

Abstract
We examined the effect of two rhesus papillomavirus 1 (RhPV) oncogenes on cytokine-induced signal transduction pathways leading to the possible activation of Ras protein (p21ras) and phosphatidylinositol kinase. p21ras in both the activated (GTP-bound) and inactivated (GDP-bound) states were quantitated. NIH 3T3 cell lines expressing the RhPV 1 E5 gene or epidermal growth factor receptor cDNA had about a sixfold higher ratio of p21ras-bound GTP to p21ras-bound GDP as compared with parental NIH 3T3 cells or a cell line expressing the RhPV 1 E7 gene under normal culture conditions, yet expressed similar levels of p21ras. Quiescent cells had dramatically reduced levels of activated p21ras, except those containing RhPV 1 E7. Levels were restored by stimulation with epidermal growth factor or platelet-derived growth factor. Both epidermal growth factor and platelet-derived growth factor receptor of RhPV 1 E5- and E7-containing cells responded to cytokine stimulation. Endogenous phosphatidylinositol-3′-kinase was up-regulated in NIH 3T3 cells transformed with the E5 genes of RhPV 1 and bovine papillomavirus 1. These results suggest that E5 genes of papillomaviruses play a major role in the regulation of transduction pathways.
Keywords: GTP-binding protein, signal transduction, receptors
The p21ras protooncogene encodes a membrane-bound, guanine nucleotide-binding protein. Activated p21ras proteins have both increased transforming activity and binding of GTP rather than GDP in vitro (1) and in vivo (2–5). Only GTP-bound-p21ras is biologically active (5, 6), inducing a cascade of protein kinases that enhance cellular proliferation (7, 8), perhaps by the binding of its effector domain to the serine/threonine kinase, Raf (9). p21ras protein or the ligand (GTP or GDP)-induced conformation has low intrinsic GTPase activity (10) and is regulated by GTPase-activating protein (GAP) or guanine nucleotide-releasing proteins (GNRP) such as Son of Sevenless (11–13) that replace GDP with GTP. GAP and GNRPs are in turn regulated by platelet-derived growth factor (PDGF) receptors (PDGFRs) and by epidermal growth factor (EGF) receptors (EGFRs), which have intrinsic tyrosine kinase activity and respond to mitogenic stimulation by their ligands.
Mitogenic activity of the EGFR is linked to the generation of mono- and polyphosphoinositides and the mobilization of Ca2+ that is mediated through a series of cytoplasmic proteins, which become transiently associated with the activated EGFR (14). Basal cells metabolize phosphatidylinositol (PI) through production of PI-4′-phosphate. Under stimulation by EGF, perhaps through a direct interaction with p21ras (15), it is the activation of PI-3′-kinase (PI-3′-K) that leads to enhanced PI turnover (16) through transient binding of PI-3′-K to the SH2 domains of the PDGFR and colony-stimulating factor 1 receptor resulting in alterations of cytoplasmic Ca2+ levels (17, 18), increased expression of c-myc and c-fos (19), and increased DNA synthesis and cellular proliferation (19–22).
The E5 gene product is the major transforming gene of bovine papillomavirus type 1 (BPV-1) (23, 24), and stimulates the transforming activity of EGFR, colony-stimulating factor 1 receptor (25), and PDGFR (26). E5 up-regulates EGFR (25) by enhancing phosphorylation, increasing the half-life of the receptor, and preventing its dimerization, endocytosis, and recycling (27). In human keratinocytes, human papillomavirus (HPV) type 16 E5 prevents degradation of the EGFR and enhances recycling of the receptor to the surface (28). The BPV-1 E5 protein binds to 16-kDa protein that is a component of the hydrogen-ATPase pump that is important for cellular GAP-junction-like membrane complexes, which play a crucial role in the conduction of signals from cell to cell (29, 30), and may control degradation of EGFR in endosomal compartments (28). Expression of EGFR is increased both in cell lines derived from cervical cancer (HeLa and Caski cells) as well as in papillomavirus-associated tumors of the human genital tract (31). While BPV-1 E5 activates human PDGFR or EGFR transfected into NIH 3T3 cells and coimmunoprecipitates with human PDGFR or EGFR that has been transfected into COS cells (32), a more recent report indicates binding and activation only for PDGFR-β (33). In contrast, E7, the major oncoprotein in HPV 16, shares homology with regions of the adenovirus E1a gene product and simian virus 40 large tumor antigen (34), and all bind to the retinoblastoma protein, which normally depresses cellular proliferation, suggesting common pathways of activation of cellular DNA replication for several types of viruses (35). It remains to be determined what the relative roles are for the E5 and E7 gene products, and whether or not these cellular events alone or in combination with other events are sufficient for malignant transformation.
We have identified and isolated from a rhesus monkey penile carcinoma a novel papillomavirus, rhesus papillomavirus type 1 (RhPV 1), that is closely related to HPV 16 (36, 37), which is associated with human genital neoplasms. The apparent sexual transmission and high oncogenic potential of RhPV 1 closely mimics the natural disease associated with highly oncogenic human genital papillomaviruses (38–40). Using RhPV 1 as the closest animal model system for genital human papillomaviruses, we have determined the effects of the RhPV 1 E5 or E7 oncoproteins on the activation of cellular p21ras and PI-3′-K and the regulation of transduction pathways.
MATERIALS AND METHODS
Plasmids.
Plasmids expressing RhPV 1 E5 (pLNCE5) and E7 (pLNCE7) genes have been described previously (39). A BPV-1 E5 expression clone was produced from a PstI fragment of BPV-1 (bp 3406–4177) that was modified to produce HindIII ends for cloning into pLNCX (41).
