

Abstract
Large conductance Ca2+- and voltage-activated K+ (BK) channels, composed of pore-forming α-subunits and auxiliary β-subunits, play important roles in diverse physiological processes. The differences in BK channel phenotypes are primarily due to the tissue-specific expression of β-subunits (β1–β4) that modulate channel function differently. Yet, the molecular basis of the subunit-specific regulation is not clear. In our study, we demonstrate that perturbation of the voltage sensor in BK channels by mutations selectively disrupts the ability of the β1-subunit—but not that of the β2-subunit—to enhance apparent Ca2+ sensitivity. These mutations change the number of equivalent gating charges, the voltage dependence of voltage sensor movements, the open-close equilibrium of the channel, and the allosteric coupling between voltage sensor movements and channel opening to various degrees, indicating that they alter the conformation and movements of the voltage sensor and the activation gate. Similarly, the ability of the β1-subunit to enhance apparent Ca2+ sensitivity is diminished to various degrees, correlating quantitatively with the shift of voltage dependence of voltage sensor movements. In contrast, none of these mutations significantly reduces the ability of the β2-subunit to enhance Ca2+ sensitivity. These results suggest that the β1-subunit enhances Ca2+ sensitivity by altering the conformation and movements of the voltage sensor, whereas the similar function of the β2-subunit is governed by a distinct mechanism.
INTRODUCTION
Large conductance Ca2+- and voltage-activated K+ (BK) channels are composed of α-subunits (1,2) encoded by a single Slo1 gene and four types of auxiliary β-subunits (β1–β4) (3–8). They are activated by membrane depolarization and elevated intracellular Ca2+ concentration ([Ca2+]i). Due to a large single channel conductance, their opening effectively hyperpolarizes the membrane resulting in the closure of voltage-dependent Ca2+ channels and, hence, a decrease in [Ca2+]i. Thus, BK channels provide a negative feedback mechanism to control membrane potential and cytosolic [Ca2+]i. Based on this mechanism, BK channels modulate various physiological processes including action potential repolarization, neurotransmitter release, smooth muscle contraction, and hearing (9–12). BK currents show various phenotypes in different tissue types, which are primarily due to the tissue-specific expression of β-subunits (β1–β4) that modulate BK channel function. For example, the β1-subunit, expressed primarily in smooth muscles, increases Ca2+ sensitivity of the channel by shifting voltage-dependent activation to a more negative voltage range in [Ca2+]i (3,13,14). Thus, the β1-subunit is essential in regulating tone in the vasculature (15), trachea (16), urinary bladder (17), and uterus (18). In contrast, the β2-subunit, expressed in the β-cells of the pancreas, ovaries, brain, and adrenal chromaffin cells, also increases the Ca2+-dependent conductance-voltage (G-V) shift and confers inactivation on the BK channel (5–7,19).
BK channel β-subunits share similar topology that includes two transmembrane segments (TM1 and TM2), a relatively large extracellular loop, and short cytosolic N- and C-termini (3) (Fig. 1). Nevertheless, the coexpression of different β-subunits with Slo1 produces various phenotypes, suggesting that different molecular mechanisms are employed by these β-subunits to modify channel function. Most interestingly, the β1- and β2-subunits share 43% identical, or 66% conservative, residues (7), and both enhance Ca2+ sensitivity of channel activation by increasing a Ca2+-dependent shift in the G-V relationship. A previous study on the biophysical properties of the channel, however, suggested that these two β-subunits modulate channel activation by different mechanisms (20). By studying the effects of these β-subunits on ionic and gating currents of mSlo1 and fitting the results to the allosteric conceptual framework provided by Horrigan, Cui, and Aldrich (denoted here as the HCA model (21)), it has been suggested that the β1-subunit may affect the apparent Ca2+ sensitivity by altering voltage-dependent activation (20,22–24). In contrast, the β2-subunit may affect the apparent Ca2+ sensitivity by altering both voltage-dependent activation and Ca2+ binding (20). However, a recent study by Savalli et al. (25) suggests that the β2-subunit may also affect BK channel activation by primarily altering voltage sensor movements. This study and others (23,26–29) have provided a solid characterization of the functional effects of β-subunits and important insights on the mechanisms of their action. Nevertheless, the molecular identities of residues/domains in α-subunits that are directly involved in the regulation of β-subunits are still not known fully. This lack of knowledge limits our ability to interpret results from biophysical studies and undermines our understanding of the physiological role of BK channels in various tissue types. The identifications of the molecular components that are specifically important for the function of different β-subunits will help remove these limitations and provide molecular targets to alter physiological and pathophysiological functions of BK channels in specific tissues.
FIGURE 1.

Cartoons of mSlo1 α- and β-subunits. All residues mutated in this study are shown. D153, R167, D186, and R213 (stars) are voltage-sensing residues. Y163 and R207 (squares) are nonvoltage-sensing residues in S2 and S4, respectively. E321 and E386 (circles) shift the G-V relationship to negative voltages without altering the Ca2+ sensitivity or the β1-induced enhancement of Ca2+ sensitivity.
