJ, Shieh WR, Rhee SG, Yin HL, Chen CS. Lipid products of phosphoinositide 3-kinase bind human profilin with high affinity. Biochemistry. 1996;35(44):14027–34. doi: 10.1021/bi961878z. [DOI] [PubMed] [Google Scholar]
- 107.Bose A, Cherniack AD, Langille SE, et al. G(alpha)11 signaling through ARF6 regulates F-actin mobilization and GLUT4 glucose transporter translocation to the plasma membrane. Mol Cell Biol. 2001;21(15):5262–75. doi: 10.1128/MCB.21.15.5262-5275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Biswas RS, Baker D, Hruska KA, Chellaiah MA. Polyphosphoinositides-dependent regulation of the osteoclast actin cytoskeleton and bone resorption. BMC Cell Biol. 2004;5(1):19. doi: 10.1186/1471-2121-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Caroni P. New EMBO members’ review: actin cytoskeleton regulation through modulation of PI(4,5)P(2) rafts. Embo J. 2001;20(16):4332–6. doi: 10.1093/emboj/20.16.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fernandis AZ, Srivastava R, Sinha RK, Subrahmanyam G. A type II phosphatidylinositol 4-kinase associates with T cell receptor zeta chain in Con A stimulated splenic lymphocytes through tyrosyl phosphorylation-dependent mechanisms. Mol Immunol. 2005;42(5):561–8. doi: 10.1016/j.molimm.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 111.Purushothaman SS, Wang B, Cleary PP. M1 protein triggers a phosphoinositide cascade for group A Streptococcus invasion of epithelial cells. Infect Immun. 2003;71(10):5823–30. doi: 10.1128/IAI.71.10.5823-5830.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Patel N, Rudich A, Khayat ZA, Garg R, Klip A. Intracellular segregation of phosphatidylinositol-3,4,5-trisphosphate by insulin-dependent actin remodeling in L6 skeletal muscle cells. Mol Cell Biol. 2003;23(13):4611–26. doi: 10.1128/MCB.23.13.4611-4626.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tsakiridis T, Vranic M, Klip A. Disassembly of the actin network inhibits insulin-dependent stimulation of glucose transport and prevents recruitment of glucose transporters to the plasma membrane. J Biol Chem. 1994;269(47):29934–42. [PubMed] [Google Scholar]
- 114.Peyrollier K, Hajduch E, Gray A, et al. A role for the actin cytoskeleton in the hormonal and growth-factor-mediated activation of protein kinase B. Biochem J. 2000;352(Pt 3):617–22. [PMC free article] [PubMed] [Google Scholar]
- 115.Brozinick JT, Jr, Hawkins ED, Strawbridge AB, Elmendorf JS. Disruption of cortical actin in skeletal muscle demonstrates an essential role of the cytoskeleton in glucose transporter 4 translocation in insulin-sensitive tissues. J Biol Chem. 2004;279(39):40699–706. doi: 10.1074/jbc.M402697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Martin SS, Haruta T, Morris AJ, Klippel A, Williams LT, Olefsky JM. Activated phosphatidylinositol 3-kinase is sufficient to mediate actin rearrangement and GLUT4 translocation in 3T3-L1 adipocytes. J Biol Chem. 1996;271:17605–17608. doi: 10.1074/jbc.271.30.17605. [DOI] [PubMed] [Google Scholar]
- 117.Gual P, Shigematsu S, Kanzaki M, et al. A Crk-II/TC10 signaling pathway is required for osmotic shock-stimulated glucose transport. J Biol Chem. 2002;4:4. doi: 10.1074/jbc.M203042200. [DOI] [PubMed] [Google Scholar]
- 118.JeBailey L, Rudich A, Huang X, Di Ciano-Oliveira C, Kapus A, Klip A. Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol Endocrinol. 2004;18(2):359–72. doi: 10.1210/me.2003-0294. Epub 2003 Nov 13. [DOI] [PubMed] [Google Scholar]
- 119.Khayat ZA, Tong P, Yaworsky K, Bloch RJ, Klip A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J Cell Sci. 2000;113(Pt 2):279–90. doi: 10.1242/jcs.113.2.279. [DOI] [PubMed] [Google Scholar]