Cell Lines.
The G54 cell line (42) was a kind gift of Takaya Satoh and Yoshito Kaziro (DNAX), and is a derivative of Swiss 3T3 cells obtained by the transfection of a vector expressing normal c-Ha-ras cDNA. G54 cells do not display a transformed phenotype, but p21ras levels are 30-fold greater than Swiss 3T3 cells by immunoblotting (42). Transformed NIH 3T3 cell lines expressing RhPV 1 E5 and RhPV 1 E7 have been previously described (39). NIH 3T3 cells were also transfected as previously described (39) with plasmid pCO11 (43), which expressed normal human EGFR in a neomycin resistance-containing plasmid, with pCO12 (43) containing the normal human EGFR and cotransfected with pSVNeo, or with the parent expression vector, pLNCX, referred to in this text simply as NIH 3T3 cells. The level of total EGFR in pCO11 cells was about four times that of the parent NIH 3T3 cells (data not shown).
Cell Culture.
G54, RhPV 1 E5, RhPV 1 E7, NIH 3T3, and pCO11 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO/BRL) supplemented with 10% (vol/vol) fetal bovine serum (GIBCO/BRL) at 37°C (steady-state growth conditions). To test the effect of growth factors on c-Ha-ras activation, cells were brought to a quiescent state and labeled with [32P]orthophosphate (DuPont/New England Nuclear catalog no. NEX-053) as described (44). Briefly, cells grown in 10% (vol/vol) (G54) FBS or 6% (vol/vol) (NIH 3T3, RhPV 1 E5, RhPV 1 E7, and pCO11) FBS were seeded at a density of 5.4 × 103 per cm2 (G54 cells) or 2.8 × 103 per cm2 (NIH 3T3, RhPV 1 E5, RhPV 1 E7, and pCO11), and the medium was replaced with DMEM containing 6% (vol/vol) (G54) FBS or 4% (vol/vol) (all others) FBS on the next day. After 5 days without changing the medium, the cells were arrested in the quiescent state. Although G54 cells constitutively express ras cDNA, 1 μM CdCl2 was added during labeling to fully activate the inducible metallothionein promoter that expresses the transfected c-Ha-ras gene. Cells were then either incubated without further additions or treated with 100 ng/ml mouse EGF [Collaborative Biomedical Products (Bedford, MA) catalog no. 40001] or 50 ng/ml human PDGF-BB recombinant homodimer (Upstate Biotechnology catalog no. 01–305) for 30 min to restore DNA synthesis (42).
Analysis of p21-Bound GDP and GTP.
The cells were disrupted, and the amounts of GTP to GDP bound to immunoprecipitated p21ras were analyzed as previously described (42). Molar ratios of p21ras-bound GTP to total bound nucleotide were calculated as follows with a factor of 1.5 to account for the different number of phosphates in GDP and GTP: % GTP = 100(GTP cpm)/[1.5 × (GDP cpm) + (GTP cpm)]. Assays were performed two to five times to ensure reproducibility.
Activation of the PI-3′-K in Papillomavirus E5-Transformed Cell Lines.
Subconfluent cells (70–80%) were incubated for 24 hr in serum-free DMEM, and then either harvested for protein or pulsed for 10 min with EGF (100 ng/ml) to activate their receptors, lysed in a buffer containing a nonionic detergent, and centrifuged (45). The supernatant protein (300 μg) was immunoprecipitated with monoclonal antibody specific for EGFR [Transduction Laboratories (Lexington, KY) catalog no. E12020, clone 13] or a non-specific antibody (rabbit anti-rat IgG, Cappel catalog no. 0113–0082) followed by precipitation with protein A (Immunoprecipitin, BRL) (45). Immunoprecipitates were resuspended in kinase buffer containing [γ-32P]ATP and sonicated PI (Serdary Research Laboratories, Englewood Cliffs, NJ) (45, 46). Reactions were incubated at 30°C for 5 min and stopped by the addition of 1 M HCl. Lipids were extracted with chloroform/methanol (1:1 vol/vol), and then chromatographed by spotting onto a preadsorbent loading strip of thin-layer silica plates (Baker Si250-PA) using a chloroform/methanol/4.0 M ammonium hydroxide (9:7:2) developing solvent, and visualized by autoradiography. Intensities were quantified with a densitometer. Distinction between PI-3′-K (type I) and the closely related PI-4′-K (type II) was determined by the former enzyme’s sensitivity in the PI kinase assay to 0.6% (vol/vol) Nonidet P-40, a nonionic detergent (47).
PAGE and Immunoblotting.
p21ras immunoprecipitates to be resolved directly by PAGE were prepared from equal amounts of extracted protein, and Western blot analyses were performed (40). Blots were incubated sequentially with Y13-259, goat anti-rat antibody conjugated to alkaline phosphatase, and treated with a color substrate (BCIP/NBT, Vector Laboratories).
