

Abstract
YY1 is a mammalian zinc-finger transcription factor with unusual structural and functional features. It has been implicated as a positive and a negative regulatory factor that binds to the CCATNTT consensus DNA element located in promoters of many cellular and viral genes. A mammalian cDNA that encodes a YY1-binding protein and possesses sequence homology with the yeast transcriptional factor RPD3 has been identified. A Gal4 DNA binding domain–mammalian RPD3 fusion protein strongly represses transcription from a promoter containing Gal4 binding sites. Association between YY1 and mammalian RPD3 requires a glycine-rich region on YY1. Mutations in this region abolish the interaction with mammalian RPD3 and eliminate transcriptional repression by YY1. These data suggest that YY1 negatively regulates transcription by tethering RPD3 to DNA as a cofactor and that this transcriptional mechanism is highly conserved from yeast to human.
Keywords: transcription factor, corepressor, protein–protein interaction
Transcriptional activation and repression are now well embedded in our understanding of gene regulation in mammalian cells. However, while great advances have been made toward understanding activation, much less is known concerning the mechanism of repression. For example, during the last two decades, a large number of genes have been identified that encode mammalian DNA binding transcription factors that activate the initiation of mRNA synthesis. In contrast, relatively few mammalian proteins have been found that negatively regulate transcription. In addition, while a great deal of effort has been devoted in understanding how protein–protein interactions mediate transcriptional activation, there are gaps in our knowledge concerning protein–protein interactions that mediate transcriptional repression.
YY1 (also known as δ, NF-E1, UCRBP, or CF1) (1–5) is a mammalian zinc-finger transcription factor that binds to and regulates positively or negatively a variety of cis DNA elements located in viral and cellular promoters. Repression of transcription by YY1 may play an important role in cell growth and differentiation (6, 7) and, therefore, provides an ideal system in which to study repression mechanisms in mammalian cells. Previously, we and a number of other groups have used the yeast two-hybrid screen along with biochemical methods to identify cellular proteins that interact with YY1. YY1-binding proteins so far identified include transcription factor Sp1 (8, 9), the oncoprotein c-myc (10), the nucleolar phosphoprotein B23 (11), cyclophilin A (12), FK506-binding protein (12), p300 (13), TAFII55 (14), ATF/CREB (15), and basal transcription factors TATA-binding protein and TFIIB (refs. 14 and 16 and unpublished data). Some of these interacting-cellular proteins can relieve YY1-induced transcriptional repression, while others inhibit the activator functions of YY1. These results suggest that a possible mechanism for transcriptional regulation by YY1 is through interaction with these cellular proteins.
We have now sequenced and characterized additional clones from our earlier two-hybrid screen for encoded proteins capable of binding to YY1. Sequence analysis revealed that one of these clones, is a mouse cDNA that bears striking sequence homology with the yeast RPD3 protein. RPD3 was first cloned in a genetic screen in yeast for negative transcriptional regulators of TRK2, a gene encoding the low-affinity potassium transporter (17, 18). It was independently identified by Nasmyth et al. (19) as SDI2, which is required for the endonuclease HO to be fully SWI5-dependent (19, 20). In yeast, RPD3 has both positive and negative effects on many genes and is thought to be a global regulator required for target genes to achieve maximal transcriptional states (17). Although RPD3 has global effects on transcription, it does not contain any known DNA-binding motifs.
Given our finding that the DNA-binding protein YY1 interacts with the non-DNA binding activator/repressor RPD3, we wondered whether one possible mechanism of YY1 is to recruit the mammalian RPD3 protein to the promoter region. Specifically, since YY1 contains an acidic activation domain but no obvious repression domain and RPD3 is primarily a negative regulator in yeast, we hypothesize that the mammalian RPD3 protein may provide the repression function when bound to YY1. In this study, the mammalian RPD3 protein was expressed as a Gal4 chimera and effectively repressed transcription of a target gene containing Gal4-binding sites. More important, we showed that mammalian RPD3 can repress transcription through a natural YY1 binding site and that transcriptional repression by YY1 requires interaction with the mammalian RPD3 protein.
