

Abstract
The complement system is an important component of the innate immune response to virus infection. The role of human complement pathways in the in vitro neutralization of three closely related paramyxoviruses, Simian Virus 5 (SV5), Mumps virus (MuV) and Human Parainfluenza virus type 2 (HPIV2) was investigated. Sera from ten donors showed high levels of neutralization against HPIV2 that was largely complement-independent, whereas nine of ten donor sera were found to neutralize SV5 and MuV only in the presence of active complement pathways. SV5 and MuV neutralization proceeded through the alternative pathway of the complement cascade. Electron microscopy studies and biochemical analyses showed that treatment of purified SV5 with human serum resulted in C3 deposition on virions and the formation of massive aggregates, but there was relatively little evidence of virion lysis. Treatment of MuV with human serum also resulted in C3 deposition on virions, however in contrast to SV5, MuV particles were lysed by serum complement and there was relatively little aggregation. Assays using serum depleted of complement factors showed that SV5 and MuV neutralization in vitro was absolutely dependent on complement factor C3, but was not dependent on downstream complement factors C5 or C8. Our results indicate that even though antibodies exist that recognize both SV5 and MuV, they are mostly non-neutralizing and viral inactivation in vitro occurs through the alternative pathway of complement. The implications of our work for development of paramyxovirus vectors and vaccines are discussed.
Keywords: Paramyxovirus
Introduction
The complement system is an important component of the innate immune response to virus infection (Biron and Sen, 2007; Blue et al., 2004). Complement serves to link innate and adaptive immunity through a large number of activities, including recognition of viruses, direct neutralization of infectivity, recruitment and stimulation of leukocytes at sites of infection, opsonization by immune cells, and activation of T and B cells in adaptive immune responses (Kemper and Atkinson, 2007; Carroll, 2004; Blue et al., 2004; Gasque, 2004). These activities of the complement cascade can have important implications for pathogenesis and viral dissemination, as well as the design of more effective vaccine vectors (Bergmann-Leitner et al., 2006; Blue et al., 2004; Delgado and Polack, 2004; Morrison et al., 2007; Reis et al., 2006). The overall goal of the work described here was to determine the mechanisms and contributions of complement in the in vitro neutralization of three closely related paramyxoviruses, simian virus 5 (SV5), human parainfluenza virus type 2 (HPIV2) and mumps virus (MuV).
The complement proteolytic cascade can be initiated through three main pathways: the classical pathway, lectin pathway and alternative pathway (Carroll, 2004; Gasque, 2004; Roozendaal and Carroll, 2006). Classical pathway activation involves either binding of the C1q component to virus-antibody complexes or association of C1q by itself to virus particles. Examples of viruses that activate the classical pathway include human T cell lymphotropic virus (HTLV; Ikeda et al., 1998), Human Immunodeficiency Virus (HIV; Ebenbichler et al., 1991) and vesicular stomatitis virus (VSV; Beebe and Cooper, 1981). The lectin pathway is activated through recognition of carbohydrate signatures on viral glycoproteins by the cellular mannan-binding lectin (MBL), and this is an important pathway in neutralization of hepatitis C virus (Ishii et al., 2001), Herpes Simplex virus 2 (Gadjeva et al., 2004) and influenza virus (Hartshorn et al., 1993). Compared to the classical and lectin pathways, the signals that activate the alternative pathway are less well understood, but they are thought to involve recognition of foreign surfaces by an antibody-independent mechanism (Pangburn et al., 1981; Gasque, 2004). The extent of sialic acid modification on microbial surfaces may contribute to induction of the alternative pathway (e.g., Madico et al., 2007; McSharry et al., 1981; Hirsch et al., 1986). Examples of viruses that activate the alternative pathway include Epstein–Barr virus (Mold et al., 1988) and Sindbis virus (Hirsch et al.,1980). Finally, West Nile virus is an example of a virus that activates all three pathways, with each pathway making a contribution to the immune response and control of infection (Mehlop and Diamond, 2006).
All three complement pathways converge on a central component C3 which is cleaved into distinct forms with specific downstream targets and functions (reviewed in Carroll, 2004; Gasque, 2004; Kerr, 1980). C3 components can be directly conjugated to viral proteins, leading to neutralization or enhanced opsonization of particles. Alternatively, C3 cleavage can activate the downstream C5 convertase, and together with components C6 through C9 this can lead to formation of the membrane attack complex (MAC) which is capable of lysing virus particles or infected cells. Thus, virus particles can be neutralized by direct binding by the upstream C1q or C3 components or by virion lysis after the cascade has terminated at the MAC stage. DNA viruses have been shown to employ a wide range of approaches to block the complement pathway (Blue et al., 2004; Zipfel et al., 2007). However, the factors that determine the extent of complement activation by RNA viruses are not completely understood.
A number of paramyxoviruses have been shown to be neutralized by either the classical or alternative pathways (McSharry et al., 1981; Hirsch et al., 1986; Sissons et al., 1980; Devaux et al., 2004). For example, both Newcastle Disease virus (NDV) and HPIV3 are neutralized through the classical pathway, however NDV neutralization was antibody-independent while HPIV3 neutralization depended on the presence of antibody (Welsh, 1977; Vasantha et al.,1988). Measles virus (MeV) particles activate the classical pathway and MeV-infected cells activate the alternative pathway resulting in the lysis of infected cells. However, neither of these activities was strictly dependent on antibodies (Devauxet al., 2004; Joseph et al., 1975). When antibodies are present, lysis of MeV-infected cells was attributed to recognition of the cell-associated hemagglutinin or the fusion protein by the alternative pathway components (Devaux et al., 2004; Ehrnst, 1977; Sissons et al., 1979).
SV5, HPIV2 and MuV are closely related viruses belonging to the rubulavirus genus of paramyxoviruses (Lamb and Parks, 2007). It has been previously shown that complement neutralization of MuV proceeds through the alternative pathway, with the activity of the HN protein being an important factor in induction of the complement cascade (Hirsch et al., 1986). The degree of activation of the alternative pathway by SV5 and MuV were shown previously to be inversely related to sialic acid concentrations on either the particle or infected cells due to the presence of viral neuraminidase activity (McSharry et al., 1981; Hirsch et al., 1986). However, the complement components and mechanisms of neutralization of SV5 and MuV have not been completely defined, and it is also unclear what viral determinants initiate the complement cascade. In this study, we tested serum from ten human donors for the contribution of complement to in vitro neutralization of SV5, MuV and HPIV2. Our results demonstrate high levels of neutralizing antibody against HPIV2 with little contribution of complement to neutralization. By contrast, SV5 and MuV were neutralized only in the presence of active complement pathways and neutralization was strictly dependent on C3. Strikingly, the outcome of complement activation differed between these closely related viruses, with SV5 neutralization involving formation of massive aggregates and MuV neutralization including virion lysis.
Results
Role of complement in the in vitro neutralization of SV5, MuV and HPIV2
Human serum from ten donors was analyzed for the ability to neutralize SV5, MuV and HPIV2. In these assays, 100 PFU of virus was incubated with diluted sera from individual donors and remaining infectivity was measured by plaque assay. As shown in Fig. 1, sera from all ten donors reduced SV5 and HPIV2 infectivity to background levels (grey bars in panels A and B). By contrast, some sera such as those from donors 4, 8 and 10 were ineffective at neutralizing MuV infectivity, and ~40–70% of the infectivity remained after incubation with serum.
Fig. 1.
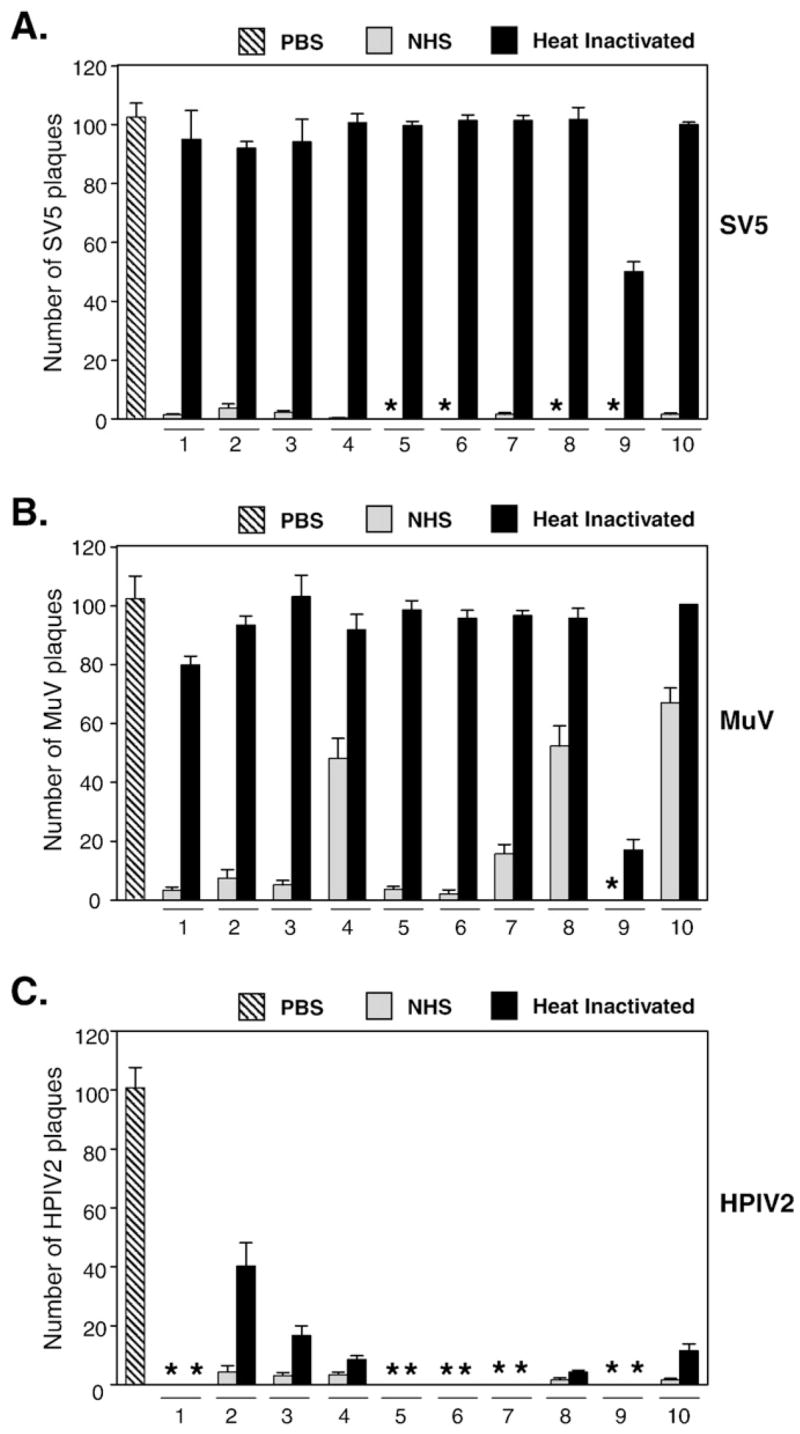
Role of complement in the in vitro neutralization of SV5, MuV and HPIV2 by human serum. One hundred PFU of SV5 (panel A), MuV (panel B) or HPIV2 (panel C) were incubated for 1 h at 37 °C with PBS (left hatched bars) or with either normal human serum (NHS, grey bars) or heat-inactivated serum (HI, black bars). Remaining infectious titers were determined by plaque assays. Results represent the average of six experiments, with error bars representing standard deviations. Numbers denote donor number. (*) indicates that no plaques were detected in these samples. For panels A and B, differences between NHS- and HI-treated samples were statistically significant, p < 0.001.