For detection of growth factor receptors, cells in triplicate were starved overnight in DMEM and pulsed for 15 min at 37°C with 1% bovine serum albumin (BSA), 1% BSA/40 ng/ml recombinant human PDGF-BB (Upstate Biotechnology catalog no. 01–105), or 1% BSA/100 ng/ml EGF (Collaborative Research catalog no. 4001) in DMEM. Cells were lysed in cold modified RIPA buffer [50 mM Tris·HCl, pH 7.4/1% (wt/vol) Nonidet P-40/0.25% (wt/vol) sodium deoxycholate/150 mM NaCl/1 mM EGTA/1 mM phenylmethylsulfonyl fluoride/1 μg/ml aprotinin and leupeptin/1 mM sodium vanadate/1 mM NaF], passed 3–5 times through a no. 22 needle, and centrifuged. Equal amounts of protein were analyzed by Western blot. PDGFR were detected using 500 ng/ml rabbit anti-PDGFR-B (Upstate Biotechnology catalog no. 06–131), followed by 1 μCi/ml (1 Ci = 37 GBq) 125I-protein A (ICN catalog no. 68038) and PhosphorImager analysis (26). This blot was then stripped (48) and checked for removal of label. Phosphotyrosines were detected by treating this blot sequentially with 100 ng/ml mouse anti-phosphotyrosine (Upstate Biotechnology catalog no. 05–321), 1.1 μg/ml rabbit anti-mouse antibody (Sigma catalog no. 6024), and 125I-protein A. From PhosphorImager analysis, ratios of phosphotyrosine to PDGFR for the each lane were calculated for each blot. An identical blot was also tested for EGFR using 1 ng/ml sheep anti-human EGFR (Upstate Biotechnology catalog no. 06–129), which cross-reacts with mouse EGFR, followed by 1:1000 dilution of rabbit anti-sheep antibody (Pierce catalog no. 31240H), and 125I-protein A.
RESULTS
We have previously established that both the E7 and E5 genes of RhPV 1 were capable of transforming NIH 3T3, or could cooperate with activated Ha-ras to transform primary epithelial cells (39). Activation of p21ras during the course of a natural papillomavirus infection could enhance progression to a malignant state. E5 proteins have been found associated with growth factor receptors (26), and control of p21ras activation is an event downstream of intermediate p21ras modulators (e.g., GAP and GNRPs) controlled by tyrosine kinase activity of growth factor receptors. We examined the activation of p21ras in cells in the presence and absence of various papillomavirus oncogenes to determine whether (i) normal p21ras is activated in transformed cells, (ii) the activation is gene specific, and (iii) stimulation by EGF, PDGF, or serum were important cofactors for the observed effects. Two NIH 3T3-derived cell lines with transformed phenotypes were compared with the parental cells. The RhPV 1 E5 and RhPV 1 E7 cell lines are transformants of RhPV 1 E5 and RhPV E7 genes, respectively (39). We also created a transformed NIH 3T3 cell line (pCO11) that overexpressed normal human EFGR cDNA. As a control of a previously studied cell line, we examined G54 cells derived from Swiss 3T3 cells that overexpressed normal cellular p21ras, but have a normal phenotype (42).
Ras Activity Under Steady-State Conditions.
Lysates of 32P-labeled cells under steady-state conditions (10% serum) (Table 1 and Fig. 1A) were prepared and p21ras/GTP and p21ras/GDP complexes were immunoprecipitated with a monoclonal antibody specific for p21ras (Y13-259). The p21ras-bound nucleotides were extracted from the immunoprecipitates, chromatographed on thin layer plates, and analyzed for the amount of radioactivity comigrating with GTP and GDP. The relative proportion of p21ras-bound GTP was approximately 6-fold higher in RhPV 1 E5-transformed cells than in the parental NIH 3T3 cells and fivefold higher than in NIH 3T3 cells transformed with the RhPV 1 E7 oncogene under identical conditions (Table 1). The level of activation of p21ras by RhPV 1 E5 was approximately the same as that observed in cells overexpressing normal human EGFR (pCO11), and was less than that observed with Swiss 3T3 cells overexpressing normal p21ras (G54) (Table 1). An equivalent number of cells from each cell line was lysed and immunoprecipitated with anti-p21ras monoclonal antibody. Western blot analysis using the same antibody showed that the levels of endogenous p21ras under these conditions are similar in NIH 3T3, RhPV 1 E5, RhPV 1 E7, and pCO11 cells (Fig. 1B).
Table 1.
Activation of p21ras
| Growth conditions* | Cell lines
|
||||
|---|---|---|---|---|---|
| NIH 3T3 | RhPV 1 E7 | RhPV 1 E5 | G54 | pCO11 | |
| Steady state | |||||
| Average (N) SEM† | 1.18 (3) 0.09 | 1.33 (4) 0.31 | 6.88 (5) 0.59 | 9.47 (2) 0.16 | 7.20 (3) 1.13 |
| Relative activation‡ | 1.0 | 1.1 | 5.8 | 8.1 | 6.1 |
| Quiescent | |||||
| Average (N) SEM | 0.10 (2) 0.02 | 1.38 (4) 0.53 | 0.61 (4) 0.22 | 1.10 (2) 0.10 | 1.30 (3) 0.53 |
| Relative activation‡ | 1.0 | 13.8 | 6.1 | 11 | 13 |
| EGF-stimulated | |||||
| Average (N) SEM | 2.87 (4) 0.52 | 2.94 (4) 0.17 | 6.16 (5) 0.65 | 8.31 (2) 1.82 | 7.53 (3) 0.82 |
| Relative activation‡ | 1.0 | 1.0 | 2.1 | 2.9 | 2.6 |
| Stimulation effect§ | 28.7 | 2.1 | 10.1 | 7.6 | 5.8 |
| PDGF-stimulated | |||||
| Average (N) SEM | 18.09 (5) 2.77 | 34.71 (3) 1.23 | 23.01 (3) 2.27 | 3.22 (2) 0.27 | 14.65 (3) 0.68 |
| Relative activation‡ | 1.0 | 1.9 | 1.3 | 0.2 | 0.8 |
| Stimulation effect§ | 180.9 | 25.2 | 37.7 | 2.9 | 11.3 |
Growth conditions tested were normal culture conditions (steady state), quiescent cells without EGF, and quiescent cells that had been stimulated with EGF or PDGF for 30 min.