- 120.Terada S, Hirokawa N. Moving on to the cargo problem of microtubule-dependent motors in neurons. Curr Opin Neurobiol. 2000;10(5):566–73. doi: 10.1016/s0959-4388(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 121.Apodaca G. Endocytic traffic in polarized epithelial cells: role of the actin and microtubule cytoskeleton. Traffic. 2001;2(3):149–59. doi: 10.1034/j.1600-0854.2001.020301.x. [DOI] [PubMed] [Google Scholar]
- 122.Stamnes M. Regulating the actin cytoskeleton during vesicular transport. Curr Opin Cell Biol. 2002;14(4):428–33. doi: 10.1016/s0955-0674(02)00349-6. [DOI] [PubMed] [Google Scholar]
- 123.Schafer DA. Coupling actin dynamics and membrane dynamics during endocytosis. Curr Opin Cell Biol. 2002;14(1):76–81. doi: 10.1016/s0955-0674(01)00297-6. [DOI] [PubMed] [Google Scholar]
- 124.Patki V, Buxton J, Chawla A, et al. Insulin action on GLUT4 traffic visualized in single 3T3-l1 adipocytes by using ultra-fast microscopy. Mol Biol Cell. 2001;12(1):129–41. doi: 10.1091/mbc.12.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Semiz S, Park JG, Nicoloro SM, et al. Conventional kinesin KIF5B mediates insulin-stimulated GLUT4 movements on microtubules. Embo J. 2003;22(10):2387–99. doi: 10.1093/emboj/cdg237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Olson AL, Eyster CA, Duggins QS, Knight JB. Insulin promotes formation of polymerized microtubules by a phosphatidylinositol 3-kinase-independent, actin-dependent pathway in 3T3-L1 adipocytes. Endocrinology. 2003;144(11):5030–9. doi: 10.1210/en.2003-0609. Epub 2003 Aug 13. [DOI] [PubMed] [Google Scholar]
- 127.Wang Q, Bilan PJ, Tsakiridis T, Hinek A, Klip A. Actin filaments participate in the relocalization of phosphatidylinositol3-kinase to glucose transporter-containing compartments and in the stimulation of glucose uptake in 3T3-L1 adipocytes [published erratum appears in Biochem J 1999 Aug 1;341(Pt 3):861] Biochem J. 1998;331(Pt 3):917–28. doi: 10.1042/bj3310917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kanzaki M, Furukawa M, Raab W, Pessin JE. Phosphatidylinositol 4,5-bisphosphate regulates adipocyte actin dynamics and GLUT4 vesicle recycling. J Biol Chem. 2004;279(29):30622–33. doi: 10.1074/jbc.M401443200. Epub 2004 Apr 28. [DOI] [PubMed] [Google Scholar]
- 129.Kandror KV, Pilch PF. gp160, a tissue-specific marker for insulin-activated glucose transport. Proc Natl Acad Sci U S A. 1994;91(17):8017–21. doi: 10.1073/pnas.91.17.8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kandror KV, Yu L, Pilch PF. The major protein of GLUT4-containing vesicles, gp160, has aminopeptidase activity. J Biol Chem. 1994;269(49):30777–80. [PubMed] [Google Scholar]
- 131.Mastick CC, Aebersold R, Lienhard GE. Characterization of a major protein in GLUT4 vesicles. Concentration in the vesicles and insulin-stimulated translocation to the plasma membrane. J Biol Chem. 1994;269(8):6089–92. [PubMed] [Google Scholar]
- 132.Keller SR, Scott HM, Mastick CC, Aebersold R, Lienhard GE. Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles. J Biol Chem. 1995;270(40):23612–8. doi: 10.1074/jbc.270.40.23612. [DOI] [PubMed] [Google Scholar]
- 133.Malide D, St-Denis JF, Keller SR, Cushman SW. Vp165 and GLUT4 share similar vesicle pools along their trafficking pathways in rat adipose cells. FEBS Lett. 1997;409(3):461–8. doi: 10.1016/s0014-5793(97)00563-2. [DOI] [PubMed] [Google Scholar]
- 134.Ross SA, Herbst JJ, Keller SR, Lienhard GE. Trafficking kinetics of the insulin-regulated membrane aminopeptidase in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 1997;239(1):247–51. doi: 10.1006/bbrc.1997.7459. [DOI] [PubMed] [Google Scholar]
