

Abstract
Mycobacterium tuberculosis produces three classes of mycolic acids that differ primarily in the presence and nature of oxygen-containing substituents in the distal portion of the meromycolate branch. The methoxymycolate series has a methoxy group adjacent to a methyl branch, in addition to a cyclopropane in the proximal position. Using the gene for the enzyme that introduces the distal cyclopropane (cma1) as a probe, we have cloned and sequenced a cluster of genes coding for four highly homologous methyl transferases (mma1–4). When introduced into Mycobacterium smegmatis, this gene cluster conferred the ability to synthesize methoxymycolates. By determining the structure of the mycolic acids produced following expression of each of these genes individually and in combination, we have elucidated the biosynthetic steps responsible for the production of the major series of methoxymycolates. The mma4 gene product (MMAS-4) catalyzes an unusual S-adenosyl-l-methionine-dependent transformation of the distal cis-olefin into a secondary alcohol with an adjacent methyl branch. MMAS-3 O-methylates this secondary alcohol to form the corresponding methyl ether, and MMAS-2 introduces a cis-cyclopropane in the proximal position of the methoxy series. The similarity of these reactions and the enzymes that catalyze them suggests that some of the structural diversity of mycolic acids results from different chemical fates of a common cationic intermediate, which in turn results from methyl group addition to an olefinic mycolate precursor.
Keywords: methyl transferase, mycolic acids, tuberculosis, cation, cyclopropane synthase
Mycobacterium tuberculosis is directly responsible for more human deaths than any other single infectious agent (1). The intrinsic resistance of this bacterium to many standard chemotherapeutic agents severely limits options for treatment (2). Isoniazid, which targets cell wall biosynthesis, is one of the most effective therapies available, yet it requires prolonged treatment regimens, lowering patient compliance and fostering drug resistance (3). The mycobacterial cell wall is the primary permeability barrier responsible for the intrinsic resistance to many hydrophobic antibiotic substances (4, 5). Understanding the biosynthesis, structure, and function of the mycobacterial cell wall is important, both from the perspective of providing novel drug targets and designing strategies for circumventing intrinsic resistance.
The cell wall contains a very high proportion of lipid in the form of mycolic acid, whose physical organization has recently been explored in detail (6, 7). Mycolic acids are long-chain, two-branched, 3-hydroxy fatty acids that range in size in mycobacteria from ≈60 to 80 carbons and contain a variety of functional groups on the long (mero) chain (8). These functional groups are either nonpolar moieties, such as olefins, methyl branches, and cyclopropanes, or polar moieties, such as ketones, methoxy groups, epoxides, and esters. The distribution of functional groups among species is distinct and useful for taxonomic identification of species (9, 10). In M. tuberculosis, three distinct mycolate species are produced, α mycolates containing two cis-cyclopropane rings and a methoxy and keto series, each of which contains a cis- or trans-cyclopropane in the proximal position, in addition to the distal oxygen function with an adjacent methyl branch (8, 11).
In M. tuberculosis and related organisms, methoxy- and ketomycolates are generally of lower abundance than α mycolates, although the exact ratios are dependent upon growth conditions (12). Ketomycolates occur in a variety of mycobacterial species (8), while methoxymycolates have only been isolated from M. tuberculosis (11), M. bovis BCG strain Moreau (but not strains Glaxo, Prague, or Pasteur; ref. 13), M. microti (12), M. marinum and M. ulcerans (14), M. asiaticum, M. gastri, M. gordonae, M. kansasii, M. szulgai, and M. africanum (10, 15). These studies were undertaken to explore the biosynthetic pathway and function of methoxymycolates in M. tuberculosis.
EXPERIMENTAL PROCEDURES
General Molecular Biological Techniques.
DNA manipulations and preparations from Escherichia coli were performed according to standard protocols (16) and as previously described (17). Mycobacterial chromosomal DNA was prepared according to the method of Bose et al. (18). Plasmid and cosmid DNA from Mycobacterium smegmatis was prepared following an initial lysis of a 1-ml culture by bead-beating (BioSpec Products, Bartlesville, OK) for 2–3 min with 0.1-mm glass beads (300 mg) in 0.3 ml of 50 mM Tris/10 mM EDTA, pH 8.0, with 100 μg of RNase A per ml.
Radiolabeling, Purification, and Analysis of Mycolic Acids.