Activation of p21ras was measured in independent assays. The average % GTP of these assays is presented with the number (N) of assays and the standard error of the mean (SEM).
Activation was compared to parental NIH 3T3 cells.
Relative p21ras activation of EGF- or PDGF-stimulated cells compared to their quiescent counterparts.
Figure 1.
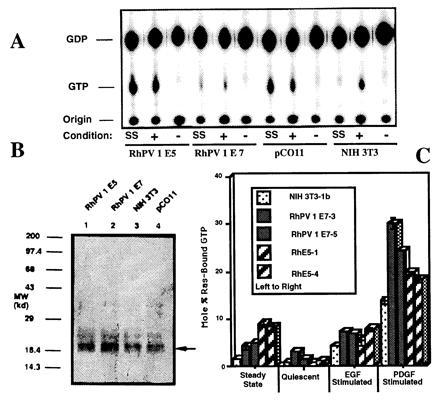
Determination of p21ras-bound guanine nucleotides from immunoprecipitates of normal and transformed NIH 3T3 cells. (A) Labeled cells were disrupted following growth under steady-state (SS), EGF-stimulated (+), or quiescent (−) conditions. Anti-p21ras antibody was used to immunoprecipitate p21ras and bound nucleotide. Following dissociation, nucleotides were separated using polyethyleneimine-cellulose chromatography and autoradiographed. The positions of the origin, GDP, and GTP are shown. (B) Anti-p21ras antibody Western blot of anti-p21ras immunoprecipitated cell lysates. Arrow indicates position of p21ras. (C) Independent additional clones of NIH 3T3 (clone 1b), RhPV 1 E7 (clones 3 and 5), and RhPV 1 E5 (clones 1 and 4) cells were tested in duplicate assays for Ras activation under various conditions as indicated with standard error bars shown.
Ras Activity Under Quiescent and Ligand-Stimulated Conditions.
All cell lines were brought to a quiescent state wherein DNA synthesis was arrested (44). We compared p21ras activation in each cell line to the same cell line under both quiescent or ligand-stimulated conditions, and to NIH 3T3 cells under similar conditions (Table 1). Under quiescent conditions, the average level of activation of p21ras was severely depressed (12-fold = 1.18/0.10) in parental NIH 3T3 cells (Table 1) as compared with its level when grown under normal steady-state conditions (Table 1). Activation in RhPV 1 E5-transformed cells in the quiescent state was reduced by about 11-fold, 7-fold in G54 cells, and 5-fold to 6-fold in pCO11-transformed cells. In cells transformed by RhPV 1 E7, activation of p21ras was not reduced (1.33/1.38). When cells were subsequently stimulated with EGF, the ratio of p21ras/GTP to p21ras/GDP generally increased to levels comparable to that of cells in the steady state. Compared with the respective quiescent cells, p21ras activation was about 10 times higher in stimulated RhPV 1 E5-transformed cells, six times higher in pCO11 cells, two times higher in RhPV 1 E7-transformed cells, and about 29 times higher in NIH 3T3 cells. As previously reported (44), G54 cells under steady-state or stimulated conditions activated p21ras compared with the quiescent state. We found this level of activation approximately equivalent to that found with RhPV 1 E5. Either in steady-state NIH 3T3 cells or in G54 cells that are in steady state or EGF-stimulated, the degree of stimulation compared with quiescent cells were only slightly higher than that which was previously reported (approximately 1–3%, and 4-fold, respectively) (42, 44). Finally, we examined the degree of activation p21ras following stimulation with PDGF-BB. p21ras in G54 cells was activated about 3-fold, comparable to that previously reported (42, 44). NIH 3T3, RhPV 1 E7, RhPV 1 E5, and pCO11 cell lines had much larger increases in p21ras activation. Compared with the quiescent state, p21ras in these cell lines was activated by 181, 25, 38, and 11 times, respectively. The mole percent of Ras-bound GTP in PDGF-stimulated cells was about equal for NIH 3T3 and pCO11 cells, but was about 1.3 and 1.9 times higher for RhPV 1 E5 and E7 cells. Thus, while the activation of p21ras in RhPV 1 E5 cells is elevated in steady-state or quiescent conditions, either E5- or E7-transformed cells can be stimulated with PDGF to a higher absolute level than NIH 3T3 cells.
An independent clone of vector-containing NIH 3T3 cells and two new independent clones of RhPV 1 E7 and E5 were each tested in duplicate under all of the conditions described above to examine the effect of clonal differences (Fig. 1C). Results were generally similar but two differences were noted. First, under steady-state conditions, both new RhPV 1 E7 clones had three times more activated Ras than NIH 3T3, but still about half that of two new RhPV 1 E5 clones. Second, under EGF-stimulated conditions, Ras activation was about equal for RhPV 1 E5 and RhPV 1 E7 cells, and was higher for both than for NIH 3T3 cells. However, relative to their respective quiescent states, ligand stimulated activation was still one and one-half to two times greater for RhPV 1 E5 cells than for RhPV1 E7 cells. Both E7 and E5 can activate Ras to different levels under various conditions.
Phosphatidylinositol Kinase (PIK) Is Up-Regulated in Papillomavirus-Transformed NIH 3T3 Cells.