In our study, we investigate the role of the voltage sensor of Slo1 in the regulation of β-subunits. Like all voltage-dependent K+ channels, BK channels are composed of four Slo1 proteins (30) (Fig. 1). Transmembrane segments S5–S6 form the pore domain, with part of S6 forming the activation gate, whereas S1–S4 form the voltage-sensing domain (VSD) (31–34). We hypothesize that if the β1- or the β2-subunit modulates BK channel activation by altering voltage sensor movements, a change in voltage sensor movements per se by mutations should affect the function of the β-subunits. A previous study by Ma et al. (34) demonstrated that mutations of charged residues in the VSD of Slo1 alter voltage sensor movements. In addition, the ionic currents of these mutations in various voltages and [Ca2+]i have been recorded, and the experimental data have been fitted to the HCA allosteric model (21,35). It was found that these mutations change various aspects of voltage-dependent gating of BK channels, including the number of equivalent gating charges (zJ and zT), the voltage dependence of voltage sensor movements (VHC), the open-close equilibrium of the channel (L0), and the allosteric coupling between voltage sensor movements and channel opening to various degrees (D) (34) (see Table 1 for definition of the parameters). We studied some of these mutations (Fig. 1) to examine if they also affect the ability of the β-subunits to enhance Ca2+ sensitivity. We chose these mutations because they affect all parameters of voltage-dependent gating (Table 1) to various degrees, so that the entire spectrum of perturbations of conformation and movements of the voltage sensor and the activation gate by these mutations were covered. We found that these mutations specifically reduced the ability of the β1-subunit to shift the voltage dependence of activation in [Ca2+]i but not that of the β2-subunit. These results indicate that the β1- and β2-subunits affect distinct molecular processes during Ca2+- and voltage-dependent activation, resulting in specific alterations of channel function.
TABLE 1.
Abbreviation and gating parameters (35)
| C, O | Closed and open conformation of the channel. |
| R, A | Resting and activated conformation of voltage sensor. |
| L | C–O equilibrium constant (unliganded channel, resting voltage sensors). |
| L0 | The zero voltage value of L (L = L0exp(zLV/kT)). |
| zL | The partial charge of L. |
| J | R–A equilibrium constant (closed, unliganded channel). |
| zJ | The partial charge of J. |
| zT | Total gating charge (zT = zL + 4zJ). |
| QC, QO | Steady-state gating charge distribution for closed or open channels. |
| VHC, VHO | Half-activating voltage of QC and QO, respectively. |
| D | Allosteric factor describing interaction between channel opening and voltage sensor activation (D = exp [−zJ(VHO − VHC)/kT]). |
EXPERIMENTAL PROCEDURES
Mutagenesis and channel expression
All α-subunit constructs were made from the mbr5 clone of mSlo1 (36). Human β1 and β2 (KCNMB1 and KCNMB2; GenBank/EMBL/DDBJ accession nos. U25138 and AF209747) cDNAs were subcloned into pcDNA3.1(+). The β2 with N-terminus deleted (β2ND)-subunit was created by removing amino acids from position 2 through 20. All mutations of mSlo1 and β2ND constructs were made using polymerase chain reaction with Pfu polymerase (Stratagene, La Jolla, CA). The polymerase chain reaction–amplified regions of all constructs were verified by sequencing. mRNA was transcribed in vitro with T3 polymerase for all mSlo1 constructs and with T7 polymerase for β1- and β2ND-subunits (Ambion, Austin, TX). A total of 0.05–20 ng of mSlo1 mRNA or a mixture of 5–15 ng of mSlo1 and 25–50 ng of β-subunit mRNAs was injected into each Xenopus laevis oocyte 2–6 days before recording.
Electrophysiology
Macroscopic currents were recorded from inside-out patches formed with borosilicate pipettes of 1 ∼ 2 MΩ resistance. Data were acquired using a patch-clamp amplifier (Axopatch 200-B; Axon Instruments, Union City, CA) and data acquisition software (Pulse; Heka Electronik, Lambrecht/Pfalz, Germany). Records were digitized at 20-μs intervals and low-pass filtered at 10 kHz with Axopatch's internal filter. The pipette solution contained the following (in mM): 140 potassium methanesulfonic acid, 20 HEPES, 2 KCl, and 2 MgCl2 (pH 7.20). The basal internal solution contained the following (in mM): 140 potassium methanesulfonic acid, 20 HEPES, 2 KCl, and 1 HEDTA (pH 7.20). Increasing EGTA concentration of the basal internal solution to 5 mM gave a [Ca2+]i of ∼0.5 nM. For other [Ca2+]i, CaCl2 was added to internal solutions to give the appropriate free [Ca2+]i. Actual free [Ca2+]i was measured using a Ca2+-sensitive electrode (Orion Research, Cambridge, MA). A total of 50 μM 18-crown-6-tetracarboxylic acid (Sigma-Aldrich, St. Louis, MO) was added to internal solutions to prevent Ba2+ block. Experiments were conducted at room temperature (22°C ∼ 24°C).
Gating currents were recorded with inside-out patches (37). The pipette solution contained (in mM): 127 tetraethylammonium (TEA), 125 HMeSO3, 2 HCl, 2 MgCl2, and 20 HEPES (pH 7.2). The internal solution contains (in mM): 141 N-ethyl-D-glucamine, 135 HMeSO3, 6 HCl, 20 HEPES, and 5 EGTA (pH 7.2); MgCl2 was added to the internal solution to reach 10 mM [Mg2+]i. Voltage commands were filtered at 20 kHz with an eight-pole Bessel filter (Frequency Devices, Ottawa, IL) to prevent the saturation of fast capacitive transients (37). Data were sampled at 100 kHz with an 18-bit analog/digital converter (ITC-18; Instrutech, Port Washington, NY) and filtered at 10 kHz with an internal filter (Axopatch; Axon Instruments). Capacitive transients and leak currents were subtracted using a P/5 protocol with a holding potential of −120 mV.
Data analysis and statistics
Relative conductance was determined by measuring tail current amplitudes at negative voltages as indicated for the wild-type (WT) and mutant mSlo1 channels. The gating charge movements were determined by integrating the area under the rising phase and single exponential fits to the decaying phase of IgON at various voltages. The G-V relationship or the charge-voltage (Qc-V) relations of the WT and mutant channels were fitted with the Boltzmann equation as follows:
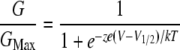 |
(1) |
or
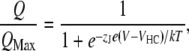 |
(2) |
where z is the number of equivalent charges, V1/2 is the voltage for channel in half activation, e is the elementary charge, k is the Boltzmann constant, and T is the absolute temperature. In Eq. 2, zJ is the gating charge associated with voltage sensor movement, VHC is the voltage for half of the gating charge movements at the closed conformation of the channel, and the other parameters have the same meaning as in Eq. 1. Curve fittings were done with Igor Pro software (WaveMetrics, Lake Oswego, OR) using the Levenberg-Marquardt algorithm to perform nonlinear least squares fits. The means of the data were obtained by averaging from 4 to 30 patches and error bars represent mean ± SE. Statistics were performed using SigmaStat 3.5 software (Systat Software, San Jose, CA); one-way analysis of variance with an all pairwise multiple comparison procedure (Tukey test) was performed. A p value < 0.05 was considered significant.