MATERIALS AND METHODS
Cloning and Sequencing of Mouse and Human RPD3.
Clone Y17 cDNA, derived from a previous yeast two-hybrid screen (11), was used as a probe to rescreen a mouse lymphoma library and a λgt10 HeLa cell library. Positive clones were subcloned into pGEM7Zf (Promega), and sequences of subcloned cDNA were obtained using synthetic primers by the dideoxynucleotide method. The final sequence was determined from both DNA strands. The predicted human and mouse RPD3 (mRPD3) amino acid sequences were compared with the protein sequence database using the blast network service at the National Center for Biotechnology Information.
Plasmids.
pGEM7Zf-mRPD3 was constructed by taking a BglII fragment containing the full-length mRPD3 cDNA and ligating it to a BamHI-digested pGEM7Zf(−) vector. Glutathione S-transferase (GST) was expressed from pGSTag (21). pGST-YY1(1–414) contained the YY1 cDNA (nt 241-1513, NcoI–BamHI fragment from the cDNA clone of human YY1 in pGEM7Zf) in pGSTag. Different YY1 deletions were generated by restriction enzyme digestions of pGST-YY1(1–414) and religations. Gal4-mRPD3 was expressed from pGal4-mRPD3, which was constructed by joining the mRPD3 coding region in-frame with the Gal4 DNA-binding domain downstream of the simian virus 40 (SV40) early promoter in pSG424 (22). mRPD3 and YY1 were expressed from pCMV-mRPD3 and pCMV-YY1, respectively, which were constructed by inserting the mRPD3 and YY1 cDNA downstream of the cytomegalovirus (CMV) promoter in pcDNAI/Amp (Invitrogen). Gal4-YY1(dl174-200) was expressed from pGal4-YY1(dl174-200), which was constructed by NotI/SmaI digestion of pGal4-YY1 (1) and religation in the presence of an oligodeoxynucleotide, 5′-GGCCGCGTCAA-3′, and its complement to produce NotI and SmaI ends. Gal4-YY1(170–200) was expressed from pGal4-YY1(170–200), which was constructed by inserting a YY1 NotI–SmaI cDNA fragment (nt 745–836) downstream and in-frame with the Gal4 DNA-binding domain of the SV40 early promoter in pSG424. All constructs were verified by dideoxynucleotide sequencing. Effector plasmids that express Gal4-VP16 and Gal4-E1A have been described (23, 24). pCAT-Control, which contains the SV40 promoter and enhancer sequences upstream of the chloramphenicol acetyltransferase (CAT) reporter gene, was obtained from Promega. The reporter plasmids pG5BCAT-SP, pBCAT-SP, pGal4-tkCAT, pSVECAT, pP5-60SVECAT, and pP5-60(mt2)SVECAT have also been described (1, 25).
In Vitro Protein–Protein Interaction Assay.
35S-labeled mRPD3 proteins were prepared from pGEM7Zf-mRPD3 using T7 RNA polymerase and the coupled transcription–translation rabbit reticulocyte lysate system (Promega). Equal molar quantities of either GST or GST–YY1 fusion proteins on glutathione-Sepharose beads were incubated with 5 μl of mRPD3 in incubation buffer [50 mM Tris·HCl, pH 8/10 mM NaCl/5 mM dithiothreitol/bovine serum albumin (5 mg/ml; BSA)] at room temperature for 60 min. The beads were pelleted by centrifugation and rinsed six times in phosphate-buffer saline. Bound proteins were separated from the beads by boiling in sample buffer (50 mM Tris·HCl, pH 6.8/0.3 M 2-mercaptoethanol/2% SDS/0.1% bromophenol blue/10% glycerol) and analyzed by electrophoresis in a SDS/10% polyacrylamide gel followed by autoradiography.
Transfection and CAT Assay.
