

Abstract
Estrogen modulates tight junctional resistance through estrogen receptor-α-mediated remodeling of occludin. The objective of the study was to understand the mechanisms involved. Experiments using human normal vaginal-cervical epithelial cells showed that human normal vaginal-cervical epithelial cells secrete constitutively matrix-metalloproteinase-7 (MMP-7) into the luminal solution and that MMP-7 is necessary and sufficient to produce estrogen decrease of tight junctional resistance and remodeling of occludin. Treatment with estrogen stimulated activation of the pro-MMP-7 intracellularly and augmented secretion of the activated MMP-7 form. Steady-state levels of MMP-7 mRNA and protein were not affected by estrogen. Estrogen modulated phosphorylation of the MMP-7, but the changes were most likely secondary to changes in cellular MMP-7 mass. Estrogen increased coimmunoreactivity of MMP-7 with the Golgi protein GPP130. Tunicamycin and brefeldin-A had no effect on cellular MMP-7 but monensin (inhibitor of Golgi traffic) blocked estrogen effects, suggesting estrogen site of action is at the Golgi system. Estrogen increased generalized secretory activity, including of luminal exocytosis of polycarbohydrates. However, estrogen increased coimmunoreactivity of MMP-7 with synaptosomal-associated protein of 25 kDa in apical membranes, suggesting soluble N-ethylmaleimide sensitive fusion factor attachment protein receptor-facilitated exocytosis of MMP-7. Treatment with the vesicular-ATPase inhibitor bafilomycin A1 inhibited activation of MMP-7. These data suggest that estrogen up-regulates activation of the MMP-7 intracellularly, at the level of Golgi, and augments secretion of activated MMP-7 through soluble N-ethylmaleimide sensitive fusion factor attachment protein receptor-dependent exocytosis. On the other hand, estrogen acidification of the luminal solution would tend to alkalinize exocytotic vesicles and may lead to decreased activation of the MMP-7. These mechanisms acting in concert could be important for regulation and control of estrogen modulation of paracellular permeability in vivo.
The major route of transport in epithelia is the paracellular (intercellular) pathway, and transport in the paracellular pathway is controlled predominantly by the resistance of the tight junctions (RTJ). The RTJ is determined by tight junctional complexes that are usually located at apical regions of the cells. Tight-junction complexes extend in a belt-like manner around the perimeter of each epithelial cell and connect neighboring cells. The tight junction complexes are composed of cytoplasmic and integral transmembrane proteins (1). Cytoplasmic tight-junction proteins couple the transmembrane tight-junctional proteins to the cortical actin and mediate signal-dependent modifications of the RTJ (2). Transmembrane tight-junctional proteins are linear polymers with extracellular stretches that associate laterally with tight-junction strands in the apposing membrane of adjacent cells to seal the intercellular space (3).
In epithelia, transmembrane tight-junctional proteins are composed of claudins and occludins. Claudins are the main building units of the tight junction and confer the baseline gating properties of the intercellular space (4). Until recently the role of occludins was debated after conflicting reports whether occludins are necessary (1, 5–8) or sufficient (9, 10) to gate the intercellular space. In the low-resistance human, vaginal-cervical epithelia occludin is present in two main forms: the full-length 65-kDa wild-type isoform and a truncated 50-kDa form (11, 12). A shift from 65- to 50-kDa dominance, e.g. that induced by treatment with estrogen, was associated with a reversible decrease in RTJ (13–16), suggesting that occludin is important for gating the intercellular space. The data also suggest that estrogen regulates epithelial permeability through remodeling of occludin.
The main objective of the present study was to better understand the mechanism of estrogen modulation of occludin. Previous experiments using cultured human vaginal-cervical epithelia showed that the truncated 50-kDa occludin form is not a distinct translated product but probably a calpain-mediated proteolytic fragment (11, 12). Protease digestion analysis suggested that estrogen, in an effect mediated by the nuclear estrogen receptor (ER)- α induces proteolysis of occludin at the middle of extracellular loop-2, resulting in formation of a truncated 50-kDa isoform (11). Because occludin extracellular loops are important for gating the intercellular space, estrogen up-regulation of a defective truncated 50-kDa form can explain the estrogen decrease in RTJ.
Expression of occludin can be regulated (1, 17–21), but until recently no studies were published about hormone regulation of occludin. Similarly, only few others groups studied hormone regulation of RTJ (22–27), but the results did not provide in-depth understanding of the mechanisms involved. Remodeling of proteins at extracellular sites in normal cells usually involves matrix metalloproteinases (MMPs) (28, 29), whereas cysteine, aspartyl, and serine proteinases play a similar role in cancer cells or in cells exposed to bacterial and viral infections (e.g. 30, 31). MMPs actions in uterine epithelial and stromal cells are relatively well understood (e.g. Ref. 32), but until recently little was known about the expression, regulation, and actions of MMPs in normal human vaginal-cervical epithelial cells. Twenty-six vertebrate MMPs and 23 human homologues have been identified (29). Preliminary experiments in the laboratory showed that of those, only MMP-7 (matrilysin) was expressed, and estrogen regulated in human normal vaginal-cervical epithelial cells.
MMP-7, a hydrolyzing protease of proteoglycans and extracellular matrix glycoproteins, exists as an inactive pro from, which becomes activated by proteolytic cleavage. It is the smallest member of the MMPs, containing only the common catalytic domain and the Zn2+ binding region but missing the hemopexin-like domain common to the other MMPs (33). MMP-7 is widely expressed and is the main MMP of epithelial cells (34), including of the female reproductive tract (32, 35, 37). MMP-7 cleavage sites involve the Gly-Leu and Asp-Leu peptide bonds (38). Interestingly, occludin extra-cellular loop-2 has a Gly226-Leu227 sequence that can potentially serve as a MMP-7 cleavage site (39), and cleavage of occludin at the Gly226-Leu227 site would generate a segment of about 50 kDa. Based on these considerations, the hypothesis of the present study was that the 50-kDa occludin form found in estrogen-treated cells is the result of MMP-7-mediated cleavage of occludin at the 226–227 position, suggesting that MMP-7 mediates estrogen remodeling of occludin and the decrease in RTJ.
The results of experiments using cultured human normal vaginal-ectocervical epithelial cells (hEVECs) support the hypothesis, and suggest three novel mechanisms of estrogen modulation of occludin and the RTJ: 1) estrogen facilitates processing and activation of the MMP-7 in the Golgi, leading to augmented secretion of already activated MMP-7; 2) estrogen facilitates secretion of the pro-MMP-7 and the activated MMP-7 by soluble N-ethylmaleimide-sensitive fusion factor attachment protein receptor (SNARE)-dependent exocytosis; and 3) estrogen-dependent luminal acid secretion mediated by the apically located vesicular (V)-ATPase would tend to alkalinize cells and the exocytotic vesicles and may lead to decreased activation of the MMP-7.
Materials and Methods
Chemicals and supplies
All chemicals and supplies were obtained from Sigma Chemicals (St. Louis, MO) unless specified otherwise. [14C]glucosamine and [32P]orthophosphate were from PerkinElmer Life Sciences (Boston MA). Trans35S-label (Translabel) was obtained from ICI (Irvine, CA). Plasmin, leupeptin, and aprotinin were from Boehringer (Petersburg, VA); MMP-2 was from Chemicon (Temecula, CA). Recombinant human promatrilysin (r-pro-MMP-7, specific activity > 3100 U/mg) was from OYC Inc. (Andover, MA). Activation of MMP-2 and MMP-7 included pretreatment with 0.5 mM 4-aminophenylmercuric acetate (APMA; Aldrich Chemical Co., Milwaukee, WI) and subsequent dialysis to eliminate the APMA. The fluorescent dye FM1–43 was from Molecular Probes (currently Invitrogen, http://www.invitrogen.com). Anocell (Anocell-10) filters were obtained from Anotec (Oxon, UK, through Sigma). Millicell (Millicell-CM) filters were from Millipore (Bedford, MA). Centricon filter tubes were obtained from Amicon (Beverly, MA).
Primary antibodies
Mouse monoclonal antioccludin antibody was from Zymed Laboratory Inc. (San Francisco, CA) (no. 33–1500, which recognizes antigenic domains at the human C-terminal of the protein). Anti MMP-7 antibodies were from Research Diagnostics Inc. [RDI; Concord MA; and included the mouse antihuman MMP-7 (pro) monoclonal antibody (no. RDI-MMP7abm-B2, which binds the human pro-MMP-7 29- but not the active/latent 21-kDa MMP-7); the mouse antihuman MMP-7 monoclonal antibody (no. RDI-MMP7abm-F12, which binds the human active MMP-7 21 kDa but not the pro-MMP-7 29 kDa); and the rabbit polyclonal anti-MMP-7 antibody (no. RDI-MMP7Nab), which binds to human MMP-7, including the pro-MMP-7 29 kDa and the activated 21 kDa MMP-7 but does not cross-react with other MMP family members]. The mouse monoclonal anti-cis-Golgi integral membrane phosphoprotein 130-kDa (GPP130) antibody (A1/118, ALX-804–603) was from AXXORA (San Diego, CA). The mouse monoclonal antisynaptosomal-associated protein of 25 kDa (SNAP25) antibody was from Transduction Laboratories (BD Biosciences, San Diego, CA).
Cell culture techniques
Secondary/tertiary cultures of hEVECs were generated from minces of histologically normal ectocervix/vagina as described (40). The discarded tissues were collected by the Cooperative Human Tissue Network at University Hospitals of Cleveland and CASE University according to the institutional review board protocol 03-90-TG. Tissues were collected from a total of nine premenopausal women aged 37–46 yr who underwent hysterectomy for medical indications unrelated to the present study. None of the women were treated with steroid sex hormones for at least 3 months before surgery. hEVECs (also referred to in the past as hECE cells) were previously characterized as phenotypically resembling the stratified squamous vaginal-ectocervical epithelial cells (40). Cells chosen for experiments were those obtained from tissues reported as human papillomavirus negative, and cultures were routinely tested for mycoplasma. Cells were grown and maintained in culture dish at 37 C in DMEM/Ham’s F12 (3:1) supplemented with nonessential amino acids, adenine (1.8·10−4 M), penicillin (100 U/ml), streptomycin (100 mg/ml), gentamicin (50 ng/ml), L-glutamine (2 mM), insulin (5 μg/ml), hydrocortisone (1·10−6M), transferrin (5 μg/ml), triiodothyronine (2·10−9 M), epidermal growth factor (0.2 nM), and 8% fetal calf serum (Sigma) in 91%O2-9%CO2 humidified incubator. For experiments using cells attached on filters, cells were plated on either Anocell or Millicell filters. Filters were coated on their upper (luminal) surface with 3–5 μg/cm2 collagen type IV as described (40). Unless stated differently, the filter experiments involved treatments with drugs added to both the luminal and subluminal solutions. For assays, cultures were shifted to modified Ringer solution, composed of (in millimoles) NaCl (120), CaCl2 (1.2), MgSO4 (1.2), KCl (5), NaHCO3 (10 mM, before equilibration with 95% O2-5% CO2), HEPES (10), glucose (5), and 0.1% BSA plus 0.2 nM epidermal growth factor.
Primary cultures of human normal cervical stromal fibroblasts were generated from discarded ectocervix/vaginal tissues after the surface epithelium was dissected to generate the hEVEC cultures. Tissues were immersed in Hanks’ balanced salt solution (HBSS) plus 2.5% collagenase for 30 min at 37 C. The subepithelial surface was gently scraped with scalpel; the resulting suspension of cells was incubated for 15 min at 37 C in HBSS plus 1.5% trypsin and 5 mM EDTA followed by two washes and incubation for 5 min at 37 C in medium composed of DMEM/Ham’s F-12 (3:1) supplemented with 5% calf serum, L-glutamine (2 mM), penicillin (100 U/ml), streptomycin (100 μg/ml), and gentamicin (50 ng/ml). Cells were plated in the same medium, and the resulting secondary-tertiary cultures were composed of stromal fibroblasts as determined by morphology, expression of vimentin, and lack of expression of involu-crin and epithelial-type cytokeratins.
For experiments with estrogen-deficient conditions, cells on filters were shifted to steroid-free medium (SFM) for 3 d. This medium was composed of phenol-red-deficient (DMEM)/Ham’s F12 or RPMI 1640 (Sigma) containing 8% heat-inactivated fetal bovine serum that was previously treated with charcoal to remove steroids. Preparation of charcoal-treated serum was described (13); briefly, dextran-coated charcoal (Sigma) was dissolved at 8% in 0.15 M NaCl, autoclaved, mixed by stirring, spun, and the pellet resuspended as 1 g per 1.25 ml in H2O. Fetal bovine serum (Hyclone, Logan, UT) was mixed with the activated charcoal-dextran at 20:1 (vol/vol) and incubated for 45 min at 55 C. At the completion of incubation, the mixture was spun twice at 800 × g for 20 min, and the supernatant (serum) was decanted and collected.