A very different profile of virus neutralization was seen using serum that had been complement-inactivated by heating at 56 °C for 30 min. As shown in Fig. 1A (black bars), treatment of SV5 with heat-inactivated (HI) serum from human donors did not decrease the number of PFU, and ~90–100 PFU still remained in samples after incubation with HI serum. An exception was seen with donor 9 serum where ~40% of the infectivity was retained following incubation with HI serum. The sensitivity of SV5 to complement neutralization could not be accounted for by growth in a specific host cell, since SV5 derived from three different host cells (Vero, A549 and MDBK) was equally sensitive to complement neutralization (data not shown). Similar results were seen when MuV was treated with HI serum (Fig. 1B, (black bars), where HI serum from 9 of 10 donors was ineffective at neutralization. Thus, neutralization of both SV5 and MuV is eliminated when complement is inactivated. By contrast, HI serum was still highly effective at reducing HPIV2 infectivity (Fig. 1C), as evident by the low number of plaques remaining after treatment with HI serum. Serum from donor 2, 3 and 4 showed ~10–40% infectivity of control samples, indicating that for these three donors, complement can influence neutralization.
To determine the level of antibody to SV5, MuV and HPIV2, serum from each donor was analyzed by ELISA using purified virus as the target antigen. As shown in Table 1, all ten donors had very high antibody titers against HPIV2, with some individual samples (e.g., donor 1, 6, and 9) having titers of at least 2 million. The mean anti-HPIV2 titer (log10) for all ten donors was 6.1. By contrast, ELISA titers against purified SV5 and MuV were lower than that seen for HPIV2 for each donor, with mean titers against SV5 and MuV being 5.1 and 5.2, respectively. Interestingly, serum from donor 9 had the highest ELISA titers against SV5 and MuV (Table 1), and also showed the least dependence on complement for neutralization (Figs. 1A and B).
Table 1.
Antibody titer log10
| Donor | SV5 | MuV | HPIV2 |
|---|---|---|---|
| 1 | 5.1 (3.0) | 5.3 | 6.3 (3.3) |
| 2 | 4.7 (2.9) | 5.0 | 6.0 (3.0) |
| 3 | 5.4 (2.9) | 5.2 | 6.2 (3.3) |
| 4 | 4.9 | 5.0 | 5.8 |
| 5 | 5.4 (2.9) | 5.3 | 6.0 (3.9) |
| 6 | 5.1 (1.0) | 5.0 | 6.4 (1.2) |
| 7 | 4.7 | 5.0 | 5.3 |
| 8 | 4.6 | 5.0 | 5.5 |
| 9 | 5.6 | 5.6 | 6.3 |
| 10 | 4.7 | 4.6 | 6.1 |
| Mean titer | 5.1 | 5.2 | 6.1 |
Elisa plates were coated with purified SV5, MuV or HPIV2 virions, and then incubated with dilutions of serum for each donor, followed by HRP-conjugated secondary goat anti-human IgG.
Data are expressed as the log10 of the dilution of serum that yielded an absorbance of two times that obtained with controls using secondary antibody only. Values in parentheses represent the ratio of IgG1 to IgG2 titers.
IgG subclasses can differ in their ability to fix complement (Valim and Lachmann, 1991). Using subclass-specific ELISAs, serum from donors 1, 2, and 3 were found to have similar anti-SV5 and anti-HPIV2 ratios of IgG1 to IgG2a of ~3 (Table 1). Donor 5 had an anti-HPIV2 ratio that was slightly higher at ~4. Donor 6 differed by having an IgG1 to IgG2a ratio for both anti-SV5 and anti-HPIV2 antibodies of ~1. Thus, anti-SV5 and anti-HPIV2 responses in human serum had similar IgG subclass ratios, a result that suggests antibody-mediated complement fixation is not responsible for differences between SV5 and HPIV2 in their dependence on complement for neutralization.
Together, these data indicate that within our donor population, serum samples have high ELISA titers against HPIV2, and these sera neutralize HPIV2 in vitro by a mechanism that is largely independent of complement. By contrast, these same samples have lower ELISA titers against SV5 and MuV and strikingly, complement but not neutralizing antibody is a major factor in the in vitro neutralization of these two paramyxoviruses. The following experiments are focused on the mechanisms of complement neutralization of SV5 and MuV, but not on HPIV2.
SV5 and MuV neutralization is mediated by the alternative pathway of complement activation
It has been well established that the classical and lectin pathways of complement activation are dependent on Ca+ ions for activity, and can be selectively blocked by the addition of EGTA containing Mg2+ (Des Prez et al., 1975), while EDTA inactivates all three pathways. To test the hypothesis that SV5 and MuV were neutralized by the alternative pathway, 100 PFU of SV5 or MuV were incubated with serum that had been pretreated with either EGTA (8 mM) containing Mg2+ (2 mM) or with EDTA (20 mM), and remaining infectivity was measured by plaque assay. As shown in Fig. 2, > 90% of the infectivity for SV5 (black bars) and MuV (gray bars) was lost by treatment with NHS or serum supplemented with EGTA and Mg2+, whereas there was no loss of infectivity following treatment with EDTA-treated serum. Treatment of virus with EGTA or EDTA by themselves did not have any effect on the viruses at the concentrations used in this assay (data not shown). These data are consistent with the proposal that complement-mediated neutralization of both SV5 and MuV proceeds through the alternative pathway.
Fig. 2.
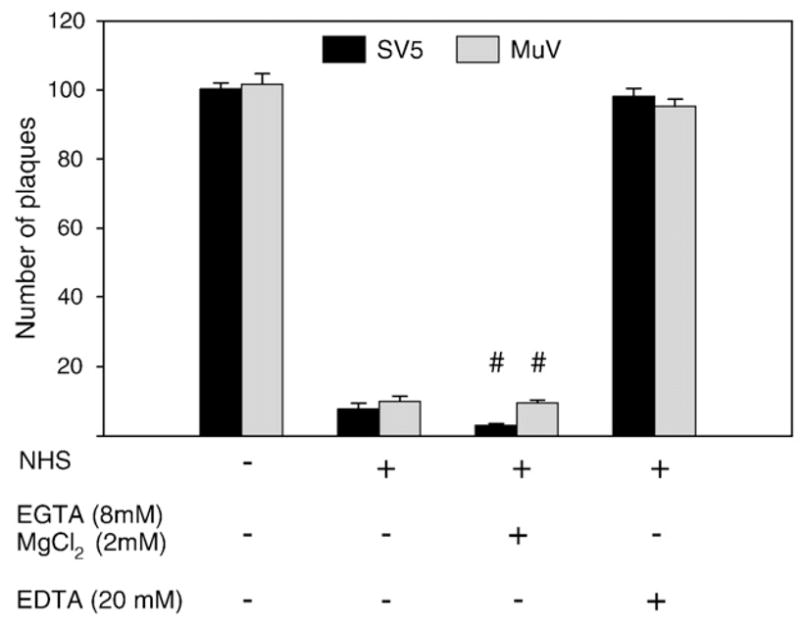
Complement-mediated neutralization of SV5 and MuV is by the alternative pathway. One hundred PFU of SV5 and MuV was incubated with either NHS, with EGTA and Mg2+-treated serum which inactivates the classical and lectin pathways, or with EDTA-treated serum which inactivates all complement pathways. Remaining infectious titers were determined by plaque assays. Results represent the average of six experiments using serum from donor number 1, with error bars representing standard deviations. Number sign, differences between EGTA-treated and EDTA-treated samples were highly significant (p < 0.001).