Methylmycolates were prepared and precipitated as previously described (17, 19). Analytical one-dimensional normal and argentation TLC were performed as described (20). For NMR analysis, mycolates were prepared from 500-ml cultures labeled with 20 μCi of (1 Ci = 37 GBq) [1-14C]acetic acid. Preparative TLC plates were analyzed using a PhosphorImager (Molecular Dynamics). Excised bands were diethyl ether-extracted thrice and filtered through 0.45-μm polyvinylidene difluoride (PVDF) filters (Gelman). After removal of the ether, mycolates were reprecipitated before NMR. Samples were analyzed using a Varian Unity Plus 500-MHz NMR spectrophotometer in deuterochloroform (Chemir–Polytech Laboratories, St. Louis). Electron-impact mass spectra were collected at 70 eV for mycolate methyl esters on a VG analytical ZAB 2-SE high field mass spectrometer (M-Scan, West Chester, PA). Mass spectral analysis of all mycolates conformed to published spectra (8, 11, 12). Compound 1 (see Fig. 2), which has not been previously characterized, displayed the following mass spectrum: (m/z [relative abundance]) 824 [2], 796 [8], 768 [9], 741 [3], 543 [4], 515 [7], 487 [4], 459 [2], 382 [100], 368 [7], 339 [18], 284 [11], 256 [14].
Figure 2.
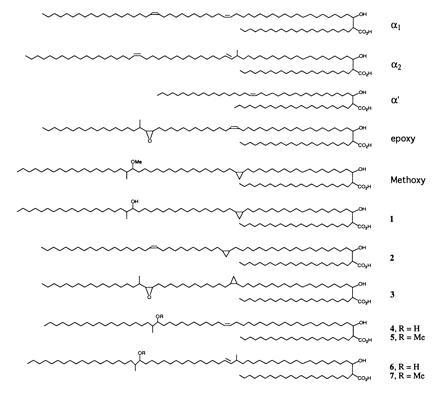
Structures of mycolic acids.
Vectors and Constructs.
For all subcloned constructs, the insert orientation and identity were confirmed by partial nucleotide sequence determination using primers that flank the polylinker of pMV206. mma1 was subcloned as a 1.2-kb NaeI–BamHI fragment from the 6.4-kb EcoRI insert into pMV206 and pMV261_Hyg [both vectors have been previously described (20)]. To subclone mma2 alone, a 1.1-kb StuI–NaeI fragment was gel-purified, HindIII linkers were ligated, and the resulting insert was cloned into both pMV206 and pMV261. To subclone mma3 alone, a 1.37-kb NaeI–HpaI fragment was excised from the EcoRI insert and blunt end-ligated into pMV206 digested with EcoRV to form pMV206_mma3. In addition, HindIII linkers were ligated to this insert, which was then ligated into pMV261. For mma4, a 1.74-kb fragment resulting from NcoI digestion of the pMV206 polylinker and insert sites was ligated to NcoI-cut pMV206 to form pMV206_mma4.
mma2 was excised as a 1.1-kb HindIII fragment from pMV206_mma2 and ligated into pMV206_mma4 to make pMV206_mma2+4. To produce the construct containing mma3 and mma4, pMV206_mma4 was digested with HindIII, and mma3 was ligated into this as a HindIII fragment from pMV206_mma3. To reconstitute the entire locus, we digested pMV206_mma2+4 with EcoRI and inserted the mma3 gene following the ligation of EcoRI linkers.
RESULTS
Identification of DNA Sequences Homologous to cma1.
Recently we identified two genes encoding the enzymes responsible for construction of the two cis-cyclopropanes in the major (α) mycolate series of M. tuberculosis (17, 20). Southern blot analysis using the cma1 gene as a probe revealed the presence of a 1-kb BamHI fragment of chromosomal DNA, which hybridized to this sequence even under stringent conditions (17). This fragment was cloned by size-fractionation of a BamHI digest of total chromosomal DNA. E. coli transformants containing pBluescript (Stratagene) ligated with such inserts were screened by hybridization, and three clones containing the 1-kb BamHI insert were isolated. Sequencing confirmed this homology, and this insert was then used as a probe to screen an M. tuberculosis H37Ra cosmid library in E. coli. Positive cosmids were isolated and rescreened by Southern analysis following BamHI and EcoRI digestion. Two independent cosmids containing the 1-kb cma1 cross-hybridizing fragment were identified.
Phenotype Associated with Heterologous Expression in M. smegmatis.