CV1 or HeLa cells were cotransfected with plasmids directing the synthesis of various effector proteins plus a CAT reporter, using the calcium phosphate method. Each transfection contained each effector and reporter DNA at 5 μg and all transfections were normalized to equal amounts of DNA with parental expression vectors. Forty-eight hours after transfection, cells were collected and CAT activity was determined in 1-h reactions.
Electrophoretic Mobility Shift Assay (EMSA).
Single-stranded oligodeoxynucleotides (5′-CGGAGGACTGTCCTCCG-3′ and 5′-CGGAGGACAGTCCTCCG-3′) were labeled individually with [γ-32P]ATP and T4 polynucleotide kinase, heated together at 65°C, and allowed to anneal by slow cooling to room temperature. Each reaction contained 20 fmol of labeled DNA, 12 mM Hepes (pH 7.9), 10% glycerol, 5 mM MgCl2, 60 mM KCl, 1 mM DTT, BSA at 50 μg/ml, 0.5 mM EDTA, 0.05% Nonidet P-40, 1 μg of poly(dI-dC), and approximately 10 μg of HeLa cell extract prepared from transfected cells. Reactions were incubated for 10 min at room temperature, separated on 4% nondenaturing polyacrylamide gel (0.0225 M Tris borate/0.0005 M EDTA), dried, and subjected to autoradiography.
RESULTS
Identification of a YY1-Binding Protein with Homology to the Yeast RPD3 Protein.
To gain a deeper understanding of the mechanism of YY1 action, we have used (11) the two-hybrid interaction screen to identify proteins capable of interacting with YY1. Fourteen mouse cDNAs that encode specific YY1-binding activities were analyzed. We have now completely sequenced one of the cDNA clones previously designated Y17 and the predicted amino acid sequence was determined by theoretical translation of the cDNA clone open reading frame (Fig. 1). Interestingly, the predicted amino acid sequence of this mouse cDNA clone is 58% identical (75% similar) to the Saccharomyces cerevisiae RPD3 protein.
Figure 1.

Predicted amino acid sequences of mouse and human RPD3 and their homology to RPD3 from other species. The deduced amino acid sequence of mRPD3 and human RPD3 (hRPD3) are aligned with the sequences of the yeast RPD3 [yRPD3; National Center for Biotechnology Information S22284 (ref. 17)], the Xenopus laevis RPD3 (xRPD3; gi:576995; unpublished sequence), and the Caenorhabditis elegans RPD3 (cRPD3; X78454; unpublished sequence). Amino acid positions are boxed where there is identity among all five species.
Using the mRPD3 cDNA as a probe, we obtained a human RPD3 (hRPD3) clone that also contains 58% identity in predicted amino acid sequences to yeast RPD3 (Fig. 1). The RPD3 protein sequence is also highly homologous to two sequences found in C. elegans and in Xenopus laevis, although it has not yet been established whether these sequences are part of a transcription factor or whether they play an essential role in transcription. Nevertheless, this high conservation of DNA and protein sequences suggests that, like yeast RPD3, the mammalian RPD3 protein may also be a key regulatory protein and perhaps an important component of the transcriptional machinery.
Analysis of Interaction Between YY1 and Mammalian RPD3.
To confirm a physical interaction between YY1 and mRPD3, we employed a YY1 affinity matrix to capture mRPD3 protein. A bacterially expressed GST–YY1 fusion protein was bound to glutathione-Sepharose beads and incubated with 35S-labeled mRPD3 protein produced by in vitro translation in a reticulocyte lysate. The beads were washed, boiled in sample buffer with detergent, and analyzed by electrophoresis in an SDS/polyacrylamide gel. As shown in Fig. 2, mRPD3 was captured by the GST–YY1 fusion protein but not by the GST polypeptide alone (compare lanes 1 and 2). Analysis of YY1 deletion segments fused to GST indicated that the interaction with mRPD3 occurred through YY1 amino acid residues 170–200 (lanes 2–13). A GST fusion of this minimal fragment (residues 170–200) was capable of interacting with mRPD3 (lane 15). Interestingly, this region of YY1 is glycine-rich (35%) and is within a previously identified transcriptional repression domain (26).