Throughout the paper the term estrogen-depleted cells describes cells shifted to SFM for 3 d, whereas estrogen-treated cells refers to estrogen-depleted cells that were treated for 2 d before experiments with a physiological concentration (10 nM) of the naturally occurring 17β-estradiol.
Measurements of transepithelial electrical resistance (RTE)
Before experiments, filters containing cells were washed three times and preincubated for 15 min at 37 C in a modified Ringer buffer composed of (in millimoles) NaCl (120), KCl (5), NaHCO3 (10, before saturating with 95% O2-5% CO2), CaCl2 (1.2), MgSO4 (1), glucose (5), HEPES (10) (pH 7.4), and 0.1% BSA in volumes of 4.7–5.2 ml in the luminal and subluminal compartments. Changes in paracellular permeability were determined as changes in the RTE across filters mounted vertically in a modified Ussing chamber from successive measurements of the transepithelial potential difference (ΔPD, lumen negative) and the transepithelial electrical current (ΔI, obtained by measuring the current necessary to clamp the offset potential to zero and normalized to the 0.6 cm2 surface area of the filter) as RTE = ΔPD/ΔI. The experimental design of the electrophysiological measurements, including calibrations and controls, the significance of the ΔPD and ΔI and the conditions for optimal determinations of RTE across low resistance epithelia, e.g. hEV-ECs, have been described and discussed (41). In some experiments, determinations of RTE were done using the EVOM epithelial voltohm-meter (WPI, Sarasota, FL) (40). Those assays were done in a pediatric incubator to maintain 95% O2-5% CO2 and 37 C atmosphere, with a total volume of 0.2 and 1.0 ml in the luminal and subluminal solutions, respectively (40).
Determinations of the dilution potential (Vdil)
The experiments were performed in the Ussing chamber as described (41). Transepithelial Vdils were determined by measuring the effect of lowering NaCl in the luminal solution on changes in voltage generated across the epithelial culture. This was done by replacing the Ringer’s buffer in the luminal compartment (130 mM NaCl) with low (10 mM) NaCl solution. The latter buffer was similar to the Ringer’s solution except that it lacked the 120 mM NaCl and was supplemented with 240 mM sucrose to compensate for osmolarity. The methods of electrophys-iological data evaluation were previously described (41). Vdil was the measured potential difference (voltageSL − voltageL) after lowering NaCl in the luminal solution, corrected for the potential-electrodes asymmetry, where the subscripts SL and L are the subluminal and luminal solutions. The Henderson diffusion equation for monocations and monoanions was used to interpret the transepithelial dilution potential in terms of ionic permeabilities. With the assumption that Na+ and Cl− are the major permeant ionic species, the relative mobilities of Na+ and Cl− in the intercellular space uCl and uNa can be determined as beta = uCl/uNa = (K + |Vdil|)/(K − |Vdil|), where K + (R·T/F).ln-(NaSL/NaL) = 68.5 mV at the given [Na+ SL] = 130 mM and [Na+ L] = 10 mM (41). For simultaneous measurements of changes in RTE and Vdil, filters containing cells were mounted in the Ussing chamber in modified Ringer’s solution. After 10 min stabilization, the luminal solution was replaced with modified Ringer’s solution containing low NaCl (10 mM) plus 240 mM sucrose (to compensate for osmolarity). Drugs were added and determinations of changes in RTE and Vdil in real time were made and analyzed as described (41).
Real-time fluoroscopy of FM1–43-loaded cells attached on filters
Cells on filters were incubated with 5 μM FM1–43 for 5 min at 37 C and loaded with the drug using electroporation (Cell-Porator Electroporation system; Gibco, Los Angeles, CA). This method results in less than 5% dead cells, an adequate intracellular incorporation of the dye and minimal photobleaching and leakage of the dye (not shown). The fluorescent styrylpyridium dye FM1–43 labels recycling populations of secretory vesicles within cells. It is incorporated into the internal membrane of endosomes but does not cross the bilayer so that only the dye bound to internal membranes is retained. The dye is more fluorescent in hydrophobic than in hydrophilic environments, such that surface exposed dye is washed out with dye-free solutions. Subsequently decreases in FM1–43 fluorescence in cells attached on filters reflect dye loss by exocytosis. Changes in FM1–43 fluorescence were determined in a custom-designed fluorescence chamber as previously described (42) with some modifications. Filters containing FM1–43-loaded cells attached on filters were housed in a custom-designed fluorescence chamber. The chamber was constructed as a modified Ussing chamber to allow separation of the subluminal solution that bathes the basolateral membranes from the luminal solution that bathes the apical membranes. A bifurcated custom-designed fiberoptics was placed over the apical surface of the cells and served to send the excitation signal and capture the emitted signal from a constant horizontal field (cross-section) of about 2·104 cells (42). For assays, filters containing confluent cultures of FM1–43-loaded cells attached on filters were housed in the fluorescence chamber. Cells were washed with modified Ringer buffer and maintained for 3 h at 37 C in darkness with 15 sec fluorescent illuminations (488/565 nm excitation/emission) every 10 min. Changes in intracellular FM1–43 were determined relative to the signal strength at time 0.
Molecular biology techniques
The method for generation of ERα-antisense oligonucleotide (ASO; 5′-TCA TGG TCA TGG TCC GT-3′) and random coil control (RC) oli-gonucleotide (5′-AGA ACG TTA CTT ACA CTG-3′), and their usefulness in blocking ER′-dependent actions in human vaginal-cervical cells was recently described (43). MMP-7 expression was modulated by using MMP-7 antisense nucleotide (5′-GTA TAT GAT ACG ATC-3′) or the RC nucleotide (5′-GTA TTA GTA TCG AAC-3′) (44). RT-PCR assays used the following primers and conditions: human MMP-7 (matrilysin) (NM_002423) forward, 5′-TCT TTG GCC TAC CTA TAA CTG G-3′, reverse, 5′-CTA GAC TGC TAC CAT CCG TC-3′ (product size 352 bp, cycles 29); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (XM_227696) forward, 5′-CCA TGT TCG TCA TGG GTG TGA ACC A-3′, reverse, 5′-GCC AGT AGA GGC AGG GAT GAT GTT C-3′ (product size 229 bp, cycles 25).
Real-time quantitative RT-PCR was done as described (11) using i-Cycler (Bio-Rad Laboratories, Hercules, CA) following the manufacturer’s instructions and with the above primers. PCR conditions were: 95 C for 2 min, 50 cycles of 30 sec denaturation at 95 C, 30 sec of annealing at 60 C, 1 min of extension at 70 C, 80 cycles of 30 sec reaction at 55 C including data collection and real-time analysis, and cooling to 4 C. Results were calculated using the comparative threshold cycle method of relative quantitation.
Protein assays
Cell-fractionation by the freeze/thaw method and preparation of cytosol and plasma membrane-enriched fractions were described (45). Western immunoblotting were described (45). Aliquots, normalized to 15 μg protein, were fractionated by 6–10% SDS-PAGE and blotted by Western analysis, and bands were analyzed semiquantitatively by densitometry. Immunoprecipitation/immunoblotting were described (45). For phosphorylation assays, cells on filters were shifted to phosphate-free DMEM containing 10 mM HEPES (pH 7.4) at 37 C and treated with 100 μCi/ml [32P]orthophosphate plus 1 μg/ml microcystin L-R (Calbiochem, San Diego, CA). After treatments, cells were washed with ice-cold PBS, lysed in lysis buffer, and processed by immunoprecipitation with the indicated antibody. Samples containing equal amounts of protein were resolved on 6–10% polyacrylamide gels and dried under vacuum. Radioactive bands were visualized by PhosphorImager [Molecular Dynamics (Amersham), Piscataway, NJ] and exposure to x-ray film (45).
Translabel labeling
Cultures were washed with HBSS and incubated in methionine-deficient medium enriched with 0.45% glucose and 10% regular medium plus 10 μCi/ml Translabel for 18 h.
[14C]glucosamine labeling
Cultures were washed with HBSS and incubated in medium plus 10 μCi/ml [14C]glucosamine for 4 d during which time the medium was replaced once. Cultures were then washed with HBSS and incubated for an additional 4–6 h in serum-free medium plus 10 μCi/ml of the radiolabeled carbohydrate. At the end of incubation, the medium was collected for assay and stored at 4 C (not more than 1 h).
Separation of secreted molecules by molecular weight (MW)
Collected conditioned medium was transferred to microcentrifuge tubes and spun briefly in a high speed microcentrifuge to remove cell debris. The medium was then spun to separate molecules by MW, using Centricon (San Diego, CA) filter tubes as described (40). The medium was layered on top of a Centricon-100 filter and the tube was spun at 4 C in a Beckman (Fullerton, CA; model J2–21) centrifuge (JA20 Rotor) at 5000 × g for 3–5 h. Upon completion of spinning, 0.5 ml of 5 mM Tris/HCl (pH 7.4) was added to the top of the filter for an additional 40 min spin and the latter step repeated. The remaining fluid in the upper compartment (about 60 μl), containing secreted molecules with MW greater than 100,000 was collected and used for assays.
Zymography
Protease activity was analyzed by substrate gel electrophoresis (zymography) in polyacrylamide gels containing 1 mg/ml pig gelatin. Samples (10 μl of total homogenates or concentrated aliquots of conditioned medium) were dissolved in modified Laemmli sample buffer [containing 2.5% (vol/vol) sodium dodecyl sulfate without β-mercaptoethanol] and electrophoresed, without prior boiling, at 4 C. After removal of the sodium dodecyl sulfate by washing in 2.5% (vol/vol) Triton X-100 in 50 mM Tris/HCl (pH 7.5) for 1 h, the gels were incubated overnight at 37 C for 18 h with gentle shaking in a buffer containing 40 mM Tris/HCl (pH 7.5), 10 mM CaCl2, and protease inhibitors (50 μM each of carbobenzoxy-L-Phe-chloromethyl ketone, L-1-tosylamino-2-phenyl-ethylchloromethyl ketone, Nalpha-tosyl-L-lysine chloromethyl ketone, and 4-[2-aminoethyl]benzenesulfonyl fluoride; Calbiochem). Gels were stained with 0.1% Coomassie Brilliant Blue (Bio-Rad) in 40% MeOH and 10% acetic acid for 45 min and destained with 7% acetic acid to allow the identification of gelatinolytic activity as clear zones in a blue background (46).
Surface (apical) protein biotinylation
hEVECs cultured on Millicell filters were washed with ice-cold PBS for 5 min and incubated with biotinylation buffer [composed of 1.25 mg/ml N-hydroxysuccinimide S-S-biotin (Pierce Chemical Co., Milwaukee, WI) freshly diluted in 10 mM triethanolamine, 2 mM CaCl2, 150 mM NaCl (pH 7.5)], added to the luminal compartment facing the apical membranes. After 20 min incubation at 4 C with gentle stirring, cells were rinsed with PBS complemented with 0.1 mM CaCl2, 1 mM MgCl2, and 100 mM glycine (pH 7.5) for 20 min at 4 C to quench unreacted biotin. The cells were then rinsed twice with ice-cold PBS complemented with 0.1 mM CaCl2 and 1 mM MgCl2, scraped in ice-cold PBS, and pelleted by centrifugation at 800 × g at 4 C. The pellets were solubilized for 45 min in 20 μl of lysis buffer [1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 50 mM Tris (pH 7.5)] supplemented with a cocktail of proteinase inhibitor (Pierce) and 10 μM phenylmethylsulfonyl fluoride. The lysates were spun at 14,000 × g for 10 min at 4 C, and the supernatants were incubated overnight with streptavidin-agarose beads to recover biotinylated proteins. Samples from the streptavidin beads were collected in 2× sample buffer containing 10% β-mercaptoethanol and incubated for 20 min at room temperature. Samples were heated at 95 C for 3 min, separated by SDS-PAGE, and subjected to Western blot analysis with the appropriate antibodies.
Tissue inhibitor of metalloproteinase (TIMP)-1-TIMP-2 quantification
Commercially available ELISA kits were used to measure TIMP-1 (MTM100; R&D, Minneapolis MN) or TIMP-2 (RPN2618; Amersham, http://proteomics.amershambiosciences.com) using 200–400 μg of protein per assay according to suppliers’ protocols. The TIMP-1 ELISA detects approximately 80% of the TIMP-1 complexed with pro-MMP-9 and approximately 60% when complexed with active MMP-9, and it does not recognize other TIMPs. The TIMP-2 ELISA recognizes free TIMP-2 (but not TIMPs-1 or TIMPs-3) and TIMP-2 complexed with the active form of MMPs (with varying increased efficiencies) but does not detect TIMP-2 complexed with pro-MMP-2. Samples and standards were incubated in duplicate in the plates precoated with specific antibodies for 2 h at room temperature, washed, and incubated with horse-radish peroxidase-conjugated antibody to form an immobilized complex. The resulting color was read at 405 nm. TIMP-1 was measured in the range of 3–50 ng/ml (sensitivity of 1.25 ng/ml). TIMP-2 was measured in the range of 8–18 ng/ml (sensitivity of 3 ng/ml).