C3 plays a significant role in the in vitro neutralization of SV5 and MuV
Complement component C3 acts as a central molecule in the complement cascade (Gasque, 2004; Pangburn et al., 1981). Human serum that had been depleted of C3 was used to test the hypothesis that C3 was an essential factor in complement-mediated in vitro neutralization of SV5 and MuV. As shown in Fig. 3A, C3-depleted serum had very little hemolytic activity (open circles), but activity was restored to levels approaching that of NHS when C3-depleted serum was supplemented with physiological concentrations of C3 (closed triangles). One hundred PFU of SV5 or MuV was incubated with NHS, C3-depleted serum or C3-depleted serum reconstituted with C3, and remaining infectivity measured by plaque assay. As shown in Figs. 3B and C, virus infectivity was reduced by > 90% with NHS (black bars), but C3-depleted serum (cross-hatched bars) had no effect on infectivity of either SV5 (panel B) or MuV (panel C). Importantly, treatment with C3-depleted serum that had been reconstituted with C3 resulted in a marked decrease in the number of SV5 and MuV plaques to levels comparable to that seen with neutralization by NHS (Figs. 3B and C).
Fig. 3.

C3 activity is required to neutralize SV5 and MuV. A) Total hemolytic complement activity of NHS, C3-depleted serum, and C3-depleted serum reconstituted with C3 was determined as described in Materials and methods. B) and C) 100 PFU of SV5 (panel B) or MuV (panel C) were incubated with PBS (hatched bars), NHS (black bars), with a 1:25 dilution of C3-depleted serum (striped bars), or 1:25 dilution of C3-depleted serum reconstituted with C3 (gray bars). Remaining infectious titers were determined by plaque assays. Results are the average of six experiments, with error bars representing standard deviations. Number signs denote p < 0.001 comparing C3-depleted samples to C3 reconstituted samples. D) C3 deposition on SV5 and MuV. Sucrose gradient-purified virus was left untreated or treated with 1:25 diluted NHS or C3-depleted serum, and complement C3 deposition on the particles was detected using anti-C3 antibody followed by 12 nm colloidal gold goat anti-mouse antibody. Samples were analyzed by EM at a magnification of 55,000 × (bar represents 0.05 μm).
The above results showing that C3 was important for in vitro neutralization raised the question of whether C3 was deposited on SV5 and MuV particles. To address this question, purified SV5 or MuV particles were collected on carbon coated gold grids and incubated for a short period (5 min, RT) with PBS, NHS, or C3-depleted serum. Samples were then probed with anti-C3 monoclonal antibody and gold-labeled secondary antibody before visualizing by electron microscopy (EM). As shown in Fig. 3D, C3 deposition was detected for both SV5 and MuV particles treated with NHS, but not for control samples in which particles were treated with C3-depleted serum. While it can be common to see lysed particles due to artifacts of the EM grid, comparison of the NHS-treated particles to the untreated particles did not show marked lysis or distortion in the viral structure. However as shown below, this was due to the short incubation period and the approach of carrying out the reactions on EM grids. Taken together, these data indicate that C3 plays a critical role in the in vitro neutralization of both SV5 and MuV and that C3 can form deposits on virions.
Complement induces aggregation of SV5 particles
The EM studies described above involved collecting virus particles on EM grids prior to exposure to human serum. We determined the effect of complement on SV5 particles in solution. Purified SV5 particles were mixed with NHS in solution at 37 °C, and at various times (0, 5, 15, 30 and 60 min) samples were placed on ice to stop the reaction. Samples were then placed on EM grids and subjected to negative staining. As shown in Fig. 4A, virus incubated without serum for 60 min showed intact particles that were loosely scattered about the grid (top left panel). By contrast, incubation of SV5 virions with NHS for as little as 5–15 min resulted in aggregation of particles into small groups, and by 30–60 min virions were seen in large aggregates (Fig. 4A, note differences in scales for micrographs). HI serum did not induce virion aggregation (not shown). In some but not all aggregate-associated virions, the viral membrane was discontinuous, indicating that limited lysis was occurring within the aggregates.
Fig. 4.
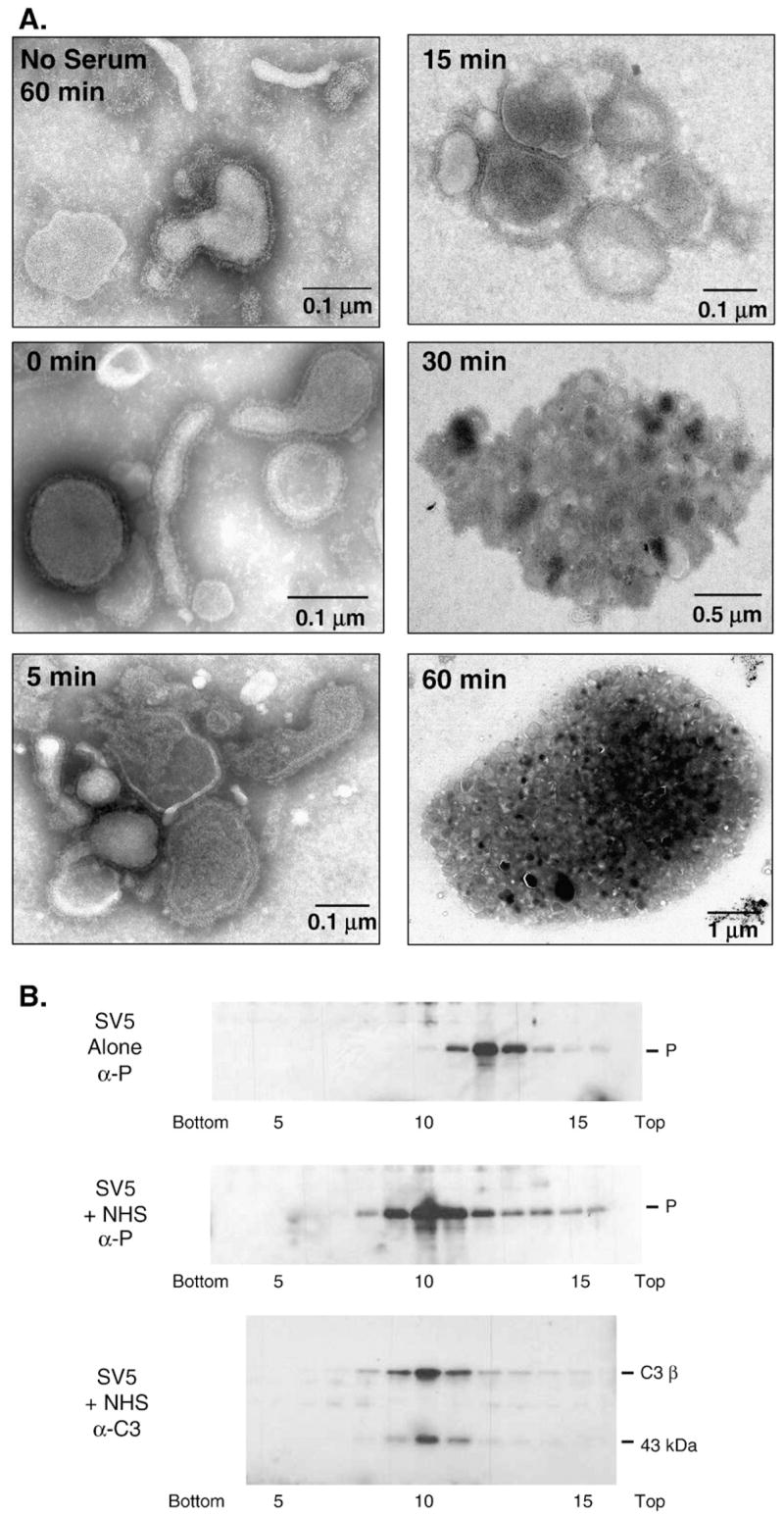
Complement induces aggregation of SV5 particles. A) EM analysis. Purified SV5 particles were incubated for the indicated times at 37 °C with a 1:25 dilution of NHS and then analyzed by negative staining and EM. Note the aggregation of SV5 particles and the differences in scale of magnifications for samples taken over time. The no serum sample represents control virions incubated at 37 °C for 60 min without serum. B) Sucrose gradient centrifugation. Purified SV5 was incubated alone or with NHS for 60 min at 37 °C and then analyzed by centrifugation through 15–60% sucrose gradients. Fractions were collected and analyzed for viral proteins by western blotting with antiserum specific for the SV5 P protein (top and middle panels). Fractions 5–15 from the NHS-treated virions were also analyzed by western blotting for the presence of C3 (bottom panel). The position of the beta chain of C3 and the 43 kDa fragment of iC3b are indicated.
The aggregation of SV5 particles by NHS was confirmed by analyzing virions by gradient centrifugation. Purified SV5 particles were left untreated or were mixed with NHS, incubated for 60 min at 37 °C, layered on top of a 15–60% sucrose gradient and subjected to ultracentrifugation. Fractions were collected and the sedimentation of viral particles was analyzed by Western blotting with antiserum specific for the viral P protein. As shown in Fig. 4B, untreated SV5 virions migrated as a peak centered between fractions 11–13 (top panel). By contrast, NHS-treated virions migrated further down the gradient with a peak centered between fractions 9–11 (middle panel), consistent with large aggregates or particles with higher density. No viral proteins were detected in the top fractions (data not shown), consistent with the above EM data showing only very low levels of virion lysis. Analysis of fractions from the NHS-treated sample with anti-human C3 antibody showed co-sedimentation of the C3 fragments in the same fractions that contained viral proteins (Fig. 4B, bottom panel). The presence of the beta chain and 43 kDa protein subunits of iC3b (Sahu et al., 1998) along in the same fractions that contained SV5 proteins indicates that C3 has been activated. Likewise, analysis of the SV5-NHS fractions for antibody demonstrated that all immunoglobulin protein was at the top of the gradient and not in the faster-sedimenting fractions that contained C3 protein (data not shown). When the virions were treated with C3 deficient serum there was no aggregation, however upon reconstitution with C3 aggregation was seen (data not shown). Thus, EM and biochemical evidence indicate that NHS treatment of SV5 virions in solution results in association with C3 and aggregation, with only a relatively small amount of virion lysis.