- 135.Waters SB, D’Auria M, Martin SS, Nguyen C, Kozma LM, Luskey KL. The amino terminus of insulin-responsive aminopeptidase causes Glut4 translocation in 3T3-L1 adipocytes. J Biol Chem. 1997;272(37):23323–7. doi: 10.1074/jbc.272.37.23323. [DOI] [PubMed] [Google Scholar]
- 136.Tojo H, Kaieda I, Hattori H, et al. The Formin family protein, formin homolog overexpressed in spleen, interacts with the insulin-responsive aminopeptidase and profilin IIa. Mol Endocrinol. 2003;17(7):1216–29. doi: 10.1210/me.2003-0056. Epub 2003 Apr 3. [DOI] [PubMed] [Google Scholar]
- 137.Paul DS, Harmon AW, Winston CP, Patel YM. Calpain facilitates GLUT4 vesicle translocation during insulin-stimulated glucose uptake in adipocytes. Biochem J. 2003;376(Pt 3):625–32. doi: 10.1042/BJ20030681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Turner MD, Cassell PG, Hitman GA. Calpain-10: from genome search to function. Diabetes Metab Res Rev. 2005;18:18. doi: 10.1002/dmrr.578. [DOI] [PubMed] [Google Scholar]
- 139.Bedi D, Clarke KJ, Dennis JC, et al. Endothelin-1 inhibits adiponectin secretion through a phosphatidylinositol 4,5-bisphosphate/actin-dependent mechanism. Biochem Biophys Res Commun. 2006;345(1):332–9. doi: 10.1016/j.bbrc.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 140.Valentijn JA, Valentijn K, Pastore LM, Jamieson JD. Actin coating of secretory granules during regulated exocytosis correlates with the release of rab3D. Proc Natl Acad Sci U S A. 2000;97(3):1091–5. doi: 10.1073/pnas.97.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J Cell Biol. 1995;128(4):589–98. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sontag JM, Aunis D, Bader MF. Peripheral actin filaments control calcium-mediated catecholamine release from streptolysin-O-permeabilized chromaffin cells. Eur J Cell Biol. 1988;46(2):316–26. [PubMed] [Google Scholar]
- 143.Lelkes PI, Friedman JE, Rosenheck K, Oplatka A. Destabilization of actin filaments as a requirement for the secretion of catecholamines from permeabilized chromaffin cells. FEBS Lett. 1986;208(2):357–63. doi: 10.1016/0014-5793(86)81049-3. [DOI] [PubMed] [Google Scholar]
- 144.Matter K, Dreyer F, Aktories K. Actin involvement in exocytosis from PC12 cells: studies on the influence of botulinum C2 toxin on stimulated noradrenaline release. J Neurochem. 1989;52(2):370–6. doi: 10.1111/j.1471-4159.1989.tb09131.x. [DOI] [PubMed] [Google Scholar]
- 145.Li G, Rungger-Brandle E, Just I, Jonas JC, Aktories K, Wollheim CB. Effect of disruption of actin filaments by Clostridium botulinum C2 toxin on insulin secretion in HIT-T15 cells and pancreatic islets. Mol Biol Cell. 1994;5(11):1199–213. doi: 10.1091/mbc.5.11.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Morita K, Oka M, Hamano S. Effects of cytoskeleton-disrupting drugs on ouabain-stimulated catecholamine secretion from cultured adrenal chromaffin cells. Biochem Pharmacol. 1988;37(17):3357–9. doi: 10.1016/0006-2952(88)90650-8. [DOI] [PubMed] [Google Scholar]
- 147.O’Konski MS, Pandol SJ. Cholecystokinin JMV-180 and caerulein effects on the pancreatic acinar cell cytoskeleton. Pancreas. 1993;8(5):638–46. doi: 10.1097/00006676-199309000-00018. [DOI] [PubMed] [Google Scholar]
