

Abstract
The objective of the study was to understand how estrogen modulates the rigidity of the cytoskeleton in epithelial cells. Estrogen depletion decreased, and treatment with 17β-estradiol increased deformability of cervical-vaginal epithelial cells. Estrogen also induced redistribution of nonmuscle myosin II-B (NMM-II-B); lesser interaction of NMM-II-B with actin; increased phosphorylation of NMM-II-B-heavy chains at threonine and serine residues; and decreased filamentation of NMM-II-B in vitro. The effects of 17β-estradiol were time and dose related and could be mimicked by diethylstilbestrol. The effects of estrogen were blocked by cotreatment with antisense oligonucleotide for the estrogen receptor-α and inhibited by ICI-182,780 and tamoxifen; omission of epithelial growth factor (EGF) from the culture medium; and cotreatments with the EGF receptor inhibitor AG1478, the ERK-MAPK inhibitor PD98059, the casein kinase-II (CK2) inhibitor 5,6-dichloro-1-β-(D)-ribofuranosylbenzimidazole, the Rho-associated kinase inhibitor Y-27632, and the nonspecific phosphatase inhibitor okadaic acid. Coadministration of 5,6-di-chloro-1-β-(D)-ribofuranosylbenzimidazole plus okadaic acid blocked the 17β-estradiol effect. H-89 or LY294002 did not significantly affect estrogen effects. Treatment with estrogen increased activation of ERK1/2 and CK2 activity. These data suggest a novel pathway of estrogen regulation of the cytoskeleton in epithelial cells. The effect is mediated by estrogen receptor-α and involves in part the EGF-EGF receptor and ERK-MAPK cascades as proximal signaling networks and the CK2 and Rho-associated kinase-regulated myosin heavy chain phosphatase as terminal effectors. Augmented phosphorylation of NMM-II-B can block filamentation and induce disassociation of the myosin from the cortical actin, and disruption of the actomyosin ring can increase cell deformability. This mechanism can explain estrogen regulation of paracellular permeability in cervical-vaginal epithelia in vivo.
One of the important roles of estrogen in females is the regulation of reproductive tract epithelial permeability. The major route of transport in secretory epithelia, including the female reproductive tract, is the paracellular (intercellular) pathway. Free movement of water and solutes in the paracellular pathway is restricted by the resistance of the tight junctions (RTJ) and the resistance of the lateral intercellular space (RLIS) in series (1, 2). The RTJ determines the overall paracellular resistance, but in leaky epithelia such as the vaginal and cervical epithelia, the contributions of the RLIS to the total resistance can be significant (3). Estrogen increases the permeability of vaginal and cervical epithelia by two known mechanisms: modulation of the tight junctional protein occludin, which decreases the RTJ (4, 5), and actin depolymerization, which decreases the RLIS (6–9). The two effects of estrogen have different time course: estrogen modulation of the RLIS by modulation of actin polymerization is observed 1–6 h after treatment, whereas modulation of occludin and the RTJ requires 24–48 h of treatment (10–12).
The RLIS is determined by the geometry of the intercellular space and therefore reciprocally by the width and shape of the epithelial cells that define this space. Our understanding of the importance of the RLIS in regulation of paracellular permeability stems from works of Spring and Hope (13), who showed that hydrostatic gradients in the subluminal → luminal direction induce fluid accumulation in the intercellular space at basolateral regions of the epithelium and dilate the intercellular space. The effect is limited to basolateral regions of the intercellular space, i.e. those in proximity to the sublumen (the blood compartment) because of the near-apical location of the tight and adherence junctions, which prevent expansion of the intercellular space at near-apical domains (Fig. 1B). This fundamental mechanism explains how surface epithelia maintain lubrication of the lumen: blood pressure in the capillary bed generates a subluminal → luminal hydrostatic gradient that expands the basolateral intercellular space and decreases the RLIS. This allows plasma transudation from the blood into the lumen through the paracellular route.
Fig. 1.
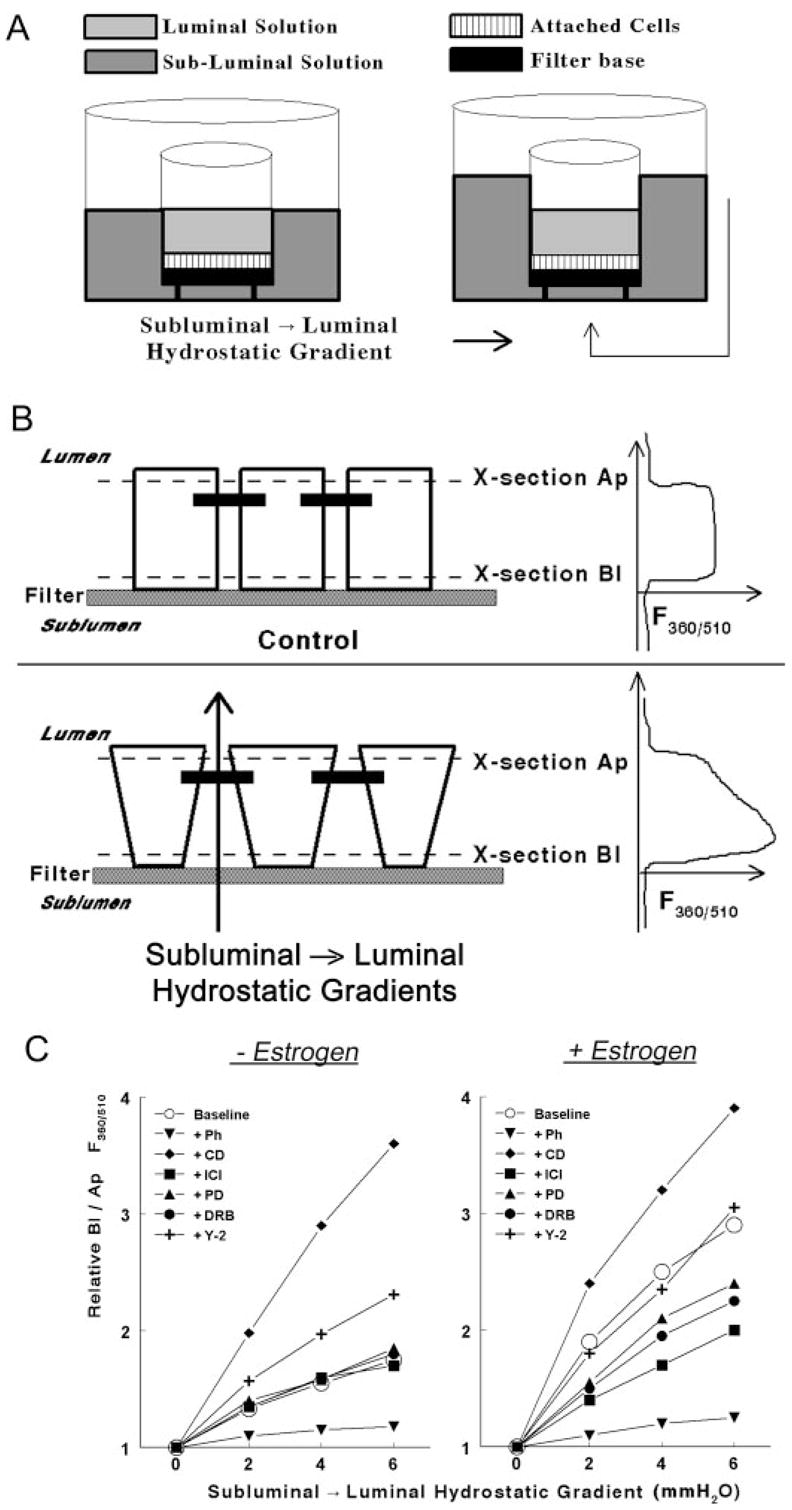
Methodology to determine rigidity of the cytoskeleton in epithelial cells attached on filters. A, A filter insert with cells (squares) attached on the filter base (filled square) was placed in a fluorescence chamber in which the luminal (light gray) and subluminal (darker gray) solutions could be independently perfused. subluminal → luminal hydrostatic gradients were established by increasing the volume of the subluminal solution above that of the luminal solution. B, Schema and cross-section of epithelial cells attached on filters. Neighboring cells are joined by junctional and adherens junctions that localize near the apical border of the cells (filled horizontal bars). In the lower panel, hydrostatic gradients in the subluminal → luminal direction were established as described above. The panels in the right parts of B are actual recordings of the F360/510 along the longitudinal vertical (Z)-axis of the cultures. See Materials and Methods for details. C, Modulation of the Bl to Ap F360/510 ratio in hEVECs attached on filters by subluminal → luminal hydrostatic gradients. Cells were shifted for 3 d to steroid free medium and treated for 2 d before the experiment with the vehicle (− Estrogen) or 10 nM 17β-estradiol (+ Estrogen). Additional cotreatments were as follows: phalloidin (Ph, 10 ng/ml, 60 min); DRB (10 μM, 6 h); Y-27632 (Y-2, 5 μM, 6 h); cytocha-lasin-D (CD, 10 μg/ml, 20 min); ICI-182,780 (ICI, 10 μM, 1 d); and PD98059 (PD, 10 μM, 6 h). Each line represents means of three to four experiments. Data were normalized to maximal increases of the Bl to Ap F360/510 ratio (arbitrary value of 4) as determined in cells treated with cytochalasin-D. Variability ranged 3–7%. Similar experiments were done using CaSki cells (not shown).
The ability of epithelial cells to change their shape in response to hydrostatic subluminal → luminal gradients depends on the elasticity of the cytoskeleton: cells expressing a rigid cytoskeleton will change their shape less readily in response to stimuli than cells expressing a dynamic, flexible cytoskeleton. One mechanism, mentioned above, is actin polymerization: formation of polymerized F-actin-dominated cytoskeleton will tend to increase the RLIS, whereas formation of depolymerized G-actin-dominated cytoskeleton will decrease the RLIS (9). Interestingly, by comparing the time course of estrogen modulation of actin status with estrogen modulation of paracellular resistance, it became apparent that the changes in actin status cannot explain entirely the estrogen regulation of RLIS because estrogen increase in G-actin preceded estrogen decrease in RLIS by about 24 h (6, 9). One of the conclusions was that estrogen modulation of the RLIS involves an additional mechanism to that of actin polymerization.
In addition to actin polymerization, the rigidity of the cytoskeleton can be also modulated by regulation of myosin filaments (14). Myosins are proteins that interact with actin and hydrolyze ATP to generate force. In nonmuscle cells including epithelial cells, myosins interact with actin and regulate actin organization into filaments that maintain the shape of cellular structures or coordinate contraction and cell division (15). Loss of myosin-II, the prototypical two-headed myosin, can disrupt actin polymerization and lead to cell detachment (15). The nonmuscle myosin-II, a subclass of myosin-II (16), was shown to play a role in epithelial cell attachment and intercellular communications (15, 17). In the human, the state of myosin-II activation regulates paracellular permeability in kidney cells (18, 19), and abrogation of paracellular resistance leads to backleak of glomerular filtrate (15). In patients with inflammatory bowel disease, colonic epithelial myosin light-chain kinase expression and enzymatic activity were found to be increased, suggesting that myosin light-chain kinase up-regulation may contribute to barrier dysfunction and the pathogenesis of inflammatory bowel disease (20).
In cervical epithelial cells, the main myosin component of the cortical actomyosin frame is nonmuscle myosin-II-B (NMM-II-B) (present data). Preliminary results from the lab showed that NMM-II-B filaments are constitutively phosphorylated, and the phosphorylation is regulated by casein kinase-II (CK2) and Rho-associated kinase (ROCK)-dependent modulation of myosin heavy-chain phosphatase. We also found that increased phosphorylation of NMM-II-B filaments abrogates filamentation and decreases myosin-actin interaction (not shown). These data could be important for our understanding of regulation of permeability because myosin-actin interaction determines the rigidity of the cytoskeleton and hence the RLIS. The present study tested the hypothesis that estrogen modulation of the RLIS involves modulation of NMM-II-B filamentation. We found that estrogen augments phosphorylation of nonmuscle myosin-II heavy-chain (NMMHC)-II-B filaments and decreases NMM-II-B filamentation. We also found that the effect of estrogen involves the estrogen receptor (ER)-α and in part also the epithelial growth factor (EGF)-EGF-receptor (EGFR) and ERK-MAPK cascades as proximal signaling networks, whereas the terminal effectors are the CK2 and the myosin heavy-chain phosphatase. These findings suggest a novel mechanism of estrogen regulation of the RLIS through modulation of NMM-II-B-actin interaction.