RESULTS
Mutations of voltage-sensing residues disrupt the β1-induced enhancement of Ca2+ sensitivity
Among all charged residues in the VSD of Slo1 channels, neutralization of D153, R167, D186, or R213 reduces the number of equivalent gating charge. Thus, these residues are proposed as voltage-sensing residues (34). The mutations of these residues also alter other parameters (VHC, L0, and D) for voltage-dependent gating, indicating that these mutations profoundly affect the conformation and movements of the voltage sensor and the activation gate. Hence, we first studied the effects of these mutations on the function of the β1- and β2-subunits.
The coexpression of the β1-subunit induces three major effects on the ionic currents of WT BK channels. First, the β1-subunit slows the activation and deactivation kinetics in 0 (20) and saturating 100 μM [Ca2+]i (Fig. 2, A and C). Second, the β1-subunit reduces the steepness of the steady-state G-V relationship (Fig. 2 B). The equivalent charge (z) that is proportional to the steepness of the G-V relationship decreases from 1.28 ± 0.02 to 0.72 ± 0.01 in 0 [Ca2+]i and from 1.32 ± 0.05 to 0.88 ± 0.03 in 100 μM [Ca2+]i. Third, the β1-subunit increases the shift of G-V relationships to more negative voltages (a leftward shift) in response to increasing [Ca2+]i; the change in G-V relationship is −83.6 ± 5.7 mV more with than without the coexpression of the β1-subunit in 100 μM [Ca2+]i.
FIGURE 2.

Mutations of the voltage-sensing residues in BK channels abolish β1-induced G-V shifts in 100 μM [Ca2+]i. (A) Macroscopic current traces in 100 μM [Ca2+]i for WT and mutant mSlo1 (upper panels) and WT and mutant mSlo1 +β1 channels (lower panels). Test pulses were with 20-mV increments in the same voltage range as in panel B for corresponding constructs. Repolarization potentials for WT, WT + β1, D153C, D153C + β1, R167A, R167A + β1, D186A, D186A + β1, R213C, and R213C + β1 channels were −100, −160, −80, −80, −100, −80, −100, −100, −80, and −80 mV, respectively. (B) Mean G-V relationships of WT and mutant mSlo1 channels expressed with and without β1-subunit in 0 and 100 μM [Ca2+]i. The G-V relationships are fit with the Boltzmann relation (solid line, see Experimental Procedures). (C) The β1-subunit prolongs the activation time course of WT and mutant mSlo1 channels. Activation time constants (τact) were obtained by fitting current traces at various voltages with a single exponential function.
Individual mutation of the four voltage-sensing residues (D153C, R167A, D186A, and R213C) reduces the zT by 25% to 55% and shifts the VHC in mSlo1 channels (34). Consequently, the voltage dependence of channel opening, measured by the position and the steepness of the G-V relationship, is changed (Fig. 2 B). Among the four mutations, D153C and R213C shift the G-V relationship to more positive voltages (a rightward shift) to such an extent, their macroscopic currents can only be measured in [Ca2+]i > 10 μM to obtain reliable G-V relations (34). Therefore, we first examined if the mutations affect β1 function at the saturating [Ca2+]i of 100 μM.
For the four mutant channels, the G-V relationship with the β1-subunit in 100 μM [Ca2+]i is either at a similar or a more positive voltage range than without the β1-subunit (Fig. 2 B, open and solid squares). However, the β1-subunit still slows the rate of activation and deactivation of the mutant channels (Fig. 2, A and C), indicating that these mutations do not abolish the association with the β1-subunit. In addition, the β1-subunit reduces the slope of the G-V relationship of D153C (z-values change from 1.31 ± 0.06 to 0.94 ± 0.03) and D186A (z-values change from 1.05 ± 0.06 to 0.52 ± 0.07) channels in 100 μM [Ca2+]i in a manner similar to the WT channel (z-values change from 1.32 ± 0.05 to 0.88 ± 0.03) (Fig. 2 B). For mutations R167A and R213C, the β1-subunit causes a relatively small reduction in the slope of the G-V relationship in 100 μM [Ca2+]i (z-values change from 1.06 ± 0.06 to 0.85 ± 0.03 and 0.90 ± 0.05 to 0.72 ± 0.02, respectively). The G-V relationships of R167A + β1, D186A + β1 in 0 [Ca2+]i, and R213C + β1 in 100 μM [Ca2+]i does not reach saturation at 300 mV, and so the actual z-values would be even smaller than those calculated from these G-V relations. Therefore, the mutations of the voltage-sensing residues specifically disrupt the β1-induced shift of the G-V relationship in high [Ca2+]i without a clear effect on the ability of the β1-subunit to change activation kinetics or the steepness of the G-V relationship.