Both positive cosmids were transformed into M. smegmatis. Purified methyl mycolates labeled with [2-14C]acetic acid from these clones revealed a very complex pattern when analyzed by two-dimensional (2D) TLC (Fig. 1C; ref. 20). This 2D TLC system resolves all the major mycolate species of M. smegmatis, which are labeled α1, α2, α′, and e (epoxy) in Fig. 1A. The corresponding mycolate structures are shown in Fig. 2. These assignments were verified by purifying each mycolate species by a combination of preparative normal phase and argentation TLC. The 500-MHz 1H-NMR parameters for each of these species are shown in Table 1. In contrast, the pattern of mycolates observed from M. tuberculosis H37Rv is shown in Fig. 1B. These mycolates have been identified previously (8) and have been shown to be the α mycolates (Fig. 1B, α), methoxymycolates (Fig. 1B, m), and ketomycolates (Fig. 1B, k). The 1H-NMR spectral data for purified methyl methoxymycolate from M. tuberculosis are shown in Table 1. Introduction of the cosmid DNA containing the cma1-homologous fragment resulted in the TLC pattern shown in Fig. 1C. The new mycolate labeled m in this figure comigrated with authentic methoxymycolate from M. tuberculosis. This mycolate was purified by preparative argentation TLC and was identical by 1H-NMR and mass spectrometry to authentic methoxymycolate.
Figure 1.

2D TLC analysis of M. smegmatis (A), M. tuberculosis (B), M. smegmatis transformed with a cosmid carrying the mma locus (C), and M. smegmatis transformed with a plasmid carrying only the 6.4-kb EcoRI fragment with cma1 homology (D). The corresponding mycolate structures are shown in Fig. 2. From left to right, silica gel without silver ion impregnation (developed twice with 9:1 hexanes:ethyl acetate), from top to bottom, with silver ions (developed thrice with the same solvent). e, Epoxymycolate; k, ketomycolate (structure not shown); m, methoxymycolate; α, α mycolate from M. tuberculosis (structure not shown).
Table 1.
1H NMR parameters for methylmycolate species
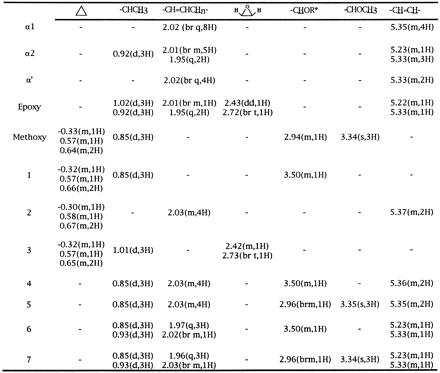
Spectra (500 MHz) were collected in deuterochloroform, and shift values are reported in ppm relative to internal CHCl3 (δ7.26 ppm). Methylmycolic acids were purified as described. The listed peaks are in addition to the signals present in all samples of methylmycolic acids; δ0.86 (−(CH2)nCH3, t, 6H), δ1.0–1.5 (−CH2(CH2)nCH2−, br s), δ1.56 (−CH2CH(OH)−, m, 2H), δ1.70 (−CH2CH(CO2Me)−, m, 2H), δ2.41 (−CH(CO2Me)−, m, 1H), δ3.63 (−CH(OH)−, br m, 1H), δ3.69 (−CO2CH3), s, 3H). α1 and α2 (21), as well as epoxy (22), assignments agree with those previously reported. t, Triplet; br, broad; s, singlet; m, multiplet; q, quartet; dd, doublet of doublets.
R = H for compounds 4 and 6, and R = Me for compounds 5 and 7.
Subcloning, Phenotype, and Growth Characteristics Associated with mma Expression.