Materials and Methods
Antibodies, chemicals, and supplies
Mouse monoclonal antihuman β-actin antibody, antioccludin antibody, and antiglyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody were from Zymed Laboratory Inc. (San Francisco, CA). Rabbit polyclonal antihuman NMMHC-II-A antibody (PRB-440P) and its antigenic peptide and the antihuman NMMHC-II-B antibody (PRB-445P) were from Covance Research Products (Berkeley, CA). Mouse monoclonal antihuman phosphoserine, phosphothreonine, and phosphotyrosine (PY20 and P99) antibodies were from Transduction Laboratories (BD Biosciences, San Jose, CA). Mouse monoclonal antibody recognizing tyrosine phosphorylated ERK1/2, rabbit polyclonal antibody recognizing total ERK1/2, and goat polyclonal anti-CK2α antibody were from Santa Cruz Biotechnology (Santa Cruz, CA). The antibodies were used according to the manufacturers’ instructions. All chemicals, unless specified otherwise, were obtained from Sigma Chemical (St. Louis, MO). 5,6-Dichloro-1-β-(D)-ribofuranosylbenzimidazole (DRB) and Y-27632 ware obtained from Calbiochem (La Jolla, CA).
Cell-culture techniques
The experiments used two types of cells: normal human epithelial vaginal-ectocervical cells (hEVECs) and CaSki cells. Secondary/tertiary cultures of hEVECs were generated from minces of histologically normal ectocervix/vagina as described (3). The discarded tissues were collected by the Cooperative Human Tissue Network at University Hospitals of Cleveland and Case Western Reserve University according to the institutional review board protocol 03–90-TG. Tissues were collected from seven premenopausal women aged 37–46 yr and six postmenopausal women aged 53–65 yr who underwent hysterectomy for medical indications unrelated to the present study. None of the women were treated with steroid sex hormones for at least 3 months before surgery. Menopause was defined as amenorrhea for 1 yr or longer or amenorrhea of at least 6 months plus climacteric symptoms and plasma levels of FSH greater than 25 mIU/ml. hEVECs (also referred to as hECE cells) were previously characterized as phenotypically resembling the stratified squamous vaginal-ectocervical epithelial cells (3). Cells chosen for experiments were those obtained from tissues reported as human papillomavirus negative, and cultures were routinely tested for mycoplasma. Experiments also used CaSki cells, which are a stable line of transformed endocervical epithelial cells (3). Culture conditions for hEVECs and CaSki cells were described (3). For experiments using cells attached on filters, cells were plated on Anocell-10 filters (Anocell, Oxon, UK, obtained through Sigma), which are ceramic-base filters, pore size of 0.02 μm width and 50 μm depth. Filters were coated on their upper (luminal) surface with 3–5 μg/cm2 collagen type IV as described (21). Filter experiments involved treatments with drugs added to both the luminal and subluminal solutions. Unless specified otherwise, the experiments used culture medium containing a saturating dose of 0.2 nM EGF that suffices to activate the EGFR (22). Some experiments used culture medium lacking EGF.
For experiments with estrogen-deficient conditions, cells on filters were shifted to steroid-free medium for 3–5 d. This medium was composed of phenol-red-deficient (DMEM)/Ham’s F12 or RPMI 1640 (Sigma) containing 8% heat-inactivated fetal bovine serum that was previously treated with charcoal to remove steroids. Preparation of charcoal-treated serum was described (6); briefly, dextrancoated charcoal (Sigma) was dissolved at 8% in 0.15 M NaCl, autoclaved, mixed by stirring, spun, and the pellet resuspended as 1 g per 1.25 ml in H2O. Fetal bovine serum (Hyclone, Logan, UT) was mixed with the activated charcoaldextran at 20:1 (vol/vol) and incubated for 45 min at 55 C. At the completion of incubation, the mixture was spun twice at 800 × g for 20 min, and the supernatant (serum) was decanted and collected.
Changes in the width of cells attached on filters
The experiments used a previously described method (23) with some modifications. Filters containing attached cells were housed in a custom-designed fluorescence chamber. The chamber was constructed as a modified Ussing chamber so that it allows separation of the subluminal solution that bathes the basolateral parts of the cells membranes, from the luminal solution that bathes the apical part of the cells membranes. A bifurcated custom-designed fiberoptics was placed over the apical surface of the cells and served to send the excitation signal and capture the emitted signal from a constant horizontal field (cross-section) of about 2 × 104 cells (23). Changes in cell width were determined in cells loaded with fura-2 by measurements of changes in intracellular fura-2 fluorescence at the isosbestic wavelengths [360/510 nm excitation/emission (F360/510)]. Because in hEVECs and CaSki cells photobleaching or leakage of fura-2 are minimal under the conditions of the experiment (23), changes in F360/510 reflect changes in the intracellular concentrations of fura-2 across the monitored cross-section and therefore changes in cell width. The bifurcated fiberoptic sleeve resting above the apical surface of the cells was attached to a stand that allowed fine (micrometers) vertical movements of the fixed fiberoptics sleeve and therefore to monitor fluorescence from different horizontal cross-sections along the vertical axis of the cells. Changes in F360/510 were determined in response to transepithelial hydrostatic gradients. Subluminal → luminal transepithelial hydrostatic gradients of 0–6 mm H2O, which prevail in the extracellular space in vivo (24, 25), were generated by increasing the fluid level of the subluminal solution above that of luminal solution by adding aliquots of culture medium to the subluminal container to predetermined levels (Fig. 1A).
A schematic representation of the system and an actual result are shown in Fig. 1. In cells bathed under normostatic conditions (Fig. 1A, left panel), a positive signal was recorded at the basal part of the cells (above the filter proper, Fig. 1B, control, upper right panel, cross-section Bl), and the signal terminated when the focus plane was above the apical part of the cell (Fig. 1B, control, upper right panel, cross-section Ap). When transepithelial hydrostatic gradients were produced in the subluminal → luminal direction (Fig. 1A, right panel, and Fig. 1B, lower panel), the F360/510 signal in plane Bl increased significantly, compared with the signal in plane Ap. The interpretation is that the intracellular concentration of fura-2 in plane Bl increased relative to that in plane Ap, i.e. the average cell width in plane Bl decreased relative to that in plane Ap. Changes in cell width, reflecting cell deformability, were determined in terms of changes in the ratio of the F360/510 signal in plane Bl relative to the F360/510 signal in plane Ap (Bl/Ap F360/510 ratio).
Protein assays
Immunostaining, light microscopy, and fluorescence microscopy were described (26). Briefly, after treatments, cells on coverslips or filters were washed with cold PBS and fixed in methanol; blocked with 3% BSA/PBS/0.2% Triton X-100 for 30 min; incubated with primary antibodies overnight at 4 C; washed three times with PBS; and incubated with secondary antibodies attached with Alexa Fluor 488 (donkey antirabbit IgG) or Alexa Fluor 594 (goat antimouse). Nuclei were stained with 4′,6′-diamino-2-phenylindole (Vector Laboratories, Burlingame, CA). In some experiments cells were incubated with horseradish peroxidase-labeled goat antirabbit IgG and heavy- and light-chain peroxidase, and the reaction was visualized by Fast-Red (Dako, http://www.dakocytomation.com). Cell fractionation by the freeze/thaw method and Western immunoblotting were described (26). Confirmation of the separation efficiency of the membrane-enriched fraction was determined by blotting with antioccludin antibody (tight junction plasma-membrane protein). Likewise, confirmation of separation efficiency of the cytosolic fraction was determined by blotting with anti-GAPDH antibody. Aliquots, normalized to 15 μg protein, were fractionated by 6–10% SDS-PAGE and blotted by Western analysis, and bands were analyzed semiquantitatively by densitometry. Immunoprecipitation/immunoblotting assays were described (26). Phosphorylation assays were described (26). Briefly, cells on filters were shifted to phosphate-free DMEM containing 10 mM HEPES (pH 7.4) at 37 C and treated with 100 μCi/ml [32P]orthophosphate (PerkinElmer Life Sciences, Boston MA) plus 1 μg/ml microcystin L–R (Calbiochem). After treatments, cells were washed with ice-cold PBS, lysed in lysis buffer, and processed by immunoprecipitation with anti-NMMHC-II-B antibody. Samples containing equal amounts of protein were resolved on 10% polyacrylamide gels and dried under vacuum. Radioactive bands were visualized by PhosphorImager [Molecular Dynamics (Amersham), Piscataway, NJ] and exposure to x-ray film.
Extraction and purification of NMM-II-B filaments
The experiments used a modification of existing methods (27, 28). All steps were done at 4 C. Total cells homogenates were incubated overnight with antinonmuscle myosin-II heavy-chain-A (NMMHC-II-A) antibody to immunoprecipitate the nonmuscle myosin-II-A (NMM-II-A). Pansorbin was added to the immunoprecipitates for 2 h, and the pansorbin-anti-NMMHC-II-A antibody-NMM-II-A complexes were pelleted by centrifugation at 25,000 × g for 5 min. The supernatants were washed three times with buffer containing 0.5 M NaCl, 50 mM 3[N-morpholino]propanesulfonic acid (pH 7.4), and 0.1 mM EGTA to remove nonspecifically bound proteins. Supernatants were resuspended in 0.2 ml of wash buffer containing 50-fold molar excess of the peptide used to generate the antihuman NMMHC-II-A antibody (GKADGAEAK-PAE, corresponding to the C terminus of the human NMMHC-II-A), and after 2 h of incubation on ice, samples were spun at 35,000 × g for 5 min and the supernatants containing the released NMM-II-B were dialyzed against several changes of wash buffer and concentrated to a volume of about 250 μl by washing in Centricon 100 (Amicon, Houston, TX). This method of purification yields a single 200-kDa band that is immuno-detected by the anti-NMMHC-II-B antibody (not shown).
In vitro NMM-II-B filament assembly assay
Purified NMM-II-B filaments at concentrations of 1–500 μg/ml (about 20 nM to 10 μM) were incubated with 2.5 mM MgSO4 and 10 mM NaCl at 4 C in the presence of 10 mM imidazole-HCl (pH 7.5) and 0.1 mM Ca2+ to induce filament assembly. At the completion of incubations, samples were centrifuged at 35,000 × g for 30 min at 4 C, and the protein content in the supernatant (i.e. nondimerized NMM-II-B) was measured and expressed per total protein measured before centrifugation as the percent nondimerized nonmuscle myosin-II-B. To ascertain complete recovery of nondimerized filaments, aliquots from the supernatant were centrifuged at 90,000 × g for 20 min. Assays in which sediment was found, indicating inappropriate separation of the monomers from the oligomers, were omitted from analysis (less than 3% of assays). NMM-II-B filament assembly was expressed in terms of the critical concentration of NMM-II-B necessary to induce dimerization from the curve of percent nondimerized NMM-II-B vs. added protein. The critical concentration of NMM-II-B necessary for dimerization was defined as the concentration of NMM-II-B that induced maximal oligomerization.
Determinations of cellular total protein
The method was described (29). Protein stains of Western blots were done using Ponceau (Ponceau S solution, P7170; Sigma) according to the manufacturer’s instructions.
Antisense oligonucleotides (ASO) assays
ER-specific ASO and random control oligonucleotide (CLO) were designed from the published sequences of the human ERα (30) (NM_012389); the human ERβ (31) (AJ002602); and the G protein-coupled receptor GPR30 (a membrane receptor responsive to the classical ligands of the ERα and ERβ (32) (AF027956) by using HYBsimulator (Advanced Gene Computing Technologies, Irvine, CA) and choosing the melting temperature of the oligonucleotides as 34 C. Possible sequences that would have the given melting temperature were identified from the target mRNA, and the cross-hybridization against the whole sequence in the GenBank database of each oligonucleotide was calculated. The oligomers ASO that would hybridize to the coding region of the ERα, ERβ, and GPR30 mRNAs were selected as 5′-TCA TGG TCA TGG TCC GT-3′, 5′-CAT CAC AGC AGG GCT ATA-3′; and 5′-TTG GGA AGT CAC ATC CAT-3′, respectively. The CLO was composed of a random 18-mer sequence AGA ACG TTA CTT ACA CTG with GC content (32%) similar to the antisense oligonucleotides. The CLO was designed such that no cross-hybridization against the ER genes would occur.