We have modified a biochemical assay to measure the activity of PI-3′-K using anti-EGFR (Fig. 2A) sera with extracts of NIH 3T3 cells transfected with constructs of the E5 ORF from RhPV 1 and BPV-1. NIH 3T3 cells not containing E5 had low PIK activity that was increased by about 45% upon EGF stimulation as determined by densitometry (Fig. 2A). No activity was observed if a nonspecific antibody was used instead of anti-EGFR. NIH 3T3 cells containing an expression vector for EGFR (pC012) also showed increased PIK activity upon stimulation with EGF, although to a greater degree than for NIH 3T3 cells. Unstimulated BPV-1 E5 and RhPV 1 E5 cell lines had 2.3–7.7 times as much PIK activity as NIH 3T3 cells. PIK activity increased in BPV-1 E5 cells by 79% and in two independent clones of RhPV 1 E5 cells by 18% and 300% following stimulation with EGF. The range may represent clonal variations. This indicates that BPV-1 E5 and RhPV1 E5 transfected cells activate PIK but retain some variable amount activation upon stimulation (Fig. 2A).
Figure 2.
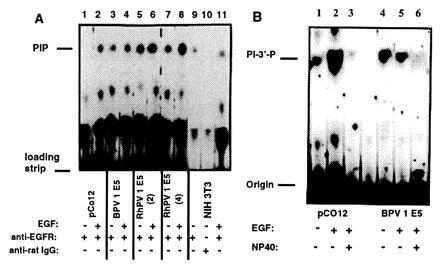
Activation of the PIK in papillomavirus E5-transformed cell lines. Confluent cells were maintained in serum-free media for 24 hr and then harvested for protein, with or without stimulation by EGF. (A) The total cellular protein was immunoprecipitated with an anti-EGFR antibody or a nonspecific antibody (rabbit anti-rat IgG), and protein A, and the PIK assay performed. Results are shown for two independent clones RhPV 1 E5(2) and RhPV 1 E5(4). (B) Distinction between PI-3′-K (type I) and the closely related PI-4′-K (type II) was determined by the former enzyme’s sensitivity to 0.6% Nonidet P-40. The position of polyphosphoinositides and the loading strip are indicated. Intermediate bands represent polyphosphatidylinositols (49).
Identification of the Up-Regulated Kinase as a Type I Kinase.
We performed kinase assays in the absence and presence of Nonidet P-40 (0.6% vol/vol), which destroys the activity of the type I kinase, characteristic of PI-3′-K (47) (Fig. 2B). The results demonstrated that the product of this kinase assay is the PI-3′-phosphate species, and not another biphosphoinsositide species such as PI-4′-phosphate. In this particular experiment, increased PIK activity from EGF stimulation was not observed for BPV-1 E5 as it had been for other experiments.
Determination of the Levels and Activation of EGFR and PDGFR Proteins.
It would be important to determine the effect of RhPV 1 E5 on both the number of receptors and state of activation of EGFR and PDGFR in these cells. PDGFR is the predominant growth factor receptor in NIH 3T3 cells [1 × 105 per cell (32)]. EGFRs are present in much lower levels and have been estimated at <1-2 × 104 per cell (32, 43, 50). We examined total levels of PDGFR and EGFR by Western blot analysis. PDGFR has been shown to affect the activation of Ras, and, conversely, Ras activation has been shown to affect PDGFR tyrosine kinase activity. In triplicate assays, RhPV 1 E5 and NIH 3T3 cells were starved overnight and treated with recombinant PDGF-BB, EGF, or media lacking either of these reagents for 15 min at 37°C, and whole cell lysates from each were prepared. Equal amounts of protein were subjected to Western blot analysis for total PDGFR (Fig. 3A), tyrosine-activated receptors (Fig. 3B), or EGFR (Fig. 4A). The phosphotyrosine band comigrated only with the mature form of PDGFR at 185 kDa and not with the much weaker immature form of PDGFR at 158 kDa (51) not easily seen in this exposure. The level of endogenous unstimulated tyrosine phosphorylation of EGFR (168 kDa) was not generally detectable by this method, but could be observed only after EGF stimulation (Fig. 3B). To confirm that this was EGFR, the anti-EGFR Western blot from which Fig. 4A was derived was stripped and reincubated with anti-phosphotyrosine antibody producing results identical to Fig. 3B. The phosphotyrosine-containing band at 168 kDa comigrated with EGFR from the original autoradiography (data not shown). The amount of EGFR phosphorylation under EGF stimulation was about the same for all three cell lines.
Figure 3.
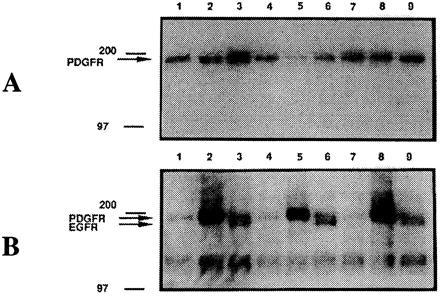
Sample Western blot analyses for PDGFR and phosphotyrosine-PDGFR. Starved cells were either mock-stimulated (lanes 1, 4, and 7), stimulated with hrPDGF-BB (lanes 2, 5, and 8), or EGF (lanes 3, 6, and 9). Cells were lysed and equal amounts of protein loaded onto a SDS/7.5% polyacrylamide gel. After Western transfer, the blots were tested successively for PDGFR-B (A) and phosphotyrosine (B) analysis. Lanes: 1–3, NIH 3T3; 4–6, RhPV 1 E7; 7–9, RhPV 1 E5. Molecular weight markers in kDa are shown on the left and the receptor locations indicated.
Figure 4.