Data presentation and analysis
Data are presented as means (± SD), and significance of differences was estimated by ANOVA using GB-STAT (Dynamic Microsystems Inc., Silver Spring, MD).
Results
Modulation of the paracellular permeability
Treatment of hEVECs attached on filters with 10 nM 17β-estradiol for 2 d decreased the RTE from 50 Ω·cm2 to about 25 Ω·cm2 (Fig. 1A). The dose and duration of treatment were chosen to produce near maximal effect, based on previous studies (13–16). The effect of estrogen involved a decrease in the RTJ because treatment with estrogen also increased the paracellular ionic mobility of Cl− relative to Na+ (UCl/UNa) (Fig. 1B).
Fig. 1.
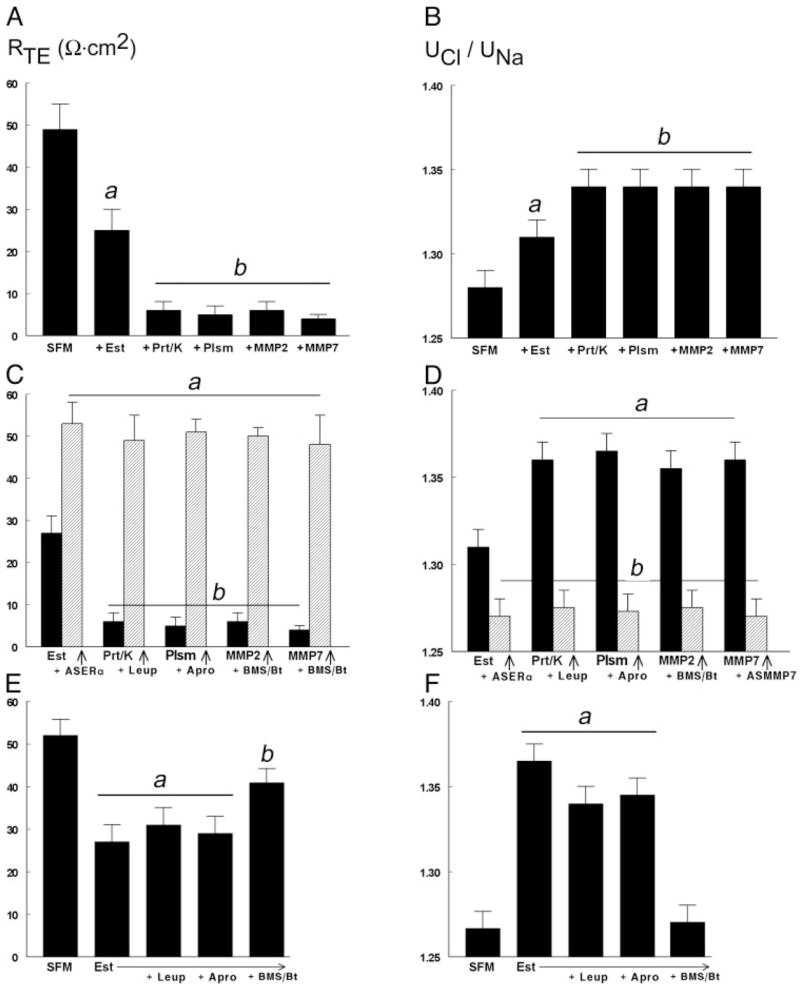
Modulation of RTE (A, C, and E) and the uCl/uNa (B, D, and F). Confluent cultures of hEVECs attached on filters were shifted to SFM for 3 d and treated for 2 d before assays with the vehicle (SFM), 10 nM 17β-estradiol (Est), or 10 nM 17β-estradiol plus 10 μM ERα anti-sense oligonucleotide (Est+ASERα). Additional treatments were one of the following proteases, alone or in combination with an inhibitor, added 15–30 min before assays to the luminal solution: proteinase-K (Prt/k, 20 μg/ml) ± leupeptin (Leup, 10 μg/ml); plasmin (Plsm, 10 μg/ml) ± aprotinin (Apro, 25 μg/ml); MMP-2 (10 μg/ml, plus 0.5 mM APMA) ± BMS-275291 (BMS, 5 μM) and batimastate (Bt, 5 μM); and MMP-7 (10 pg/ml, plus APMA) ± BMS-275291 and batimastate. Shown are means (± SD) of three to five repeats. A, P < 0.01, compared with SFM (A, B, E, and F) or Est (C and D); b, P < 0.04–0.01, compared with SFM and +Est (A, and B); Est+ASERα (C, and D); and SFM and Est+Leup and Est+Apro (E).
Similar to estrogen, treatments with extracellular proteases decreased the RTE as well. Using amounts and durations of drugs that produce near maximal effects (based on preliminary experiments), it was found that treatments with proteinase-K, plasmin, MMP-2, and MMP-7 decrease the RTE (Fig. 1A) by abrogation of the RTJ (Fig. 1B). The effects of estrogen on RTE and RTJ could be blocked by cotreatment with the ERβ antisense oligonucleotide (Fig. 1, C and D) but not with the antisense oligonucleotide for the ERβ or the antisense oligonucleotide for the G protein-coupled receptor GPR30 (not shown). The effects of proteinase-K could be blocked by cotreatment with leupeptin, those of plasmin by aprotinin, and those of MMP-2 and MMP-7 by BMS-275291 plus batimastate (Fig. 1, C and D). Cotreatments with leupeptin or aprotinin did not affect estrogen effects on RTE (Fig. 1E) or RTJ (Fig. 1F). In contrast, cotreatment with BMS-275291 plus batimastate inhibited estrogen decrease in RTE (Fig. 1E) and blocked estrogen increase in RTJ (Fig. 1F).
MMP-7 mediates estrogen modulation of RTJ
To ascertain the specificity of MMP-7 modulation of permeability, hEVECs were cotreated with the rabbit polyclonal anti-MMP-7 antibody MMP-7 Nab, which reacts with both the pro-MMP-7 and the activated MMP-7. As is shown in Fig. 2A, coincubation with the MMP-7 Nab blocked MMP-7 decrease in RTE.
Fig. 2.
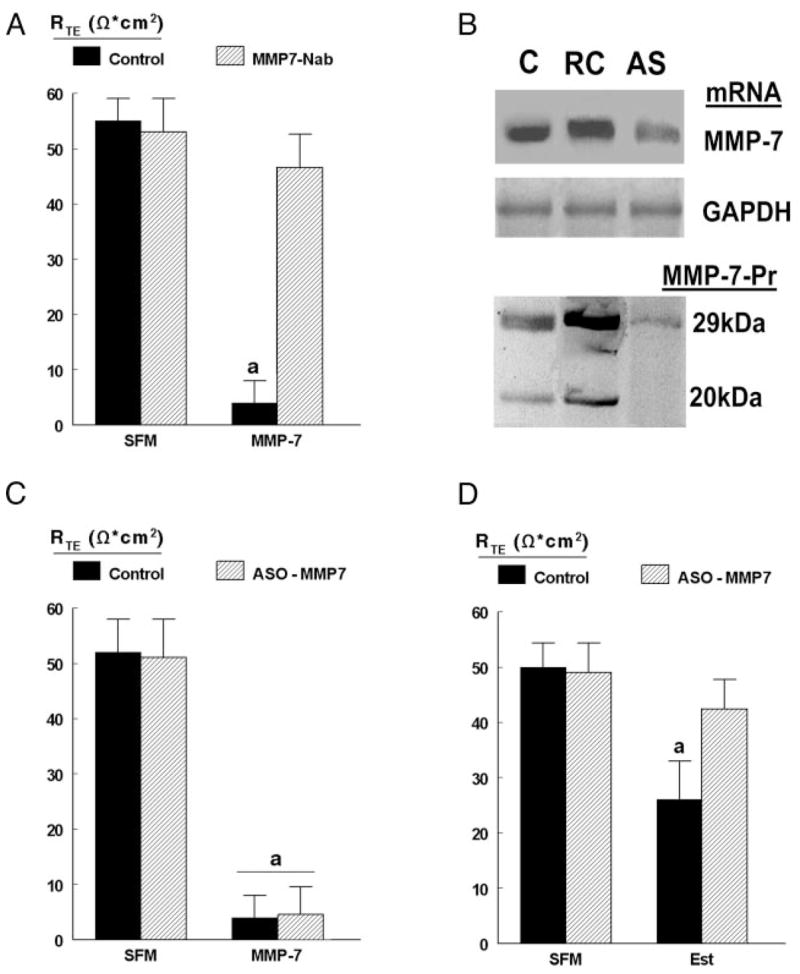
MMP-7 modulation of RTE. A, Estrogen-depleted cells attached on filters were treated with 10 pg/ml MMP-7 (plus APMA) for 15 min in the absence (filled bars) or presence (hatched bars) of excess MMP7Nab-pAb 1 μg/ml added to the luminal solution in a total volume of 0.2 ml 30 min before assays. Levels of RTE were determined using the EVOM epithelial voltohmmeter. B, Modulation of MMP-7 mRNA and MMP-7 protein with antisense oligonucleotides. hEVECs attached on filters were treated for 2 d with 10 μM of the MMP-7 antisense nucleotide (AS) or the RC nucleotide. C, Control (no transfections). Western blots used the MMP7Nab-pAb. C, Estrogen-depleted cells attached on filters were treated for 2 d with the vehicle (C, control, filled bars) or 10 μM MMP-7 ASO nucleotide (hatched bars). Some cultures were also treated with 10 pg/ml MMP-7 (plus APMA) for 15 min before assays. D, Estrogen-depleted (SFM) or estrogen-treated cells (Est) attached on filters were cotreated for 2 d before experiments with the vehicle (C, control, filled bars) or 10 μM MMP-7 ASO nucleotide (hatched bars). In C and D, levels of RTE were determined in the Ussing chamber. Shown are means (± SD) of three to six repeats. a, P < 0.01, compared with SFM control.
Treatment of hEVECs with the antisense oligonucleotide of MMP-7 alone had no significant effect on RTE, but it inhibited expression of MMP-7 mRNA and both the pro-MMP-7 (29 kDa) and activated MMP-7 (20 –21 kDa) proteins (Fig. 2B). Treatment with the MMP-7 antisense oligonucleotide did not affect decreases in RTE induced by cotreatment with MMP-7 (Fig. 2C). However, in estrogen-treated cells, cotreatment with the MMP-7 antisense oligonucleotide blocked estrogen decrease in RTE (Fig. 2D). Collectively, the data in Figs. 1 and 2 show that MMP-7 are sufficient and necessary for estrogen decrease in RTJ and therefore suggest that MMP-7 mediates estrogen decrease in RTJ.
MMP-7 effects on occludin
The reported mechanism by which estrogen modulates the RTJ is through remodeling of occludin (11). The result in Fig. 3A confirms (11) that treatment with estrogen augments the expression of a truncated 50-kDa form of occludin, which based on previous studies is a proteolytic fragment of the protein (11, 12). Treatment with the ERα antisense oligonucleotide alone had no significant effect, but cotreatment with the ERα antisense oligonucleotide blocked estrogen effect (Fig. 3A), suggesting that the ERα mediates estrogen effect.
Fig. 3.