Figure 2.
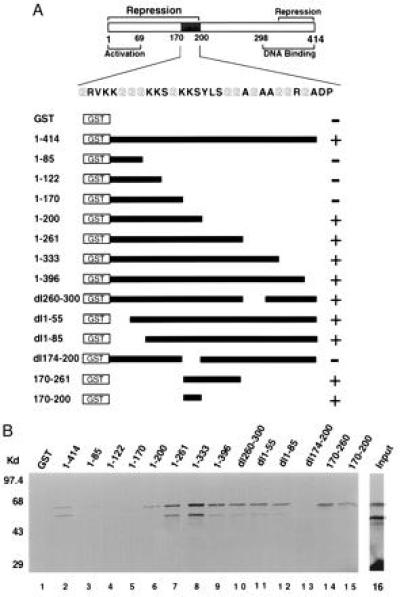
Identification of a glycine-rich mRPD3-interacting domain in YY1. (A) Schematic drawing of YY1 and GST–YY1 fusion constructs. The glycine-rich region (mRPD3 binding domain) is shaded in gray. The location of activation, repression, and DNA binding domains on YY1 is from refs. 1, 7, and 26–28. The ability of each GST–YY1 fusion protein to bind mRPD3 is indicated (+ or −). (B) Representative autoradiogram of in vitro translated mRPD3 protein captured by GST–YY1 fusion proteins. Three experiments yielded consistent results. The input lane (lane 16) was loaded with one-third the amount of mRPD3 used in the binding reactions. The sizes of molecular weight markers are indicated to the left.
The Cloned Mammalian RPD3 Protein Represses Transcription.
To determine whether the protein encoded by the mRPD3 cDNA has any effect on transcription when bound near a promoter, we constructed a mRPD3 chimeric protein with an added DNA-binding specificity to distinguish it from endogenous mammalian RPD3 activity. When Gal4–mRPD3 was cotransfected into CV1 cells with the target plasmid pG5BCAT-SP, containing five Gal4-binding sites, a 21-fold repression of CAT activity was observed (Fig. 3B, compare lanes 1 and 3). Repression was dependent on the presence of the Gal4-binding sites because CAT expression was not affected when pBCAT-SP, lacking Gal4-binding sites, was used as a target (Fig. 3B, compare lanes 4 and 5). Repression was also dependent on the Gal4 DNA-binding domain, as mRPD3 without the DNA-binding domain failed to repress (Fig. 3B, lane 2). Finally, similar repression was observed in HeLa cells with a minimal Gal4–thymidine kinase promoter, indicating that repression was independent of cell type or basal promoter (Fig. 3B, compare lanes 6 and 7).
Figure 3.
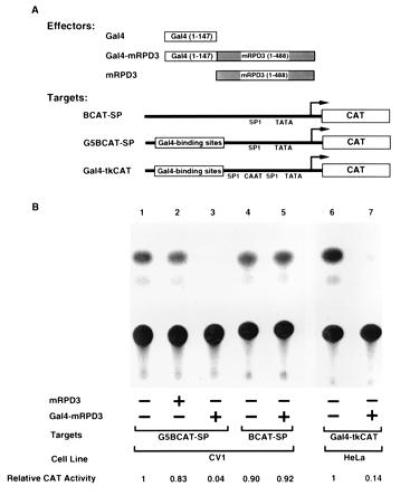
Gal4–mRPD3 fusion protein represses transcription from promoters with Gal4-binding sites. (A) Schematic drawing of plasmids used in transfections. (B) Representative CAT assays showing that mRPD3 can repress transcription. Seven experiments yielded consistent results. Plasmids included in each transfection are indicated (+).
Repression by YY1 Depends on Interaction with Mammalian RPD3.