Southern blot analysis showed that the 1-kb BamHI fragment hybridized with an EcoRI fragment of ≈6.4-kb common to both cosmids (data not shown). This fragment was cloned into the EcoRI site of pMV206 and used to transform M. smegmatis (Fig. 1D). The major species produced by transformants carrying this fragment was methoxymycolate. The next most abundant mycolate was identified by its 1H-NMR spectra and TLC mobility as the wild-type α′ mycolate. The final mycolate species produced (Fig. 1D, 1) was determined to correspond to the structure shown in Fig. 2 based on four observations; (i) the presence of a single proton multiplet at δ3.50 ppm in the 1H-NMR spectrum corresponding to a methine proton attached to a carbon bearing a hydroxyl group and the absence of signals related to the methyl ether of authentic methoxy; (ii) the presence of a three-proton doublet at δ0.85 ppm corresponding to a methyl branch on a carbon adjacent to a carbon bearing a hydroxyl group; (iii) the much lower TLC mobility of this mycolate species correlating with the presence of a polar functional group; and (iv) the appearance of an abundant fragment ion in the mass spectrum at m/z 339 corresponding to the expected favorable fragmentation adjacent to a secondary hydroxyl group. The loss of both α series and the appearance of methoxy- (Fig. 1D, m) and hydroxymycolates (Fig. 1D, 1) was accompanied by an alteration of the surface morphology of the recombinant organism (data not shown). Colonies carrying this plasmid were wetter and more domed appearing than the control organisms and formed much smaller colonies. This phenotype appeared to be very unstable in M. smegmatis and was lost quickly upon passage.
Mycolates containing secondary hydroxyl groups have previously been identified as resulting from the acid-catalyzed hydrolysis of the epoxymycolates of Mycobacterium fortuitum (23). The mycolates shown in Fig. 1 do not appear to have been derived from epoxymycolates, as epoxymycolates are still present in these samples and preparation of methyl mycolates using the milder aqueous tetrabutyl ammonium hydroxide hydrolysis (24) without subsequent acidification gave the same TLC pattern (data not shown).
Sequencing and Identification of the mma Gene Cluster.
The DNA sequence of the entire 6393-bp EcoRI fragment was determined and deposited in GenBank (accession no. U66108U66108). Open reading frame analysis of this region was performed as described (17). This analysis revealed the presence of six open reading frames as shown in Fig. 3A. The first of these showed significant homology to the yeast ABC1 protein, while the second open reading frame was most homologous to carboxylic acid esterases, two of which appear to be found in association with O-methyltransferases involved in the production of anthracyclines and daunorubicin (25, 26).
Figure 3.

(A) Open reading frame and restriction map of the 6.4-kb EcoRI fragment. (B) Multiple alignment of the protein sequences of CMAS-1, CMAS-2, MMAS-1, MMAS-2, MMAS-3, MMAS-4, and cyclopropane fatty acid synthase (CFAS).
All four of the remaining proteins were closely related to each other, cyclopropane mycolic acid synthase 1 (CMAS-1), CMAS-2 (17, 20), and the cyclopropane fatty acid synthase protein from E. coli (27). An alignment of all seven proteins is shown in Fig. 3B. These proteins all share a highly conserved S-adenosyl-l-methionine (SAM) binding domain around amino acids 98–111 (numbering according to the alignment shown in Fig. 3B), which can be represented for the family of mycobacterial enzymes as PGMTLLD(I/V)GCGWG. Several other highly conserved regions are shared by these enzymes, so that the minimum identity between any two members of this family is 52% at the amino acid level. These genes have been designated mma1–4 to indicate their involvement in methoxymycolic acid biosynthesis.
Subcloning Individual mma Activities.
To understand the functions of the individual members of the mma family, we subcloned each activity separately. A 1.2-kb fragment that contained only mma1 was cloned into pMV261 and pMV206. When transformed into M. smegmatis, this construct did not alter the phenotype of mycolates produced (data not shown). In the pMV261 construct, the orientation of the insert DNA is such that expression is driven by the mycobacterial HSP60 promoter (28). Likewise, subcloning of the 1.4-kb NaeI–HpaI fragment containing only mma3 into pMV206 and pMV261 did not alter the phenotype of the mycolates produced by transformed M. smegmatis.
A 1.1-kb fragment that contained only mma2 was also cloned into pMV206, and methyl mycolates from the resulting M. smegmatis transformants were analyzed by 2D TLC (Fig. 4A). Preparative normal and argentation TLC allowed the isolation and identification of the new mycolate species produced as compound 2 (Fig. 2; NMR parameters are shown in Table 1). Notably, this compound is the same mycolate produced by cis-cyclopropanation of the proximal position of the α mycolate series by cma2 in M. smegmatis (20). As with the CMAS-2 activity previously reported, mma2 produced a more polar mycolate (3), which was identified by a comparison of the 1H-NMR parameters of compound 3 and authentic epoxy mycolate, as shown in Table 1.
Figure 4.