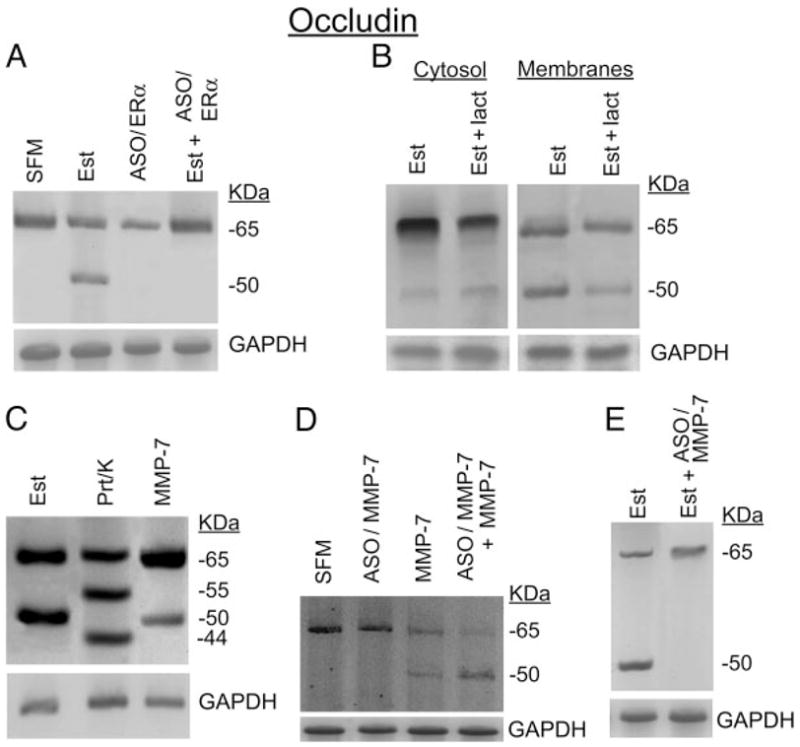
Modulation of occludin: occludin immunoblots of homogenates of hEVECs attached on filters. A, Estrogen-depleted (SFM) or estrogen-treated cells (Est) were co-treated for 2 d before assays with 10 μM ERα antisense oligonucleotide (ASO/ERα or Est+ASO/ERα). B, Estrogen-treated cells were cotreated for 6 h before assays with either the vehicle (Est) or lactacystin (lact., 10 μM, Est+lact). Cells were harvested, fractionated into cytosol- and plasma membrane-enriched fractions, and immunoblotted with antioccludin antibody. Similar results (not shown) were obtained with treatment with chloroquine (15 μM, 6 h). C, Cells were treated with 17β-estradiol (Est, 10 nM, 2 d); proteinase-K (Prt/k, 20 μg/ml, 30 min); or MMP-7 (10 pg/ml, plus 0.5 mM APMA, 15 min). Proteinase-K, MMP-7, and APMA were added to the luminal solution. D, Estrogen-depleted cells (SFM) were treated for 2 d before experiments with 10 μM MMP-7 antisense oligonucleotides (ASO/MMP-7) in the absence or presence of MMP-7 [10 pg/ml, plus 0.5 mM APMA, added 15 min before assays (both added to the luminal solution)]. E, Estrogen-treated cells were cotreated for 2 d before assays with the vehicle (Est) or 10 μM ERα ASO (ASO/ERα) plus MMP-7 [10 pg/ml, plus 0.5 mM APMA, added 15 min before assays (both added to the luminal solution)]. In A–E, after blotting with the antioccludin antibody, membranes were reprobed for immunoreaction with anti GAPDH antibody. The experiments were repeated three to four times with similar results.
To determine whether remodeling of occludin occurred intracellularly, estrogen-treated cells were cotreated for 6 h before assays with lactacystin (proteasome inhibitor) or chloroquine (inhibitor of lysosomes). Cytosolic and plasma-membrane-enriched fractions were prepared in the presence of protease inhibitors (to prevent recycling into the cytosol of occludin molecules degraded by extracellular proteinase) and were immunoblotted with antioccludin antibody. Figure 3B shows occludin immunoreactivity in both the cytosolic and membrane fractions, but the 50-kDa form was found predominantly in the membrane fraction. Cotreatment with lactacystin had no significant effect (Fig. 3B). Similar results were obtained in cells cotreated with chloroquine (not shown). These data suggest that occludin remodeling does not occur intracellularly.
The extracellular effects of MMP-7 on occludin expression are shown in Fig. 3C. Similar to estrogen, treatment with MMP-7 also induced expression of a 50-kDa occludin form. In contrast, proteinase-K induced de novo expression of 55-and 44-kDa bands (Fig. 3C and Ref. 11). Previous studies showed that plasmin induces expression of 55- and 48-kDa bands, and MMP-2 induces expression of 60-, 58-, 53-, and 45-kDa forms (11).
Pretreatment of estrogen-depleted cells with the MMP-7 antisense oligonucleotide did not affect occludin expression and did not affect MMP-7 induction of the occludin 50-kDa form (Fig. 3D). However, in estrogen-treated cells, cotreatment with the MMP-7 antisense oligonucleotide blocked expression of the occludin 50-kDa form (Fig. 3E). Collectively, the data in Fig. 3 show that MMP-7 are sufficient and necessary for estrogen remodeling of occludin and suggest that MMP-7 mediates estrogen remodeling of occludin.
Estrogen modulation of MMP-7 secretion and expression
Conditioned media were obtained from the subluminal and luminal compartments of hEVECs attached on filters, and MMP-7 expression was determined using anti-MMP-7 antibodies. No MMP-7 immunoreactivity was found in conditioned media obtained from the subluminal compartment (not shown). In contrast, immunoblots using luminal conditioned media showed significant MMP-7 immunoreactivity (Fig. 4, A–C). In those samples the anti-MMP-7 Nab pAb reacted with 29- and 21-kDa forms, which presumably represent the pro-MMP-7 and the activated MMP-7, respectively. The data in Fig. 4, A–C, also show a consistent pattern of estrogen regulation of MMP-7 secretion: treatment with estrogen decreased the density of the 29-kDa form (Fig. 4, A and B) and increased the density of the 21-kDa form (Figs. 4, A and C). Densitometry of three experiments showed that treatment with estrogen decreased the density of the 29-kDa form by about 4-fold and increased the density of the 21-kDa form by about 4-fold (Fig. 4, A–C). Cotreatment with the ERα antisense oligonucleotide blocked estrogen effects (Fig. 4, A–C).
Fig. 4.
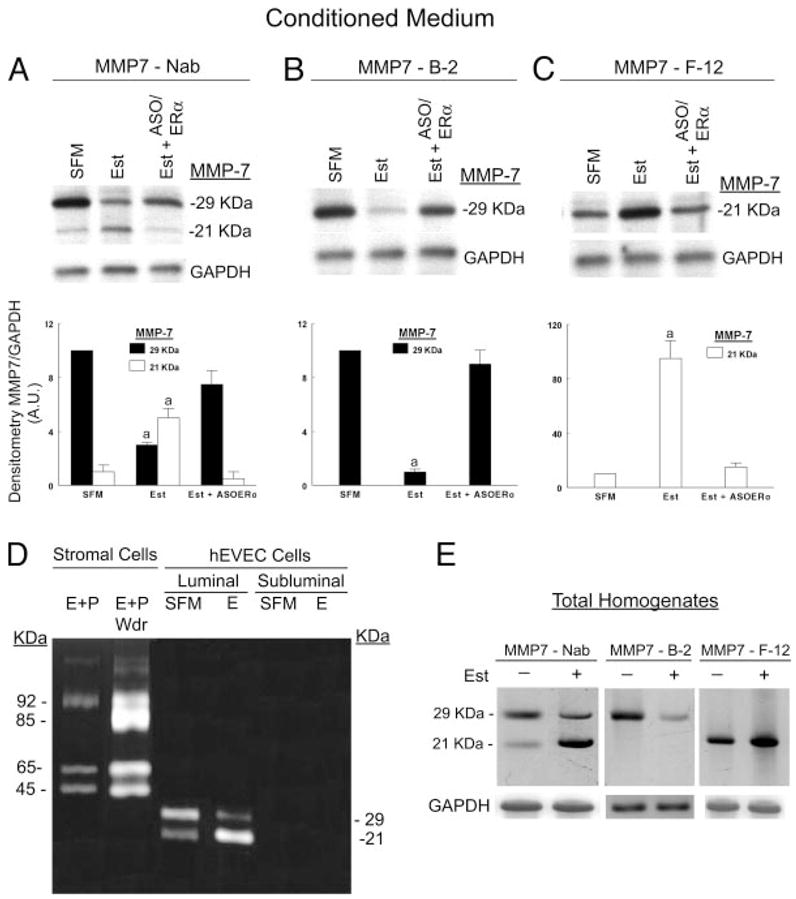
Estrogen modulation of MMP-7 secretion and expression. A–C, Conditioned media were obtained from the luminal compartment of estrogen-depleted cells (SFM); estrogen-treated cells (Est); or estrogen-treated cells that were cotreated for 2 d with 10 μM ERα ASO before assays (Est+ASO/ERα). Samples were concentrated by centrifugation, and aliquots containing equal amounts of proteins were separated by gel electrophoresis and immunoblotted with MMP-7 Nab antibody (A), MMP-7 B-2 antibody (B), or MMP–7 F-12 antibody (C). Results of three to six experiments are summarized in the graphs; densitometries of MMP-7 per GAPDH were normalized to an arbitrary unit (A.U.) of 10 of the SFM category in A. a, P < 0.01, compared with SFM and Est+ASO ERα. D, Gelatin zymography of secreted proteins from cervical stromal fibroblasts and hEVECs. Stromal cells (grown in cultures plates) and hEV-ECs (grown on filters) were shifted to SFM for 3 d. Stromal cells were treated with 10 nM 17β-estradiol plus 1 μM progesterone for 2 d (E+P) or 10 nM 17β-estradiol plus 1 μM progesterone for 2 d and then with SFM for additional 24 h[E+P/withdrawal (Wdr) to stimulate secretion of MMPs]. hEVECs were treated with 10 nM 17β-estradiol for 3 d (E). Aliquots of conditioned medium (10–20 μl) containing equal amounts of proteins were assayed by gelatin zymography under nonreducing conditions. Conditioned media of the stromal cells revealed the presence of 92/85- and 65/45-kDa bands, corresponding, respectively, to secreted pro-MMP-9/activated MMP-9 and pro-MMP-2/activated MMP-2. Conditioned media of hEVECs revealed the presence of 29/21-kDa bands, corresponding, respectively, to secreted pro-MMP-7/activated MMP-7. E, Total homogenates of the hEVECs from which conditioned media of D were obtained were fractionated by gel electrophoresis and immunoblotted with the MMP-7 Nab, MMP-7 B-2 or MMP–7 F-12 antibodies. The experiments in D and E were repeated three times with similar trends.
Estrogen modulation of activity of the secreted MMP-7 was determined by gelatin zymography under nonreducing conditions. Positive controls were conditioned media of human normal cervical stromal cells (fibroblasts). Conditioned media of cervical stromal cells treated with estrogen plus progesterone showed a significant gelatinolytic activity at 92, 65, and 45 kDa (Fig. 4D). In conditioned media of stromal cells treated with estrogen plus progesterone and then deprived of the hormones, the gelatinolytic activity was even greater, including an activity at 85 kDa (Fig. 4D). The 92/85-and 65/45-kDa gelatinolytic activities correspond, respectively, to secreted pro-MMP-9/activated MMP-9, and pro-MMP-2/activated MMP-2 (47–50).
No gelatinolytic activity was found in subluminal conditioned media of hEVEC cultures attached on filters; in contrast, a significant gelatinolytic activity at 29 and 21 kDa was found in luminal conditioned media (Fig. 4D). In estrogen-depleted cells, the 29-kDa gelatinolytic activity was more pronounced, whereas treatment with estrogen reversed the pattern of the effect (Fig. 4D). Homogenates of the cells from which the conditioned media were obtained showed a similar pattern of immunoreactivity with the MMP-7 antibodies (Fig. 4E) of the pro-MMP-7/activated MMP-7 as described in Fig. 4, A–C, for the conditioned media.
Collectively, the data in Fig. 4, A–E, suggest that hEVECs secrete MMP-7 to the luminal compartment and that treatment with estrogen stimulates activation of the pro-MMP-7 intracellularly to an activated MMP-7 form and augments secretion of the activated MMP-7.
Mechanisms of estrogen modulation of MMP-7 secretion
Steady-state mRNA and protein levels
Figure 5A (left panel) shows that steady-state levels of MMP-7 mRNA were not affected by estrogen treatment. Evaluation of estrogen effects on intracellular MMP-7 protein was done by labeling cells with Trans35S-label and immunoprecipitation of total cells homogenates with the MMP-7 Nab antibody. Figure 5A (right panel) shows that steady-state levels of MMP-7 protein were also not affected by estrogen treatment. Collectively, the data in Fig. 5A ruled out estrogen regulation of MMP-7 through transcription, posttranscriptional mRNA stabilization, and protein translation and stability. Instead, the results suggest posttranslational regulation of the MMP-7.
Fig. 5.