To assess the effects of the ASOs and CLO on ERα, ERβ, or GPR30 mRNA expression, hEVEC and CaSki cells were treated with or without ASO or CLO at concentrations of 10 μM. RT-PCR assays were carried out after 2 d, and assay experiments after 3–4 d. RT-PCR assays used the following primers and conditions: ERα forward, 5′-TTA CGA AGT GGG CAT GAT GA-3′; reverse, 5′-ATC TTG TCC AGG ACT CGG TG-3′ (product size 711 bp, 35 cycles); ERβ forward, 5′-GAG GCC TCC ATG ATG ATG TC-3′; reverse, 5′-TCT CCA GCA GCA GGT CAT AC-3′ (product size 610 bp, 35 cycles); GPR30 forward, 5′-TAA TAA GTC GAC GGG TCT CTT CCT-3′, reverse, 5′-ATT ATT GGA TCC TAC ACG GCA CTG C-3′ (product size, 652 bp, 35 cycles) (AF027956); and GAPDH (XM_227696) forward, 5′-CCA TGT TCG TCA TGG GTG TGA ACC A-3′; reverse, 5′-GCC AGT AGA GGC AGG GAT GAT GTT C-3′ (product size 229 bp, cycles 25). End points for successful transfections were 50% or greater reduction in ERα, ERβ, or GPR30 mRNA, compared with GAPDH mRNA; there were no significant effects on GAPDH mRNA and no significant effects of the CLO (not shown).
Determinations of CK2 activity
CK2 activity in cell lysates was determined by the ability to phosphorylate the decapeptide RRREEETEEE, which is a specific phosphate acceptor for CK2 (33), using a previously described method (34). Assays were carried out in 30 μl of buffer [composed of 50 mM Tris-HCl (pH 8.2), 50 mM NaCl, 25 mM KCl, 10 mM MgCl2, 10 μM Naorthovanadate] plus 15 μM [γ-32P]ATP (2 Ci/mmol) and 0.15 mM peptide substrate. Reactions were initiated by the addition of 1 μl of lysate (10–15 μg total protein) and were allowed to proceed for 15 min at 30 C (to reach maximal velocity). Phosphorylation was stopped by adding 20 μl of 40% trichloroacetic acid to each sample. After centrifugation, 10 μl of the supernatant were applied to P-81 phosphocellulose paper. The paper was washed in 100 mM phosphoric acid to remove excess [γ-32P]ATP, and the radioactivity was measured by scintillation counting. In parallel tubes, reactions were carried out in the presence of 10 μg/ml alkaline phosphatase (which inactivates CK2), added 15 min before the assay. CK2 activity (in nanomoles Pi per minute per milligram protein) was determined in terms of alkaline-phosphatase-sensitive change in the accumulated phosphoprotein (t15 min – baseline). Cellular expression of CK2 mRNA was determined by RT-PCR using primers designed based on the published sequence of the human CK2α (X70251): forward, 5′-GAA GTA TTT GAA GCC ATC A-3′; reverse, 5′-ATC ATA ATT GTC ATG TCC AT-3′ (product size 350 bp, 30 cycles).
Statistical analysis of the data
Data are presented as means (± SD), and significance of differences among means was estimated by ANOVA using GB-STAT (Dynamic Microsystems Inc., Silver Spring, MD).
Results
Estrogen effects on cell deformability
Hydrostatic gradients
It was previously shown that luminal → subluminal hydrostatic gradients have no effect on epithelial permeability (3). In contrast, subluminal → luminal hydrostatic gradients increase permeability by decreasing the RLIS secondary to dilatation of the lateral intercellular space at basolateral domains (Fig. 1B, lower panel, and Ref. 3). To quantify the effect, cells attached on filters were exposed to hydrostatic gradients in the subluminal →luminal direction (Fig. 1A, right panel), and changes in cell width were determined in terms of changes in F360/510 along the longitudinal axis of the cells (z-axis). As is shown in Fig. 1B, the F360/510 ratio in basolateral regions of the cells was greater in cells exposed to subluminal → luminal hydrostatic gradient than cells bathed in normostatic conditions.
The effects of estrogen on the F360/510 ratio are summarized in Fig. 1C. Compared with estrogen-depleted cells (Fig. 1C, left panel), treatment with 17β-estradiol at the physiological concentration of 10 nM facilitated a greater increase in the Bl to Ap F360/510 ratio in response to subluminal → luminal hydrostatic gradients (Fig. 1C, right panel). This result suggests that estrogen facilitated a decrease in cell width in basolateral regions of the cells in response to subluminal → luminal hydrostatic gradients.
Effects of modulators of actin and myosin
Changes in the Bl to Ap F360/510 ratio in response to subluminal → luminal hydrostatic gradients were determined in cells treated with agents that modulate cytoskeletal actin and the actomyosin status. Drugs were used at doses that produced submaximal effects, and lengths of treatments were chosen so that on removal of the drugs (by washes), F360/510 levels returned to baseline within 1–2 h (not shown). The reversibility of the effects assured that observed changes in F360/510 were not the result of toxic effects of the drugs.
Regardless of estrogen status, treatment with the F-actin-disrupting agent cytochalasin-D facilitated the largest increase in Bl to Ap F360/510 ratio in response to the hydrostatic gradients, whereas treatment with the F-actin depolymerization-inhibitor phalloidin facilitated the smallest increase in the Bl to Ap F360/510 ratio (Fig. 1C).
In estrogen-depleted cells, treatment with the ERα blocker ICI-182,780, the ERK/MAPK inhibitor PD98059, or the CK2 inhibitor DRB had no effect on the Bl to Ap F360/510 responses to subluminal → luminal hydrostatic gradients (Fig. 1C, left panel). In contrast, treatment with the ROCK inhibitor Y-27632 facilitated an increase in the Bl to Ap F360/510 ratio (Fig. 1C, left panel), but the Bl to Ap F360/510 ratios did not reach levels seen in estrogen-treated cells (Fig. 1C, right panel).
In estrogen-treated cells, cotreatments with ICI-182,780, PD98059, or DRB resulted in smaller increases in the Bl to Ap F360/510 ratio than cells treated only with estrogen; however, the Bl to Ap F360/510 ratios were higher than in estrogen-depleted cells. In estrogen-treated cells, cotreatment with Y-27632 had no additional effect on the Bl to Ap F360/510 ratio to that seen in cells treated only with estrogen (Fig. 1C, right panel).
Collectively, the results in Fig. 1 suggest that estrogen modulation of the responses to subluminal → luminal hydrostatic gradients involves modulation of the cytoskeletal actomyosin and cell deformability.
Estrogen effects on actin and on NMM-II isoforms
Immunostaining
Estrogen depletion resulted in actin confinement to cell membrane regions in the form of a narrow cortical frame (Fig. 2Ai and Al), whereas treatment with estrogen induced rounding of the cortical actin and the cells (Fig. 2A, f,o).
Fig. 2.
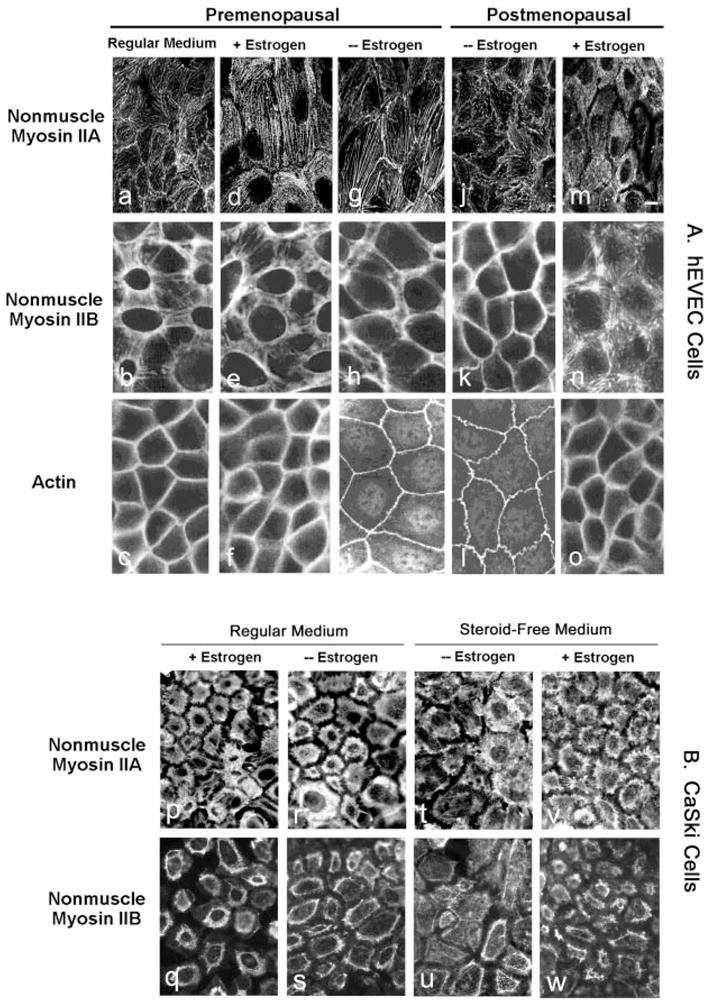
Immunofluorescence of NMM-II-A and NMM-II-B in hEVECs (A) and CaSki cells (B). A, Tertiary cultures of hEVECs on filters from a premenopausal woman (a–i) and a postmenopausal woman (j-o) were maintained in regular culture medium or shifted for 3 d to steroid-free medium in the absence (− Estrogen) or presence of 10 nM 17β-estradiol (+ Estrogen). Cells were stained with the anti-NMMHC-II-A antibody (a, d, g, j, and m), anti-NMMHC-II-B antibody (b, e, h, k, and n), or anti-β-actin antibody (c, f, i, l, and o). Bar (m), 15 μm. B, Immunofluorescence of NMM-II-A (p, r, t, and v) and NMM-II-B (q, s, u, and w) in CaSki cells. Cells on filters were maintained in regular culture medium (p, r, q, and s) or shifted for 3 d to steroid-free medium (t, v, u, and w) in the absence (− Estrogen, r, s, v, and w) or presence of 10 nM 17β-estradiol (+ Estrogen, p, q, t, and u). The experiments were repeated twice with similar trends.
In hEVECs of premenopausal and postmenopausal women (Fig. 2A, a,d,g,j,m) and CaSki cells (Fig. 2B, p,r,t,v), NMM-II-A staining localized mainly in the cytoplasm and perinuclear regions. Depletion of estrogen or treatment with estrogen had no significant effects on the immunostaining with the anti NMMHC-II-A antibody.
In hEVECs of premenopausal and postmenopausal women (Fig. 2A, b,e,h,k,n) and CaSki cells (Fig. 2B, q,s,u,w), NMM-II-B staining localized mainly in plasma membrane regions, and estrogen status had marked effects on the immunostaining with the anti-NMMHC-II-B antibody. In estrogen-deprived hEVECs of pre- and postmenopausal women, NMM-II-B staining was similar to actin staining and was confined mainly in the peripheral cortical frame (Fig. 2A, h,k). Treatment with estrogen resulted in disassociation of the myosin from the cortical actin and its sequestration into cytoplasmic and perinuclear areas (Fig. 2A, e,n). Similar trends were observed in CaSki cells (Fig. 2B). In hEVECs and CaSki cells maintained in regular culture medium, the cellular distribution of NMM-II-B was similar to that observed in estrogen-treated cells, and estrogen treatment had no additional effect (Fig. 2, Ab, Bq, and Bs). The most likely explanation is that regular culture medium contains estrogenic activity that suffices to activate the estrogen receptor mechanism (6).