Previous studies have shown that YY1 can repress transcription when directed to the upstream sequence of a minimal thymidine kinase promoter in HeLa cells (1). To determine the effects of mRPD3 on YY1-induced transcriptional repression, we targeted YY1 to promoters containing Gal4-binding sites and cotransfected a plasmid expressing mRPD3. As shown in Fig. 4B, overexpression of mRPD3 consistently increased YY1 repression activity (lanes 4 and 7), although the extent to which Gal4–YY1 repressed transcription varied depending on the combination of reporter constructs and cell type. Neither the SV40 early promoter used to control production of Gal4–YY1 nor the Gal4–YY1 protein level was affected by mRPD3 (Fig. 4 C, compare lanes 1 to 2 and 3 to 4, and D, compare lanes 2 to 3 and 4 to 5), thus ruling out the possibility that mRPD3 increased the amount of Gal4–YY1 fusion protein in the transfected cells. Furthermore, the mRPD3 protein did not effect transcription of two Gal4 fusion activators (Fig. 4E, compare lanes 3 to 4 and 5 to 6). These results indicate that repression by YY1 is mediated, at least in part, by interaction with mammalian RPD3 protein.
Figure 4.

mRPD3 protein increases Gal4–YY1-induced transcriptional repression through a glycine-rich domain in YY1. (A) Schematic drawing of plasmids used in transfections. (B, C, and E–G) Representative CAT assays showing that mRPD3 can further repress transcription through a Gal4–YY1 glycine-rich domain. Each CAT assay shown is only a representative of at least three experiments. Plasmids included in each transfection are indicated (+). Reporter plasmid is pCAT-Control for C and pGal4-tkCAT for E–G. (D) Mouse RPD3 has no effect on Gal4–YY1 as determined by EMSA. No protein was added in reaction presented in lane 1. Arrow indicates position of Gal4–YY1–DNA complex.
Since amino acids 170–200 in YY1 are required for YY1–mRPD3 interaction, we next asked whether this sequence is important for repression. As shown in Fig. 4F, deletion of amino acids 174–200 rendered the YY1 protein incapable of repression (compare lanes 1 and 4), whereas a minimal region containing amino acids 170–200 of YY1 conferred transcriptional repression when directed to a promoter by the Gal4 DNA-binding domain (lane 3). A Gal4–YY1(dl174-200) fusion is readily detectable in transfected cells by Western blot analysis using an antibody directed to the DNA-binding domain of Gal4 (data not shown). Furthermore, the mRPD3 protein can repress transcription through a Gal4 fusion protein containing only amino acids 170–200 of YY1 (Fig. 4G, lane 3). Thus, our data strongly suggest that the glycine-rich domain in YY1 is not only important for binding to mRPD3 in vitro but is also critical for transcriptional repression in vivo. This finding is consistent with earlier work that demonstrated that amino acids 1–201 of YY1 inhibited transcription nearly as well as a fusion protein containing the complete YY1 sequence (26).
A Nonfusion Mammalian RPD3 Protein Represses Transcription from a Promoter Containing YY1-Binding Sites.
To be certain that the mRPD3 repression effect observed is not an artificial phenomenon derived from fusion of the mRPD3 protein to Gal4, we cotransfected a mRPD3 expression plasmid with a target plasmid containing natural YY1 binding sites. As shown in Fig. 5, a modest repression of CAT activity was observed (compare lanes 5 and 7). This repression was dependent on the presence of YY1-binding sites because CAT activity was not affected when the reporter constructs lacked YY1-binding sites (compare lanes 1 and 3) or contained mutated YY1-binding sites (compare lanes 9 and 11). More important, when a third plasmid that expresses YY1 was included in the transfection, a more dramatic repression was seen only from the target that contains the wild-type YY1-binding sites (lanes 4, 8, and 12).
Figure 5.

Nonfusion mRPD3 protein represses transcription from promoters with YY1-binding sites. Representative CAT assays showing that mRPD3 can further repress transcription through YY1. Each CAT assay shown is a representative of at least three experiments. All relative CAT activities are normalized with control β-galactosidase expressions. Plasmids included in each transfection are indicated (+). Effector plasmid is pCMV-YY1 and pCMV-mRPD3. Reporter plasmid is pSVECAT for no YY1-binding site, pP5-60SVECAT for wild-type YY1-binding sites, and pP5-60(mt2)SVECAT for mutated YY1-binding sites.