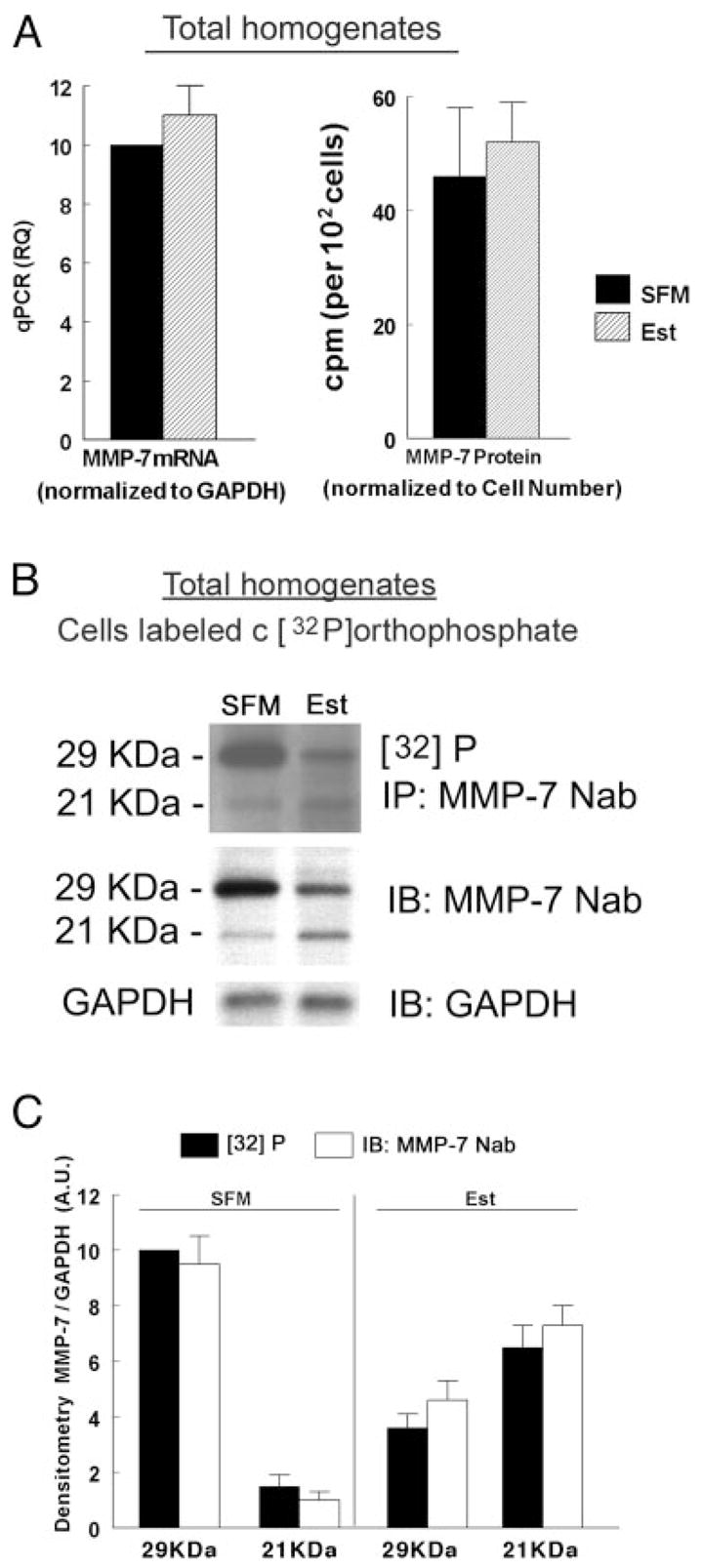
Estrogen effects on MMP-7 expression and phosphorylation. A, Left panel, Quantitative PCR analysis of MMP-7 mRNA in homogenates of estrogen-depleted (SFM) and estrogen-treated cells (Est). Data were normalized to GAPDH mRNA. Right panel, MMP-7 protein analysis. Estrogen-depleted and estrogen-treated cells were labeled with Trans35S-label. Total cells homogenates were immonoprecipitated (IP) with the MMP-7 Nab antibody, and radioactivity was determined in aliquots containing equal amounts of proteins. Data were normalized to cell number per dish. Shown are means (± SD) of three experiments. B and C, Effects on MMP-7 phosphorylation. Estrogen-depleted or estrogen-treated cells were labeled with [32P]orthophosphate. Total homogenates were immonoprecipitated with the MMP-7 Nab antibody and fractionated by gel electrophoresis. After visualization of the radioactive bands (upper panel), gels were immunoblotted (IB) with the MMP-7 Nab antibody, reblotted, and immunoblotted with anti GAPDH antibody (lower panels). The experiment was repeated three times with similar trends. C, Densitometry of the data in B (MMP-7 per GAPDH normalized to an arbitrary unit (A.U.) of 10 for the [32]P-29kDa-SFM category in panel B).
Phosphorylation
Figures 5, B and C, show that treatment with estrogen decreased the density of the radiolabeled 29-kDa band and increased the density of the radiolabeled 21-kDa band. However, those changes paralleled the changes in total 29/21-kDa MMP-7 proteins induced by estrogen. Therefore, most likely the data in Fig. 5B do not represent estrogen modulation of MMP-7 phosphorylation, and the changes are secondary to changes in cellular MMP-7 mass.
Processing in the endoplasmic reticulum (ER) and Golgi
The experiments in Fig. 6, A–D, tested estrogen modulation of MMP-7 processing and activation in the endoplasmic reticulum/Golgi. In Fig. 6A, total homogenates of estrogen-depleted and of estrogen-treated hEVECs were immunoprecipitated with the MMP-7 B-2 or MMP-7 F-12 antibodies and immunoblotted with the anti cis-Golgi protein GPP130 antibody. Treatment with estrogen increased the coimmunoreactivity of MMP-7 29 and 21 kDa with GPP130, without affecting cellular GAPDH. These results suggest that estrogen increases activation of the pro-MMP-7 in the Golgi network. To gain better understanding of the site of estrogen effects, cells were treated with tunicamycin (inhibitor of N-acetylglucosamine addition to dolicholphosphate, the first step in the formation of the core oligosaccharide in N-linked glycosylation); brefeldin-A (agent that fuses Golgi with endoplasmic reticulum and effectively blocks endoplasmic reticulum processing); or monensin (inhibitor of intra-Golgi traffic). Neither of the agents had any significant effect on cellular expression of the pro-MMP-7 when administered alone (Fig. 6, B–D). Also, when coadministered in estrogen-treated cells, neither tunicamycin nor brefeldin-A had any significant effect on the estrogen increase in cellular pro-MMP-7 (Fig. 6, B and C). In contrast, cotreatment with monensin blocked estrogen increase in cellular pro-MMP-7 (Fig. 6D). These results suggest that the estrogen site of action is at the Golgi system.
Fig. 6.
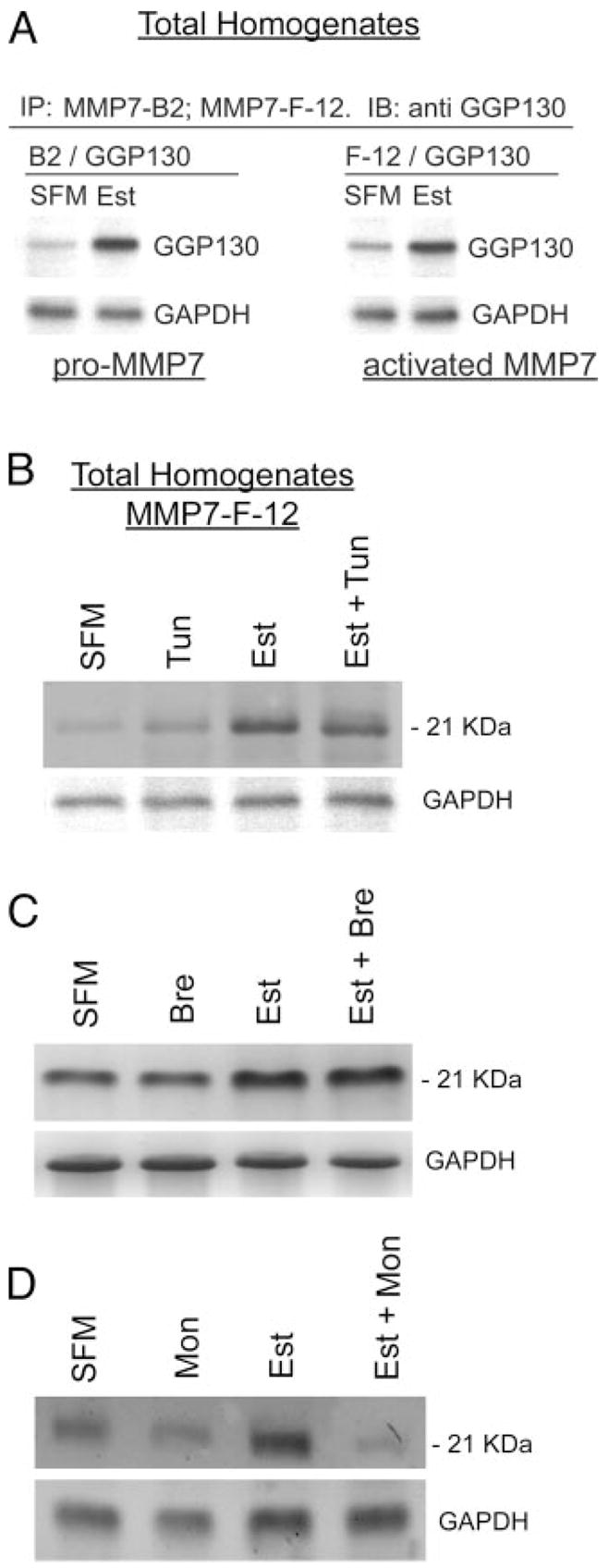
Estrogen modulation of MMP-7 processing. A, Total homogenates of estrogen-depleted (SFM) or estrogen-treated cells (Est) were fractionated by gel electrophoresis and immunoprecipitated (IP) with the MMP-7 B-2 or with the MMP-7 F-12 antibodies and immunoblotted (IB) with the anti-GPP130 antibody. B–D, Estrogen-depleted or estrogen-treated cells were cotreated with tunicamycin (B, tun; 10 μg/ml for 72 h), brefeldin-A (C, Bre; 10 μM for 120 min), or monensin (D, Mon; 10 μM for 150 min). Total homogenates were fractionated by gel electrophoresis and immunoblotted with the anti-MMP-7 F-12 antibody. Parallel fractions were immunoblotted with the anti-GAPDH antibody. The experiments were repeated three times with similar trends.
Exocytosis
The experiments in Fig. 7, A–C, tested estrogen regulation of MMP-7 secretion through exocytosis. Using fractions of apical membranes obtained by surface biotinylation, it was found that treatment with estrogen increased the coimmunoreactivity of MMP-7 29- and 21-kDa forms with SNAP25 (Fig. 7A). SNAP25 is part of the heterotrimeric SNARE complex that also includes the v-SNARE and syntaxin (51) and is the minimal machinery required for the exocytotic membrane fusion process. The results in Fig. 7A therefore suggest that estrogen facilitates exocytosis of MMP-7.
Fig. 7.
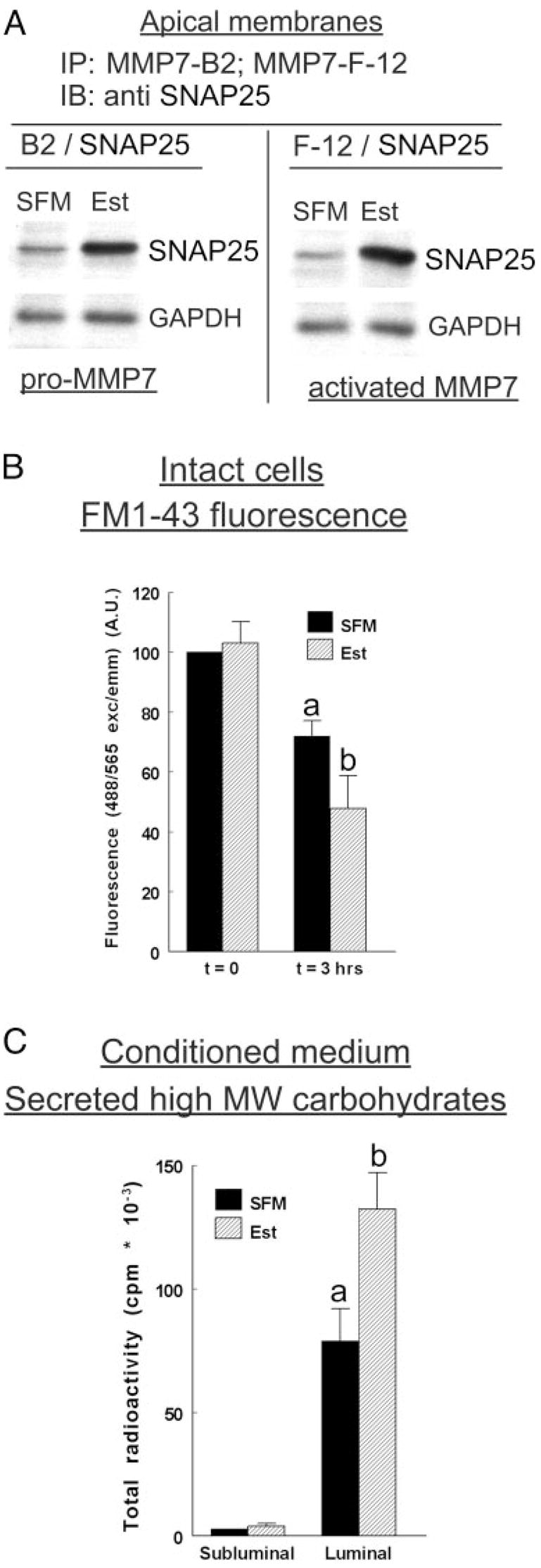
Estrogen modulation of MMP-7 exocytosis. A, Apical membranes were obtained by biotinylation from estrogen-depleted and estrogen-treated cells. Fractions were immunoprecipitated (IP) with the MMP-7 B-2 or the MMP-7 F-12 antibodies and immunoblotted (IB) with the anti-SNAP25 antibody. Parallel fractions were immunoblotted with the anti-GAPDH antibody. The experiments were repeated four times with similar trends. B, Effects on FM1–43 fluorescence. Estrogen-depleted or estrogen-treated cells attached on filters were loaded with FM1–43. The luminal and subluminal solutions were flushed continuously, and intracellular fluorescence (488/565 nm exc/emm) was determined at the indicated times. Data were normalized to fluorescence at t = 0 in the SFM category. a, P < 0.01, compared with t = 0, SFM or Est. b, P < 0.01, compared with t = 3 h, SFM category. C, Effects on secretion of high MW carbohydrates. Estrogen-depleted or estrogen-treated cells attached on filters were labeled with [14C]glucosamine. Conditioned media were obtained from the luminal or subluminal compartments. Samples were concentrated by centrifugation and separated by gel electrophoresis, and total radioactivity was counted in aliquots containing equal amounts of proteins. a, P < 0.01, compared with subluminal; b, P < 0.01, compared with SFM luminal. B and C show means (± SD) of three to four experiments.