2D TLC analysis of M. smegmatis recombinants with various subclones of the mma locus. (A) pMV206_mma2 (MMAS-2 only). (B) pMV206_mma4 (MMAS-4 only). (C) pMV206_mma2+4 (MMAS-2 + MMAS-4). (D) pMV206_mma3+4 (MMAS-3 + MMAS-4). TLC conditions were the same as those in Fig. 1, and mycolate species are identified in Fig. 2.
The 2-kb NcoI–EcoRI fragment, which contained only mma4 as an intact reading frame, was also cloned into pMV206. Mycolic acids from the resulting M. smegmatis transformants showed a complete absence of the α1 and α2 series of mycolates (Fig. 4B), and such organisms showed an altered surface morphology. Two abundant, more polar mycolate species were evident in addition to the mycolate identified as α′. Both of these mycolates had proton NMR signals at δ3.50 and at δ0.85 ppm (Table 1), suggesting that the distal position was equivalent to that observed in compound 1. Mycolate 4 contained olefinic resonances at δ5.36 ppm identical to the cis-olefinic linkage observed in the α1 series, thus this mycolate corresponds to structure 4 in Fig. 2. Mycolate 6 contained the more complex olefinic proton pattern consisting of signals at δ5.23 and δ5.33 ppm, which is associated with the trans linkage and adjacent methyl branch of the α2 series. In addition, mycolate 6 also displayed a second branched methyl group at δ0.93 identical to the methyl group observed in the α2 series. Thus this mycolate corresponds to structure 6 in Fig. 2.
Pairwise Reconstruction and Reconstitution of the mma Locus.
Since mma1 and mma3 appeared to have no independent function when cloned separately into wild-type M. smegmatis, we attempted to reconstitute the locus pairwise. First, mma4 was ligated into a pMV206 construct containing only mma2. Resulting M. smegmatis transformants produced the mycolate pattern shown in Fig. 4C. Purification of mycolates from this recombinant confirmed that species 1 was in fact the proximally cis-cyclopropanated, distally hydroxy, and methyl-branched species shown in Fig. 2. This result is consistent with the “CMAS-2”-like activity of mma2 acting upon a meromycolate precursor, which was also acted upon by an mma4-encoded activity. In support of this, the other major mycolate species produced in this recombinant is the cis-cyclopropanated epoxy species, compound 3.
To simultaneously express both mma4 and mma3, a DNA fragment containing mma3 was ligated into restricted pMV206_mma4. Mycolates isolated from M. smegmatis transformants displayed the complex phenotype shown in Fig. 4D. Mycolates 4 and 6 are still visible in this profile, as are the wild-type M. smegmatis mycolic acids α1, α2, α′, and epoxy. Two new species, which are slightly more polar than the α-series but are retarded to differing degrees by silver ion impregnation, are visible. The fastest moving of these in the silver dimension (7) displayed a clear singlet at δ3.34 ppm, indicating the presence of a methyl ether and the associated methyl branch doublet at δ0.85 ppm. In addition to these resonances, this mycolate also displayed olefinic resonances at δ5.23 and δ5.33 ppm, which appeared to be associated with a trans double bond with an adjacent methyl branch at δ0.92 ppm. The second of the new mycolate species (5) from this recombinant also possessed resonances characteristic of the methoxymycolate distal portions. In addition, this mycolate possessed olefinic resonances at δ5.35 ppm consistent with a cis double bond, which is also consistent with its slower mobility on silver TLC. Thus this mycolate corresponds to structure 5 in Fig. 2, the methoxy, methyl-branched derivative of the α1 series.
The final reconstitution experiment involved cloning mma3 back into the construct already expressing mma2 and mma4 (pMV206_mma2+4) to reconstitute the whole mma locus. This construct, when transformed back into M. smegmatis, was sufficient to fully restore the phenotype observed using the 6.4-kb EcoRI fragment.