For the WT channels, the enhanced leftward G-V shift induced by the β1-subunit becomes less pronounced in reduced [Ca2+]i (Figs. 2 B and 3). In 0 [Ca2+]i, the β1-subunit even induces a rightward G-V shift. This β1-induced, greater leftward shift of the G-V relationship in response to increasing Ca2+ concentrations has been referred to as an enhanced Ca2+ sensitivity of channel activation (20,22,23,27,38,39). Fig. 3 A shows the V1/2-[Ca2+]i relationship for R167A and WT mSlo1 channels in six different [Ca2+]i. Although the β1-induced leftward G-V shift for WT channels increases with increasing [Ca2+]i, mutation R167A completely abolishes the β1-induced G-V shift in all [Ca2+]i. In contrast, the β1-induced G-V shifts in varying [Ca2+]i for D186A mutant channels are not that simple (Fig. 3 B). If we simply compare V1/2-values at each individual [Ca2+]i for D186A, the β1-induced leftward shift of the G-V relationship seems to be abolished by this mutation, because the β1-subunit causes either a large rightward shift or no significant shift in any specific [Ca2+]i. However, if we compare the V1/2 difference between 0 and 100 μM [Ca2+]i (ΔV1/2-Ca), i.e., the total shift of the G-V relationship caused by Ca2+ binding, it is −71.6 ± 3.1 mV greater with the β1-subunit than without it. As a comparison, the β1-subunit enhances ΔV1/2-Ca by −128.0 ± 4.7 mV for the WT channels. Therefore, the mutation D186A does not completely abolish the β1-induced enhancement of Ca2+ sensitivity but reduces it by 45%.
FIGURE 3.

Mutations of voltage-sensing residues significantly reduced the β1-induced enhancement of Ca2+ sensitivity. (A and B) V1/2-[Ca2+]i relationships of WT, R167A, and D186A mSlo1 channels with and without β1-subunit. (C) V1/2-[Ca2+]i relationship of E386A and D153C:E386A mSlo1 channels with and without β1-subunit. The G-V relationship of D153C:E386A is shifted too far rightward to record any measurable currents at 0.5 nM [Ca2+]i. (D) V1/2–[Ca2+]i relationship of E321A and R213C:E321A mSlo1 channels with and without β1-subunit.
Because mutations D153C and R213C shift the G-V relationship to extremely positive voltages, it is difficult to measure the effects of the β1-subunit in [Ca2+]i lower than 100 μM. Therefore, we do not know whether the lack of the G-V shift in 100 μM [Ca2+]i is because the β1-subunit loses its ability to increase Ca2+ sensitivity of the mutant channels or is simply because the β1-subunit shifts the G-V relationship of the mutant channels to more positive voltages in all [Ca2+]i. Thus, comparing V1/2-values in one specific [Ca2+]i might not be sufficient to represent the β1-induced changes of the apparent Ca2+ sensitivity. To bring back the G-V relationship of these two mutant channels to less positive voltages, we made double mutations D153C:E386A and R213C:E321A. E321 is located at the COOH-terminus of S6 and E386 is located at the cytoplasmic domain (Fig. 1). Compared with WT channels, the single-site mutation E321A or E386A shifts the G-V relationship to more negative voltages by ∼−85 or −65 mV, respectively, at all [Ca2+]i-values between 0 and 100 μM (Fig. 3, C and D). Most importantly, they have no significant effect on the β1-induced enhancement of Ca2+ sensitivity (Fig. 3, C and D; p > 0.05). Fig. 3, C and D, also show the V1/2-[Ca2+]i relationship for D153C:E386A and R213C:E321A mutant channels. The β1-subunit fails to alter the G-V shift of D153C:E386A double mutant at any [Ca2+]i between 2 to 100 μM [Ca2+]i, consistent with the results of the single-site mutant D153C in 100 μM [Ca2+]i (Figs. 2 B and 3 C ). In the case of R213C:E321A, there is a parallel rightward shift of the G-V relationship with the β1-subunit at all [Ca2+]i, which is also consistent with the result of the single-site mutant R213C in 100 μM [Ca2+]i (Figs. 2 B and 3 D). Similar to D153C and R213C currents, the activation and deactivation kinetics of the currents of double mutants slowed in the presence of the β1-subunit (data not shown), indicating that the β1-subunit still associated with the double mutant channels. Therefore, D153C and R213C mutations abolish the β1-subunit's effect of increasing Ca2+ sensitivity.
Mutations of voltage-sensing residues do not reduce the β2-induced enhancement of Ca2+ sensitivity
The coexpression of the β2-subunit with WT BK channels induces inactivation and, similar to the β1-subunit, a larger leftward shift of the G-V relationship in response to increasing Ca2+ concentrations, which is referred to as the enhancement of Ca2+ sensitivity (20,38–40). To examine the role of voltage-sensing residues in the β2-induced Ca2+ sensitivity enhancement, we coexpressed the N-terminus deleted β2-subunit (β2ND-subunit, see Experimental Procedures), which removes the inactivation peptide (4,20,26,41), with the mutations of all voltage-sensing residues, D153C, R167A, D186A, and R213C, respectively. Similar to the effects of the β1-subunit on the WT channels, the β2ND-subunit slows down the activation and deactivation kinetics of these mutant channels (Fig. 4 A). The β2ND-subunit also induced a leftward shift of the G-V relationship for each of the mutant channels in 100 μM [Ca2+]i (Fig. 4, B and C). Fig. 4 D plots ΔV1/2-Ca between 0 and 100 μM [Ca2+]i for WT and mutant channels with and without the β2ND-subunit. Because mutations D153C and R213C shift the G-V relationship to extremely positive voltages, it is difficult to measure the effects of the β2-subunit in 0 [Ca2+]i. Therefore, we studied the D153C:E321A and R213C:E321A channels, for which we could record currents in both 0 and 100 μM [Ca2+]i with and without the β2ND-subunit (Fig. 4 D). The β2ND can enhance the Ca2+ sensitivity for all four mutant channels, as it does for WT channels. Taken together, the disruption of the VSD movements by mutating voltage-sensing residues does not significantly influence the effect of β2ND on Ca2+ sensitivity. The differential effects of the mutations on the β1- and β2-subunits suggest that these two subunits modulate Ca2+-dependent activation of BK channels through different molecular mechanisms.
FIGURE 4.