Complement induces lysis of MuV particles
To determine if complement also induced MuV aggregation, purified MuV particles were left untreated or were incubated with NHS and then analyzed by negative staining and EM. Untreated and 0 time point samples showed loosely scattered but intact particles on the grid (Fig. 5), and when MuV was incubated with NHS there was evidence of only a very low degree of aggregation. However, as early as 5 min after incubationwith NHS virions began to show signs of lysis, and by 15 min of NHS treatment most virions were distorted and showed leakage of internal components (Fig. 5). After 30 min incubation with NHS, MuV virions showed structures resembling nucleocapsids oozing out of the particles. At later times, dark shadowy regions were seen on the grid consistent with massive complement deposition around lysed particles, and this was confirmed by immunogold labeling of these areas with anti-C3 antibody (data not shown). Thus, NHS treatment of MuV results in virion lysis, unlike the aggregation seen with NHS-treated SV5 particles.
Fig. 5.
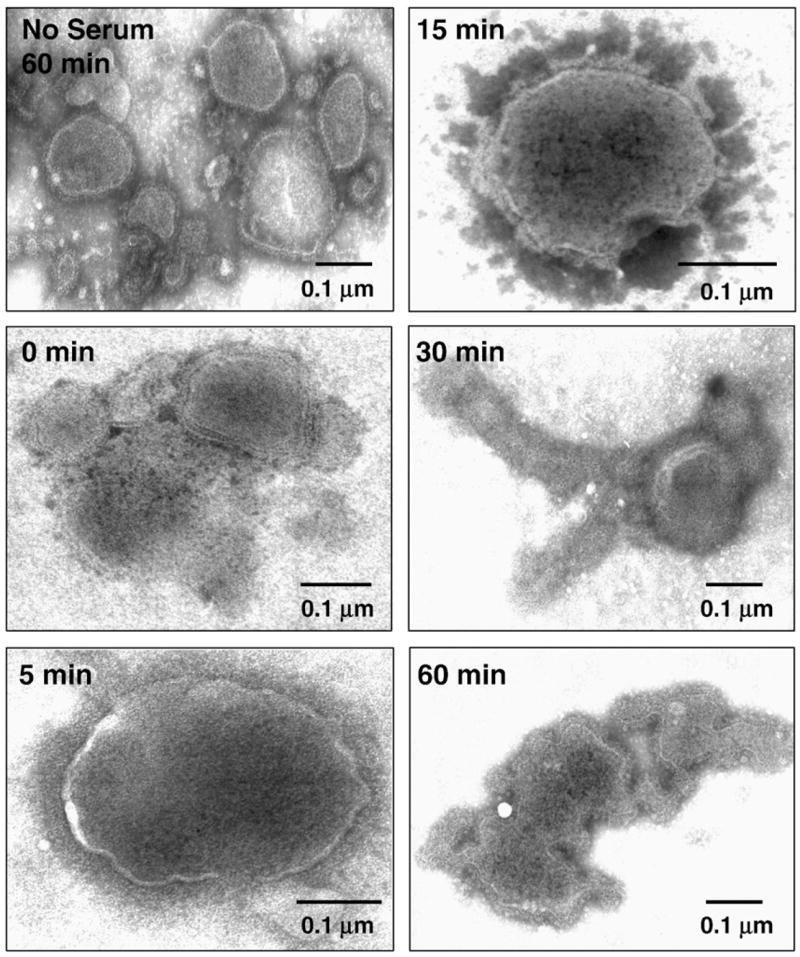
Complement induces lysis of MuV particles. Purified MuV particles were incubated for the indicated times at 37 °C with NHS and then analyzed by negative staining and EM. Note that MuV particles are not massively aggregated as seen in Fig. 4 for SV5. The solid bar in each figure represents 0.1 μm. The no serum sample represents control virions incubated at 37 °C for 60 min without serum.
The lysis of MuV particles by NHS was confirmed by sucrose gradient analysis as described in Fig. 4B for SV5. After treatment of MuV with NHS and centrifugation, gradient fractions were analyzed by Western blotting with antiserum to the SV5 P protein, which cross-reacts with the MuV P protein. In the absence of NHS, virion-associated P protein was detected as a rapidly migrating peak in fractions 10–15 (Fig. 6A), similar to that seen with untreated SV5 particles (Fig. 4B). By contrast, the P protein in NHS-treated MuV samples was detected in two distinct regions of the gradient: fractions 5–10 near the bottom of the gradient and fractions 35–40 at the top of the gradient (Fig. 6B). Interestingly, in the top fractions P protein was detected as an unexpectedly high molecular weight species, whereas P protein in the bottom fractions was of the expected size of ~40 kDa (Fig. 6B). To determine if complement proteins were associated with both the fast and slower migrating peaks, fractions 2–10 from the bottom of the gradient and fractions 30–40 at the top the of the gradient were probed with anti-C3 antibody. As shown in Figs. 6C and D, small amounts of complement iC3b (beta chain and 43 kDa proteins) were detected in the same bottom fractions that contained P protein (fractions 5–9). Importantly however, much larger amounts were detected in the top fractions 35–40 which also contained the high molecular weight P complex. Of the C3-containing proteins in fractions 37–40, a high molecular weight species was detected (arrow Fig. 6D) which was not in marker lane samples of purified C3 or C3b, and this protein species had the same apparent size as the P-associated high molecular weight protein seen in panel B. Thus, the fractions at the top of the gradient contain a new large protein species that reacts with anti-P and anti-C3 antiserum. Taken together, the above EM and biochemical studies on NHS-treated MuV particles are consistent with lysis of the MuV virions, and with formation of a high MW protein species that contains at least in part P protein complexed with an activated C3 product. Thus, NHS treatment of purified virions results in aggregation in the case of SV5 (Fig. 4), but lysis of particles in the case of MuV (Figs. 5 and 6).
Fig. 6.
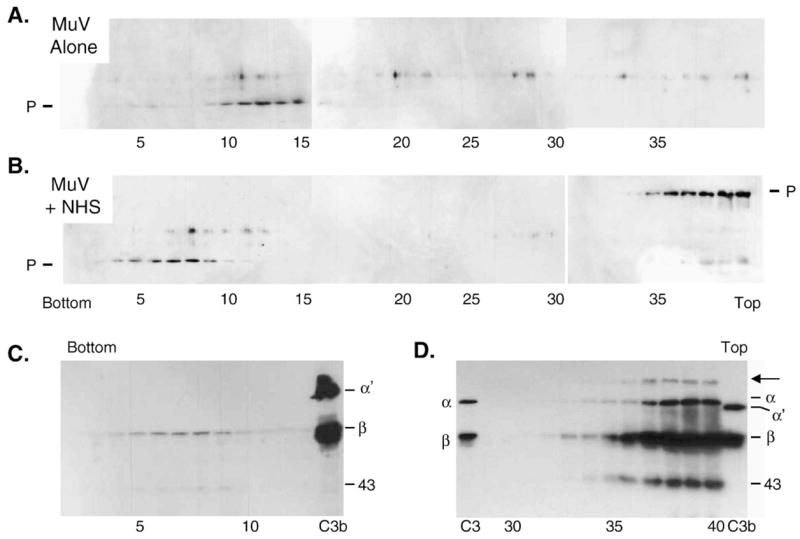
Sucrose gradient analysis of NHS-treated MuV particles. Purified MuV was incubated alone (panel A) or with NHS (panel B) for 60 min at 37 °C and then analyzed by centrifugation through 15–60% sucrose gradients. Fractions were collected and analyzed for virion proteins by western blotting with antiserum raised against the SV5 P protein which cross-reacts with MuV P protein (top and middle panels). A cross-reactive band of unknown origin is seen above the position of the P protein. C) Fractions 2–13 and 30–40 from the NHS-treated virion gradient were also analyzed by Western blotting for the presence of C3. The arrow indicates a high molecular weight protein that is not found in purified proteins in the marker lanes containing fragments of C3 and C3b.
C5 but not C8 contributes to in vitro neutralization of SV5 and MuV by complement
Complement component C5 acts as a convergence point for all the three complement activation pathways (Hammer et al., 1975, Podack et al., 1976) and is a key component in formation of the MAC, but C5 can also directly contribute to virus neutralization (Friedman et al., 2000; Hirsch et al.,1980). The role of C5 in SV5 and MuV neutralization was examined using C5-depleted serum. As shown in Fig. 7A, C5-depleted serum had very little hemolytic activity (open circles), but activity was restored to levels approaching that of NHS with physiological concentrations of C5 (closed triangles). One hundred PFU of SV5 or MuV was incubated with NHS or with C5-depleted serum, and remaining infectivity measured by plaque assay. As shown in Figs. 7B and C, C5-depleted serum by itself was capable of neutralizing both SV5 and MuV (striped bars), but this was most evident at high concentrations of C5-depleted serum (e.g., 1:15 and 1:25 dilutions). However, at lower concentrations C5 deficient serum had less of an effect on neutralization, and reconstituting C5 resulted in more effective neutralization for SV5 (compare 1:75 dilution of C5-depleted and C5 reconstituted serum, respectively). MuV neutralization was more sensitive to serum dilution, but C5 reconstitution had only a modest effect on neutralization (Fig. 7C).
Fig. 7.