- 148.Randhawa VK, Bilan PJ, Khayat ZA, et al. VAMP2, but Not VAMP3/Cellubrevin, Mediates Insulin-dependent Incorporation of GLUT4 into the Plasma Membrane of L6 Myoblasts. Mol Biol Cell. 2000;11(7):2403–2417. doi: 10.1091/mbc.11.7.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tong P, Khayat ZA, Huang C, Patel N, Ueyama A, Klip A. Insulin-induced cortical actin remodeling promotes GLUT4 insertion at muscle cell membrane ruffles. J Clin Invest. 2001;108(3):371–81. doi: 10.1172/JCI12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Torok D, Patel N, Jebailey L, et al. Insulin but not PDGF relies on actin remodeling and on VAMP2 for GLUT4 translocation in myoblasts. J Cell Sci. 2004;117(Pt 22):5447–55. doi: 10.1242/jcs.01421. Epub 2004 Oct 5. [DOI] [PubMed] [Google Scholar]
- 151.Braiman L, Alt A, Kuroki T, et al. Activation of protein kinase C zeta induces serine phosphorylation of VAMP2 in the GLUT4 compartment and increases glucose transport in skeletal muscle. Mol Cell Biol. 2001;21(22):7852–61. doi: 10.1128/MCB.21.22.7852-7861.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Elmendorf JS. Fluidity of insulin action. Mol Biotechnol. 2004;27(2):127–38. doi: 10.1385/MB:27:2:127. [DOI] [PubMed] [Google Scholar]
- 153.Sechi AS, Wehland J. The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J Cell Sci. 2000;113(Pt 21):3685–95. doi: 10.1242/jcs.113.21.3685. [DOI] [PubMed] [Google Scholar]
- 154.Chen F, Ma L, Parrini MC, et al. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr Biol. 2000;10(13):758–65. doi: 10.1016/s0960-9822(00)00571-6. [DOI] [PubMed] [Google Scholar]
- 155.Missy K, Van Poucke V, Raynal P, et al. Lipid products of phosphoinositide 3-kinase interact with Rac1 GTPase and stimulate GDP dissociation. J Biol Chem. 1998;273(46):30279–86. doi: 10.1074/jbc.273.46.30279. [DOI] [PubMed] [Google Scholar]
- 156.Clodi M, Vollenweider P, Klarlund J, et al. Effects of general receptor for phosphoinositides 1 on insulin and insulin-like growth factor I-induced cytoskeletal rearrangement, glucose transporter-4 translocation, and deoxyribonucleic acid synthesis. Endocrinology. 1998;139(12):4984–90. doi: 10.1210/endo.139.12.6351. [DOI] [PubMed] [Google Scholar]
- 157.Settleman J. Getting in shape with Rho. Nat Cell Biol. 2000;2(1):E7–9. doi: 10.1038/71390. [DOI] [PubMed] [Google Scholar]
- 158.Lee NA, Reasner CA. Beneficial effect of chromium supplementation on serum triglyceride levels in NIDDM. Diabetes Care. 1994;17(12):1449–52. doi: 10.2337/diacare.17.12.1449. [DOI] [PubMed] [Google Scholar]
- 159.Rabinovitz H, Friedensohn A, Leibovitz A, Gabay G, Rocas C, Habot B. Effect of chromium supplementation on blood glucose and lipid levels in type 2 diabetes mellitus elderly patients. Int J Vitam Nutr Res. 2004;74(3):178–82. doi: 10.1024/0300-9831.74.3.178. [DOI] [PubMed] [Google Scholar]
- 160.Vladeva SV, Terzieva DD, Arabadjiiska DT. Effect of chromium on the insulin resistance in patients with type II diabetes mellitus. Folia Med (Plovdiv) 2005;47(3–4):59–62. [PubMed] [Google Scholar]
- 161.Pattar GR, Tackett L, Liu P, Elmendorf JS. Chromium picolinate positively influences the glucose transporter system via affecting cholesterol homeostasis in adipocytes cultured under hyperglycemic diabetic conditions. Mutation Research. 2006 doi: 10.1016/j.mrgentox.2006.06.018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Guclu F, Ozmen B, Hekimsoy Z, Kirmaz C. Effects of a statin group drug, pravastatin, on the insulin resistance in patients with metabolic syndrome. Biomed Pharmacother. 2004;58(10):614–8. doi: 10.1016/j.biopha.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 163.Sonmez A, Baykal Y, Kilic M, et al. Fluvastatin improves insulin resistance in nondiabetic dyslipidemic patients. Endocrine. 2003;22(2):151–4. doi: 10.1385/endo:22:2:151. [DOI] [PubMed] [Google Scholar]