DISCUSSION
The biosynthesis of methoxymycolates was previously thought to proceed through reduction of the corresponding ketomycolates or their precursors, with the methyl group of methionine being incorporated into both methoxy and methyl branch carbons (12, 14, 29). The isolation of long-chain fatty acids containing between 44 and 49 carbons possessing both cyclopropyl and methoxy substituents suggests that these modifications occur to the growing meromycolate chain, but the precise substrate is unknown (30). The nature of the enzymes responsible for ketomycolate biosynthesis is unknown. Ketomycolates could result from an oxidative transformation of the hydroxymycolate precursor by an as yet undescribed enzyme. While ketomycolates and unsaturated mycolates have been found to not be biosynthetically interconvertable at the level of intact mycolic acids in Mycobacterium aurum (31), the relationship between keto- and methoxymycolate precursors has not been determined. The intermediacy of ketomycolates (or ketone-containing mycolate precursors) in methoxymycolate biosynthesis is not supported by the present findings. MMAS-4 alone is capable of transforming the distal position of an olefinic meromycolate precursor into a secondary hydroxyl group with an adjacent methyl branch. MMAS-3 appears to operate on this newly formed hydroxyl group and O-methylates using a second SAM-derived methyl group. Finally, MMAS-2 appears to act upon the proximal cis-olefin and results in cis-cyclopropane formation. The reason for the apparent duplication of cis-cyclopropane synthase activities for the proximal position is unclear. One possible explanation is the preference of MMAS-2 over CMAS-2 for oxygenated mycolates (data not shown), suggesting that MMAS-2 may operate in vivo only on oxygenated mycolate series. In fact, in the presence of MMAS-4, MMAS-2 activity appears to be enhanced, since all of the mycolate species produced have only cis-cyclopropanes (Fig. 4C). This argues that MMAS-4 activity may precede MMAS-2 activity in the normal biosynthetic course, although this is obviously not a strict requirement, as MMAS-2 is functional in the absence of MMAS-4. The most likely course for the biosynthetic pathway is shown in Fig. 5, in which the action of MMAS-4 on an olefinic precursor that may be (at the proximal position) cis-olefin, trans-olefin, or not yet formed yields the distally hydroxy and methyl-substituted mycolate precursor. This product is then O-methylated by MMAS-3, and the proximal cis-cyclopropane is then introduced by MMAS-2.
Figure 5.
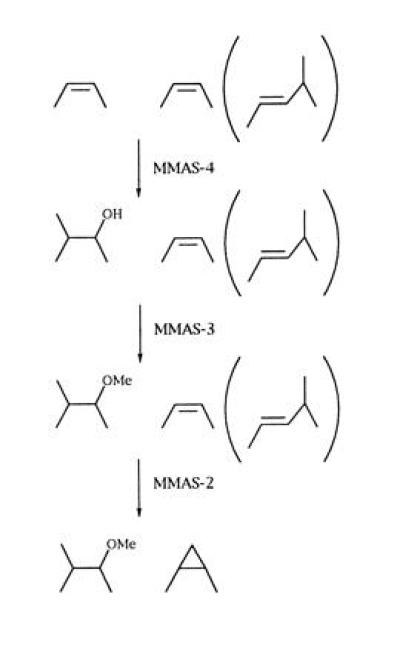
Proposed biosynthetic pathway for methoxymycolic acids.
The function of MMAS-1 remains cryptic at this point. Neither expression alone nor in various combinations with other mma genes resulted in an observable phenotype when introduced into M. smegmatis. MMAS-1 appeared to be expressed in M. smegmatis as assessed by the appearance of a band of the appropriate size by Coomassie brilliant blue staining of polyacrylamide gels (data not shown). MMAS-1 may be functional but lack the appropriate substrate to catalyze methyl transfer.
Since MMAS-4 has a typical SAM binding site, this reaction presumably proceeds through addition of water to the intermediate cation formed upon methyl group addition to the cis-olefinic precursor. Although this is an uncommon outcome for a SAM-dependent methylase, it does suggest a simplifying hypothesis for mycolate biosynthetic modification reactions as shown in Fig. 6. This scheme proposes that both CMAS and MMAS enzymes catalyze the SAM-dependent formation of a high-energy cationic intermediate. The actual product formed would then depend upon the nature of residues near the active site, which may be positioned to abstract a proton from the incoming methyl group (CMAS-1 and -2 and MMAS-2) or facilitate water addition to the cation (MMAS-4). This also predicts the existence of an enzyme whose activity would serve to interconvert the α1 and α2 series by methyl addition in M. smegmatis.
Figure 6.