In view of the marked effects of estrogen on cellular NMM-II-B and the relative little effect on cellular NMM-II-A, all subsequent experiments focused on NMM-II-B.
Cellular content and distribution of NMM-II-B
Treatment with 10 nM 17β-estradiol increased cellular NMM-II-B 2-fold over the 48-h period of treatment (Figs. 3, A and B). The effect was dose related (Fig. 3, A and C), reaching saturation at 5–25 nM. Cell-fractionation assays confirmed that treatment with estrogen significantly changed the cellular distribution of NMM-II-B. As early as 6–12 h after treatment, the amount of NMM-II-B recovered in plasma membrane-enriched fractions decreased, reaching trace amounts at 24–48 h, whereas the amount of the myosin recovered in the cytosol increased inversely (Fig. 4).
Fig. 3.
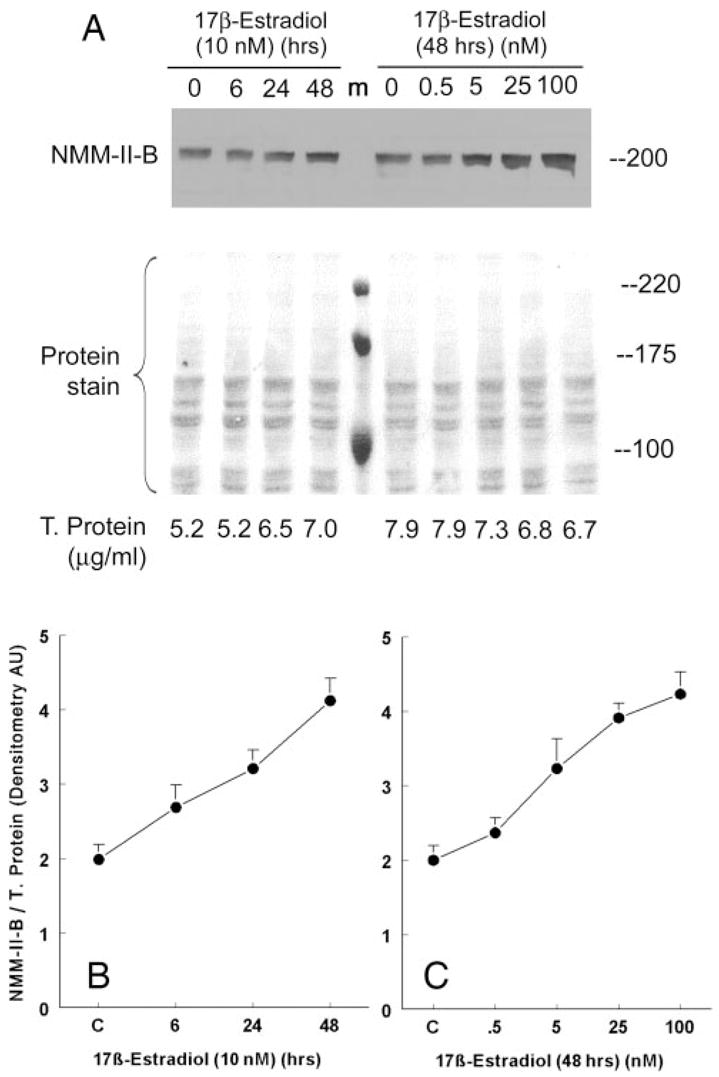
Estrogen effects on cellular content of NMM-II-B. CaSki cells were shifted for 3 d to steroid-free medium and treated with 17β-estradiol at the concentrations and for the durations indicated in the figure. Proteins were resolved on 6% PAGE and Western blotted using anti-NMMHC-II-B antibody. T. protein, Total protein. A, Western blots with the corresponding Ponceau protein stains. Shown are also the amounts of total proteins layered in each lane. m, Protein markers. B and C, Results of densitometry analysis of the Western blots in A relative to total cellular protein (means ± SD of three experiments).
Fig. 4.
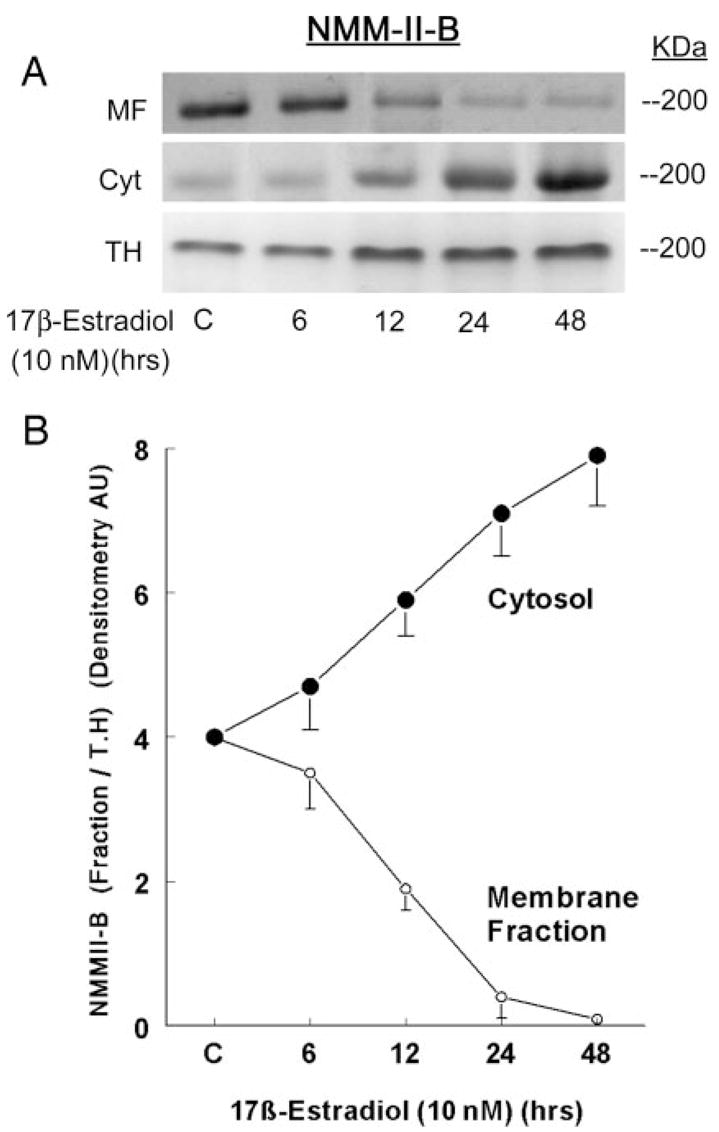
Estrogen effects on cellular distribution of NMM-II-B. CaSki cells were shifted for 3 d to steroid-free medium and treated with 10 nM 17β-estradiol for the durations indicated in the figure. Cell fractions [membrane-enriched fraction (MF), cytosol (Cyt), or total homogenates (TH)] were resolved on 6% PAGE and Western blotted using anti-NMMHC-II-B antibody. The lower panel shows densitometry of the NMM-II-B bands in the MF and Cyt relative to the TH (means ± SD of three experiments).
Actin-NMM-II-B interaction
Effects of estrogen status on actin-NMM-II-B interaction were determined using the coimmunoprecipitation technique. Estrogen depletion had no significant effect on cell content of actin, but it decreased NMM-II-B content and increased the degree of NMM-II-B-actin coimmunoprecipitation (Fig. 5, A and B). Treatment with estrogen also had no significant effect on cell content of actin, but it reversed the effects of estrogen depletion on cell NMM-II-B content and actin-NMM-II-B interaction. Treatment with estrogen increased the content of NMM-II-B and decreased the degree of NMM-II-B-actin coimmunoprecipitation (Fig. 5, A and B). Densitometry analysis of the data in Fig. 5A showed that, compared with cells maintained in regular medium, estrogen depletion decreased the ratio of NMM-II-B to actin by about half, and treatment with estrogen increased it by about half (Fig. 5B). Similarly, depletion of estrogen increased the ratio density of the coimmunoprecipitated product of actin-NMM-II-B relative to cellular actin by about 3-fold, and treatment with estrogen decreased it 5-fold (Fig. 5B). Fig. 5, A and B, also shows that cotreatment with the ERα antagonist ICI-182,780 inhibited the estrogen increase in cellular NMM-II-B and increased the ratio density of the coimmunoprecipitated product of actin-NMM-II-B relative to cellular actin. These results suggest that the estrogen increase in cellular NMM-II-B and the estrogen decrease in the coimmunoprecipitated product of actin-NMM-II-B relative to cellular actin involve the ERα.
Fig. 5.
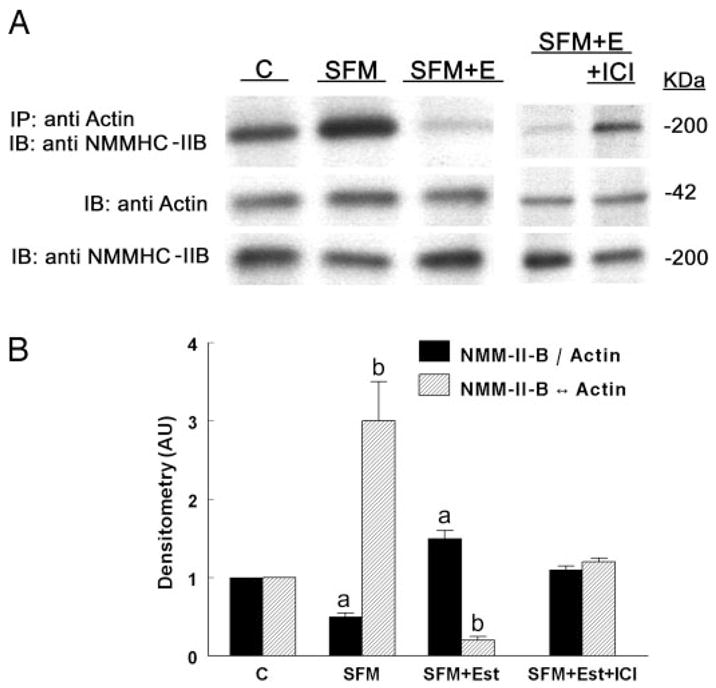
Estrogen effects on actin-NMM-II-B coimmunoprecipitation. Assays involved CaSki cells maintained in regular culture medium (C, control); cells shifted for 3 d to steroid-free medium (SFM); cells maintained in SFM and treated with 10 nM 17β-estradiol for 2 d (SFM + Est); and SFM + E cells cotreated with ICI-182,780 (ICI, 10 μM) for 24 h before assays. A, Cell lysates were fractionated on 6% PAGE (for NMM-II-B assays) or 10% PAGE (for actin assays). Upper panel, Immunoprecipitations (IP) with anti-βactin antibody and Western immunoblots (IB) with the anti-NMMHC-II-B antibody. Middle panel, Western immunoblots with anti-β-actin antibody. Lower panel, Western immunoblots with the anti-NMMHC-II-B antibody. B, Densitometry analysis of the data (means ± SD of three experiments). AU, Arbitrary units. a and b, P < 0.01, compared with C.
Estrogen modulation of NMM-II-B phosphorylation
One of the mechanisms that regulate NMM-II-B function is phosphorylation (15). To understand whether estrogen modulates NMM-II-B phosphorylation status, the intracellular ATP pool of CaSki cells was labeled by incubation of cells with [32P]orthophosphate, and cell lysates were immunoprecipitated with the anti-NMMHC-II-B antibody. Autoradiography revealed that in estrogen-depleted cells, NMM-II-B is phosphorylated at baseline conditions, and treatment with estrogen increased the phosphorylation (Fig. 6A). When expressed relative to cellular actin, treatment with 10 nM 17β-estradiol for 2 d increased NMM-II-B content 4-fold, whereas the density ratio of the phosphorylated NMM-II-B band relative to actin increased 9-fold (Fig. 6B). Cotreatment with DRB attenuated estrogen increase in NMM-II-B phosphorylation by about half, and cotreatment with ICI-182,780 blocked estrogen effect (Fig. 6, A and B).
Fig. 6.