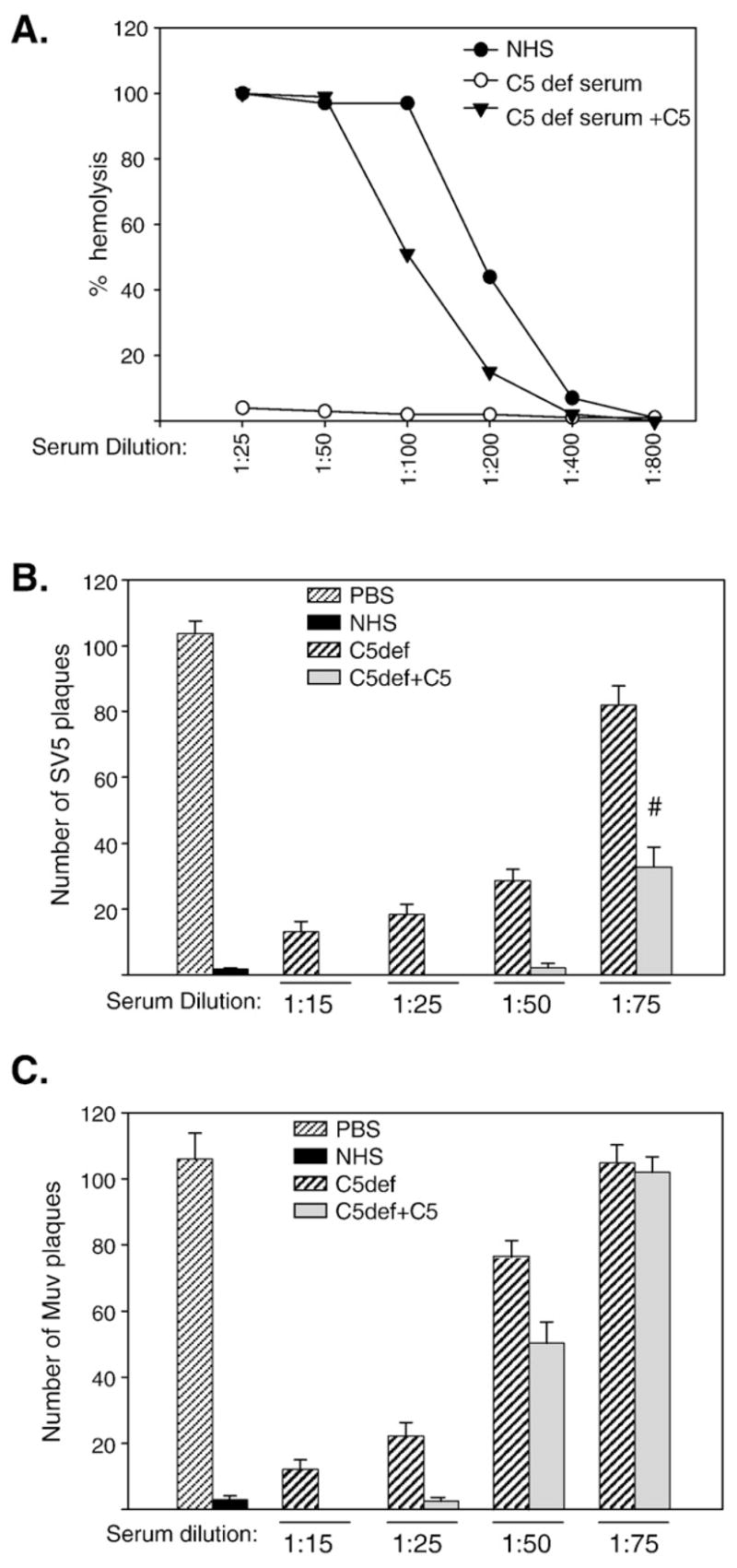
C5 can contribute to the neutralization of SV5 and MuV. A) Total hemolytic complement activity of NHS, C5-depleted serum, and C5-depleted serum reconstituted with C5 was determined as described in Materials and methods. B) and C) One hundred PFU of SV5 (panel B) or MuV (panel C) were incubated with PBS, 1:25 dilution of NHS, with the indicated dilutions of C5-depleted serum, or C5-depleted serum reconstituted with C5. Remaining infectious titers were determined by plaque assays. Results are the average of six experiments, with error bars representing standard deviations. Number sign denotes p < 0.001 comparing C5-depleted samples to C5 reconstituted samples.
Because C8 forms one of the major constituents of the MAC, the effect of C8 depletion on SV5 and MuV neutralization was examined. As shown in Fig. 8A, C8-deficient serum by itself had negligible hemolytic activity, which was recovered upon reconstituting with physiological C8 concentrations. C8-deficient serum was capable of neutralizing both SV5 and MuV, with a very similar decrease in the ability of C8-deficient and C8-reconstituted serum to neutralize as the dilution of serum was increased (Fig. 8B and C). These data indicate that by comparison to C3, neither C5 nor C8 is essential for in vitro neutralization of SV5 or MuV. However, C5 can enhance neutralization of SV5 and MuV at low concentrations of serum. EM studies demonstrated that MuV particles were not lysed using C5- or C8-deficient serum (not shown). Thus, MuV neutralization is complement-dependent and relies on C3, but does not require virion lysis by the MAC involving C5 and C8.
Fig. 8.
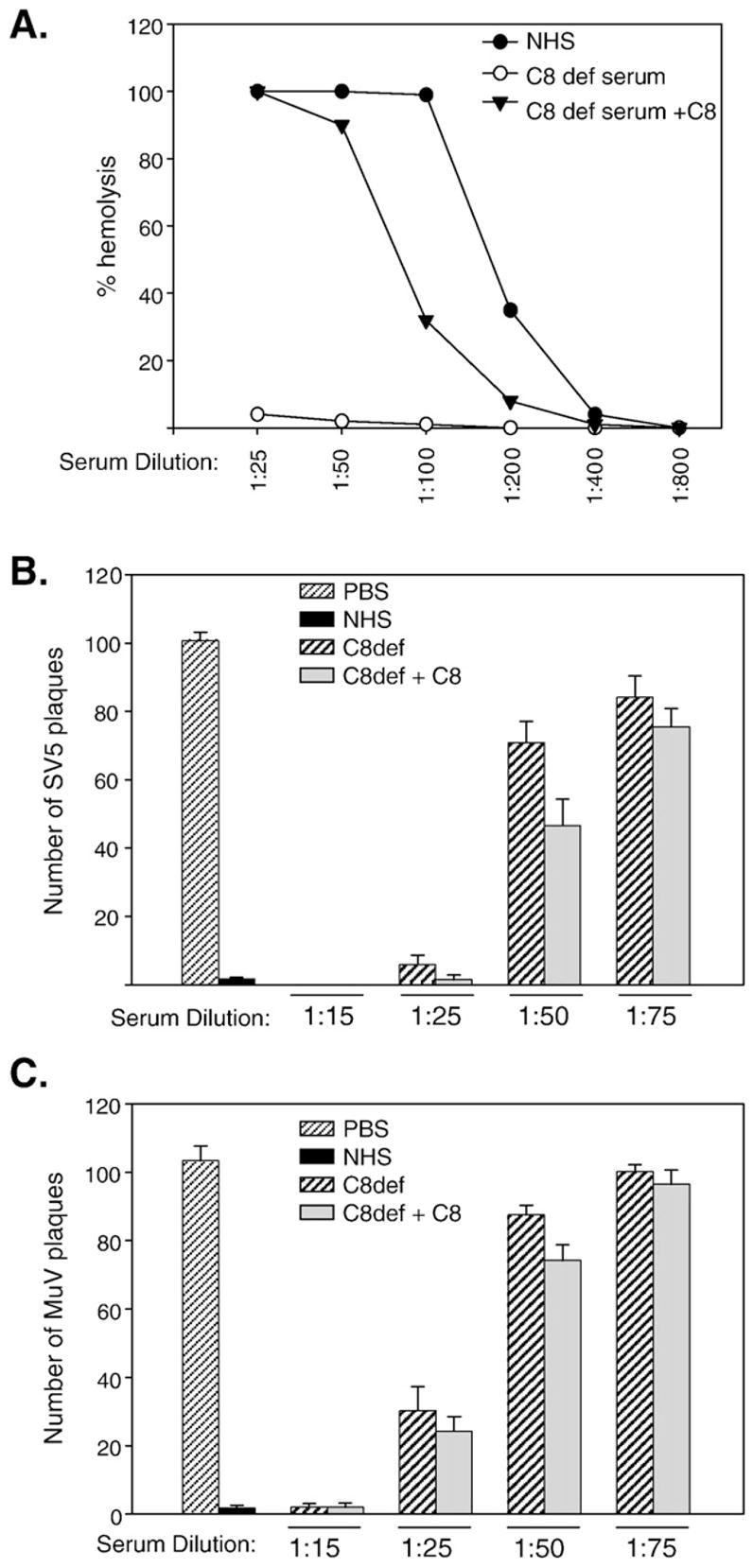
C8 is not required to inactivate SV5 or MuV. A) Total hemolytic complement activity of NHS, C8-deficient serum, and C8-deficient serum reconstituted with C8 was determined as described in Materials and methods. B) and C) One hundred PFU of SV5 (panel B) or MuV (panel C) were incubated with PBS, 1:25 dilution of NHS, with the indicated dilutions of C8-deficient serum, or C8-deficient serum reconstituted with C8. Remaining infectious titers were determined by plaque assays. Results are the average of six experiments, with error bars representing standard deviations.