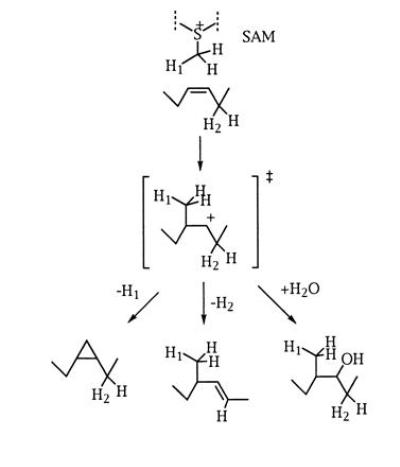
The fate of a common cationic intermediate determines the mycolic acid product formed. The addition of a methyl group from SAM generates a cationic intermediate that can be deprotonated to form a cyclopropane or a trans-olefin with an adjacent methyl group or can react with water to form the hydroxy- (methoxy-) mycolate series.
The function of each class of mycolate in the cell wall is unknown. In the proximal position, it has been suggested that the ratio of cis-to-trans geometries may determine the packing density of carbon chains in the inner leaflet of the asymmetric lipid bilayer and directly influence cell wall permeability (5, 8, 32). Cyclopropanation at the distal position has been shown to enhance resistance to oxidative killing (17), and cyclopropanation at the proximal position appears to affect cell wall fluidity (20). A potential chemical role for non-α mycolates containing oxygen functions in the distal position might involve providing hydrogen-bonding opportunities for ancillary cell wall-associated lipids (8). This role is supported in part by the observation that expressing MMAS-4 alone, MMAS-3 and MMAS-4 together, or the entire gene cluster together has dramatic effects on the surface morphology of recombinant M. smegmatis. These effects can be partially explained by the increased association of hydrophilic molecules with the exposed hydroxy or methoxy group in the distal position of the recombinant meromycolate.
The proposal for a common intermediate generated by related enzymes involved in mycolate modification has important implications. First, the mycolic acid modification pathways described appear to represent an exception to the rule that new enzyme activities are recruited by optimizing substrate binding from an existing array of enzymatic species catalyzing defined chemical transformations (33). Within the set of mycobacterial enzymes described here, mutation of a preexisting enzyme with defined substrate binding capabilities results in the generation of novel chemical transformations, and hence results in structural and functional diversity within mycolic acids. The variety of roles played by these different structural classes of mycolic acid constituents in mediating the interaction of mycobacteria with their environment further suggests that targeting such reactions represents a viable chemotherapeutic approach (4). Developing chemotherapeutics that simultaneously target the transition state of multiple enzymes proceeding through such an intermediate would circumvent the ability of the organism to acquire resistance through single step mutational events, dramatically decreasing the opportunity for the emergence of drug resistance.
Acknowledgments
We would like to thank Harlan Caldwell, Khisimuzi Mdluli, and Witold Cieplak for reviewing the manuscript; Yaqi Zhu and Deborah Crane for technical assistance; and George Kenyon and Robert Belland for helpful conversations.
Footnotes
References
- 1.Dolin P J, Raviglione M C, Kochi A. Bull W H O. 1994;72:213–220. [PMC free article] [PubMed] [Google Scholar]
- 2.Jarlier V, Nikaido H. FEMS Microbiol Lett. 1994;123:11–18. doi: 10.1111/j.1574-6968.1994.tb07194.x. [DOI] [PubMed] [Google Scholar]
- 3.Iseman M D. Proc Natl Acad Sci USA. 1994;91:2428–2429. doi: 10.1073/pnas.91.7.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barry C E, III, Mdluli K. Trends Microbiol. 1996;4:275–281. doi: 10.1016/0966-842x(96)10031-7. [DOI] [PubMed] [Google Scholar]
- 5.Brennan P J, Nikaido H. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 6.Nikaido H, Kim S-H, Rosenberg E Y. Mol Microbiol. 1993;8:1025–1030. doi: 10.1111/j.1365-2958.1993.tb01647.x. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Rosenberg E Y, Nikaido H. Proc Natl Acad Sci USA. 1995;92:11254–11258. doi: 10.1073/pnas.92.24.11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minnikin D E. In: The Biology of the Mycobacteria. Ratledge C, Stanford J, editors. San Diego: Academic; 1982. pp. 95–184. [Google Scholar]
- 9.Minnikin D E, Minnikin S M, Parlett J H, Goodfellow M, Magnusson M. Arch Microbiol. 1984;139:225–231. doi: 10.1007/BF00402005. [DOI] [PubMed] [Google Scholar]
- 10.Minnikin D E, Minnikin S M, Hutchinson I G, Goodfellow M, Grange J M. J Gen Microbiol. 1984;130:363–367. doi: 10.1099/00221287-130-2-363. [DOI] [PubMed] [Google Scholar]
- 11.Minnikin, D. E. & Polgar, N. (1967) Chem. Commun., 1172-1174.