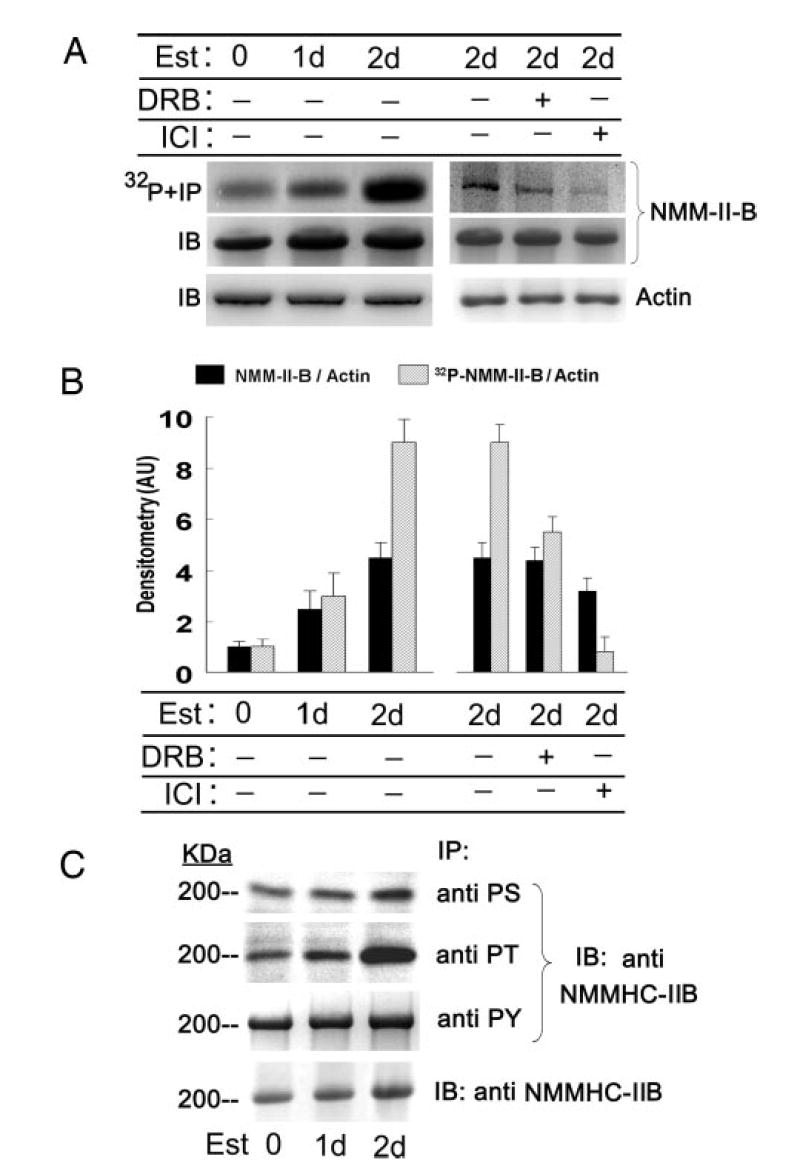
Estrogen (Est) effects on phosphorylation of NMM-II-B. CaSki cells were shifted for 3 d to steroid-free medium and treated with 10 nM 17β-estradiol for the durations indicated in the figure. In some experiments cells were also cotreated with DRB (10 μM, 6 h before assays) or ICI-182,780 (ICI, 10 μM, 1 d before assays). A, upper and middle panels, Cells were labeled with [32P]orthophosphate; lysates were fractionated on 6% PAGE and immunoprecipitated (IP) with the anti-NMMHC-II-B antibody. Lower panel, Lysates were fractionated on 10% PAGE and immunoblotted with anti-β-actin antibody. B, Densitometry of the data in A (means ± SD of three experiments). AU, Arbitrary units. C, Experiments were done as in A, except that cell lysates were immunoprecipitated with antiphosphoserine (PS), antiphosphothreonine (PT), and antiphosphotyrosine antibodies and immunoblotted (IB) with the anti-NMMHC-II-B antibody. Lower panel shows Western immunoblots (IB) of cell lysates with anti-NMM-II-HC-B antibody. The experiment was repeated twice with similar trends.
To determine what residues of the myosin are phosphorylated, cell lysates were immunoprecipitated with antiphospho-antibodies and immunoblotted with the anti-NMMHC-II-B antibody. Treatment with estrogen increased mainly phosphoserine and phosphothreonine immunoreactivities, with lesser effect on the phosphotyrosine immunoreactivity (Fig. 6C).
Estrogen effects on the assembly of NMM-II-B filaments
Optimal binding of NMM-II-B to actin requires NMM-II-B in its homodimeric form (27, 28, 35–37). To understand whether the estrogen effects shown in Figs. 2–6 involve modulation of oligomerization of NMM-II-B filaments, NMM-II-B filaments were extracted from lysates of estrogen-depleted and estrogen-treated cells, and changes in filaments assembly were determined in terms of the critical concentration of NMM-II-B filaments necessary to induce oligomerization in vitro. Figure 7A demonstrates the methodology and end point evaluation, and Figs. 7B, 8, and 9 show the results.
Fig. 7.
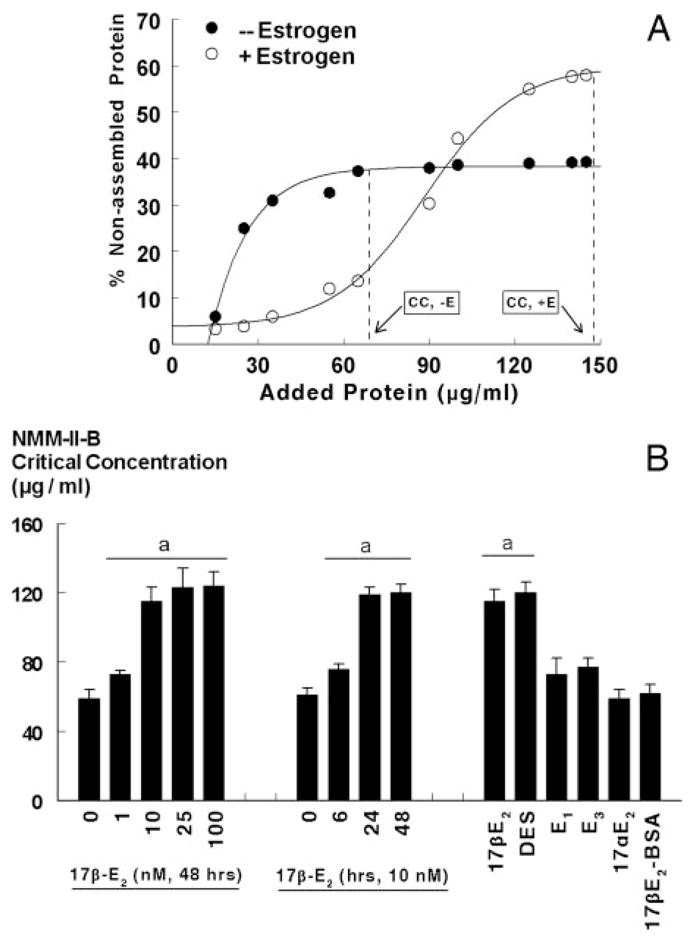
Estrogen modulation of NMM-II-B filaments assembly. A, CaSki cells were shifted for 3 d to steroid-free medium and treated with the vehicle (− Estrogen) or 10 nM 17β-estradiol for 2 d (+ Estrogen). Changes in the assembly (homodimerization) of NMM-II-B filaments were determined in vitro using purified preparations of NMM-II-B filaments in terms of the critical concentration (CC) of NMM-II-B filaments as determined from the x-intersection of the vertical broken lines (initial deflection of the percent nonassembled protein). B, Time effect (left panel), dose effect (middle panel), and specificity (right panel) of estrogen modulation of NMM-II-B filaments assembly. hEVECs were shifted for 3 d to steroid-free medium and treated with the vehicle, 17β-estradiol (17β-E2) for the durations or the indicated doses, or one of the following estrogens (all for 48 h): diethylstilbestrol (DES, 10 nM); estrone (E1, 100 nM); estriol (E3, 100 nM); 17α-estradiol (17αE2, 10 nM); and 17β-estradiol-6-[o-carboxymethyl]oxime-BSA (17β-E2-BSA, 1 μM). Changes in the assembly of NMM-II-B filaments were determined in terms of the NMM-II-B critical concentration. Shown are means ± SD of three to six experiments. Similar effects of 17β-estradiol were also obtained in CaSki cells (not shown). a, P < 0.01–0.04, compared with no treatment with estrogen.
Fig. 8.
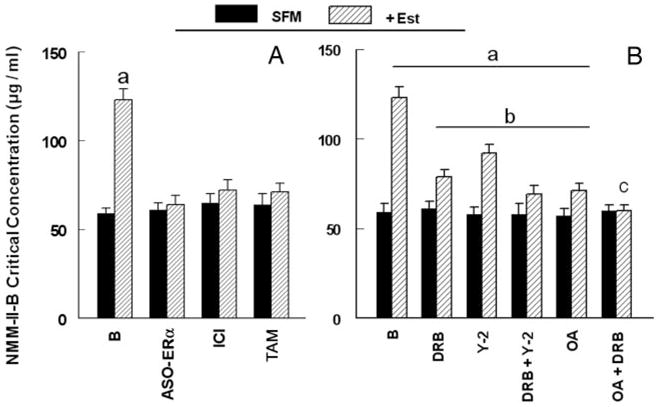
Estrogen modulation of NMM-II-B filaments assembly. A, Involvement of the ERα. hEVECs were shifted for 3 d to steroid-free medium (SFM) and treated with the vehicle or 10 nM 17β-estradiol (Est) for 2 d alone or in combination with one of the following agents/drugs: ASO-ERα (10 μM, 2 d); ICI-182,780 (ICI, 10 μM, 1 d); or tamoxifen (TAM, 10 μM, 1 d). Changes in the assembly of NMM-II-B filaments were determined in terms of the NMM-II-B critical concentration. Shown are means ± SD of three to six experiments. a, P <0.01, compared with B (baseline). B, Effects of DRB, Y-27632, and okadaic acid. hEV-ECs were shifted for 3 d to steroid-free medium (SFM) and treated with the vehicle or 10 nM 17β-estradiol (Est) for 2 d alone or in combination with one of the following agents: DRB (10 μM, 6 h); Y-27632 (Y-2, 5 μM, 6 h); and okadaic acid (OA, 10 μM, 6 h). Shown are means ± SD of three to six experiments. a, P < 0.01, compared with B (baseline) SFM. b, P < 0.01, compared with B (baseline) + Est. c, P < 0.05, compared with OA. Similar results (not shown) were obtained in CaSki cells.
Fig. 9.