- 164.Suzuki M, Kakuta H, Takahashi A, et al. Effects of atorvastatin on glucose metabolism and insulin resistance in KK/Ay mice. J Atheroscler Thromb. 2005;12(2):77–84. doi: 10.5551/jat.12.77. [DOI] [PubMed] [Google Scholar]
- 165.Chen KL, Lu JJ, Lien TF, Chiou PW. Effects of chromium nicotinate on performance, carcase characteristics and blood chemistry of growing turkeys. Br Poult Sci. 2001;42(3):399–404. doi: 10.1080/00071660120055403. [DOI] [PubMed] [Google Scholar]
- 166.Musi N, Hirshman MF, Nygren J, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51(7):2074–81. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- 167.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 169.Thyfault JP, Cree MG, Zheng D, et al. Contraction of insulin-resistant muscle normalizes insulin action in association with increased mitochondrial activity and fatty acid catabolism. Am J Physiol Cell Physiol. 2007;292(2):C729–39. doi: 10.1152/ajpcell.00311.2006. [DOI] [PubMed] [Google Scholar]
- 170.Huang C, Thirone AC, Huang X, Klip A. Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J Biol Chem. 2005;280(19):19426–35. doi: 10.1074/jbc.M412317200. Epub 2005 Mar 11. [DOI] [PubMed] [Google Scholar]
- 171.Golub T, Pico C. Spatial control of actin-based motility through plasmalemmal PtdIns(4,5)P2-rich raft assemblies. Biochem Soc Symp. 2005;(72):119–27. doi: 10.1042/bss0720119. [DOI] [PubMed] [Google Scholar]
- 172.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414(6865):782–7. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 173.Zierath JR, Krook A, Wallberg-Henriksson H. Insulin action and insulin resistance in human skeletal muscle. Diabetologia. 2000;43(7):821–35. doi: 10.1007/s001250051457. [DOI] [PubMed] [Google Scholar]
- 174.Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab. 2000;11(9):351–6. doi: 10.1016/s1043-2760(00)00323-4. [DOI] [PubMed] [Google Scholar]
- 175.Deshmukh A, Coffey VG, Zhong Z, Chibalin AV, Hawley JA, Zierath JR. Exercise-Induced Phosphorylation of the Novel Akt Substrates AS160 and Filamin A in Human Skeletal Muscle. Diabetes. 2006;55(6):1776–1782. doi: 10.2337/db05-1419. [DOI] [PubMed] [Google Scholar]
- 176.Garvey WT, Olefsky JM, Marshall S. Insulin receptor down-regulation is linked to an insulin-induced postreceptor defect in the glucose transport system in rat adipocytes. J Clin Invest. 1985;76(1):22–30. doi: 10.1172/JCI111950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kozka IJ, Clark AE, Holman GD. Chronic treatment with insulin selectively down-regulates cell-surface GLUT4 glucose transporters in 3T3-L1 adipocytes. J Biol Chem. 1991;266(18):11726–31. [PubMed] [Google Scholar]
- 178.Kozka IJ, Holman GD. Metformin blocks downregulation of cell surface GLUT4 caused by chronic insulin treatment of rat adipocytes. Diabetes. 1993;42(8):1159–65. doi: 10.2337/diab.42.8.1159. [DOI] [PubMed] [Google Scholar]
- 179.Garvey WT, Olefsky JM, Matthaei S, Marshall S. Glucose and insulin co-regulate the glucose transport system in primary cultured adipocytes. A new mechanism of insulin resistance. J Biol Chem. 1987;262(1):189–97. [PubMed] [Google Scholar]
- 180.Garvey WT, Olefsky JM, Marshall S. Insulin induces progressive insulin resistance in cultured rat adipocytes. Sequential effects at receptor and multiple postreceptor sites. Diabetes. 1986;35(3):258–67. doi: 10.2337/diab.35.3.258. [DOI] [PubMed] [Google Scholar]
- 181.Pryor PR, Liu SC, Clark AE, Yang J, Holman GD, Tosh D. Chronic insulin effects on insulin signalling and GLUT4 endocytosis are reversed by metformin. Biochem J. 2000;348(Pt 1):83–91. [PMC free article] [PubMed] [Google Scholar]