To determine whether the effects on exocytosis are specific, two additional experiments were done. The first used the fluorescent styrylpyridium dye FM1–43, which labels recycling populations of secretory vesicles within cells (52). Using FM1–43-loaded hEVECs attached on filters, it was found that loss of FM1–43 fluorescence (i.e. exocytosis) was augmented by treatment with estrogen (Fig. 7B). The second experiment studied estrogen effect on secretion of [14C]glucosamine-labeled high MW (>100k) molecules, i.e. mucins (Ref. 40). The results confirmed (40) that hEVECs secrete high MW [14C]glucosamine-labeled molecules preferentially into the luminal compartment and that the effect is augmented by treatment with estrogen (Fig. 7C). Collectively, the data of Fig. 7, A–C, suggest that: 1) estrogen facilitates exocytosis of proteins and polycarbohydrates; 2) estrogen facilitates exocytosis of MMP-7 into the luminal compartment; 3) pro-MMP-7 and activated MMP-7 are both secreted by SNARE-mediated exocytosis; and 4) estrogen facilitates exocytosis of the MMP-7s.
Effects of bafilomycin A1 on extracellular acidity
Extracellular acidity can modulate activity of extracellular proteases (53, 54). In the woman, the luminal fluid in the vaginal and ectocervical canal is acidic, and the acidic milieu of the fluid is determined by active H+ secretion through V-ATPase located in the apical membrane of vaginal-ectocervical epithelial cells (55). Because the activity of the V-ATPase is estrogen dependent (55), the hypothesis was tested that secretion and luminal activation of the MMP-7 depend on estrogen regulation of luminal acid secretion.
It was previously shown that cotreatment of estrogen-treated cells attached on filters with the V-ATPase inhibitor bafilomycin A1 blocks acidification of the luminal fluid (55). However, estrogen-depleted cells do not significantly acidify the luminal solution, and in those cells, bafilomycin A1 does not induce a significant additional effect on luminal pH (55). Using a similar experimental design, estrogen-depleted or estrogen-treated hEVECs were cotreated with bafilomycin A1, added to the luminal solution. In estrogen-treated cells treatment with bafilomycin A1 attenuated estrogen-increase in the expression of cellular pro-MMP-7 (Fig. 8, A and B). Similarly, conditioned media of estrogen-treated hEVECs cotreated with bafilomycin A1 showed lesser expression of the 29- and 21-kDa MMP-7 forms (Fig. 8, C and D) and lesser MMP-7 gelatinolytic activity (Fig. 8E). The acidic milieu per se did not affect MMP-7 activation (Fig. 8F).
Fig. 8.
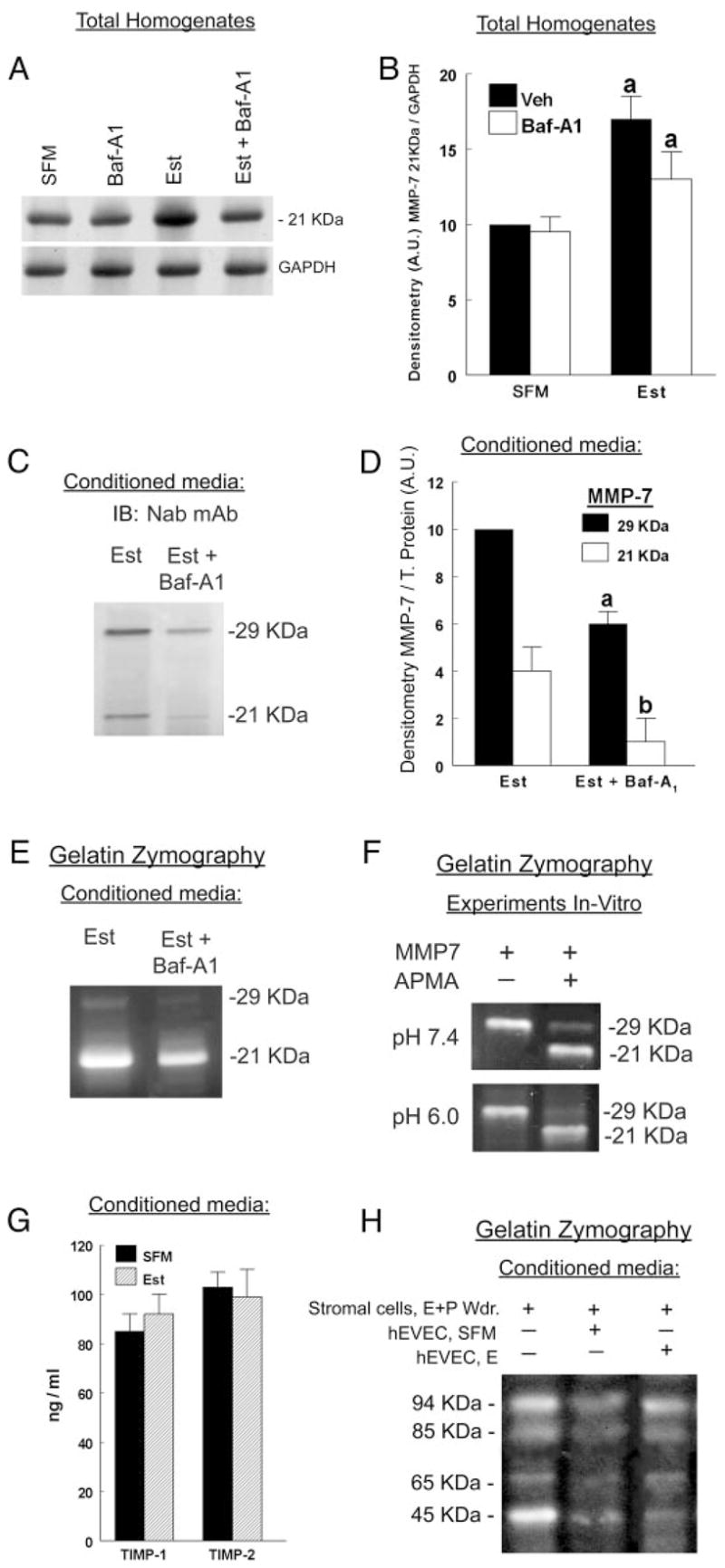
A–E, Effects of treatment with bafilomycin A1. Estrogen-depleted or estrogen-treated cells were cotreated for 30 min with bafilomycin A1 (Baf-A1, 1 μM, added to the luminal solution). A, Total homogenates were fractionated by gel electrophoresis and immunoblotted with the anti-MMP7-F-12 antibody. Parallel fractions were immunoblotted with the anti-GAPDH antibody. B, Densitometry of the data in A (means ± SD of three experiments). Veh, vehicle. Ratios of MMP-7 21 kDa per GAPDH were normalized to an arbitrary unit (A.U.) of 10 for the SFM-Veh category in A. C, Conditioned media were obtained from the luminal compartment of estrogen-only-treated cells (Est) or Est + Baf-A1-treated cells. Samples were concentrated by centrifugation, and aliquots containing equal amounts of proteins were separated by gel electrophoresis and immunoblotted (IB) with the MMP-7 Nab antibody. D, Densitometry of the data in C (means ± SD of three experiments). Ratios of MMP-7 per total (T.) protein were normalized to an arbitrary unit (A.U.) of 10 for the 29-kDa-Est category in C. a and b, P < 0.01, compared with Est. E, Samples of conditioned media from C were analyzed by gelatin zymography. Experiments were repeated three times with similar trends. F, pH effects on MMP-7 activity in vitro. MMP-7 (± APMA) were dissolved in PBS (pH 7.4 or 6.0), separated by gel electrophoresis, and assayed by gelatin zymography. The experiments were repeated three times with similar trends. G, Levels of TIMP-1 and TIMP-2 (means ± SD of three experiments) in luminal conditioned media of estrogen-depleted and estrogen-treated cells were determined by ELISA. H, Conditioned media were collected from the luminal compartments of cultures containing estrogen-depleted or estrogen-treated hEVECs and mixed with 1 μg/ml anti-MMP-7 Nab antibody (to inactivate the MMP-7). Eight microliters of the samples were mixed with 2 μl of conditioned media collected from cultures of stromal cells previously treated with estrogen plus progesterone followed by withdrawal from that treatment (as in Fig. 4). The combined mixed samples were fractionated by gel electrophoresis and assayed by gelatin zymography. The experiments were repeated three times with similar trends.
TIMPs
The results in Fig. 8G show that treatment with estrogen did not affect significantly the expression of immunoreactive TIMP-1 and TIMP-2 in the luminal conditioned medium. The experiment in Fig. 8H tested whether the estrogen increase in MMP-7 secretion and activation could be the result of blocking inhibitory control over TIMPs (56). Conditioned media were collected from cultures of human cervical stromal cells previously treated with estrogen plus progesterone followed by withdrawal from that treatment. In addition, luminal conditioned media were also collected from cultures of estrogen-depleted and estrogen-treated hEVECs and mixed with the anti MMP-7 Nab antibody to inactivate the MMP-7 (to eliminate possible proteolysis of MMP-7 on MMP-9 and MMP-2). The conditioned media of the stromal and hEVECs were mixed, fractionated by gel electrophoresis, and assayed by gelatin zymography. The objective was to determine whether estrogen stimulates secretion of inhibitors of TIMPs that would result in greater activation of the stromal cells’ MMP-9 and MMP-2 (e.g. Fig. 4D). The results in Fig. 8H show that conditioned media of hEVECs tended to decrease the gelatinolytic activity of the stromal cells’ activated MMP-2 (45 kDa) but had no effect on the pro-MMP-2 (65 kDa) or the pro-MMP-9 (94 kDa) and the activated MMP-9 (85 kDa). Treatment with estrogen did not affect those responses (Fig. 8H), suggesting that in hEVECs estrogen does not induce secretion of TIMPs inhibitors.
Discussion
The four novel findings of the present study are as follows: 1) MMP-7 is sufficient and necessary for estrogen decrease in RTJ and remodeling of occludin; 2) estrogen, via an ERα-mediated mechanism, up-regulates activation of MMP-7 in-tracellularly and its secretion into the luminal solution; 3) estrogen facilitates activation of MMP-7 in the Golgi network and augments SNARE-dependent exocytosis of pro- and activated MMP-7; and 4) treatment with bafilomycin A1 attenuates secretion of the pro-MMP-7. The results support the hypothesis that estrogen decrease in RTJ is mediated by MMP-7-dependent cleavage of occludin, possibly at the Gly226-Leu227 site of extracellular loop 2. The expected product would be a 50-kDa form, as we found (Refs. 11, 12 and present results). Formation of a truncated form would result in defective transmembrane junctional protein that fails to gate the intercellular space, leading to decreased RTJ. The results may be of physiological significance because experiments used a well-characterized system of human normal vaginal-cervical cells that maintain phenotypic characteristics of the native vaginal-ectocervical epithelia, and the doses of the natural 17β-estradiol were within the physiological range in the woman.