Mutations of voltage-sensing residues do not reduce the β2-induced enhancement of Ca2+ sensitivity. (A) Macroscopic current traces in 100 μM [Ca2+]i of WT and mutant mSlo1 + β2ND. Test pulses were with 20-mV increments. Repolarization potentials for WT + β2, D153C + β2, R167A + β2, D186A + β2, and R213C + β2 channels were −160, −100, −100, −120, and −100 mV, respectively. (B) Mean G-V relationships of WT and mutant mSlo1 channels expressed with and without the β2ND-subunit in 0 and 100 μM [Ca2+]i. G-V relationships are fit with the Boltzmann relation (solid line). (C) β2ND-induced V1/2 shifts of WT and mutant channels in 100 μM [Ca2+]i. (D) V1/2 shifts from 0 to 100 μM [Ca2+]i for WT and mutant channels with and without β2ND-subunit.
Differential effects of mutating nonvoltage-sensing residues on the β1- and β2-induced enhancement of Ca2+ sensitivity
The mutations of the voltage-sensing residues affect the voltage sensor in two major aspects: they reduce the number of zJ and shift VHC to more positive ranges. These similar effects suggest that the mutations of the voltage-sensing residues may cause similar perturbations of the conformation and movements of the voltage sensor, whereas mutations of nonvoltage-sensing residues that do not cause the same functional effects may perturb the conformation and movements of the voltage sensor differently. To examine how these mutations affect the function of β-subunits, we studied R207Q and Y163E, which do not reduce zJ but shift VHC to either negative or positive voltage ranges (34).
R207 is one of three Arg residues in S4, whereas Y163 is located in S2 (Fig. 1). Similar to the mutations of the voltage-sensing residues, in 0 [Ca2+]i, mutation R207Q reduced the steepness of the G-V relationship of mSlo1 channels (z = 0.57 ± 0.03 vs. 1.28 ± 0.02 for WT) (34,42) (Fig. 5 A). To the contrary, Y163E increased the steepness of the G-V relationship (z = 1.68 ± 0.10) (Fig. 5 B). They also shifted the G-V relationship in opposite directions at 0 [Ca2+]i. R207Q shifted V1/2 to hyperpolarizing potentials by −80.8 ± 3.6 mV, whereas Y163E resembled the mutations of the voltage-sensing residues by shifting V1/2 to depolarizing potentials by +23.7 ± 3.2 mV (Fig. 5, A and B).
FIGURE 5.

Mutations of nonvoltage-sensing residues R207 and Y163 exhibit differential effects on the β1-induced enhancement of Ca2+ sensitivity. (A–C) Mean G-V relationships of R207Q, Y163E, and Y163K channels with and without β1-subunit in 0 and 100 μM [Ca2+]i. The G-V relationships are fit with the Boltzmann relation (solid lines). (D) V1/2 shifts from 0 to 100 μM [Ca2+]i for WT, R207Q, Y163E, and Y163K channels with and without β1-subunit. (E) Gating current traces for Y163K channels. Test pulses were from −20 to 280 mV with 20-mV increments. (F) QC-V relations for WT and Y163K channels. QC was obtained by integrating the on gating current over time. Solid lines are fits to the Boltzmann relation.
The β1-subunit enhances ΔV1/2-Ca between 0 and 100 μM [Ca2+]i by 146.1 ± 5.1 mV for R207Q channels and by 55.5 ± 3.0 mV for Y163E (Fig. 5), either an increase or a decrease from the β1-induced enhancement of ΔV1/2-Ca for WT channels (128.0 ± 4.7 mV). Given these results, it seems that the β1-induced enhancement of Ca2+ sensitivity is not correlated with only a change of the shape or position of the G-V relationship or the reduction of the number of equivalent gating charges. However, all the mutations that reduce or abolish the ability of the β1-subunit to enhance Ca2+ sensitivity also shift VHC to positive voltage ranges (Figs. 3 and 5) (34).
To further test if a positive shift of VHC is correlated to the reduction of β1-induced enhancement of Ca2+ sensitivity, we studied mutation Y163K. Unlike the mutation of the same residue (Y163E), Y163K does not reduce β1-induced enhancement of Ca2+ sensitivity significantly (p > 0.05) (Fig. 5, C and D). The β1-subunit enhances ΔV1/2-Ca between 0 and 100 μM [Ca2+]i by 105.5 ± 5.0 mV, similar to that of the WT mSlo1 (128.0 ± 4.7 mV) (Fig. 5 D). The effect of Y163K on VHC has not been reported previously. Therefore, we measured the gating currents of Y163K at various voltages (Fig. 5 E) and plotted the relationship between the on gating charge and voltage (QC-V; Fig. 5 F) (21). The QC-V relation was fit with a Boltzmann equation (21) to obtain the number of equivalent gating charge (zJ = 0.65 ± 0.04) and VHC (136.1 ± 3.0 mV). Opposite to mutation Y163E, Y163K shifted VHC to a less positive voltage. Thus, for the two mutations of the same residue, a larger reduction of β1-induced enhancement of Ca2+ sensitivity is correlated with a larger positive shift of VHC. This result further suggests that β1 regulation is sensitive to a specific change of the conformation and movements of the voltage sensor.
We also coexpressed the β2ND-subunit with mutations R207Q, Y163E, and Y163K. The effects of the β2ND-subunit on gating properties of the channel were not affected significantly by these mutations (Fig. 6). Importantly, the β2ND-subunit did not reduce the ΔV1/2-Ca between 0 and 100 μM [Ca2+]i compared with the WT mSlo1 (Fig. 6 D). Thus, unlike the effects of the β1-subunit, the effects of the β2ND-subunit are not sensitive to alterations of the conformation and movements of the voltage sensor and the activation gate caused by the VSD mutations.
FIGURE 6.