- 182.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–8. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 183.Brindley DN, Abousalham A, Kikuchi Y, Wang CN, Waggoner DW. “Cross talk” between the bioactive glycerolipids and sphingolipids in signal transduction. Biochem Cell Biol. 1996;74(4):469–76. doi: 10.1139/o96-051. [DOI] [PubMed] [Google Scholar]
- 184.Guo D, Donner DB. Tumor necrosis factor promotes phosphorylation and binding of insulin receptor substrate 1 to phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. J Biol Chem. 1996;271(2):615–8. doi: 10.1074/jbc.271.2.615. [DOI] [PubMed] [Google Scholar]
- 185.Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol. 1998;18(9):5457–64. doi: 10.1128/mcb.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Stratford S, Dewald DB, Summers SA. Ceramide dissociates 3′-phosphoinositide production from pleckstrin homology domain translocation. Biochem J. 2001;354:359–368. doi: 10.1042/0264-6021:3540359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50(11):2563–71. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- 188.Kralik SF, Liu P, Leffler BJ, Elmendorf JS. Ceramide and glucosamine antagonism of alternate signaling pathways regulating insulin- and osmotic shock-induced glucose transporter 4 translocation. Endocrinology. 2002;143(1):37–46. doi: 10.1210/endo.143.1.8606. [DOI] [PubMed] [Google Scholar]
- 189.Chavez JA, Knotts TA, Wang LP, et al. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem. 2003;278(12):10297–303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- 190.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. 1991;266(8):4706–12. [PubMed] [Google Scholar]
- 191.Rossetti L. Perspective: Hexosamines and nutrient sensing. Endocrinology. 2000;141(6):1922–5. doi: 10.1210/endo.141.6.7566. [DOI] [PubMed] [Google Scholar]
- 192.McClain DA, Crook ED. Hexosamines and insulin resistance. Diabetes. 1996;45(8):1003–9. doi: 10.2337/diab.45.8.1003. [DOI] [PubMed] [Google Scholar]
- 193.Ross SA, Chen X, Hope HR, et al. Development and Comparison of Two 3T3-L1 Adipocyte Models of Insulin Resistance: Increased Glucose Flux vs Glucosamine Treatment. Biochem Biophys Res Commun. 2000;273(3):1033–1041. doi: 10.1006/bbrc.2000.3082. [DOI] [PubMed] [Google Scholar]
- 194.Hawkins M, Hu M, Yu J, et al. Discordant effects of glucosamine on insulin-stimulated glucose metabolism and phosphatidylinositol 3-kinase activity. J Biol Chem. 1999;274(44):31312–9. doi: 10.1074/jbc.274.44.31312. [DOI] [PubMed] [Google Scholar]
- 195.Buse MG, Robinson KA, Marshall BA, Hresko RC, Mueckler MM. Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. Am J Physiol Endocrinol Metab. 2002;283(2):E241–50. doi: 10.1152/ajpendo.00060.2002. [DOI] [PubMed] [Google Scholar]
- 196.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 197.Del Prato S, Leonetti F, Simonson DC, Sheehan P, Matsuda M, DeFronzo RA. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia. 1994;37(10):1025–35. doi: 10.1007/BF00400466. [DOI] [PubMed] [Google Scholar]
- 198.Rizza RA, Mandarino LJ, Genest J, Baker BA, Gerich JE. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia. 1985;28(2):70–5. doi: 10.1007/BF00279918. [DOI] [PubMed] [Google Scholar]
- 199.Henry RR, Ciaraldi TP, Mudaliar S, Abrams L, Nikoulina SE. Acquired defects of glycogen synthase activity in cultured human skeletal muscle cells: influence of high glucose and insulin levels. Diabetes. 1996;45(4):400–7. doi: 10.2337/diab.45.4.400. [DOI] [PubMed] [Google Scholar]