Discussion
Complement is an important factor in the neutralization of a large number of viruses, but the mechanisms of neutralization and signals activating the complement cascade are not completely understood for many virus systems including the paramyxoviruses. The goal of the work described here was to determine the extent and mechanisms by which complement contributes to the neutralization of three closely related rubulaviruses: SV5, MuV and HPIV2. Our data indicate that human serum contains activity that neutralizes all three of these closely related viruses. Most importantly however, the extent to which complement is involved in neutralization of SV5 and MuV differs significantly from that of HPIV2. In vitro neutralization of both SV5 and MuV was largely dependent on complement, since HI serum from only one of ten donors showed neutralization of greater than 50% of the control samples. HPIV2 differed from SV5 and MuV in this regard, since HI serum from all ten donors still effectively neutralized HPIV2 by greater than 50% of controls. These results indicate that within our sample group HPIV2-specific neutralizing antibody is widely present and in some cases the neutralization can be further enhanced by complement. In sharp contrast, antibodies against SV5 and MuV were largely non-neutralizing, at least at the dilutions and with the donor population studied here.
Our results are consistent with reports that both SV5 and MuV are neutralized by the alternative pathway (McSharry et al., 1981), but extend previous work by identifying key components in the complement pathway that are necessary for in vitro neutralization. C3 forms the central molecule of the complement cascade (Kerr, 1980; Sahu and Lambris, 2001) and the consequences of C3 activation can include steric hindrance by coating of virion surfaces and aggregation (Oldstone et al., 1974), initiation of MAC formation and virion lysis (Bartholomew et al., 1978; Cooper et al., 1976; Mills et al., 1979), or opsonization to enhance capture by antigen presenting cells. Our data show a strong dependence on C3 for the inactivation of SV5 and MuV, since C3-depleted serum had no effect on virus infectivity, while reconstitution with physiological concentrations of C3 fully restored neutralizing capacity to C3-depleted serum. EM and biochemical studies support this role for C3 in neutralization, since C3 can be found deposited on virions. In the case of MuV, our data also support direct conjugation to internal virion components such as P protein, which is consistent with complement-mediated lysis of virions.
The most striking result from our studies is that although both SV5 and MuV show dependence on C3 for neutralization and C3 deposition on virions, the outcome of in vitro complement activation for these two closely related paramyxoviruses differs significantly. Incubation of SV5 particles with human serum resulted in efficient neutralization that correlated with formation of massive aggregates that were easily visualized by EM. On sucrose gradients, serum-treated SV5 migrated to a heavier fraction of the gradient compared to untreated virus, and virion proteins were found in the same gradient fractions that included activated C3 components. Importantly, the SV5 P protein which was used to assay the sedimentation position of virions was not conjugated to C3 (see Fig. 4B), consistent with C3 reacting with the external surface of particles without virion lysis. In the case of polyoma virus, neutralization occurs by virion aggregation due to the association of antibody to the viral particles which is coordinated by the cross linking effect of activated C3 (Oldstone et al., 1974). Our results indicate that this differs for SV5, since gradient fractions that contained virus and C3 did not contain detectable immunoglobulin protein. These data are consistent with SV5 being neutralized by C3-dependent crosslinking of virions or by trapping of virions within C3-rich matrices. Future studies will distinguish between these two mechanisms by identifying SV5 proteins that interact with C3.
Strikingly and in sharp constrast to SV5, MuV virions treated with human serum did not form large aggregates but instead showed lysis of the virion envelope. Sucrose gradient analysis of serum-treated MuV particles showed the viral P protein in two regions: 1) a less abundant faster-sedimenting species corresponding to low levels of aggregated virions with low C3 content and 2) a more abundant slow-sedimenting species at the top of the gradient which was enriched in C3. Importantly, the slow-sedimenting fractions of serum-treated MuV particles showed a higher molecular weight P protein consistent with conjugation to C3. Since the viral P protein is normally found as an internal protein attached to the viral nucleocapsid beneath the virion envelope (Lamb and Parks, 2007), we interpret these results as evidence that activation of complement leads to C3 deposition on and lysis of the MuV virion. This in turn leads to conjugation of C3 with the exposed P protein into a high molecular weight species. Due to our limitation in available MuV-specific reagents, it is presently not clear whether P is the only viral target of C3 conjugation or if other viral proteins are also covalently modified.
The terminal C5- and C8-dependent pathway which constitutes the MAC component is critical for neutralization of some viruses. MAC formation often leads to lysis of virions or virus infected cells as has been shown in the case of HPIV3 and herpes virus particles (Friedman et al., 2000; Vasantha et al., 1988) or lysis of HSV infected human fibroblasts (Daniels et al., 1980). However, there are examples of virus infections that activate MAC formation, but the complex does not lyse the infected cells (Cooper et al., 1974). In our studies, C5 had a very minor effect on MuV neutralization and was not an essential factor, whereas C8 was not essential for neutralization of either SV5 or MuV. Thus, C3 is an essential factor in neutralization of both SV5 and MuV, and leads to virion aggregation or lysis, respectively. However, while MuV neutralization normally proceeds along a pathway ultimately leading to MAC formation and virion lysis, this is not an essential end-stage to the pathway and MuV can be neutralized after activation of the cascade only to the C3 level.
What factors could contribute to the differences in mechanisms of complement-mediated neutralization of SV5 and MuV by NHS? In our model, complement components recognize the surface of SV5 and MuV virions to activate the alternative pathway and C3 deposition for both viruses. However, a key difference is in the steps downstream of C3 activation, and some divergent signal dictates whether the pathway ends with either aggregation of SV5 particles or lysis of MuV particles. Aggregation without lysis has been reported in the case of influenza virus, however this occurred due to classical pathway activation and was dependent on antibody (Jayasekara et al., 2007). In addition, complement-mediated lysis of HPIV3 has been shown to involve C1q and IgM antibody (Vasantha et al., 1988). In the case of SV5 and MuV however, antibodies do not appear to play a critical role in in vitro neutralization, since no immunoglobulin was found associated with virions in gradient fractions. While it is possible that our assays are not sensitive enough to detect small amounts of IgG, other explanations are that the antibody is removed by centrifugation conditions or by complement deposition or that the majority of the antibodies detected in our ELISA are to internal components. Thus, we propose that early complement interaction occurs with some viral protein on the outer coat, independent of antibody.
The lipid bilayer of both SV5 and MuV is characterized by the presence of three proteins, the fusion protein (F), the hemagglutinin–neuraminidase protein (HN) and the SH small hydrophobic protein (Lamb and Parks, 2007). Any or all of these proteins could contribute to differences in the strength of signal or differences in signals that determine how far the complement cascade proceeds. The amount of sialic acid on viral surface derived from the host cell has been shown to act as a trigger for alternative pathway activation in the case of vesicular stomatitis virus and SV5, with lower amounts of sialic acid content being a more potent signal for activation of the alternative pathway (McSharry et al., 1981). SV5 and MuV particles display the same differences in aggregation versus lysis even when they are grown in a common host cell type such as Vero cells (not shown), suggesting that differences in complement neutralization are due to a viral component. The extent of viral neuraminidase activity can be an important factor in complement activation by different strains of MuV (Hirsch et al., 1986). Together, these results would be consistent with a model whereby differences between the neuraminidase activity of the W3A strain of SV5 and the Enders strain of MuV results in particles with differences in sialic acid modification to HN, F or host proteins that are incorporated into budding particles.
Our studies have implications for understanding immunity to vaccination as well as the use of recombinant paramyxoviruses as viral vectors. Despite wide-spread vaccination, there has been a recent resurgence in reported cases of MuV (Peltola et al., 2007), and it has also been reported that pre-existing neutralizing antibodies from previous immunization with one strain may not be able to give protection against a newly emerged strain (Nojd et al., 2001). A better understanding of the parameters that lead to MuV neutralization by complement may shed light on differences between MuV isolates and the effectiveness of different vaccine strains. Likewise, a number of paramyxoviruses are being developed as vaccines or therapeutic vectors (e.g., Von Messling and Cattaneo, 2004), and pre-existing immunity against these vectors can be of significant concern. In the case of SV5 vectors (Parks and Alexander-Miller, 2002; Parks et al., 2002; Tompkins et al., 2007), our results indicate that complement but not antibody is a major factor in neutralization at least in vitro. Thus, it may be possible to design vectors that overcome the activation of complement to be better able to disseminate for therapy.
Materials and methods
Cells and viruses
Monolayer cultures of CV-1 and A549 cells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 U/ml), streptomycin (100 μg/ml) and 200 mM L-Glutamine. Recombinant rSV5 (W3A strain), mumps virus (Enders strain, ATCC VR-1379), and HPIV2 (Greer strain) were grown in MDBK, Vero and CV-1 cells, respectively. Viruses were concentrated by centrifugation through a glycerol cushion (5 h; 25,000 rpm; SW28 rotor), and resuspended virus was further purified by centrifugation on a 30–60% sucrose gradient (2 h; 23,000 rpm; SW28 rotor). The virus band was collected, pelleted, resuspended in DMEM containing 0.75% BSA and stored at −80 °C.
Complement reagents
Normal human serum (NHS) collected from 10 healthy adult volunteers was used as a source of complement for the complement assays. The blood was allowed to clot for 1 h at 37 °C, the clot retracted and further incubated for 30 min at 37 °C and the serum was separated, aliquoted and stored at −80 °C. Complement inactivated serum was prepared by heating NHS at 56 °C for 30 min. The classical, mannan-binding lectin (MBL) or alternative pathway involvement in virus neutralization was tested using NHS that was treated with 20 mM EDTA to block all the complement pathways or with 8 mM EGTA and 2 mM Mg2+ to block the classical and the MBL pathways but not the alternative pathway. C3-depleted, C5-depleted (both Calbiochem, CA), and C8-deficient human serum (Sigma, Mo) were reconstituted with physiological concentrations of purified human C3 (1.2 mg/ml), C5 (75 μg/ml) or C8 (80 μg/ml) [Calbiochem, CA].