Occludin 65-kDa form was found both in the cytosolic and plasma membrane fraction of hEVECs (Fig. 3B). The cytosolic65-kDa occludin is most likely newly synthesized and intransit occludin, whereas that in the membrane fraction is the protein inserted into the plasma membrane junctional complex. Most of the occludin 50-kDa form was found in membrane fractions. This finding and the fact that lactacystin and chloroquine did not modulate estrogen occludin effect suggest that remodeling of occludin did not occur at intracellular sites because intracellular remodeling of proteins occurs in compartmentalized cytoplasmic proteasomes or lysosomes. MMP-7, on the other hand, exerted its effect on occludin extracellularly. Because estrogen effect is possibly mediated by MMP-7, the results suggest that estrogen modulation of occludin involves remodeling of occludin extracellular loop 2 by a direct effect at apical extracellular domains of the tight junction. MMP-7 substrates are numerous and, in addition to occludin, may include adherens junction proteins such as E-cadherin (29, 57, 58). Cleavage of E-cadherin can disrupt epithelial confluence and secondarily abrogate the RTJ (57). However, we have previously shown that in hEVECs estrogen does not affect E-cadherin (11), ruling out modulation of E-cadherin as the estrogen/MMP-7 mechanism of RTJ decrease.
One of the objectives of the study was to understand how estrogen regulates MMP-7 activity. The present results ruled out estrogen modulation of MMP-7 transcription and translation and suggest at least three posttranslational mechanisms: modulation of MMP-7 processing in the Golgi system, SNARE-dependent exocytosis, and modulation of vesicular acid secretion. Posttranslational processing of proteins, including the MMP-7, involves glycosylation and trafficking in the endoplasmic reticulum and Golgi networks before insertion in the plasma membranes or exocytosis. MMPs are synthesized as inactive zymogens with a presignal peptide at the N terminal that directs the MMP to the endoplasmic reticulum and is removed on protein maturation. Attached to the predomain is a propeptide containing cysteine switch element at the C-terminal end and a C-terminal catalytic domain. The prodomain maintains enzyme latency until it is removed or disrupted. Proteolytic cleavage of the propeptide converts inactive pro-MMP-7 into active MMP-7 through disruption of the Cys-Zn2+ interaction (29). The past theory of MMP posttranslational activation was that MMPs are constitutively secreted by exocytosis and activated in the extracellular fluid. More recent studies suggested that MMPs can be activated by intracellular signals before they reach the cell surface or are secreted (59, 60) and that MMPs secretion by exocytosis can be regulated (29). The latter hypotheses are supported by the present estrogen data.
The finding that treatment with monensin, inhibitor of intra-Golgi traffic, blocked estrogen increase in cellular pro-MMP-7 indicates that the MMP-7 is activated already in the Golgi or more proximal sites. Lack of an effect by tunicamycin (inhibitor of glycosylation) and brefeldin-A (modulator of endoplasmic reticulum-Golgi fusion) suggests that estrogen modulation of MMP-7 processing does not occur at the endoplasmic reticulum or endoplasmic reticulum-Golgi fusion sites. Whether the effect occurs at the cis-, medial-, or trans-Golgi systems is at present unknown.
The mechanism of estrogen activation of MMP-7 in the Golgi system is unknown. Treatment with ASO-ERα blocked estrogen modulation of the RTJ, occludin remodeling, and MMP-7 activation, suggesting involvement of the ERα mechanism. Lack of an effect on MMP-7 steady-state mRNA and protein levels rules out the involvement of the classical genotropic action mediated by the ERα/coactivator complex. Alternatively, estrogen acting via the ERα could increase activity of intermediate cascades that secondarily affect MMP-7 activation. Examples are genomic regulation of endothelial nitric oxide synthase leading to nongenomic modulation of activity of target proteins that control actin polymerization (16) and actin-myosin interaction (61). However, these cascades usually involve phosphorylation of the target protein, but the present results ruled out phosphorylation of the MMP-7. Therefore, one possibility would be modulation of activity of enzymes that are involved in the proteolytic cleavage of the propeptide that converts inactive pro-MMP-7 into an active MMP-7 (62).
The present results showed that estrogen enhances secretion of MMP-7 by SNARE-related exocytosis. The data also suggest that estrogen enhances secretion via other mechanisms. However, whereas the FM1–43 data are compatible with generalized enhanced secretion (63), the [14C]glucosamine-labeled high MW secretion and the SNARE-related exocytosis showed predominantly apical secretion. Because in epithelial cells membrane polarization is determined by the tight junctions, and because activated MMP-7 can disrupt occludin and decrease the RTJ (present results), the present data predict that MMP-7 proteolysis of occludin can unseal the intercellular space to diffusion of molecules. Transport of the MMP-7 and other MMPs from the luminal compartment into the subluminal compartment can disrupt basolateral aspects of the tight junctions as well as adherens molecules and desmosomes located more basally than the tight junctions. Proteolysis of tight junction proteins at apical and basolateral domains of the tight junction as well as other intercellular connecting proteins that form the intercellular bridges could lead to loss of epithelial barrier capacity. The presence of endogenous inhibitors of MMPs (TIMPs) and other control mechanisms is therefore important for maintaining the delicate balance of estrogen decrease in RTJ vs. loss of epithelial barrier capacity through overactivation and uncontrolled MMP-7 secretion. One such mechanism could be estrogen regulation of acid secretion.
The third mechanism of estrogen activation of MMP-7 could be by regulation of vesicular acidification. Treatment with bafilomycin A1 attenuated the gelatinolytic activity of secreted MMP-7. The effect was not direct because the gel-atinolytic activity of MMP-7 was not dependent on pH. More likely it was secondary to decreased intracellular activation of MMP-7 and decreased luminal secretion of the pro- and activated MMP-7. Treatment with bafilomycin A1 blocks the apically located plasma membrane V-ATPase (55), which is the driving force for apical acid secretion. However, treatment with bafilomycin A1 also blocks vesicular V-ATPases, which control the acidic vesicular milieu, including in exocytotic vesicles (64). Because treatment with bafilomycin A1 inhibited intracellular activation of MMP-7, it is possible that the presence of an acidic milieu is necessary for the continued activation of MMP-7 during its trafficking from Golgi through the exocytotic machinery.
hEVECs acidify their extracellular luminal milieu constitutively (55). This mechanism is estrogen dependent, primarily through estrogen up-regulation of apical membrane V-ATPase activity. The mechanism of estrogen action is not entirely clear. It involves the ERα but is not the result of transcription or translation regulation (55). It could be secondary to the metabotrophic effects of estrogen and its ability to increase influx of glucose and fluxes of lactate via glycolysis (65) that control the assembly of the V-ATPase complex (66) and the stoichiometry of the proton transport and AT-Pase activity (36). Whether estrogen also up-regulates the activity of vesicular V-ATPases is at present unknown. However, the present and our past data (55) suggest the opposite. Estrogen up-regulation of apical membrane V-ATPase is associated with enhanced intracellular alkalosis (Gorodeski, G. I., unpublished results). This effect would tend to increase the electrochemical gradient of the vesicular V-ATPases and may result in less vesicular acidification. Therefore, estrogen acidification of the luminal solution could lead to decreased vesicular acidification and inhibit MMP-7 activation.
The present results ruled out modulation of TIMP-1 and -2 and their activity status as the mechanism of estrogen modulation of MMP-7 activity. The possibility that estrogen modulates activity of other MMPs that can secondarily activate or block MMP-7 was not determined.
In conclusion, the present data suggest that estrogen modulation of the RTJ is mediated by MMP-7-dependent remodeling of occludin. Estrogen up-regulates activation of the MMP-7 intracellularly, at the level of Golgi, and augments secretion of activated MMP-7 through SNARE-dependent exocytosis. On the other hand, estrogen acidification of the luminal solution would tend to attenuate acidification of exocytotic vesicles and may lead to decreased activation of the MMP-7. These mechanisms acting in concert could be important for regulation and control of estrogen modulation of paracellular permeability in vivo.
Supplementary Material
Acknowledgments
The technical support of Kimberley Frieden, Brian De-Santis, and Dipika Pal is acknowledged.
This work was supported by National Institutes of Health Grants HD29924 and AG15955.
Abbreviations
- APMA
4-Aminophenylmercuric acetate
- ASO
anti-sense oligonucleotide
- ER
estrogen receptor
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HBSS
Hanks’ balanced salt solution
- hEVEC
human normal vaginal-ectocervical epithelial cell
- ΔI
change in electrical current
- MMP
matrix-metalloproteinase
- MW
molecular weight
- ΔPD
change in potential difference
- RC
random coil control
- RTE
transepithelial electrical resistance
- RTJ
resistance of the tight junctions
- SFM
steroid-free medium
- SNAP25
synaptosomal-associated protein of 25 kDa
- SNARE
soluble N-ethylmaleimide-sensitive fusion factor attachment protein receptor
- TIMP
tissue inhibitor of metalloproteinase
- V
vesicular
- Vdil
dilution potential
Footnotes
Disclosure: The author has nothing to disclose.
References
- 1.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–293. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 2.Van Itallie CM, Anderson JM. The molecular physiology of tight junction pores. Physiology. 2004;19:331–338. doi: 10.1152/physiol.00027.2004. [DOI] [PubMed] [Google Scholar]
- 3.Nusrat A, Brown GT, Tom J, Drake A, Bui TTT, Quan C, Mrsny RJ. Multiple protein interactions involving proposed extracellular loop domains of the tight junction protein occludin. Mol Biol Cell. 2005;16:1725–1734. doi: 10.1091/mbc.E04-06-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68:403–429. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- 5.McCarthy KM, Skare IB, Stankewich MC, Furuse M, Tsukita S, Rogers RA, Lynch RD, Schneeeberger EE. Occludin is a functional component of the tight junction. J Cell Sci. 1996;109:2287–2298. doi: 10.1242/jcs.109.9.2287. [DOI] [PubMed] [Google Scholar]
- 6.Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, Inazawa J, Fujimoto K, Tsukita S. Mammalian occludin in epithelial cells: its expression and subcellular distribution. Eur J Cell Biol. 1997;73:222–231. [PubMed] [Google Scholar]
- 7.Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136:399–409. doi: 10.1083/jcb.136.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balda MS, Flores-Maldonado C, Cereijido M, Matter K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J Cell Biochem. 2000;78:85–96. [PubMed] [Google Scholar]
- 9.Saitou M, Fujimoto K, Doi Y, Itoh M, Fujimoto T, Furuse M, Takano H, Noda T, Tsukita S. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol. 1998;141:397–408. doi: 10.1083/jcb.141.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubota K, Furuse M, Sasaki H, Sonoda N, Fujita K, Nagafuchi A, Tsukita S. Ca2+ -independent cell-adhesion activity of claudins, a family of integral membrane proteins localized at tight junctions. Curr Biol. 1999;9:1035–1038. doi: 10.1016/s0960-9822(99)80452-7. [DOI] [PubMed] [Google Scholar]
- 11.Zeng R, Li X, Gorodeski GI. Estrogen abrogates transcervical tight junctional resistance by acceleration of occludin modulation. J Clin Endocrinol Metab. 2004;89:5145–5155. doi: 10.1210/jc.2004-0823. [DOI] [PubMed] [Google Scholar]