DISCUSSION
Previously, this and other laboratories have identified two regions on YY1 that mediate transcriptional repression (1, 26–28). Herein, we have further delineated the repression domain in one region to amino acids 170–200. More important, we have identified a novel mammalian corepressor that interacts with this domain.
YY1, like most eukaryotic repressors described to date, seems to act directly on the general transcription machinery—a mechanism referred to as active repression (for review, see refs. 29–32). Three types of domains have been identified thus far in active transcriptional repressors (for review, see refs. 29–32): alanine-rich, glutamine-rich, and/or proline-rich. Currently, it is not known whether these repression domains function by contact with the general transcriptional machinery, and proteins that interact with these domains have yet to be identified. Inspection of the YY1 amino acid sequence has failed to reveal any resemblance to the primary sequence motifs that characterize any of the repression domains previously described. Intriguingly, we have found herein that a sequence rich in glycine can mediate YY1 repression through interaction with mammalian RPD3. Whether repression through a glycine-rich domain is limited only to YY1 or whether it is a more widespread phenomenon among other transcriptional repressors remains to be determined.
Similar to YY1, retinoic acid receptors and retinoid X receptors also activate and repress transcription (for review, see refs. 33 and 34). Interestingly, it was recently determined that the underlying mechanism for repression of these receptors is also a result of recruitment of a corepressor (35, 36). Perhaps one of the best characterized examples of transcriptional repression involving a repressor–corepressor mechanism is the yeast α2/Mcm1 proteins (37). The homeodomain protein α2 and the SRF-like protein Mcm1 binds cooperatively to operator DNA sequences. However, the binding of α2/Mcm1 to operator DNA is insufficient to bring about repression. Instead, repression takes place only when a protein complex Ssn6 (Cyc8)/Tup1 is recruited to the promoter. In the presence of Tup1, Ssn6 can also repress transcription when directed to a promoter by fusion with a bacterial DNA-binding domain. It was suggested that Ssn6 serves as an adaptor between DNA-bound repressors and corepressor Tup1 (38). Our data suggests that the repression mechanism by YY1 is similar to the α2/Mcm1 complex and the function of mammalian RPD3 is parallel to the yeast Ssn6/Tup1. But unlike mRPD3, the corepressor complex Ssn6/Tup1 interacts with α2 through Tup1’s WD repeats (39). Since mRPD3 does not contain any WD repeat, this suggests the existence of multiple mechanisms for non-DNA-binding repressors to recognize the appropriate DNA-bound proteins. Also, unlike Tup1, which contains alanine-rich regions similar to one class of repression domains previously described (38), mRPD3 does not contain any alanine-rich regions or any of the repression domains previously described.
Another example of repressor–corepressor mechanisms came from recent studies of the basic helix–loop–helix– leucine zipper myc superfamily proteins (40, 41). It was found that Mad and the related protein Mxi1 have high affinity for a mammalian homolog of the yeast protein Sin3 [also known as RPD1 (42)]. Mad, Max, and the mammalian Sin3 proteins bind DNA as a ternary complex. Transcriptional repression by Mad–Max requires interaction with the mammalian Sin3 protein suggesting that Mad–Max represses transcription by recruiting mammalian Sin3 to a promoter as a corepressor. The identification of the mammalian homologs of both RPD1 and RPD3 suggests the possibility that the functional heterologous complex formed by these proteins in yeast may be conserved in humans. Intriguingly, it has been demonstrated that c-Myc binds to and inhibits the transcriptional activation and repression activity of YY1 (10). This observation leads to the possibility that YY1 and c-Myc may participate with RPD1 and RPD3 to form a large regulatory complex that is highly conserved from yeast to human.
Note.