Mutations of nonvoltage-sensing residues R207 and Y163 do not reduce the β2-induced enhancement of Ca2+ sensitivity. (A–C) Mean G-V relationships of R207Q, Y163E, and Y163K channels with and without the β2ND-subunit in 0 and 100 μM [Ca2+]i. The G-V relationships are fit with the Boltzmann relation (solid line). (D) V1/2 shifts from 0 to 100 μM [Ca2+]i for WT and mutant channels with and without β2ND-subunit.
DISCUSSION
The fine-tuning of BK channel properties by β-subunits greatly diversifies physiological roles of BK channels in various tissues. Each type of β-subunits may have been evolved to target different parts of BK channel α-subunit to modulate various channel properties. Previous studies have shown that the N-terminus and the S0 transmembrane segment of Slo1 are important for the β1-induced G-V shift (27,28). In this study, we found that the disturbance of voltage sensor movements by mutations disrupted the β1-induced enhancement of Ca2+ sensitivity. For all the mutations, the reduction of the β1-induced enhancement of Ca2+ sensitivity is correlated quantitatively with the positive shift of VHC (Fig. 7 A). These results provide direct evidence that the effect of the β1-subunit on Ca2+ sensitivity relies on the conformation and movements of the VSD. In contrast, the β2-induced enhancement of Ca2+ sensitivity was not reduced significantly by these mutations regardless of how voltage sensor movements were altered (Fig. 7), suggesting that the voltage sensor movements are not directly linked to the function of the β2-subunit. These results suggest that the β2-subunit may interact with a different molecular domain of Slo1 to alter the responses of the channel to Ca2+ binding.
FIGURE 7.

Relationship of the β1- and β2ND-induced enhancement of Ca2+ sensitivity with the parameters of HCA model for mutant mSlo1 channels (A, VHC) (B, zJ) (C, zT) (D, L0) and (E and D). The parameters of the HCA model (Table 1) for the WT and mutant mSlo1 channels except for Y163K were adapted from Ma et al. (34). The mutations are represented by numbers (panel A) in all panels. ΔΔV1/2 is the β1- or β2-induced enhancement of Ca2+ sensitivity from 0 to 100 μM [Ca2+]i: ΔΔV1/2 = ΔV1/2 − mSlo1 + β (0Ca − 100 Ca) − ΔV1/2 − mSlo1 (0Ca − 100 Ca). ΔΔV1/2 (induced by the β1-subunit)-VHC relationship is empirically fit with the Boltzmann function (panel A, solid line,). (1*) The β1-induced ΔΔV1/2 for D153C was assumed to be 0 according to the result of D153C:E386A channels (Fig. 3 C); and the β2-induced ΔΔV1/2 was for D153C calculated from the result of D153C:E321A channels (Fig. 4 D). (7**) The β1- and β2-induced ΔΔV1/2-values for R213C were calculated from the results of R213C:E321A channels (Figs. 3 D and 4 D).
The β1- and β2-subunits share substantial structural similarities, with 43% identical or 66% conservative residues (7). They also share similar function by enhancing the apparent Ca2+ sensitivity of activation. Therefore, it is remarkable that distinct molecular mechanisms may underlie the regulation of Ca2+ sensitivity by these two β-subunits. According to the study by Orio and Latorre (20), one of the most prominent differences between the β1- and β2-subunit is that, whereas the effects of the β1-subunit can be explained by changes in voltage sensor movements, the effects of the β2-subunit can only determined if the allosteric factor for Ca2+ dependence also is altered. Similar to this result, our finding shows that some mutations of voltage-sensing residues are sufficient to abolish the effect of β1 in enhancing Ca2+ sensitivity but have a much smaller or a complete lack of impact on the β2 effect. These results suggest that the β1-subunit may affect the voltage sensor to alter Ca2+ sensitivity; whereas the β2-subunit may influence Ca2+ dependent mechanism more directly. It should be noted that such a mechanistic distinction may explain the primary effects of the two β-subunits, although each β-subunit may use both mechanisms for their total effects. For example, it has been suggested that the β1-subunit enhances Ca2+ sensitivity by affecting voltage sensor movements; in addition, however, it also changes Ca2+ binding (22,24). Likewise, it has been proposed that the β2-subunit alters voltage sensor movements. In a recent voltage clamp fluorometry study (25), Savalli et al. demonstrated that the β2-subunit induces a leftward shift on the voltage axis of the fluorescence versus voltage (F(V)) relations when the S3-S4 linker of the α-subunit was labeled with thiol-reactive fluorescent dyes. The authors suggested that this result indicated an alteration of the voltage sensor movements due to the association of the β2-subunit. Notwithstanding these complexities, our results indicate that various structural domains of both the α-subunits and the β-subunits can interact to modulate different molecular processes during channel gating and produce similar functional changes. Our results also provide the basis to further identify these structural domains and molecular processes that are important for the regulation of BK channels by β-subunits.
How does the β1-subunit alter the voltage-dependent gating to enhance Ca2+ sensitivity (i.e., the β-subunit–induced larger leftward shift of G-V relationship in response to increasing Ca2+ concentrations)? Orio and Latorre (20) found that the association of human β1 reduced the number of zJ, decreased the intrinsic L0, and increased the allosteric coupling between voltage sensor movements and channel opening (D). The reduction of zJ was proposed as the primary cause for an enhanced Ca2+ sensitivity. In a study of bovine β1, Bao and Cox (22) found that a shift of the voltage sensor movements to more negative voltages (VHC) induced by the β1-subunit was the primary reason for an enhanced Ca2+ sensitivity. More recently, Wang and Brenner (23) found that mouse β1 shifted VHC to negative voltages and reduced L0, but the enhancement of Ca2+ sensitivity was primarily due to the changes in L0. These studies have made it clear that the enhancement of Ca2+ sensitivity by the β1-subunit is accompanied by significant changes in voltage-dependent gating, suggesting that the voltage sensor is a critical structural component for β1 regulation. However, although the β1-subunits from three different species all enhance Ca2+ sensitivity, this enhancement may not be the consequence of changes in any particular voltage-dependent parameters. No single gating parameter was significantly altered by all three β1 orthologs. These results seem to suggest that the enhancement of Ca2+ sensitivity and the changes in voltage dependence are all the consequences of a change in channel conformation and movements during gating, but that none of the phenomenological changes in gating parameters is the cause of others.