- 12.Zhu L, Li X, Zeng R, Gorodeski GI. Changes in tight junctional resistance of the cervical epithelium are associated with modulation of content and phosphorylation of occludin 65 kDa and 50 kDa forms. Endocrinology. 2006;147:977–989. doi: 10.1210/en.2005-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorodeski GI. Estrogen increases the permeability of the cultured human cervical epithelium by modulating cell deformability. Am J Physiol. 1998;275:C888–C899. doi: 10.1152/ajpcell.1998.275.3.C888. [DOI] [PubMed] [Google Scholar]
- 14.Gorodeski GI. Effects of menopause and estrogen on cervical epithelial permeability. J Clin Endocrinol Metab. 2000;85:2584–2595. doi: 10.1210/jcem.85.7.6671. [DOI] [PubMed] [Google Scholar]
- 15.Gorodeski GI. Vaginal-cervical epithelial permeability decreases after menopause. Fertil Steril. 2000;76:753–761. doi: 10.1016/s0015-0282(01)02377-9. [DOI] [PubMed] [Google Scholar]
- 16.Gorodeski GI. Estrogen biphasic regulation of paracellular permeability of cultured human vaginal-cervical epithelia. J Clin Endocrinol Metab. 2001;86:4233–4243. doi: 10.1210/jcem.86.9.7817. [DOI] [PubMed] [Google Scholar]
- 17.Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol. 1997;137:1393–1401. doi: 10.1083/jcb.137.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordenonsi M, Mazzon E, De Rigo L, Baraldo S, Meggio F, Citi S. Occludin dephosphorylation in early development of Xenopus laevis. J Cell Sci. 1997;110:3131–3139. doi: 10.1242/jcs.110.24.3131. [DOI] [PubMed] [Google Scholar]
- 19.Tsukamoto T, Nigam S. Role of tyrosine phosphorylation in the reassembly of occludin and other tight junction proteins. Am J Physiol. 1999;276:F737–F750. doi: 10.1152/ajprenal.1999.276.5.F737. [DOI] [PubMed] [Google Scholar]
- 20.Clarke H, Soler AP, Mullin JM. Protein kinase C activation leads to dephosphorylation of occludin and tight junction permeability increase in LLC-PK1 epithelial cell sheets. J Cell Sci. 2000;113:3187–3196. doi: 10.1242/jcs.113.18.3187. [DOI] [PubMed] [Google Scholar]
- 21.Wachtel M, Bolliger MF, Ishihara H, Frei K, Bluethmann H, Gloor SM. Down-regulation of occludin expression in astrocytes by tumour necrosis factor (TNF) is mediated via TNF type-1 receptor and nuclear factor-kB activation. J Neurochem. 2001;78:155–162. doi: 10.1046/j.1471-4159.2001.00399.x. [DOI] [PubMed] [Google Scholar]
- 22.Fleet IR, Goode JA, Hamon MH, Laurie MS, Linzell JL, Peaker M. Secretory activity of goat mammary glands during pregnancy and the onset of lactation. J of Physiol. 1975;251:763–773. doi: 10.1113/jphysiol.1975.sp011120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neville MC, Peaker M. Ionized calcium in milk and the integrity of the mammary epithelium in the goat. J Physiol. 1981;313:561–570. doi: 10.1113/jphysiol.1981.sp013682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson GE. Cortisol and regulation of tight junctions in the mammary gland of the late-pregnant goat. J Dairy Res. 1996;63:305–308. doi: 10.1017/s0022029900031794. [DOI] [PubMed] [Google Scholar]
- 25.Stelwagen K, Farr VC, McFadden HA, Prosser CG, Davis SR. Time course of milk accumulation-induced opening of mammary tight junctions, and blood clearance of milk components. Am J Physiol. 1997;273:R379–R386. doi: 10.1152/ajpregu.1997.273.1.R379. [DOI] [PubMed] [Google Scholar]
- 26.Murphy CR. Junctional barrier complexes undergo major alterations during the plasma membrane transformation of uterine epithelial cells. Hum Reprod. 2000;3(Suppl):182–188. doi: 10.1093/humrep/15.suppl_3.182. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen DA, Parlow AF, Neville MC. Hormonal regulation of tight junction closure in the mouse mammary epithelium during the transition from pregnancy to lactation. J Endocrinol. 2001;170:347–356. doi: 10.1677/joe.0.1700347. [DOI] [PubMed] [Google Scholar]
- 28.Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM. Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol. 2000;18:1135–1149. doi: 10.1200/JCO.2000.18.5.1135. [DOI] [PubMed] [Google Scholar]
- 29.Sternlicht MD, Werb Z. How matrixmetalloproteinases regulate cell behavior. Annu ev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hooper JD, Clements JA, Quigley JP, Antalis TM. Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J Biol Chem. 2001;276:857–860. doi: 10.1074/jbc.R000020200. [DOI] [PubMed] [Google Scholar]
- 31.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 32.Rudolph-Owen LA, Slayden OD, Matrisian LM, Brenner RM. Matrix metalloproteinase expression in Macaca mulatta endometrium. Biol Reprod. 1998;59:1349–1359. doi: 10.1095/biolreprod59.6.1349. [DOI] [PubMed] [Google Scholar]
- 33.Wilson CL, Matrisian LM. Matrilysin: an epithelial matrix metalloproteinase with potentially novel functions. Int J Biochem Cell Biol. 1996;28:123–136. doi: 10.1016/1357-2725(95)00121-2. [DOI] [PubMed] [Google Scholar]
- 34.Parks WC, Lopez-Boado YS, Wilson CL. Matrilysin in epithelial repair and defense. Chest. 2001;120:36S–41S. doi: 10.1378/chest.120.1_suppl.s36. [DOI] [PubMed] [Google Scholar]
- 35.Woessner JF. Regulation of matrilysin in the rat uterus 1996. Biochem Cell Biol. 1996;74:777–784. doi: 10.1139/o96-084. [DOI] [PubMed] [Google Scholar]
- 36.Arata Y, Nishi T, Kawasaki-Nishi S, Shao E, Wilkens S, Forgac M. Structure, subunit function and regulation of the coated vesicle and yeast vacuolar [H(+)]-ATPases. Biochim Biophys Acta. 2002;1555:71–74. doi: 10.1016/s0005-2728(02)00257-8. [DOI] [PubMed] [Google Scholar]
- 37.Maymon E, Romero R, Pacora P, Gervasi MT, Edwin SS, Gomez R, Seubert DE. Matrilysin (matrix metalloproteinase 7) in parturition, premature rupture of membranes, and intrauterine infection. Am J Obstet Gynecol. 2000;182:1545–1553. doi: 10.1067/mob.2000.107652. [DOI] [PubMed] [Google Scholar]
- 38.Agnihotri R, Crawford HC, Haro H, Matrisian LM, Havrda MC, Liaw L. Osteopontin, a novel substrate for matrix metalloproteinase-3 (stromelysin-1) and matrix metalloproteinase-7 (matrilysin) J Biol Chem. 2001;276:28261–28267. doi: 10.1074/jbc.M103608200. [DOI] [PubMed] [Google Scholar]
- 39.Ando-Akatsuka Y, Saitou M, Hirase T, Kishi M, Sakakibara A, Itoh M, Yonemura S, Furuse M, Tsukita S. Interspecies diversity of the occludin sequence: cDNA cloning of human, mouse, dog and rat kangaroo homologues. J Cell Biol. 1996;133:43–47. doi: 10.1083/jcb.133.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorodeski GI, Romero MF, Hopfer U, Rorke E, Utian WH, Eckert RL. Human uterine cervical epithelial cells grown on permeable support—a new model for the study of differentiation and transepithelial transport. Differentiation. 1994;56:107–118. doi: 10.1046/j.1432-0436.1994.56120107.x. [DOI] [PubMed] [Google Scholar]
- 41.Gorodeski GI, Peterson D, De Santis BJ, Hopfer U. Nucleotide-receptor mediated decrease of tight-junctional permeability in cultured human cervical epithelium. Am J Physiol. 1996;270:C1715–C1725. doi: 10.1152/ajpcell.1996.270.6.C1715. [DOI] [PubMed] [Google Scholar]
- 42.Gorodeski GI, Whittembury J. A novel fluorescence chamber for the determination of volume changes in human CaSki cell cultures attached on filters. Cell Biochem Biophys. 1998;29:307–332. doi: 10.1007/BF02737900. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Zhou L, Gorodeski GI. Estrogen regulates epithelial cell deformability by modulation of cortical actomyosin through phosphorylation of non-muscle myosin-heavy-chain II-B filaments. Endocrinology. 2006;147:5236–5248. doi: 10.1210/en.2006-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Momiyama N, Koshikawa N, Ishikawa T, Ichikawa Y, Hasegawa S, Nagashima Y, Mitsuhashi M, Miyazaki K, Shimada H. Inhibitory effect of matrilysin antisense oligonucleotides on human colon cancer cell invasion in vitro. Mol Carcinog. 1998;22:57–63. [PubMed] [Google Scholar]
- 45.Feng YH, Wang L, Wang Q, Li X, Zeng R, Gorodeski GI. ATP ligation stimulates GRK-3–mediated phosphorylation and β-arrestin-2- and dynamin-dependent internalization of the P2X7-receptor. Am J Physiol. 2005;288:C1342–C1356. doi: 10.1152/ajpcell.00315.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woessner JF. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5:2145–2154. [PubMed] [Google Scholar]
- 47.Irwin JC, Kirk D, Gwatkin RBL, Navre M, Cannon P, Giudice LC. Human endometrial matrix metalloproteinase-2, a putative menstrual proteinase. J Clin Invest. 1996;97:438–447. doi: 10.1172/JCI118433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imada K, Ito A, Sato T, Namiki M, Nagase H, Mori Y. Hormonal regulation of matrix metalloproteinase 9/gelatinase B gene expression in rabbit uterine cervical fibroblasts. Biol Reprod. 1997;56:575–580. doi: 10.1095/biolreprod56.3.575. [DOI] [PubMed] [Google Scholar]
- 49.Salamonsen LA, Butt AR, Hammond FR, Garcia S, Zhang J. Production of endometrial matrix metalloproteinases, but not their tissue inhibitors, is modulated by progesterone withdrawal in an in vitro model for menstruation. J Clin Endocrinol Metab. 1997;82:1409–1415. doi: 10.1210/jcem.82.5.3920. [DOI] [PubMed] [Google Scholar]
- 50.Rigot V, Marbaix E, Lemoine P, Courtoy PJ, Eeckhout Y. In vivo peri-menstrual activation of progelatinase B (proMMP-9) in the human endometrium and its dependence on stromelysin 1 (MMP-3) ex vivo. Biochem J. 2001;358:275–280. doi: 10.1042/0264-6021:3580275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sørensen JB, Matti U, Wei SH, Nehring RB, Voets T, Ashery U, Binz T, Neher E, Rettig J. The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc Natl Acad Sci USA. 2002;99:1627–1632. doi: 10.1073/pnas.251673298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meffert MK, Calakos NC, Scheller RH, Schulman H. Nitric oxide modulates synaptic vesicle docking/fusion reactions. Neuron. 1996;16:1229–1236. doi: 10.1016/s0896-6273(00)80149-x. [DOI] [PubMed] [Google Scholar]
- 53.Davis GE. Identification of an abundant latent 94-kDa gelatin-degrading metalloprotease in human saliva which is activated by acid exposure: implications for a role in digestion of collagenous proteins. Arch Biochem Biophys. 1991;286:551–554. doi: 10.1016/0003-9861(91)90078-w. [DOI] [PubMed] [Google Scholar]
- 54.Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- 55.Gorodeski GI, Hopfer U, Liu CC, Margles E. Estrogen acidifies vaginal pH by upregulation of proton secretion via the apical membrane of vaginal-ectocervical epithelial cells. Endocrinology. 2005;146:816–824. doi: 10.1210/en.2004-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Salamonsen LA. Tissue inhibitor of metalloproteinases (TIMP)-1, -2 and -3 in human endometrium during the menstrual cycle. Mol Hum Reprod. 1997;3:735–741. doi: 10.1093/molehr/3.9.735. [DOI] [PubMed] [Google Scholar]
- 57.Sternlicht MD, Bissell MJ, Werb Z. The matrix metalloproteinase stromelysin-1 acts as a natural mammary tumor promoter. Oncogene. 2000;19:1102–1113. doi: 10.1038/sj.onc.1203347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davies G, Jiang WG, Mason MD. Matrilysin mediates extracellular cleavage of E-cadherin from prostate cancer cells: a key mechanism in hepatocyte growth factor/scatter factor-induced cell-cell dissociation and in vitro invasion. Clin Cancer Res. 2001;7:3289–3297. [PubMed] [Google Scholar]
- 59.Pei D, Weiss SJ. Furin-dependent intracellular activation of the human stromelysin-3 zymogen. Nature. 1995;375:244–247. doi: 10.1038/375244a0. [DOI] [PubMed] [Google Scholar]
- 60.Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 61.Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev. 2002;23:665–686. doi: 10.1210/er.2001-0045. [DOI] [PubMed] [Google Scholar]
- 62.Greenfield JP, Leung LW, Cai D, Kaasik K, Gross RS, Rodriguez-Boulan E, Greengard P, Xu H. Estrogen lowers Alzheimer β-amyloid generation by stimulating trans-Golgi network vesicle biogenesis. J Biol Chem. 2002;277:12128–12136. doi: 10.1074/jbc.M110009200. [DOI] [PubMed] [Google Scholar]
- 63.Pickett JA, Edwardson M. Compound exocytosis: mechanisms and functional significance. Traffic. 2006;7:109–116. doi: 10.1111/j.1600-0854.2005.00372.x. [DOI] [PubMed] [Google Scholar]
- 64.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar H+ -ATPase. Physiol Rev. 2003;84:1263–1314. doi: 10.1152/physrev.00045.2003. [DOI] [PubMed] [Google Scholar]
- 65.Neeman M, Degani H. Early estrogen-induced metabolic changes and their inhibition by actinomycin D and cycloheximide in human breast cancer cells: 31P and 13C NMR studies. Proc Natl Acad Sci USA. 1989;86:5585–5589. doi: 10.1073/pnas.86.14.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakamura S. Glucose activates H(+)-ATPase in kidney epithelial cells. Am J Physiol. 2004;287:C97–C105. doi: 10.1152/ajpcell.00469.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.