While this manuscript was under review, we learned that Schreiber and colleagues (43) have cloned another protein related to RPD3 based on biochemical copurification with histone deacetylase activity in human. The human RPD3 protein (HD1) reported by Schreiber’s group is 75% identical in DNA sequence and 85% identical in protein sequence compared with the human RPD3 clone reported here. Comparison of the mRPD3 sequence with human HD1 also yielded similar results (76% identical in DNA sequence and 86% identical in protein sequence).
Acknowledgments
We thank G. Chinnadurai, H. Dressler, S. Elledge, W. H. Lee, and B. Shan for generous gifts of plasmids and libraries; and P. D. Gardner, E. Lee, Y. Shi, and A. Tomkinson for discussion and critical reading of the manuscript. This work was supported by grants from the National Science Foundation, the National Cancer Institute, and the Texas Advanced Research Program.
Footnotes
References
- 1.Shi Y, Seto E, Chang L-S, Shenk T. Cell. 1991;67:377–388. doi: 10.1016/0092-8674(91)90189-6. [DOI] [PubMed] [Google Scholar]
- 2.Hariharan N, Kelley D E, Perry R P. Proc Natl Acad Sci USA. 1991;88:9799–9803. doi: 10.1073/pnas.88.21.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park K, Atchison M L. Proc Natl Acad Sci USA. 1991;88:9804–9808. doi: 10.1073/pnas.88.21.9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flanagan J R, Becker K G, Ennist D L, Gleason S L, Driggers P H, Levi B Z, Appella E, Ozato K. Mol Cell Biol. 1992;12:38–44. doi: 10.1128/mcb.12.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riggs K J, Merrell K T, Wilson G, Calame K. Mol Cell Biol. 1991;11:1765–1769. doi: 10.1128/mcb.11.3.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hahn S. Curr Biol. 1992;2:152–154. doi: 10.1016/0960-9822(92)90268-f. [DOI] [PubMed] [Google Scholar]
- 7.Shrivastava A, Calame K. Nucleic Acids Res. 1994;22:5151–5155. doi: 10.1093/nar/22.24.5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seto E, Lewis B, Shenk T. Nature (London) 1993;365:462–464. doi: 10.1038/365462a0. [DOI] [PubMed] [Google Scholar]
- 9.Lee J S, Galvin K, Shi Y. Proc Natl Acad Sci USA. 1993;90:6145–6149. doi: 10.1073/pnas.90.13.6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shrivastava A, Saleque S, Kalpana G V, Artandi S, Goff S P, Calame K. Science. 1993;262:1889–1892. doi: 10.1126/science.8266081. [DOI] [PubMed] [Google Scholar]
- 11.Inouye C J, Seto E. J Biol Chem. 1994;269:6506–6510. [PubMed] [Google Scholar]
- 12.Yang W M, Inouye C J, Seto E. J Biol Chem. 1995;270:15187–15193. doi: 10.1074/jbc.270.25.15187. [DOI] [PubMed] [Google Scholar]
- 13.Lee J S, Galvin K M, See R H, Eckner R, Livingston D, Moran E, Shi Y. Genes Dev. 1995;9:1188–1198. doi: 10.1101/gad.9.10.1188. [DOI] [PubMed] [Google Scholar]
- 14.Chiang C M, Roeder R G. Science. 1995;267:531–536. doi: 10.1126/science.7824954. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Q, Gedrich R W, Engel D A. J Virol. 1995;69:4323–4330. doi: 10.1128/jvi.69.7.4323-4330.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Usheva A, Shenk T. Cell. 1994;76:1115–1121. doi: 10.1016/0092-8674(94)90387-5. [DOI] [PubMed] [Google Scholar]