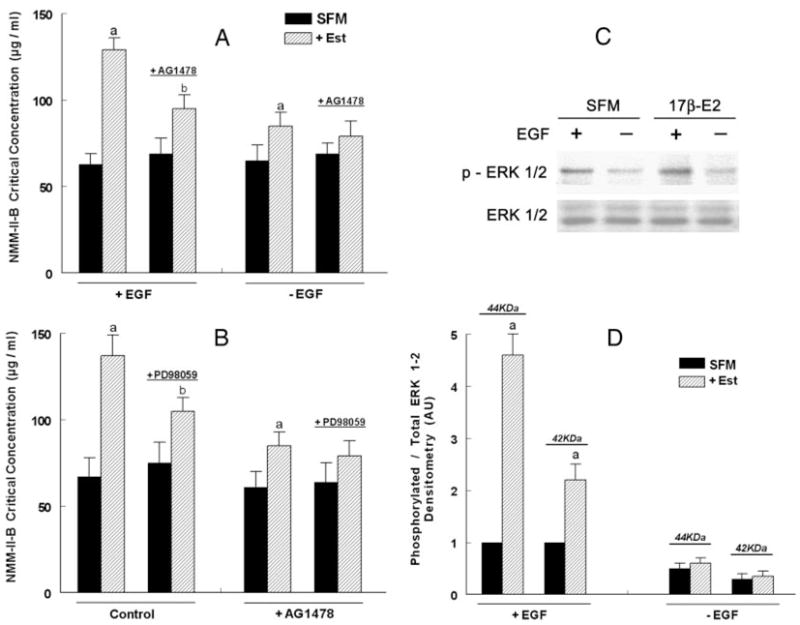
Effects of EGF, AG1478, and PD98059 on estrogen modulation of NMM-II-B filaments assembly. hEVECs were shifted for 3 d to steroid-free medium (SFM) and treated with the vehicle or 10 nM 17β-estradiol (Est) for 2 d alone or in combination with AG1478 (5 μM, 12 h) and/or PD98059 (10 μM, 6 h). A, Twenty-four hours before experiments, some cultures were shifted to medium lacking EGF (−EGF), whereas other were continued to be maintained in 0.2 nM EGF. B, Experiments were done in 0.2 nM EGF. Changes in the assembly of NMM-II-B filaments were determined in terms of the NMM-II-B critical concentration. Shown are means ± SD of three to six experiments. a, P < 0.01, compared with SFM; b, P < 0.01–0.05, compared with SFM and cells not treated with AG1478. Similar results (not shown) were obtained in CaSki cells. C, Effects of estrogen and EGF on ERK1/2 phosphorylation. hEVECs were shifted for 3 d to steroid-free medium (SFM) and treated with 10 nM 17β-estradiol (17β-E2) for 2 d. One day before experiments, some cultures were shifted to medium lacking EGF, whereas other were continued to be maintained in 0.2 nM EGF. The experiment was repeated twice with similar trends. D, Densitometry analysis of the data [density of the phosphorylated ERK1/2 (p-ERK1/2) relative to total ERK1/2 for the 44- and 42-kDa isoforms, means ± SD of three experiments]. AU, Arbitrary units. Ratio densitometry of the bands in SFM and +EGF conditions were defined as 1 U. a, P < 0.01, compared with C.
Sensitivity and specificity of estrogen effect
The critical concentration of filaments necessary for assembly was higher in estrogen-treated than estrogen-depleted cells (Fig. 7). The effects of 17β-estradiol were dose and time related (Fig. 7), with similar kinetics to the effects of estrogen on cellular content of the NMM-II-B (Fig. 3). Treatment with 10 nM diethylstilbestrol mimicked estrogen modulation of NMM-II-B assembly; in contrast, treatments with 17α-estradiol, the weak estrogenic ligands estrone and estriol, or the nonpermeable estrogen 17β-E2-BSA had no significant effects (Fig. 7B).
Involvement of ERα
Treatments with the ASO for the ERα or with ICI-182,780 and tamoxifen had no significant effect on NMM-II-B critical concentration (Fig. 8A). On the other hand, cotreatments with the ASO-ERα, ICI-182,780, or tamoxifen blocked 17β-estradiol increase in NMM-II-B critical concentration (Fig. 8A). Treatments with the ASO for the ERβ and GPR30 as well as with progesterone (1 μM, 2 d) alone had no effects on NMM-II-B critical concentration, and cotreatments of these agents/drugs with 17β-estradiol did not significantly affect estrogen action (not shown). Collectively, these data suggest the involvement of ERα as mediator of estrogen modulator of NMM-II-B filaments.
Involvement of CK2, ROCK, and myosin heavy-chain phosphatase
Treatments with DRB, Y-27632, and okadaic acid, alone or in combination, had no significant effect on NMM-II-B critical concentration (Fig. 8B). Cotreatments with DRB or Y-27632 attenuated estrogen increase in NMM-II-B critical concentration (Fig. 8B). Combined treatments of DRB plus Y-27632 attenuated estrogen modulation increase in NMM-II-B critical concentration more than each drug alone but failed to block estrogen effect (Fig. 8B). Combined treatment of DRB plus okadaic acid blocked estrogen effect (Fig. 8B). Collectively, these data suggest the involvement of CK2, ROCK, and myosin heavy-chain phosphatase in estrogen modulation of NMM-II-B filamentation.
Involvement of the EGFR
To determine whether the EGFR mechanism plays a role in estrogen modulation of NMM-II-B filamentation, assembly assays were done on NMM-II-B filaments obtained from cells grown in the presence or absence of EGF. In estrogen-depleted cells, removal of EGF from the culture medium had no substantial effect on the critical concentration of NMM-II-B. In contrast, in estrogen-treated cells lack of EGF attenuated the estrogen increase in the critical concentration of NMM-II-B (Fig. 9A).
To better understand the potential role of the EGFR mechanism, the effects of the EGFR inhibitor AG1478 on NMM-II-B filamentation were studied. Treatment of estrogen-depleted cells with the EGFR inhibitor AG1478 had no effect on the critical concentration of NMM-II-B, regardless of whether cells were grown in the presence or absence of EGF. However, cotreatment of estrogen-treated cells grown in the presence of EGF with AG1478 inhibited estrogen increase in the NMM-II-B critical concentration to levels observed in cells grown in the absence of EGF (Fig. 9A). These data suggest that the EGFR mechanism is involved in mediating estrogen modulation of NMM-II-B filamentation.
Involvement of the ERK/MAPK cascade
To determine whether the EGFR mechanism plays a role in estrogen modulation of NMM-II-B filamentation, the effects of the ERK/MAPK inhibitor PD98059 were studied. In estrogen-depleted cells, PD98059 had no substantial effect on the critical concentration of NMM-II-B. In contrast, in estrogen-treated cells, PD98059 attenuated the estrogen increase in the critical concentration of NMM-II-B (Fig. 9B). In estrogen-treated cells, combined treatments of PD98059 plus AG1478 had no additional effect to that of PD98059 (Fig. 9B). These data suggest that the ERK/MAPK cascade is involved in mediating estrogen modulation of NMM-II-B filamentation. Lack of additive inhibition also suggests that the EGFR and ERK/MAPK cascade may operate via a common signaling pathway.
Lack of effect of H-89 and LY294002
Treatment of estrogen-depleted cells grown in the presence of EGF with H-89, inhibitor of protein kinase A, or with LY294002, inhibitor of phosphatidylinositol 3-kinase-Akt, produced no significant changes in NMM-II-B critical concentration (not shown). Similarly, in estrogen-treated cells grown in the presence of EGF cotreatment with H-89 or with LY294002 had no significant effect on the estrogen increase in NMM-II-B critical concentration (not shown).
Estrogen activation of ERK1/2
The results in Fig. 9,A and B, suggested the involvement of EGFR and the ERK/MAPK cascade in estrogen modulation of NMM-II-B filamentation. Previous studies in cervical cells investigated the interaction between estrogen and EGFR signaling (e.g. Ref. 38). However, less was known in cervical cells about the interaction between estrogen and the ERK/MAPK signaling. To better understand this interaction, we studied estrogen activation of ERK in hEVECs, in terms of ERK1/2 phosphorylation. Fig. 9, C and D, shows that in the absence of EGF, the steady-state level of ERK1/2 phosphorylation was low, and estrogen did not exert a significant effect. In estrogen-depleted cells incubated in the presence of 0.2 nM EGF, ERK1/2 phosphorylation was substantial, and cotreatment with 10 nM 17β-estradiol increased it 2.5-fold (p-ERK1/2 42 kDa) or 4.5-fold (p-ERK1/2 44 kDa) without affecting significantly ERK expression (Fig. 9C). DRB, Y-27632, or okadaic acid did not produce any significant additional effect on ERK1/2 phosphorylation to that of estrogen (not shown). These results suggest that in human vaginal-cervical cells, estrogen up-regulates ERK1/2 phosphorylation.
Estrogen activation of CK2
One of the conclusions from the data in Fig. 8 is the possible involvement of CK2 as terminal mediator of estrogen effects on NMM-II-B. This novel hypothesis was tested more directly by determinations of the effects estrogen treatment on CK2 expression and activity. Neither estrogen depletion nor treatment with 17β-estradiol had any significant effect on cellular CK2 mRNA or protein expression (not shown). In contrast, estrogen status markedly affected CK2 activity. In lysates of hEVECs obtained from estrogen-treated cells, CK2 activity was 3-fold greater than in lysates obtained from estrogen-depleted cells (Fig. 10). CK2 activity in lysates of cells treated with ICI-182,780, DRB, Y-27632, or okadaic acid was similar to that in control cells. CK2 activities in lysates of cells cotreated with estrogen plus Y-27632 or estrogen plus okadaic acid were similar to that determined in lysates of cells treated with estrogen only, suggesting that treatments with Y-27632 or okadaic acid do not modulate estrogen increase in CK2 activity. In contrast, CK2 activities in lysates of cells cotreated with estrogen plus ICI-182,780 or estrogen plus DRB were smaller than that determined in lysates of cells treated with estrogen only, in the range observed in lysates obtained from estrogen-depleted cells, suggesting that treatments in vivo with ICI-182,780 and DRB blocked CK2 activity.
Fig. 10.
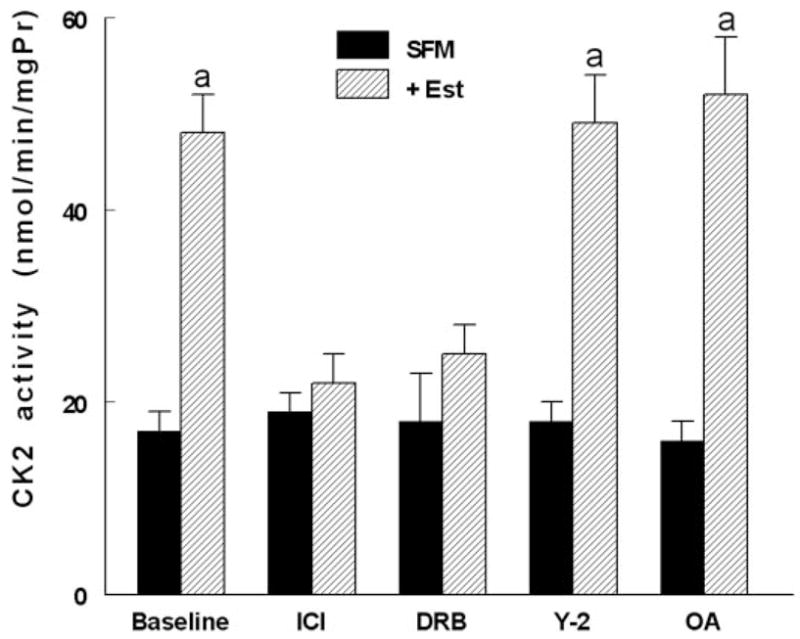
Estrogen modulation of CK2 activity. hEVECs were shifted for 3 d to steroid-free medium (SFM) and treated with the vehicle or 10 nM 17β-estradiol (Est) for 2 d, alone or in combination with one of the indicated drugs: ICI-182,780 (ICI, 10 μM, 1 d); DRB (10 μM, 6 h); Y-27632 (Y-2, 5 μM, 6 h); and okadaic acid (OA, 10 μM, 6 h). CK2 activity was determined in cells lysates in terms of the alkaline-phosphatase-sensitive accumulation of [γ-32P]RRREEETEEE (expressed as nanomoles per minute per milligram protein [mgPr]). A, P < 0.01, compared with SFM and baseline.
Discussion
The main objective of the study was to understand how estrogen modulates the cytoskeletal actomyosin frame in epithelial cells. We discovered that treatments with physiological concentrations of 17β-estradiol decreased the rigidity of the cytoskeleton; the effect involved increased phosphorylation of NMM-II-B filaments on serine and threonine residues and decreased NMM-II-B filamentation, abrogation of NMM-II-B-actin interaction, and redistribution of the myosin from submembranous regions into cytoplasmic and perinuclear domains. Estrogen also increased NMM-II-B cell content in a time- and dose-related manner. The data suggest that the effect of estrogen is mediated via the ERα and involves the EGFR and ERK/MAPK cascades as proximal mediators, whereas CK2- and ROCK-regulated myosin heavy-chain phosphatase are involved as distal mediators. Although the possibility that some of these effects are unrelated epiphenomena has not been ruled out, the present results support the hypothesis that the effects are related and represent a novel cascade of estrogen action. Accordingly, enhanced phosphorylation inhibits oligomerization of the myosin filaments, leading to decreased interaction of the NMM-II-B filaments with cortical actin. This novel hypothesis is depicted schematically in Fig. 11.