The results of this study demonstrate that mutating the voltage sensor can alter the enhancement of Ca2+ sensitivity by the β1-subunit. These mutations also change voltage dependence of channel gating (34). Therefore, our results indicate that the voltage sensor is the primary target regulated by the β1-subunit for all the functional consequences of β1 association. We compared the changes in the Ca2+ sensitivity caused by β1 to the changes in voltage dependence caused by these mutations and found that the changes in the enhancement of Ca2+ sensitivity is quantitatively correlated with the shift of VHC but not with other parameters (Fig. 7). These comparisons indicate that only a specific perturbation of the conformation and movements of the voltage sensor would disrupt the ability of the β1-subunit to enhance Ca2+ sensitivity. Such a specific perturbation also alters the stability of the voltage sensor at activated state. Further study is needed to identify the molecular mechanism for this specific perturbation and its correlation with VHC.
Acknowledgments
The authors thank Larry Salkoff for kindly providing the mSlo1 clone; Robert Brenner, who kindly provided the β1 and β2 clones; and Frank Horrigan, who kindly provided the Y163E and Y163K mutant mSlo1 clones.
This work was supported by the Epilepsy Foundation (U.S.L.) and a grant from the National Institutes of Health to J.C. (R01-HL70393). ). J.C. is an associate professor of biomedical engineering on the Spencer T. Olin Endowment.
Huanghe Yang and Guohui Zhang contributed equally to this work.
Editor: Richard W. Aldrich.
References
- 1.Adelman, J. P., K. Z. Shen, M. P. Kavanaugh, R. A. Warren, Y. N. Wu, A. Lagrutta, C. T. Bond, and R. A. North. 1992. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 9:209–216. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson, N. S., G. A. Robertson, and B. Ganetzky. 1991. A component of calcium-activated potassium channels encoded by the Drosophila SLO locus. Science. 253:551–555. [DOI] [PubMed] [Google Scholar]
- 3.Knaus, H. G., K. Folander, M. Garcia-Calvo, M. L. Garcia, G. J. Kaczorowski, M. Smith, and R. Swanson. 1994. Primary sequence and immunological characterization of beta-subunit of high conductance Ca2+-activated K+ channel from smooth muscle. J. Biol. Chem. 269:17274–17278. [PubMed] [Google Scholar]
- 4.Wallner, M., P. Meera, and L. Toro. 1999. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane beta-subunit homolog. Proc. Natl. Acad. Sci. USA. 96:4137–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia, X. M., J. P. Ding, and C. J. Lingle. 1999. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J. Neurosci. 19:5255–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uebele, V. N., A. Lagrutta, T. Wade, D. J. Figueroa, Y. Liu, E. McKenna, C. P. Austin, P. B. Bennett, and R. Swanson. 2000. Cloning and functional expression of two families of beta-subunits of the large conductance calcium-activated K+ channel. J. Biol. Chem. 275:23211–23218. [DOI] [PubMed] [Google Scholar]
- 7.Brenner, R., T. J. Jegla, A. Wickenden, Y. Liu, and R. W. Aldrich. 2000. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J. Biol. Chem. 275:6453–6461. [DOI] [PubMed] [Google Scholar]
- 8.Torres, Y. P., F. J. Morera, I. Carvacho, and R. Latorre. 2007. A marriage of convenience: β-subunits and voltage-dependent K+ channels. J. Biol. Chem. 282:24485–24489. [DOI] [PubMed] [Google Scholar]
- 9.Salkoff, L., A. Butler, G. Ferreira, C. Santi, and A. Wei. 2006. High-conductance potassium channels of the SLO family. Nat. Rev. Neurosci. 7:921–931. [DOI] [PubMed] [Google Scholar]
- 10.Ledoux, J., M. E. Werner, J. E. Brayden, and M. T. Nelson. 2006. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda). 21:69–78. [DOI] [PubMed] [Google Scholar]
- 11.Toro, L., M. Wallner, P. Meera, and Y. Tanaka. 1998. Maxi-KCa, a unique member of the voltage-gated K channel superfamily. News Physiol. Sci. 13:112–117. [DOI] [PubMed] [Google Scholar]
- 12.Fettiplace, R., and P. A. Fuchs. 1999. Mechanisms of hair cell tuning. Annu. Rev. Physiol. 61:809–834. [DOI] [PubMed] [Google Scholar]
- 13.Giangiacomo, K. M., M. Garcia-Calvo, H. G. Knaus, T. J. Mullmann, M. L. Garcia, and O. McManus. 1995. Functional reconstitution of the large-conductance, calcium-activated potassium channel purified from bovine aortic smooth muscle. Biochemistry. 34:15849–15862. [DOI] [PubMed] [Google Scholar]
- 14.Nelson, M. T., and J. M. Quayle. 1995. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 268:C799–C822. [DOI] [PubMed] [Google Scholar]
- 15.Brenner, R., G. J. Perez, A. D. Bonev, D. M. Eckman, J. C. Kosek, S. W. Wiler, A. J. Patterson, M. T. Nelson, and R. W. Aldrich. 2000. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 407:870–876. [DOI] [PubMed] [Google Scholar]
- 16.Semenov, I., B. Wang, J. T. Herlihy, and R. Brenner. 2006. BK channel beta1-subunit regulation of calcium handling and constriction in tracheal smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 291:L802–L810. [DOI] [PubMed] [Google Scholar]
- 17.Herrera, G. M., T. J. Heppner, and M. T. Nelson. 2000. Regulation of urinary bladder smooth muscle contractions by ryanodine receptors and BK and SK channels. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279:R60–R68. [DOI] [PubMed] [Google Scholar]
- 18.Khan, R. N., S. K. Smith, J. J. Morrison, and M. L. Ashford. 1993. Properties of large-conductance K+ channels in human myometrium during pregnancy and labour. Proc. Biol. Sci. 251:9–15. [DOI] [PubMed] [Google Scholar]