- 17.Vidal M, Gaber R F. Mol Cell Biol. 1991;11:6317–6327. doi: 10.1128/mcb.11.12.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vidal M, Buckley A M, Hilger F, Gaber R F. Genetics. 1990;125:313–320. doi: 10.1093/genetics/125.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nasmyth K, Stillman D, Kipling D. Cell. 1987;48:579–587. doi: 10.1016/0092-8674(87)90236-4. [DOI] [PubMed] [Google Scholar]
- 20.Stillman D J, Dorland S, Yu Y. Genetics. 1994;136:781–788. doi: 10.1093/genetics/136.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ron D, Dressler H. Biotechniques. 1992;13:866–869. [PubMed] [Google Scholar]
- 22.Sadowski I, Ptashne M. Nucleic Acids Res. 1989;17:7539. doi: 10.1093/nar/17.18.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadowski I, Ma J, Triezenberg S, Ptashne M. Nature (London) 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 24.Lillie J W, Green M R. Nature (London) 1989;338:39–44. doi: 10.1038/338039a0. [DOI] [PubMed] [Google Scholar]
- 25.Kamine J, Subramanian T, Chinnadurai G. Proc Natl Acad Sci USA. 1991;88:8510–8514. doi: 10.1073/pnas.88.19.8510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis B A, Tullis G, Seto E, Horikoshi N, Weinmann R, Shenk T. J Virol. 1995;69:1628–1636. doi: 10.1128/jvi.69.3.1628-1636.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J S, See R H, Galvin K M, Wang J, Shi Y. Nucleic Acids Res. 1995;23:925–931. doi: 10.1093/nar/23.6.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bushmeyer S, Park K, Atchison M L. J Biol Chem. 1995;270:30213–30220. doi: 10.1074/jbc.270.50.30213. [DOI] [PubMed] [Google Scholar]
- 29.Johnson A D. Cell. 1995;81:655–658. doi: 10.1016/0092-8674(95)90524-3. [DOI] [PubMed] [Google Scholar]
- 30.Herschbach B M, Johnson A D. Annu Rev Cell Biol. 1993;9:479–509. doi: 10.1146/annurev.cb.09.110193.002403. [DOI] [PubMed] [Google Scholar]
- 31.Renkawitz R. Trends Genet. 1990;6:192–197. doi: 10.1016/0168-9525(90)90176-7. [DOI] [PubMed] [Google Scholar]
- 32.Cowell I G. Trends Biochem Sci. 1994;19:38–42. doi: 10.1016/0968-0004(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 33.Glass C K. Endocr Rev. 1994;15:1503–1519. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- 34.Mangelsdorf D J, Evans R M. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 35.Kurokawa R, Soderstrom M, Horlein A, Halachmi S, Brown M, Rosenfeld M G, Glass C K. Nature (London) 1995;377:451–454. doi: 10.1038/377451a0. [DOI] [PubMed] [Google Scholar]
- 36.Chen J D, Evans R M. Nature (London) 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 37.Keleher C A, Redd M J, Schultz J, Carlson M, Johnson A D. Cell. 1992;68:709–719. doi: 10.1016/0092-8674(92)90146-4. [DOI] [PubMed] [Google Scholar]
- 38.Tzamarias D, Struhl K. Nature (London) 1994;369:758–761. doi: 10.1038/369758a0. [DOI] [PubMed] [Google Scholar]
- 39.Komachi K, Redd M J, Johnson A D. Genes Dev. 1994;8:2857–2867. doi: 10.1101/gad.8.23.2857. [DOI] [PubMed] [Google Scholar]
- 40.Ayer D E, Lawrence Q A, Eisenman R N. Cell. 1995;80:767–776. doi: 10.1016/0092-8674(95)90355-0. [DOI] [PubMed] [Google Scholar]
- 41.Schreiber-Agus N, Chin L, Chen K, Torres R, Rao G, Guida P, Skoultchi A I, DePinho R A. Cell. 1995;80:777–786. doi: 10.1016/0092-8674(95)90356-9. [DOI] [PubMed] [Google Scholar]
- 42.Vidal M, Strich R, Esposito R E, Gaber R F. Mol Cell Biol. 1991;11:6306–6316. doi: 10.1128/mcb.11.12.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taunton J, Hassig C A, Schreiber S L. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]