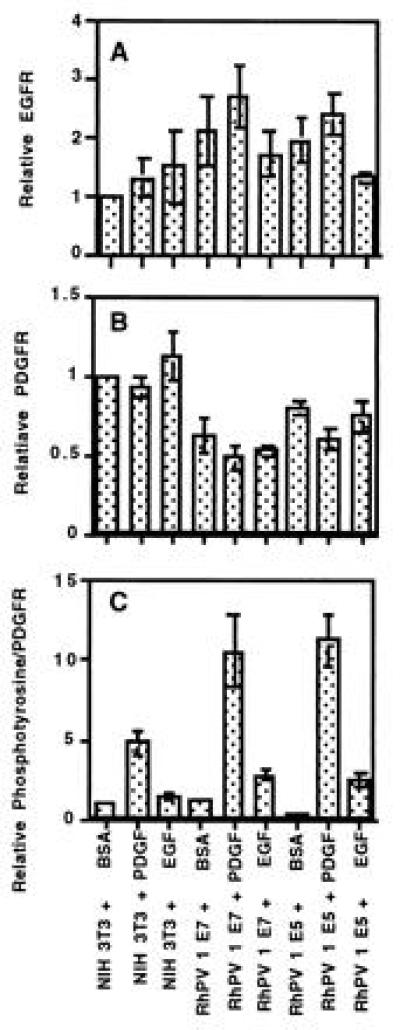
Relative EGFR, PDGFR, and phosphotyrosine to PDGFR ratios. Cells were treated as in Fig. 3 to detect EGFR (A), PDGFR (B), or phosphotyrosine and PDGFR (C) ratios. Using PhosphorImager analysis, the intensity of each band (less background) in A and B was normalized to the intensity of the unstimulated NIH 3T3 (NIH 3T3 + BSA) cells for that blot. In C, the intensity of phosphotyrosine signal comigrating with PDGFR was compared with the intensity of the PDGFR signal from the same blot and normalized to the ratio obtained from unstimulated NIH 3T3 cells. Error bars show the standard error of the mean of the ratios. Results are from three independent experiments, and blots of two were in duplicate.
Total endogenous EGFR for starved or PDGF-stimulated RhPV 1 E5 or E7 cells was about twice that of NIH 3T3 cells, but was similar for all after EGF stimulation (Fig. 4A). RhPV 1 E5 and E7 cells had 50–80% the level of endogenous PDGFR of comparable NIH 3T3 cells (Fig. 4B).
As previously reported in other cells (26), compared with unstimulated cells, NIH 3T3 cells stimulated with PDGF have increased (4.8 times) tyrosine phosphorylation of PDGFR (Fig. 4C). Unstimulated NIH 3T3 and RhPV 1 E7 cells have equivalent amounts of PDGFR activation, while RhPV 1 E5 cells have about half as much. However, PDGF-stimulated RhPV 1 E5 and E7 cells have 10 to 11 times higher phosphorylation of the PDGFR than unstimulated NIH 3T3 cells, about twice that seen for stimulated NIH 3T3 cells. The relative levels of PDGFR activation after EGF stimulation were 1.4, 2.8, and 2.5 for NIH 3T3, RhPV 1 E7, and RhPV 1 E5 cells, respectively.
DISCUSSION
Under steady-state growth conditions, there is a significantly higher level of activation of p21ras in NIH 3T3 cells transformed by the RhPV 1 E5 gene as compared with cells transformed by RhPV 1 E7 gene or the parental cell line. The result with RhPV 1 E5 cells parallels that observed with pCO11 cells that contains an increased amount of intrinsic tyrosine kinase activity from the protooncogene human EGFR. The relative levels of the p21ras protein present in these cell lines are roughly equivalent, and thus cannot be a factor in the differences in activation that we have observed. Stimulation of serum-starved cells with EGF or PDGF-BB increased the level of active p21ras/GTP complex, as has been reported for quiescent G54 cells when stimulated to initiate DNA synthesis with serum, PDGF, or EGF (44). In our experiments, the response to serum was markedly larger in cells expressing RhPV 1 E5 than RhPV 1 E7 proteins. Ras activation was greater for both E7 and E5 cells than NIH 3T3 cells under EGF stimulation (Fig. 1C), and, in one cell line, E5-induced activation was greater than E7-induced activation (Table 1). Under PDGF stimulation, activation of Ras was greatest for E7, followed by E5 and then for vector-containing cells. RhPV 1 E5 activation of p21ras in NIH 3T3 cells was similar to that observed for erb-2/neu or v-src oncogenes, which have intrinsic tyrosine kinase activity (44). In support of our finding of p21ras activation by E5, several downstream targets of p21ras have been shown to be activated by the HPV 11 E5 gene including c-jun and junB (52), and protein kinase C (53). In addition, HPV 16 E5, but not E6 or E7 increased MAP kinase activity (54).