- 200.Flores-Riveros JR, McLenithan JC, Ezaki O, Lane MD. Insulin down-regulates expression of the insulin-responsive glucose transporter (GLUT4) gene: effects on transcription and mRNA turnover. Proc Natl Acad Sci U S A. 1993;90(2):512–6. doi: 10.1073/pnas.90.2.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ricort JM, Tanti JF, Van Obberghen E, Le Marchand-Brustel Y. Alterations in insulin signalling pathway induced by prolonged insulin treatment of 3T3-L1 adipocytes. Diabetologia. 1995;38(10):1148–56. doi: 10.1007/BF00422363. [DOI] [PubMed] [Google Scholar]
- 202.Nelson BA, Robinson KA, Buse MG. High glucose and glucosamine induce insulin resistance via different mechanisms in 3T3-L1 adipocytes. Diabetes. 2000;49(6):981–91. doi: 10.2337/diabetes.49.6.981. [DOI] [PubMed] [Google Scholar]
- 203.Strawbridge AB, Elmendorf JS. Phosphatidylinositol 4,5-Bisphosphate Reverses Endothelin-1-Induced Insulin Resistance via an Actin-Dependent Mechanism. Diabetes. 2005;54(6):1698–705. doi: 10.2337/diabetes.54.6.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Strawbridge AB, Elmendorf JS. Endothelin-1 impairs glucose transporter trafficking via a membrane-based mechanism. J Cell Biochem. 2006;97(4):849–56. doi: 10.1002/jcb.20687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jiang ZY, Zhou QL, Chatterjee A, et al. Endothelin-1 modulates insulin signaling through phosphatidylinositol 3-kinase pathway in vascular smooth muscle cells. Diabetes. 1999;48(5):1120–30. doi: 10.2337/diabetes.48.5.1120. [DOI] [PubMed] [Google Scholar]
- 206.Ishibashi KI, Imamura T, Sharma PM, Huang J, Ugi S, Olefsky JM. Chronic endothelin-1 treatment leads to heterologous desensitization of insulin signaling in 3T3-L1 adipocytes. J Clin Invest. 2001;107(9):1193–202. doi: 10.1172/JCI11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wilkes JJ, Hevener A, Olefsky J. Chronic endothelin-1 treatment leads to insulin resistance in vivo. Diabetes. 2003;52(8):1904–9. doi: 10.2337/diabetes.52.8.1904. [DOI] [PubMed] [Google Scholar]
- 208.Ottosson-Seeberger A, Lundberg JM, Alvestrand A, Ahlborg G. Exogenous endothelin-1 causes peripheral insulin resistance in healthy humans. Acta Physiol Scand. 1997;161(2):211–20. doi: 10.1046/j.1365-201X.1997.00212.x. [DOI] [PubMed] [Google Scholar]
- 209.Teuscher AU, Lerch M, Shaw S, Pacini G, Ferrari P, Weidmann P. Endothelin-1 infusion inhibits plasma insulin responsiveness in normal men. J Hypertens. 1998;16(9):1279–84. doi: 10.1097/00004872-199816090-00009. [DOI] [PubMed] [Google Scholar]
- 210.Idris I, Patiag D, Gray S, Donnelly R. Tissue- and time-dependent effects of endothelin-1 on insulin-stimulated glucose uptake. Biochem Pharmacol. 2001;62(12):1705–8. doi: 10.1016/s0006-2952(01)00815-2. [DOI] [PubMed] [Google Scholar]
- 211.Juan CC, Fang VS, Huang YJ, Kwok CF, Hsu YP, Ho LT. Endothelin-1 induces insulin resistance in conscious rats. Biochem Bio-phys Res Commun. 1996;227(3):694–9. doi: 10.1006/bbrc.1996.1571. [DOI] [PubMed] [Google Scholar]
- 212.Williams MW, Bloch RJ. Extensive but Coordinated Reorganization of the Membrane Skeleton in Myofibers of Dystrophic (mdx) Mice. J Cell Biol. 1999;144(6):1259–1270. doi: 10.1083/jcb.144.6.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mulvey C, Harno E, Keenan A, Ohlendieck K. Expression of the skeletal muscle dystrophin-dystroglycan complex and syntrophinnitric oxide synthase complex is severely affected in the type 2 diabetic Goto-Kakizaki rat. European Journal of Cell Biology. 2005;84(11):867. doi: 10.1016/j.ejcb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 214.Andersen H, Nielsen S, Mogensen CE, Jakobsen J. Muscle Strength in Type 2 Diabetes. Diabetes. 2004;53(6):1543–1548. doi: 10.2337/diabetes.53.6.1543. [DOI] [PubMed] [Google Scholar]