- 19.Hicks, G. A., and N. V. Marrion. 1998. Ca2+-dependent inactivation of large conductance Ca2+-activated K+ (BK) channels in rat hippocampal neurones produced by pore block from an associated particle. J. Physiol. 508:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orio, P., and R. Latorre. 2005. Differential effects of beta 1 and beta 2 subunits on BK channel activity. J. Gen. Physiol. 125:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horrigan, F. T., J. Cui, and R. W. Aldrich. 1999. Allosteric voltage gating of potassium channels I. Mslo ionic currents in the absence of Ca2+. J. Gen. Physiol. 114:277–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao, L., and D. H. Cox. 2005. Gating and ionic currents reveal how the BKCa channel's Ca2+ sensitivity is enhanced by its beta1 subunit. J. Gen. Physiol. 126:393–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang, B., and R. Brenner. 2006. An S6 mutation in BK channels reveals beta1 subunit effects on intrinsic and voltage-dependent gating. J. Gen. Physiol. 128:731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nimigean, C. M., and K. L. Magleby. 2000. Functional coupling of the beta(1) subunit to the large conductance Ca2+-activated K+ channel in the absence of Ca2+. Increased Ca2+ sensitivity from a Ca2+-independent mechanism. J. Gen. Physiol. 115:719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savalli, N., A. Kondratiev, S. B. de Quintana, L. Toro, and R. Olcese. 2007. Modes of operation of the BKCa channel {beta}2 subunit. J. Gen. Physiol. 130:117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orio, P., Y. Torres, P. Rojas, I. Carvacho, M. L. Garcia, L. Toro, M. A. Valverde, and R. Latorre. 2006. Structural determinants for functional coupling between the beta and alpha subunits in the Ca2+-activated K+ (BK) channel. J. Gen. Physiol. 127:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrow, J. P., S. I. Zakharov, G. Liu, L. Yang, A. J. Sok, and S. O. Marx. 2006. Defining the BK channel domains required for beta1-subunit modulation. Proc. Natl. Acad. Sci. USA. 103:5096–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wallner, M., P. Meera, and L. Toro. 1996. Determinant for beta-subunit regulation in high-conductance voltage-activated and Ca2+-sensitive K+ channels: an additional transmembrane region at the N terminus. Proc. Natl. Acad. Sci. USA. 93:14922–14927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian, X., C. M. Nimigean, X. Niu, B. L. Moss, and K. L. Magleby. 2002. Slo1 tail domains, but not the Ca2+ bowl, are required for the beta 1 subunit to increase the apparent Ca2+ sensitivity of BK channels. J. Gen. Physiol. 120:829–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen, K. Z., A. Lagrutta, N. W. Davies, N. B. Standen, J. P. Adelman, and R. A. North. 1994. Tetraethylammonium block of Slowpoke calcium-activated potassium channels expressed in Xenopus oocytes: evidence for tetrameric channel formation. Pflugers Arch. 426:440–445. [DOI] [PubMed] [Google Scholar]
- 31.Yellen, G. 2002. The voltage-gated potassium channels and their relatives. Nature. 419:35–42. [DOI] [PubMed] [Google Scholar]
- 32.Bezanilla, F. 2000. The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80:555–592. [DOI] [PubMed] [Google Scholar]
- 33.Swartz, K. J. 2004. Towards a structural view of gating in potassium channels. Nat. Rev. Neurosci. 5:905–916. [DOI] [PubMed] [Google Scholar]
- 34.Ma, Z., X. J. Lou, and F. T. Horrigan. 2006. Role of charged residues in the S1—S4 voltage sensor of BK channels. J. Gen. Physiol. 127:309–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horrigan, F. T., and R. W. Aldrich. 2002. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120:267–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butler, A., S. Tsunoda, D. P. McCobb, A. Wei, and L. Salkoff. 1993. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science. 261:221–224. [DOI] [PubMed] [Google Scholar]
- 37.Horrigan, F. T., and R. W. Aldrich. 1999. Allosteric voltage gating of potassium channels II. Mslo channel gating charge movement in the absence of Ca2+. J. Gen. Physiol. 114:305–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McManus, O. B., L. M. Helms, L. Pallanck, B. Ganetzky, R. Swanson, and R. J. Leonard. 1995. Functional role of the beta subunit of high conductance calcium-activated potassium channels. Neuron. 14:645–650. [DOI] [PubMed] [Google Scholar]
- 39.Cox, D. H., and R. W. Aldrich. 2000. Role of the beta1 subunit in large-conductance Ca2+-activated K+ channel gating energetics. Mechanisms of enhanced Ca2+ sensitivity. J. Gen. Physiol. 116:411–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nimigean, C. M., and K. L. Magleby. 1999. The beta subunit increases the Ca2+ sensitivity of large conductance Ca2+-activated potassium channels by retaining the gating in the bursting states. J. Gen. Physiol. 113:425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia, X. M., J. P. Ding, and C. J. Lingle. 2003. Inactivation of BK channels by the NH2 terminus of the beta2 auxiliary subunit: an essential role of a terminal peptide segment of three hydrophobic residues. J. Gen. Physiol. 121:125–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui, J., and R. W. Aldrich. 2000. Allosteric linkage between voltage and Ca2+-dependent activation of BK-type mslo1 K+ channels. Biochemistry. 39:15612–15619. [DOI] [PubMed] [Google Scholar]