Elisa
MaxiSorp 96-well ELISA plates (Nunc) were coated with sucrose gradient-purified SV5, MuV, or HPIV2 particles at 12.5 μg/ml (50 μl/well) and UV-irradiated for 20 min at a distance of 10 cm. Plates were incubated at 4 °C overnight, washed three times with PBS/Tween (0.2%), and wells were blocked with 200 μl of PBS containing 2% BSA for 2 h at room temperature (RT). Dilutions of heat-inactivated NHS were added to wells, incubated for 1.5 h, and wells were washed with PBS/Tween before incubation for 1 h with HRP-conjugated goat anti-human IgG (Jackson ImmunoResearch Laboratories, PA) or subclass-specific secondary antibodies. The HRP substrate TMB (tetramethylbenzidine dihydrochloride, Sigma) was added to each well and incubated in the dark for 20 min. The enzymatic reaction was stopped with 2N sulfuric acid, and the absorbance was determined at 450 nm on a Labsystems Multiskan Plus plate reader (Fisher Scientific, GA). Antibody titers were defined as the dilution of serum that yielded an absorbance two times that obtained in controls with secondary antibody only.
Hemolytic activity assays and virus neutralization assays
The activity of complement-depleted serum was tested by reconstitution with the respective depleted protein and was compared to NHS for total hemolytic complement activity (CH50) by a hemolytic assay as described earlier (Sahu et al., 1998). Briefly, two fold diluted NHS or the reconstituted serum (100 μl) was mixed with 5 μl of antibody sensitized sheep erythrocytes (EA, 1 × 109 cells/ml) and the volume was made up to 250 μl with DGVB+ [Gelatin veronal buffer (Sigma, St. Louis, MO) containing 0.5 mM MgCl2 and 0.15 mM CaCl2 and 2.5% dextrose] and incubated at 37 °C for 30 min with constant vortexing. The reaction was stopped by placing the samples on ice followed by centrifugation at 4 °C for 5 min at 3000 rpm and hemolysis in 200 μl of the supernatant was measured by reading absorbance at 405 nm.
Complement-mediated neutralization assays were carried out as described previously with some modifications (Friedman et al., 1996). Briefly, 100 plaque forming units (PFU) of SV5, MuV or HPIV2 (100 PFU/100 μl) were incubated with varying dilutions of NHS which served as the complement source or heat-inactivated NHS (HI). Control samples received PBS alone. After incubation at 37 °C for 1 h, viral titers were determined by plaque assay as described previously (Wansley and Parks, 2002). Neutralization by complement was represented as the number of plaques below the initial input of 100 PFU. Each data point represented an average and standard deviation of six independent experiments. Statistical analysis was by the student’s t-test.
Electron microscopy
To stain virus particles on grids, C3 deposition on SV5 and MuV was analyzed by taking 10 μl of a 1 × 105 PFU/ml purified viral particles on carbon coated 200 mesh gold grids (Electron Microscopy Sciences, PA) and incubated at RT for 5 min. The grids were blocked with PBS containing 1% BSA. Adsorbed virus on the grids was treated with 1:25 diluted NHS, or with C3-depleted human serum and probed with mouse anti-human C3/C3b monoclonal antibody (clone 755; Cell Sciences, MA), at a dilution of 1:10. Bound antibody was detected with 12 nm colloidal gold goat anti-mouse antibody (Jackson Immunoresearch). Particles were then negative-stained with 2% phosphotungstic acid (pH 6.6) and analyzed under a Philips TEM400 transmission electron microscope (Tompkins et al., 2007).
To visualize effects of complement under physiological conditions, purified SV5 or MuV particles were mixed with NHS at a dilution of 1:2 and incubated for various time points (0, 5, 15, 30 and 60 min) at 37 °C followed by addition of the samples onto carbon coated 200 mesh gold grids. After adsorption for 5 min, grids were washed with PBS, negatively stained with 2% phosphotungstic acid and observed under Philips TEM400 transmission electron microscope.
Ultracentrifugation and western blotting
Sucrose gradient-purified SV5 or MuV was mixed with an equal volume of NHS (1:1) and incubated for 1 h at 37 °C. The reaction mixture was layered on top of a 15–60% pre-equilibrated sucrose gradient in NTE buffer as described previously (Paterson et al., 1995). After centrifugation (40,000 rpm, 1 h, SW50.1, 4 °C), 250 μl fractions were collected from the bottom of the gradient and analyzed by SDS-PAGE and Western blotting as described previously (Dillon and Parks, 2007), using rabbit antisera to the SV5 P protein (Parks et al., 2002) which cross-reacts with the MuV P protein. Alternatively, blots were probed with goat anti-human C3 polyclonal antibody at a dilution of 1:1000 (MP Biomedics, Cappell reagent, CA). Blots were visualized by enhanced chemiluminescence and exposure to film.
Acknowledgments
We thank Dr. Martha Alexander-Miller for helpful comments on the manuscript. We also thank Ken Grant for help with electron microscopy and acknowledge the excellent technical assistance of Ellen Young and Lara Smith. This work was supported in part by NIH grant AI058948 (G.P.) and AI060642 (S.B.M.).
References
- Bartholomew RM, Esser AF, Muller Eberhard HJ. Lysis of oncornaviruses by human serum Isolation of the viral complement (C1) receptor and identification as p15E. J Exp Med. 1978;147:844–853. doi: 10.1084/jem.147.3.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe DP, Cooper NR. Neutralization of vesicular stomatitis virus (VSV) by human complement requires a natural IgM antibody present in human serum. J Immunol. 1981;126:1562–1568. [PubMed] [Google Scholar]
- Bergmann-Leitner ES, Leitner WW, Tsokos GC. Complement 3d: from molecular adjuvant to target of immune escape mechanisms. Clin Immunol. 2006;121:177–185. doi: 10.1016/j.clim.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Blue CE, Spiller OB, Blackbourn DJ. The relevance of complement to virus biology. Virology. 2004;319:176–184. doi: 10.1016/j.virol.2003.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA, Sen GC. Innate responses to viral infections. In: Fields B, Knipe D, Howley P, editors. Fields Virology. 5. Lippincott Williams and Wilkins Publishers; Philadelphia, Pa: 2007. pp. 249–278. [Google Scholar]
- Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–986. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- Cooper NR, Polley MJ, Oldstone MBA. Failure of terminal complement components to induce lysis of Moloney virus transformed lymphocytes. J Immunol. 1974;112:866–868. [PubMed] [Google Scholar]
- Cooper NR, Jensen FC, Welsh RM, Jr, Oldstone MBA. Lysis of RNA tumor viruses by human serum: direct antibody-independent triggering of the classical complement pathway. J Exp Med. 1976;144:970–984. doi: 10.1084/jem.144.4.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CA, Bodner S, Trofatter KF., Jr Scanning and transmission electron microscopic studies of complement-mediated lysis and antibody-dependent cell-mediated cytolysis of herpes simplex virus-infected human fibroblasts. Am J Pathol. 1980;100:663–682. [PMC free article] [PubMed] [Google Scholar]
- Delgado MF, Polack FP. Involvement of antibody, complement and cellular immunity in the pathogenesis of enhanced respiratory syncytial virus disease. Expert Rev Vaccines. 2004;3:693–700. doi: 10.1586/14760584.3.6.693. [DOI] [PubMed] [Google Scholar]
- Des Prez RM, Bryan CS, Hawiger J, Colley DG. Function of the classical and alternate pathways of human complement in serum treated with ethylene glycol tetraacetic acid and MgCl2-ethylene glycol tetraacetic acid. Infect Immun. 1975;11:1235–1243. doi: 10.1128/iai.11.6.1235-1243.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux P, Christiansen D, Plumet S, Gerlier D. Cell surface activation of the alternative complement pathway by the fusion protein of measles virus. J Gen Virol. 2004;85:1665–1673. doi: 10.1099/vir.0.79880-0. [DOI] [PubMed] [Google Scholar]
- Dillon PJ, Parks GD. Role for the phosphoprotein P subunit of the paramyxovirus polymerase in limiting induction of host cell antiviral responses. J Virol. 2007;81:11116–11127. doi: 10.1128/JVI.01360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebenbichler CF, Thielens NM, Vornhagen R, Marschang P, Arlaud GJ, Dierich MP. Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J Exp Med. 1991;174:1417–1424. doi: 10.1084/jem.174.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnst AC. Complement activation by measles virus cytotoxic antibodies: alternative pathway C activation by hemagglutination-inhibition antibodies but classical activation by hemolysin antibodies. J Immunol. 1977;118:533–539. [PubMed] [Google Scholar]
- Friedman HM, Wang L, Fishman NO, Lambris JD, Eisenberg RJ, Cohen GH, Lubinski J. Immune evasion properties of herpes simplex virus type 1 glyco-protein gC. J Virol. 1996;70:4253–4260. doi: 10.1128/jvi.70.7.4253-4260.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HM, Wang L, Pangburn MK, Lambris JD, Lubinski J. Novel mechanism of antibody-independent complement neutralization of Herpes Simplex virus type 1. J Immunol. 2000;165:4528–4536. doi: 10.4049/jimmunol.165.8.4528. [DOI] [PubMed] [Google Scholar]