BPV-1 E5 transformation correlates with its ability to mediate increases in tyrosine phosphorylation of the PDGFR in C127 cells (26). EGFR and PDGFR have tyrosine kinase activity, and the phosphorylation of tyrosine residues of certain putative key substrates such as phospholipase C type γ (55–57), PI-3′-K (58), GAP (59–61), and Raf (62) are thought to be initial events in signal transduction pathway associated with cellular mitogenesis. In mammalian cells, an adapter protein (Sem/Grb2) appears to mediate the interaction of some GNRPs with activated receptor molecules (63) and p21ras bringing membrane-bound p21ras into a complex with tyrosine kinase receptors and other signal associated molecules (11–13, 64). As GAP is also present in many receptor complexes (65), all of the necessary components for ligand-stimulated transduction of cellular growth signals exist in close proximity. E5 protein was shown to induce DNA synthesis in serum-starved cells demonstrating that E5 could functionally replace growth factors in this assay (66, 67). Moreover, Petti et al. (26) also demonstrated that the E5 gene caused acute morphological transformation and receptor activation in C127 cells. In our experiments, the presence of RhPV 1 E5 or E7 slightly increases the amount of total endogenous EGFR. NIH 3T3 cells transfected with exogenous human EGFR showed increased levels of cells surface EGFR when transformed with BPV-1 E5 (25). We found total PDGFR in RhPV 1 E5- or E7-transformed cells to be somewhat decreased compared with parental NIH 3T3 cells. BPV-1 E5-containing FR3T3 and C127 cells have roughly equal amounts of PDGFR as their parent cells (26). It is possible that the higher level of EGFR in E5-containing cells alone may account for the increase that we observed in Ras activation. Due to the undetectable amount of phosphorylated EGFR in quiescent cells, we were not able to determine the numerical relative activation of the EGFRs following EGF stimulation, although it was clear that activation occurred upon EGF stimulation in all cell lines. RhPV 1 E5- and E7-containing NIH 3T3 cells have approximately the same basal activation of PDGFR compared with NIH 3T3 cells. All cell lines are responsive to PDGFR activation by PDGF-BB, but more so for cells containing either oncogene. This differs from other reports in BPV E5-containing FR3T3 cells where PDGFR were constitutively activated, and showed little extra response to ligand stimulation (26). It is not yet clear if E5 is activating p21ras by interacting only to increase activation of the growth factor receptors, or may also be interacting with intermediate positive and negative modulators of p21ras activity. In NIH 3T3 cells Ras activation by EGF and PDGF was not associated with increased Ras-GAP activity, but rather with increased GNRP activity, and this activity could be increased with v-Src or ErbB2 but not v-Mos or v-Raf (68).
In our study stimulation with EGF also caused a small increase in PDGFR-tyrosine phosphorylation of all cells compared with unstimulated cells. PDGFR activation alone is closely linked with Ras activation through SHPTP2 (69, 70), Grb2, and Son of Sevenless. EGF, but not PDGF, can increase transformation of C127, J23T3, or F3T3 cells by either HPV 6 or HPV 16 E5 (28).
Downstream events of activation of EGFR include activation of PIK. We demonstrated that there was an up-regulation of PI-3′-K in NIH 3T3 cells transformed by the RhPV 1 E5. Thus, E5 may affect several cytokine induction pathways downstream of growth factor receptors.
In most papillomavirus-associated malignancies, there is usually integration of the viral genome with retention of the E6 and E7 genes and commonly a loss of E5 function. However, the expression of the E5 gene and its interaction with host cytokine-induced transduction pathways may provide a key element to the initiation of malignant transformation. A recent report suggested that E5 could transform NIH 3T3 cells but was not necessary for maintenance of the transformed state (71). The results of this study lend credence to this possibility.
Acknowledgments
We thank Dr. T. Satoh for his invaluable assistance in resolving technical problems. This work was supported by grants from the National Institutes of Health (CA 26542) and the Minnesota Leukemia Fund.
Footnotes
Abbreviations: RhPV, rhesus papillomavirus; BPV, bovine papillomavirus; EGF, epidermal growth factor; EGFR, EGF receptor; PDGF, platelet-derived growth factor; PDGFR, PDGF, receptor; PI-3′-K, phosphatidylinositol-3′-kinase; PIK, phosphatidylinositol kinase; GAP, GTPase-activating protein; GNRP, guanine nucleotide-releasing protein.
References
- 1.Barbacid M. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 2.Satoh T, Endo M, Nakamura S, Kaziro Y. FEBS Lett. 1988;236:185–189. doi: 10.1016/0014-5793(88)80311-9. [DOI] [PubMed] [Google Scholar]
- 3.Hoshino M, Kawakita M, Hattori S. Mol Cell Biol. 1988;8:4169–4173. doi: 10.1128/mcb.8.10.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibbs J, Schaber M, Marshall M, Scholnick E, Sigal I. J Biol Chem. 1987;262:10426–10429. [PubMed] [Google Scholar]
- 5.Trahey M, McCormick F. Science. 1987;238:542–545. doi: 10.1126/science.2821624. [DOI] [PubMed] [Google Scholar]
- 6.Satoh T, Nakamura S, Kaziro Y. Mol Cell Biol. 1987;7:4553–4556. doi: 10.1128/mcb.7.12.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hattori S, Fukuda M, Yamashita T, Nakamura S, Gotoh Y, Nishida E. J Biol Chem. 1992;267:20346–20351. [PubMed] [Google Scholar]
- 8.Dent P, Haser W, Haystead T, Vincent L, Roberts T, Sturgill T. Science. 1992;257:1404–1407. doi: 10.1126/science.1326789. [DOI] [PubMed] [Google Scholar]