Fig. 11.
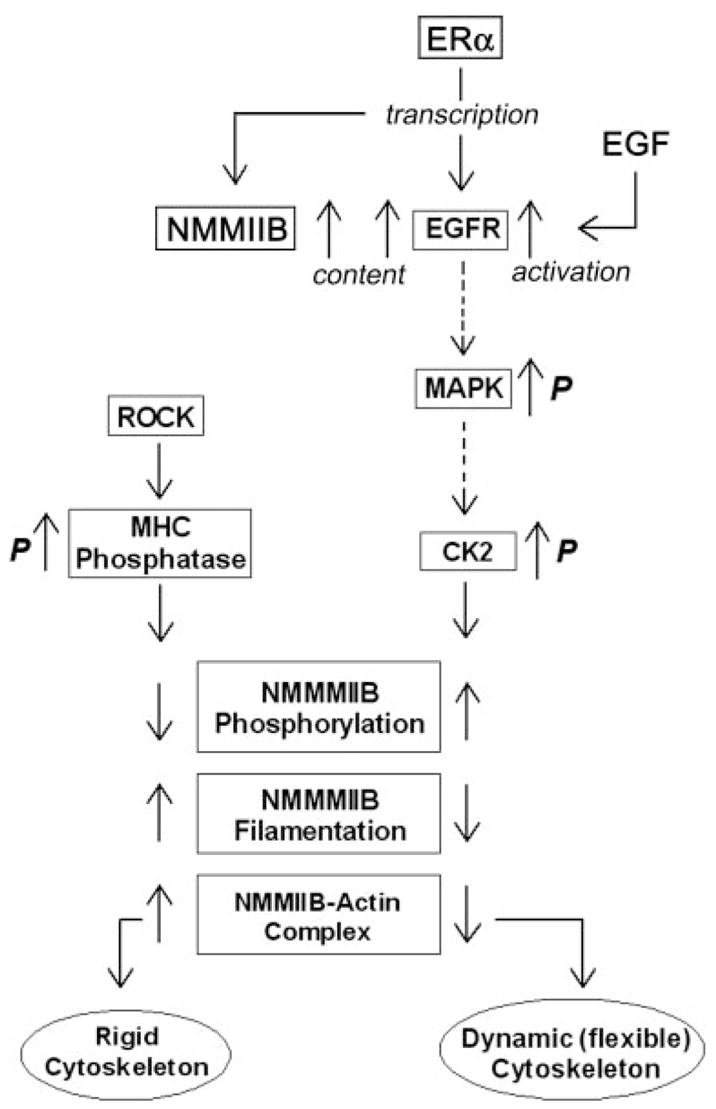
Model of estrogen regulation of NMM-II-B of the cortical actomyosin in human vaginal-cervical epithelial cells. Estrogen induces phosphorylation (p) of NMMHC-II-B filaments, abrogation of NMM-II-B filamentation, and disassociation of the myosin from the cortical actin, which lead to formation of a dynamic (flexible) cytoskeleton. The proximal cascade of estrogen effect involves ERα up-regulation of NMM-II-B and EGFR genes transcription. Midsteps involve the EGF-EGFR and ERK-MAPK signaling cascades, and the terminal effectors are the CK2 and ROCK-regulated myosin heavy-chain (MHC) phosphatase. Whether the EGFR/MAPK cascades modulate CK2 phosphorylation status or whether the effect involves other signaling networks that operate in parallel remains to be determined (broken lines).
One of our specific objectives was to understand the mechanism and signaling of estrogen effects. The data suggest that estrogen increase in NMM-II-B cell content could be explained by the involvement of the classical genotropic action mediated by the ERα/coactivator complex. In contrast, estrogen effects on NMM-II-B phosphorylation, NMM-II-B-actin interaction, and NMM-II-B filamentation cannot be explained by transcription regulation alone. The results showed that NMM-II-B filaments are constitutively phosphorylated, possibly through CK2 and ROCK regulation of myosin heavy-chain phosphatase. Accordingly, myosin heavy-chain phosphatase dephosphorylates NMMHC-II-B, thereby enhancing filamentation and increased myosin-actin interaction, and phosphorylation of the myosin heavy-chain phosphatase by ROCK blocks myosin heavy-chain phosphatase activity and decreases filamentation (Fig. 11). CK2, on the other hand, induced phosphorylation of the NMMHC-II-B, thereby inhibiting filamentation and myosin-actin interaction. One possibility is that CK2 and ROCK phosphorylate the myosin heavy-chain phosphatase in concert (28, 39). Alternatively, CK2 phosphorylates NMMHC-II-B directly because NMMHC-II-B contains phosphorylation sites for CK2 (37, 40). Estrogen up-regulated CK2 activity and treatment with the CK2 inhibitor DRB inhibited estrogen modulation of NMM-II-B filamentation. Treatment with the ROCK inhibitor Y-27632 or the nonspecific phosphatase inhibitor okadaic acid also attenuated estrogen effect, but only combined treatments of DRB plus Y-27632 blocked estrogen effect. Thus, the combined actions of CK2 and myosin heavy-chain phosphatase can account for estrogen effect (Fig. 11).
Our data support the hypothesis that CK2 mediates in part estrogen-phosphorylation of NMMHC-II-B. CK2 is a ubiquitous, highly pleiotropic and constitutively active serine/threonine protein kinase (41). Although there are more than 300 documented substrates for CK2, most are involved in gene expression, protein synthesis, and signaling but only a few in the regulation of the cytoskeleton (41), and only two reports documented the involvement of myosin II in smooth muscle cells (40, 42). The present data are the first to suggest that NMMHC-II-B of epithelial cells is also a CK2 substrate. This conclusion is of interest, given the known role of CK2 in cell survival, cell growth and proliferation (43), and the known mitogenic properties of estrogen.
The present data and our previous studies in the field also suggest involvement of ERα and the EGFR/ERK-MAPK cascades in the proximal arm of estrogen signaling. Human vaginal and cervical epithelial cells express ERα and ERβ mRNA and protein (44) and mRNA for the GPR30 (not shown). However, estrogen modulation of NMM-II-B filamentation could be blocked only with ASOs for the ERα. In addition, the ERα-specific inhibitor ICI-182,780 inhibited estrogen activation of CK2, estrogen modulation of NMM-II-B-actin coimmunoprecipitation, and estrogen modulation of NMM-II-B phosphorylation. Moreover, the pharmacology of estrogen action, i.e. the time and dose effect and the specificity pattern, supports involvement of the ERα in estrogen modulation of NMM-II-B phosphorylation, filamentation, and interaction with actin.
Treatment with estrogen had no significant effect on cellular CK2 levels, and estrogen regulation of CK2-dependent NMM-II-B activity therefore cannot be explained by the classical direct ER/coactivator gene transcription regulation of CK2. An alternative explanation is that it involves ERα transcription up-regulation of intermediary steps (9, 45) or ERα-mediated activation of kinase-initiated signaling cascades that participate in the signal cascade (46–49). Estrogen modulation of NMM-II-B filamentation depended on the presence of EGF in the culture medium, and it could be attenuated by blocking the EGFR. Because the EGFR is transcriptionally up-regulated by estrogen (50), including in human cervical cells (38), it is possible that the EGFR is mediator of estrogen action. Accordingly, the EGFR is a rate-limiting mediator involved in activation of the CK2, and ERα-up-regulation of cellular EGFR increases activity of this kinase (Fig. 11). This hypothesis is supported by findings that CK2 can be a substrate for the EGF/EGFR kinase mechanism (34). The hypothesis may be physiologically relevant because EGF is constitutively secreted by stromal cervical and vaginal cells (51); the EGFR can be activated by changes in cell size (52); and the rate-limiting effect for EGF actions in many tissues is cellular levels of the EGFR (53).
The present data also support involvement of the ERK/MAPK cascade in estrogen action. Treatment with the ERK/MAPK inhibitor PD98059 attenuated estrogen modulation of cytoskeletal deformability, NMM-II-B filamentation, and increased ERK activation. The NMM-II-B filamentation effect of estrogen was dependent on EGF but was not affected by treatments with DRB, Y-27632, or okadaic acid. A possible explanation is of a hierarchical effect, namely that the EGFR/MAPK cascades modulate CK2 phosphorylation status. Another possible explanation is the involvement of other signaling networks that operate in parallel (Fig. 11). At present relatively little is known which other signaling networks are involved, although lack of effects of H-89 and LY294002 effectively ruled out the involvement of protein kinase A and the phosphatidylinositol 3-kinase-Akt networks. A third possibility is feedback signaling regulation, namely potentiation of estrogen effect through CK2 phosphorylation of the ERα (54, 55).
The present data may be physiologically relevant for our understanding of estrogen modulation of epithelial permeability because the rigidity of the cytoskeleton determines the RLIS and thereby the permeability characteristics of the epithelium (3). Previous studies in human cervical-vaginal epithelia showed that estrogen can regulate the rigidity of the cortical actomyosin through modulation of actin polymerization (6, 9, 21). The present data suggest an additional novel mechanism, by modulation of the cortical myosin. The two mechanisms have different time courses: modulation of actin polymerization requires 1–6 h of treatment with estrogen (6–9) and modulation of NMM-II-B 6–24 h (present data). The two mechanisms are also hierarchically different because actin polymerization induces cytoskeletal changes that override NMM-II-B-related modulation of the cytoskeleton. Thus, it is possible that short-term effects of estrogen, e.g. the preovulatory increase in epithelial permeability, are mediated by actin depolymerization, whereas the long-term effects of estrogen require modulation of the myosin.
In summary, the present results suggest a novel mechanism of estrogen regulation of the cytoskeletal actomyosin frame in epithelial cells. Estrogen induces phosphorylation of NMMHC-II-B filaments and abrogation of NMM-II-B filamentation that could lead to disassociation of the myosin from the cortical actin. Disruption of the actomyosin ring increases cell deformability and may lead to decreased RLIS. The effect of estrogen is mediated by the ERα and may involve the EGF-EGFR and ERK-MAPK as proximal signaling cascades. Terminal effectors are possibly the CK2- and ROCK-regulated myosin heavy-chain phosphatase. This novel cellular mechanism can explain, in part, the estrogen increase in epithelial permeability in vivo.
Supplementary Material
Acknowledgments
The technical support of Kimberley Frieden, Brian De-Santis, and Dipika Pal is acknowledged.
This work was supported by National Institutes of Health Grants HD29924 and AG15955 (to G.I.G.).
Abbreviations
- ASO
Antisense oligonucleotide
- CLO
control oligonucleotide
- CK2
casein kinase-II
- DRB
5,6-dichloro-1-β-(D)-ribofuranosylbenzimidazole
- EGF
epithelial growth factor
- EGFR
EGF-receptor
- ER
estrogen receptor
- GAPDH
antiglyceraldehyde-3-phosphate dehydrogenase
- hEVEC
human epithelial vaginal-ectocervical cell
- NMMHC
nonmuscle myosin-II heavy chain
- NMM-II-A
nonmuscle myosin-II-A
- NMM-II-B
nonmuscle myosin-II-B
- RLIS
resistance of the lateral intercellular space
- ROCK
Rho-associated kinase
- RTJ
resistance of the tight junctions
Footnotes
Disclosure statement: the authors have nothing to disclose.