- Gadjeva M, Paludan SR, Theil S, Slavov V, Ruseva M, Eriksson K, Lowhagen GB, Shi L, Takahashi K, Ezekowitz A, Jensenius JC. Mannan-binding lectin modulates the response to HSV-2 infection. Clin Exp Immunol. 2004;138:304–311. doi: 10.1111/j.1365-2249.2004.02616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasque P. Complement: a unique innate immune sensor for danger signals. Mol Immunol. 2004;41:1089–1098. doi: 10.1016/j.molimm.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Hammer CH, Nicholson A, Mayer MM. On the mechanism of cytolysis by complement: evidence on insertion of C5b and C7 subunits of the C5b,6,7 complex into phospholipid bilayers of erythrocyte membranes. Proc Natl Acad Sci U S A. 1975;72(12):5076–5080. doi: 10.1073/pnas.72.12.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn KL, Sastry K, White MR, Anders EM, Super M, Ezekowitz RA, Tauber AI. Human mannose-binding protein functions as an opsonin for influenza A viruses. J Clin Invest. 1993;91(4):1414–1420. doi: 10.1172/JCI116345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch RL, Winkelstein JA, Griffin DE. The role of complement in viral infections III. Activation of the classical and alternative complement pathways by Sindbis virus. J Immunol. 1980;124:2507–2510. [PubMed] [Google Scholar]
- Hirsch RL, Wolinsky JS, Winkelstein JA. Activation of the alternative complement pathway by mumps infected cells: relationship to viral neuraminidase activity. Arch Virol. 1986;87:181–190. doi: 10.1007/BF01315298. [DOI] [PubMed] [Google Scholar]
- Ikeda F, Haraguchi Y, Jinno A, Iino Y, Morishita Y, Shiraki H, Hoshino H. Human complement component C1q inhibits the infectivity of cell-free HTLV-I. J Immunol. 1998;161(10):5712–5719. [PubMed] [Google Scholar]
- Ishii Y, Shimomura H, Itoh M, Miyake M, Ikeda F, Miyaike J, Fujioka S, Iwasaki Y, Tsuji HH, Suji T. Cold activation of serum complement in patients with chronic hepatitis C: study on activating pathway and involvement of IgG. Acta Med Okayama. 2001;55(4):229–235. doi: 10.18926/AMO/31989. [DOI] [PubMed] [Google Scholar]
- Jayasekara JP, Moseman EA, Carroll MC. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J Virol. 2007;81(7):3487–3494. doi: 10.1128/JVI.02128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph BS, Cooper NR, Oldstone MBA. Immunologic injury of cultured cells infected with measles virus. I Role of IgG antibody and the alternative pathway. J Exp Med. 1975;141:761–774. [PMC free article] [PubMed] [Google Scholar]
- Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev, Immunol. 2007;7(1):9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- Kerr MA. The human complement system: assembly of the classical pathway C3 convertase. Biochem J. 1980;189:173–181. doi: 10.1042/bj1890173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RA, Parks GD. Paramyxoviridae: The viruses and their replication. In: Knipe David M, Howley Peter M, editors. Fields Virology. 5. Wolters Kluwer and Lippincott Williams and Wilkins; 2007. pp. 1449–1496. [Google Scholar]
- McSharry JJ, Pickering RJ, Caliguiri LA. Activation of the alternative complement pathway by enveloped viruses containing limited amounts of sialic acid. Virology. 1981;114:507–515. doi: 10.1016/0042-6822(81)90230-0. [DOI] [PubMed] [Google Scholar]
- Madico GJ, Ngampasutadol S, Gulati U, Vogel PA, Rice Ram S. Factor H binding and function in sialylated pathogenic neisseiae is influenced by gonococcal, but not meningococcal, porin. J Immunol. 2007;178:4489–4497. doi: 10.4049/jimmunol.178.7.4489. [DOI] [PubMed] [Google Scholar]
- Mehlhop E, Diamond MS. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J Exp Med. 2006;203(5):1371–1381. doi: 10.1084/jem.20052388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills BJ, Beebe DP, Cooper NR. Antibody-independent neutralization of vesicular stomatitis virus by human complement. II. Formation of VSV-lipoprotein complexes in human serum and complement–dependent viral lysis. J Immunol. 1979;123(6):2518–2524. [PubMed] [Google Scholar]
- Mold C, Bradt BM, Nemerow GR, Cooper NR. Activation of the alternative complement pathway by EBV and the viral envelope glycoprotein, gp350. J Immunol. 1988;140(11):3867–3874. [PubMed] [Google Scholar]
- Morrison TE, Fraser RJ, Smith PN, Mahalingam S, Heise MT. Complement contributes to inflammatory tissue destruction in a mouse model of Ross River virus-induced disease. J Virol. 2007;81:5132–5143. doi: 10.1128/JVI.02799-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojd J, Tecle T, Samuelsson A, Orvell C. Mumps virus neutralizing antibodies do not protect against reinfection with a heterologous mumps virus genotype. Vaccine. 2001;19:1727–1731. doi: 10.1016/s0264-410x(00)00392-3. [DOI] [PubMed] [Google Scholar]
- Oldstone MBA, Cooper NR, Larson DL. Formation and biologic role of polyoma virus-antibody complexes. A critical role for complement. J Exp Med. 1974;140:549–565. doi: 10.1084/jem.140.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Formation of the initial C3 convertase of the alternative pathway: acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. 1981;154:856–867. doi: 10.1084/jem.154.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks GD, Alexander-Miller MA. High avidity cytotoxic T lymphocytes to a foreign antigen are efficiently activated following immunization with a recombinant paramyxovirus, simian virus 5. J Gen Virol. 2002;83:1167–1172. doi: 10.1099/0022-1317-83-5-1167. [DOI] [PubMed] [Google Scholar]
- Parks GD, Young VA, Koumenis C, Wansley EK, Layer JL, Cooke KM. Controlled cell killing by a recombinant nonsegmented negative strand RNA virus. Virology. 2002;193:192–203. doi: 10.1006/viro.2001.1298. [DOI] [PubMed] [Google Scholar]
- Paterson RG, Leser GP, Shaughnessy MA, Lamb RA. Paramyxovirus SV5 V protein binds two atoms of zinc and is a structural component of virions. Virology. 1995;208:121–131. doi: 10.1006/viro.1995.1135. [DOI] [PubMed] [Google Scholar]
- Peltola H, Kulkarni PS, Kapre SV, Paunio M, Jadhav SS, Dhere RM. Mumps outbreaks in Canada and the United States: time for new thinking on mumps vaccines. Clin Infec Dis. 2007;45:459–466. doi: 10.1086/520028. [DOI] [PubMed] [Google Scholar]
- Podack ER, Kolb WP, Muller-Eberhard HJ. The C5b-9 complex: subunit composition of the classical and alternative pathway-generated complex. J Immunol. 1976;116(5):1431–1434. [PubMed] [Google Scholar]
- Reis ES, Falcao DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulation proteins factor 1 and factor H. Scand. J Immunol. 2006;63:155–168. doi: 10.1111/j.1365-3083.2006.01729.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal R, Carroll MC. Emerging patterns in complement-mediated pathogen recognition. Cell. 2006;125:29–32. doi: 10.1016/j.cell.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Sahu A, Lambris JD. Structure and biology of complement C3, a connecting link between innate and acquired immunity. Immunol Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- Sahu A, Isaacs SN, Soulika AM, Lambris JD. Interaction of vaccinia virus complement control proteins: Factor I mediated degradation of C3b to iC3b inactivates the alternative complement pathway. J Immunol. 1998;160:5596–5604. [PubMed] [Google Scholar]
- Sissons JG, Cooper NR, Oldstone MB. Alternative complement pathway-mediated lysis of measles virus infected cells: induction by IgG antibody bound to individual viral glycoproteins and comparative efficacy of F(ab’)2 and Fab’ fragments. J Immunol. 1979;123(5):2144–2149. [PubMed] [Google Scholar]
- Sissons JG, Oldstone MBA, Schreiber RD. Antibody-independent activation of the alternative complement pathway by measles virus-infected cells. Proc Natl Acad Sci U S A. 1980;77(1):559–562. doi: 10.1073/pnas.77.1.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins SM, Lin Y, Leser GP, Kramer KA, Haas DL, Howerth EW, Xu J, Kennett MJ, Durbin RK, Durbin JE, Tripp R, Lamb RA, He B. Recombinant parainfluenza virus 5 (PIV5) expressing the influenza A virus hemagglutinin provides immunity in mice to influenza A virus challenge. Virology. 2007;362:139–150. doi: 10.1016/j.virol.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valim YML, Lachmann PJ. The effect of antibody isotype and antigenic epitope density on the complement-fixing activity of immune complexes: a systematic study using chimeric anti-NIP antibodies with human Fc regions. Clin Exp Immunol. 1991;84:1–8. doi: 10.1111/j.1365-2249.1991.tb08115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasantha S, Coelingh KL, Murphy BR, Dourmashkin RR, Hammer CH, Frank MM, Fries L. Interactions of a nonneutralizing IgM antibody and complement in parainfluenza virus neutralization. Virology. 1988;167:433–441. [PubMed] [Google Scholar]
- Von Messling V, Cattaneo R. Toward novel vaccines and therapies based on negative-strand RNA viruses. Curr Top Microbiol Immunol. 2004;283:281–312. doi: 10.1007/978-3-662-06099-5_8. [DOI] [PubMed] [Google Scholar]
- Wansley EK, Parks GD. Naturally occurring substitutions in the P/V gene convert the noncytopathic paramyxovirus simian virus 5 into a virus that induces alpha/beta interferon synthesis and cell death. J Virol. 2002;76(20):10109–10121. doi: 10.1128/JVI.76.20.10109-10121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh RM., Jr Host cell modification of Lymphocytic choriomeningitis virus and Newcastle disease virus altering viral inactivation by human complement. J Immunol. 1977;118:348–354. [PubMed] [Google Scholar]
- Zipfel PF, Wurzner R, Skerka C. Complement evasion of pathogens: common strategies are shared by diverse organisms. Mol Immunol. 2007;44:3850–3857. doi: 10.1016/j.molimm.2007.06.149. [DOI] [PubMed] [Google Scholar]