- 12.Davidson L A, Draper P, Minnikin D E. J Gen Microbiol. 1982;128:823–828. doi: 10.1099/00221287-128-4-823. [DOI] [PubMed] [Google Scholar]
- 13.Minnikin D E, Minnikin S M, Dobson G, Goodfellow M, Portaels F, Van Den Breen L, Sesardic D. J Gen Microbiol. 1983;129:889–891. doi: 10.1099/00221287-129-3-889. [DOI] [PubMed] [Google Scholar]
- 14.Daffe M, Laneele M A, Lacave C. Res Microbiol. 1991;142:397–403. doi: 10.1016/0923-2508(91)90109-n. [DOI] [PubMed] [Google Scholar]
- 15.Kaneda K, Naito S, Imaizumi S, Yano I, Mizuno S, Tomiyasu I, Baba T, Kusunose E, Kusunose M. J Clin Microbiol. 1986;24:1060–1070. doi: 10.1128/jcm.24.6.1060-1070.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 17.Yuan Y, Lee R E, Besra G S, Belisle J T, Barry C E., III Proc Natl Acad Sci USA. 1995;92:6630–6634. doi: 10.1073/pnas.92.14.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bose M, Chander A, Das R H. Nucleic Acids Res. 1993;21:2529–2530. doi: 10.1093/nar/21.10.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamid M E, Minnikin D E, Goodfellow M. J Gen Microbiol. 1993;139:2203–2213. doi: 10.1099/00221287-139-9-2203. [DOI] [PubMed] [Google Scholar]
- 20.George K M, Yuan Y, Sherman D R, Barry C E., III J Biol Chem. 1995;270:27292–27298. doi: 10.1074/jbc.270.45.27292. [DOI] [PubMed] [Google Scholar]
- 21.Danielson S J, Gray G R. J Biol Chem. 1982;257:12196–12203. [PubMed] [Google Scholar]
- 22.Minnikin D E, Minnikin S M, Goodfellow M. Biochim Biophys Acta. 1982;712:616–620. [Google Scholar]
- 23.Minnikin D E, Minnikin S M, Goodfellow M. Biochim Biophys Acta. 1982;712:616–620. [Google Scholar]
- 24.Hamid M E, Minnikin D E, Goodfellow M, Ridell M. Zentralbl Bakteriol Mikrobiol Hyg Ser A. 1993;279:354–367. doi: 10.1016/s0934-8840(11)80368-3. [DOI] [PubMed] [Google Scholar]
- 25.Niemi J, Mantsala P. J Bacteriol. 1995;177:2942–2945. doi: 10.1128/jb.177.10.2942-2945.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madduri K, Hutchinson C R. J Bacteriol. 1995;177:3879–3884. doi: 10.1128/jb.177.13.3879-3884.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang A-Y, Grogan D W, Cronan J E., Jr Biochemistry. 1992;31:11020–11028. doi: 10.1021/bi00160a011. [DOI] [PubMed] [Google Scholar]
- 28.Stover C K, de la Cruz V F, Fuerst T R, Burlein J E, Benson L A, Bennett L T, Bansal G P, Young J F, Lee M H, Hatfull G F, Snapper S B, Barletta R G, Jacobs W R, Jr, Bloom B R. Nature (London) 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 29.Gastambide-Odier M, Delaumeny J M, Lederer E. C R Hebd Seances Acad Sci. 1964;259:3404–3407. [PubMed] [Google Scholar]
- 30.Qureshi N, Takayama K, Schnoes H K. J Biol Chem. 1980;255:182–189. [PubMed] [Google Scholar]
- 31.Lacave C, Laneele M-A, Daffe M, Montrozier H, Laneele G. Eur J Biochem. 1989;181:459–466. doi: 10.1111/j.1432-1033.1989.tb14747.x. [DOI] [PubMed] [Google Scholar]
- 32.Liu, J. L., Barry, C. E., III, Besra, G. S. & Nikaido, H. N. (1996) J. Biol. Chem., in press. [DOI] [PubMed]
- 33.Babbitt P C, Mrachko G T, Hasson M S, Huisman G W, Kolter R, Ringe D, Petsko G A, Kenyon G L, Gerlt J A. Science. 1995;267:1159–1161. doi: 10.1126/science.7855594. [DOI] [PubMed] [Google Scholar]