References
- 1.Reuss L. Tight junction permeability to ions and water. In: Cereijido M, editor. Tight-junctions. Boca Raton, FL: CRC Press; 1991. pp. 49–66. [Google Scholar]
- 2.Ussing HH, Zerahn K. Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Acta Physiol Scand. 1951;23:110–127. doi: 10.1111/j.1748-1716.1951.tb00800.x. [DOI] [PubMed] [Google Scholar]
- 3.Gorodeski GI. The cultured human cervical epithelium: a new model for studying transepithelial paracellular transport. J Soc Gynecol Investig. 1996;3:267–280. doi: 10.1016/s1071-5576(96)00028-7. [DOI] [PubMed] [Google Scholar]
- 4.Zeng R, Li X, Gorodeski GI. Estrogen abrogates transcervical tight junctional resistance by acceleration of occludin modulation. J Clin Endocrinol Metab. 2004;89:5145–5155. doi: 10.1210/jc.2004-0823. [DOI] [PubMed] [Google Scholar]
- 5.Zhu L, Li X, Zeng R, Gorodeski GI. Changes in tight junctional resistance of the cervical epithelium are associated with modulation of content and phosphorylation of occludin 65-kilodalton and 50-kilodalton forms. Endocrinology. 2006;147:977–989. doi: 10.1210/en.2005-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorodeski GI. Estrogen increases the permeability of the cultured human cervical epithelium by modulating cell deformability. Am J Physiol. 1998;275:C888–C899. doi: 10.1152/ajpcell.1998.275.3.C888. [DOI] [PubMed] [Google Scholar]
- 7.Gorodeski GI. Role of nitric oxide and cGMP in the estrogen regulation of cervical epithelial permeability. Endocrinology. 2000;141:1658–1666. doi: 10.1210/endo.141.5.7473. [DOI] [PubMed] [Google Scholar]
- 8.Gorodeski GI. NO increases permeability of cultured human cervical epithelia by cGMP-mediated increase in G-actin. Am J Physiol. 2000;278:C942–C952. doi: 10.1152/ajpcell.2000.278.5.C942. [DOI] [PubMed] [Google Scholar]
- 9.Gorodeski GI. cGMP-dependent ADP-depolymerization of actin mediates estrogen increase in human cervical epithelia permeability. Am J Physiol. 2000;279:C2028–C2036. doi: 10.1152/ajpcell.2000.279.6.C2028. [DOI] [PubMed] [Google Scholar]
- 10.Gorodeski GI. Effects of menopause and estrogen on cervical epithelial permeability. J Clin Endocrinol Metab. 2000;85:2584–2595. doi: 10.1210/jcem.85.7.6671. [DOI] [PubMed] [Google Scholar]
- 11.Gorodeski GI. Vaginal-cervical epithelial permeability decreases after menopause. Fertil Steril. 2001;76:753–761. doi: 10.1016/s0015-0282(01)02377-9. [DOI] [PubMed] [Google Scholar]
- 12.Gorodeski GI. Estrogen biphasic regulation of paracellular permeability of cultured human vaginal-cervical epithelia. J Clin Endocrinol Metab. 2001;86:4233–4243. doi: 10.1210/jcem.86.9.7817. [DOI] [PubMed] [Google Scholar]
- 13.Spring KR, Hope A. Size and shape of the lateral intercellular spaces in a living epithelium. Science. 1978;200:54–58. doi: 10.1126/science.635571. [DOI] [PubMed] [Google Scholar]
- 14.Sutherland JD, Witke W. Molecular genetic approaches to understanding the actin cytoskeleton. Curr Opin Cell Biol. 1999;11:142–151. doi: 10.1016/s0955-0674(99)80018-0. [DOI] [PubMed] [Google Scholar]
- 15.Sellers JR. Myosins: a diverse superfamily. Biochim Biophys Acta. 2000;1496:3–22. doi: 10.1016/s0167-4889(00)00005-7. [DOI] [PubMed] [Google Scholar]
- 16.Bresnick AR. Molecular mechanisms of nonmuscle myosin-II regulation. Curr Opin Cell Biol. 1999;11:26–33. doi: 10.1016/s0955-0674(99)80004-0. [DOI] [PubMed] [Google Scholar]
- 17.Sutton TA, Mang HE, Atkinson SJ. Rho-kinase regulates myosin II activation in MDCK cells during recovery after ATP depletion. Am J Physiol. 2001;281:F810–F818. doi: 10.1152/ajprenal.2001.281.5.F810. [DOI] [PubMed] [Google Scholar]
- 18.Hecht G, Pestic L, Nikcevic G, Koutsouris A, Tripuraneni J, Lorimer DD, Nowak G, Guerriero V, Jr, Elson EL, Lanerolle PD. Expression of the catalytic domain of myosin light chain kinase increases paracellular permeability. Am J Physiol. 1996;271:C1678–C1684. doi: 10.1152/ajpcell.1996.271.5.C1678. [DOI] [PubMed] [Google Scholar]
- 19.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 20.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 21.Gorodeski GI. Aging and estrogen effects on transcervical-transvaginal epithelial permeability. J Clin Endocrinol Metab. 2005;90:345–351. doi: 10.1210/jc.2004-1223. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Feng YH, Gorodeski GI. EGF facilitates epinephrine inhibition of P2X7-receptor mediated pore formation and apoptosis: a novel signaling network. Endocrinology. 2005;146:164–174. doi: 10.1210/en.2004-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorodeski GI, Whittembury J. A novel fluorescence chamber for the determination of volume changes in human CaSki cell cultures attached on filters. Cell Biochem Biophys. 1998;29:307–332. doi: 10.1007/BF02737900. [DOI] [PubMed] [Google Scholar]
- 24.Lentner C. In: Heart and circulation Geigy scientific tables. Lentner C, editor. Vol. 5. 1990. pp. 220–229. [Google Scholar]
- 25.Gorodeski GI, De Santis BJ, Goldfarb J, Utian WH, Hopfer H. Osmolar changes regulate the paracellular permeability of cultured human cervical epithelium. Am J Physiol. 1995;269:C870–C877. doi: 10.1152/ajpcell.1995.269.4.C870. [DOI] [PubMed] [Google Scholar]
- 26.Feng YH, Wang L, Wang Q, Li X, Zeng R, Gorodeski GI. ATP ligation stimulates GRK-3-mediated phosphorylation and β-arrestin-2-and dynamin-dependent internalization of the P2X7-receptor. Am J Physiol. 2005;288:C1342–C1356. doi: 10.1152/ajpcell.00315.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murakami N, Elzinga M, Singh SS, Chauhan VP. Direct binding of myosin II to phospholipid vesicles via tail regions and phosphorylation of the heavy chains by protein kinase C. J Biol Chem. 1994;269:16082–16090. [PubMed] [Google Scholar]
- 28.Murakami N, Singh SS, Chauhan VP, Elzinga M. Phospholipid binding, phosphorylation by protein kinase C, and filament assembly of the COOH terminal heavy chain fragments of nonmuscle myosin II isoforms MIIA and MIIB. Biochemistry. 1995;34:16046–16055. doi: 10.1021/bi00049a019. [DOI] [PubMed] [Google Scholar]
- 29.Gorodeski GI, Eckert RL, Utian WH, Rorke EA. Maintenance of in vivo-like keratin expression, sex steroid responsiveness and estrogen receptor expression in cultured human ectocervical epithelial cells. Endocrinology. 1990;126:399–406. doi: 10.1210/endo-126-1-399. [DOI] [PubMed] [Google Scholar]
- 30.Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G, Waterfield M, Chambon P. Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci USA. 1985;82:7889–7893. doi: 10.1073/pnas.82.23.7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. The complete primary structure of human estrogen receptor β (hER β) and its heterodimerization with ER α in vivo and in vitro. Biochem Biophys Res Commun. 1998;243:122–126. doi: 10.1006/bbrc.1997.7893. [DOI] [PubMed] [Google Scholar]
- 32.Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- 33.Sommercorn J, Mulligan JA, Lozeman FJ, Krebs EG. Activation of casein kinase II in response to insulin and to epidermal growth factor. Proc Natl Acad Sci USA. 1987;84:8834–8838. doi: 10.1073/pnas.84.24.8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ackerman P, Glover CV, Osheroff N. Stimulation of casein kinase II by epidermal growth factor: relationship between the physiological activity of the kinase and the phosphorylation state of its beta subunit. Proc Natl Acad Sci USA. 1990;87:821–825. doi: 10.1073/pnas.87.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinard JH, Pollard TD. The effect of heavy chain phosphorylation and solution conditions on the assembly of Acanthamoeba myosin-II. J Cell Biol. 1989;109:1529–1535. doi: 10.1083/jcb.109.4.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murakami N, Chauhan VP, Elzinga M. Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry. 1998;37:1989–2003. doi: 10.1021/bi971959a. [DOI] [PubMed] [Google Scholar]
- 37.Murakami N, Kotula L, Hwang YW. Two distinct mechanisms for regulation of nonmuscle myosin assembly via the heavy chain: phosphorylation for MIIB and mts 1 binding for MIIA. Biochemistry. 2000;39:11441–11451. doi: 10.1021/bi000347e. [DOI] [PubMed] [Google Scholar]
- 38.Jacobberger JW, Sizemore N, Gorodeski GI, Rorke EA. Transforming growth factor β regulation of epidermal growth factor receptor in ectocervical epithelial cells. Exp Cell Res. 1995;220:390–396. doi: 10.1006/excr.1995.1330. [DOI] [PubMed] [Google Scholar]
- 39.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 40.Kelley CA, Adelstein RS. The 204-kDa smooth muscle myosin heavy chain is phosphorylated in intact cells by casein kinase II on a serine near the carboxyl terminus. J Biol Chem. 1990;265:17876–17882. [PubMed] [Google Scholar]
- 41.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 42.Pinna LA, Meggio F, Sarno S. Casein kinase-2 and cell signaling. In: Papa S, Tager JM, editors. Biochemistry of cell membranes. Basel: Birkhauser Verlag; 1995. pp. 15–27. [Google Scholar]
- 43.Unger GM, Davis AT, Slaton JW, Ahmed K. Protein kinase CK2 as regulator of cell survival: implications for cancer therapy. Curr Cancer Drug Targets. 2004;4:77–84. doi: 10.2174/1568009043481687. [DOI] [PubMed] [Google Scholar]
- 44.Gorodeski GI, Pal D. Involvement of estrogen receptors α and β in the regulation of cervical permeability. Am J Physiol. 2000;278:C689–C696. doi: 10.1152/ajpcell.2000.278.4.C689. [DOI] [PubMed] [Google Scholar]
- 45.Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev. 2002;23:665–686. doi: 10.1210/er.2001-0045. [DOI] [PubMed] [Google Scholar]
- 46.Manolagas SC, Kousteni S. Perspective: nonreproductive sites of action of reproductive hormones. Endocrinology. 2001;142:2200–2204. doi: 10.1210/endo.142.6.8221. [DOI] [PubMed] [Google Scholar]
- 47.Kousteni S, Han L, Chen J-R, Almeida M, Plotkin LI, Bellido T, Manolagas SC. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. 2003;111:1651–1664. doi: 10.1172/JCI17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Losel R, Wehling M. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol. 2003;4:46–56. doi: 10.1038/nrm1009. [DOI] [PubMed] [Google Scholar]
- 49.Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–376. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- 50.Levin ER. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol. 2003;17:309–317. doi: 10.1210/me.2002-0368. [DOI] [PubMed] [Google Scholar]
- 51.Hom YK, Young P, Wiesen JF, Miettinen PJ, Derynck R, Werb Z, Cunha GR. Uterine and vaginal organ growth requires epidermal growth factor receptor signaling from stroma. Endocrinology. 1998;139:913–921. doi: 10.1210/endo.139.3.5817. [DOI] [PubMed] [Google Scholar]
- 52.Lezama R, Diaz-Tellez A, Ramos-Mandujano G, Oropeza L, Pasantes-Morales H. Epidermal growth factor receptor is a common element in the signaling pathways activated by cell volume changes in isosmotic, hyposmotic or hyperosmotic conditions. Neurochem Res. 2005;30:1589–1597. doi: 10.1007/s11064-005-8837-5. [DOI] [PubMed] [Google Scholar]
- 53.Bazley LA, Gullick WJ. The epidermal growth factor receptor family. Endocr Relat Cancer. 2005;1:S17–S27. doi: 10.1677/erc.1.01032. [DOI] [PubMed] [Google Scholar]
- 54.Kuiper GG, Brinkmann AO. Steroid hormone receptor phosphorylation: is there a physiological role? Mol Cell Endocrinol. 1994;100:103–107. doi: 10.1016/0303-7207(94)90287-9. [DOI] [PubMed] [Google Scholar]
- 55.Tzeng DZ, Klinge CM. Phosphorylation of purified estradiol-liganded estrogen receptor by casein kinase II increases estrogen response element binding but does not alter ligand stability. Biochem Biophys Res Commun. 1996;223:554–560. doi: 10.1006/bbrc.1996.0933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.