- 9.Vojtek A, Hollenberg S, Cooper J. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- 10.Kaziro Y. Biochim Biophys Acta. 1978;505:95–127. doi: 10.1016/0304-4173(78)90009-5. [DOI] [PubMed] [Google Scholar]
- 11.Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P, Bar-Sagi D, Margolis B, Schlessinger J. Nature (London) 1993;363:85–88. doi: 10.1038/363085a0. [DOI] [PubMed] [Google Scholar]
- 12.Rozakis-Adcock M, Fernley R, Wade J, Pawson T, Bowtell D. Nature (London) 1993;363:83–85. doi: 10.1038/363083a0. [DOI] [PubMed] [Google Scholar]
- 13.Egan S, Giddings B, Brooks M, Buday L, Sizeland A, Weinberg R. Nature (London) 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell R. Cell Calcium. 1982;3:429–440. doi: 10.1016/0143-4160(82)90028-8. [DOI] [PubMed] [Google Scholar]
- 15.Kodaki T, Woscholski R, Hallberg B, Rodriguez-Viciana P, Downward J, Parker P. Curr Biol. 1994;4:798–806. doi: 10.1016/s0960-9822(00)00177-9. [DOI] [PubMed] [Google Scholar]
- 16.Scheffner M, Munger K, Byrne J, Howley P. Proc Natl Acad Sci USA. 1991;88:5523–5527. doi: 10.1073/pnas.88.13.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moolenaar W, Tsien R, Vander-Saag P, De Laat S. Nature (London) 1983;304:645–647. doi: 10.1038/304645a0. [DOI] [PubMed] [Google Scholar]
- 18.Coughlin S, Escobedo J, Williams L. Science. 1989;24:1191–1194. doi: 10.1126/science.2466336. [DOI] [PubMed] [Google Scholar]
- 19.Müller R, Bravo R, Burckhardt J, Curran T. Nature (London) 1984;312:716–720. doi: 10.1038/312716a0. [DOI] [PubMed] [Google Scholar]
- 20.Armelin H. Proc Natl Acad Sci USA. 1973;70:2702–2706. doi: 10.1073/pnas.70.9.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollenberg M, Cuatrecasas P. Proc Natl Acad Sci USA. 1973;70:2964–2968. doi: 10.1073/pnas.70.10.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ushiro H, Cohen S. J Biol Chem. 1980;255:8363–8365. [PubMed] [Google Scholar]
- 23.Schiller J, Vass W, Vousden K, Lowy D. J Virol. 1986;57:1–6. doi: 10.1128/jvi.57.1.1-6.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiMaio D, Guralski D, Schiller J. Proc Natl Acad Sci USA. 1986;83:1797–1801. doi: 10.1073/pnas.83.6.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin P, Vass W, Schiller J, Lowy D, Velu T. Cell. 1989;59:21–32. doi: 10.1016/0092-8674(89)90866-0. [DOI] [PubMed] [Google Scholar]
- 26.Petti L, Nilson L, DiMaio D. EMBO J. 1991;10:845–855. doi: 10.1002/j.1460-2075.1991.tb08017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cochet C. In: Growth Factors and Oncogenes. Bolla M, Chambaz E, Vrousos C, editors. Montrouge, France: John Libbey Eurotext Ltd.; 1989. pp. 13–24. [Google Scholar]
- 28.Straight S, Hinkle P, Jewers R, McCance D. J Virol. 1993;67:4521–4532. doi: 10.1128/jvi.67.8.4521-4532.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein D, Schlegel R. EMBO J. 1990;9:137–145. doi: 10.1002/j.1460-2075.1990.tb08089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstein D, Finbow M, Andresson T, Mclean P, Smith K, Bubb V, Schlegel R. Nature (London) 1991;352:347–349. doi: 10.1038/352347a0. [DOI] [PubMed] [Google Scholar]
- 31.McGlennen R, Ostrow R, Carson L, Stanley M, Faras A. Am J Obstet Gynecol. 1991;165:696–705. doi: 10.1016/0002-9378(91)90312-f. [DOI] [PubMed] [Google Scholar]
- 32.Cohen B, Goldstein D, Rutledge L, Vass W, Lowy D, Schlegel R, Schiller J. J Virol. 1993;67:5303–5311. doi: 10.1128/jvi.67.9.5303-5311.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldstein D, Li W, Wang L, Heidaran M, Aaronson S, Shinn R, Schlegel R, Pierce J. J Virol. 1994;68:4432–4441. doi: 10.1128/jvi.68.7.4432-4441.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phelps W, Yee C, Münger K, Howley P. Cell. 1988;53:539–547. doi: 10.1016/0092-8674(88)90570-3. [DOI] [PubMed] [Google Scholar]
- 35.Dyson N, Howley P, Munger K, Harlow E. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 36.Kloster B, Manias D, Ostrow R, Shaver M, McPherson S, Rangen S, Uno H, Faras A. Virology. 1988;166:30–40. doi: 10.1016/0042-6822(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 37.Bernard H, Chan S, Delius H. Curr Top Microbiol Immunol. 1994;186:33–54. doi: 10.1007/978-3-642-78487-3_3. [DOI] [PubMed] [Google Scholar]
- 38.Ostrow R, McGlennen R, Shaver M, Kloster B, Houser D, Faras A. Proc Natl Acad Sci USA. 1990;87:8170–8174. doi: 10.1073/pnas.87.20.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ostrow R, Liu Z, Schneider J, McGlennen R, Forslund K, Faras A. Virology. 1993;196:861–867. doi: 10.1006/viro.1993.1547. [DOI] [PubMed] [Google Scholar]
- 40.Ostrow R, Coughlin S, McGlennen R, Johnson A, Ratterree M, Scheffler J, Yaegashi N, Galloway D, Faras A. J Gen Virol. 1995;76:293–299. doi: 10.1099/0022-1317-76-2-293. [DOI] [PubMed] [Google Scholar]
- 41.Miller A, Rosman G. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 42.Satoh T, Masami E, Nakafuku M, Nakamura S, Kaziro Y. Proc Natl Acad Sci USA. 1990;87:5993–5997. doi: 10.1073/pnas.87.15.5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velu T, Beguinot L, Vass W, Zhang K, Merlino G, Pastan I, Lowy D. J Cell Biochem. 1989;39:153–166. doi: 10.1002/jcb.240390207. [DOI] [PubMed] [Google Scholar]
- 44.Satoh T, Masami E, Nakafuku M, Akiyama T, Yamamoto T, Kaziro Y. Proc Natl Acad Sci USA. 1990;87:7926–7929. doi: 10.1073/pnas.87.20.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bjorge J, Chan T, Antczak M, Kung H, Fujita D. Proc Natl Acad Sci USA. 1990;87:3816–3820. doi: 10.1073/pnas.87.10.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitman M, Downes P, Keeler M, Keller T, Cantley L. Nature (London) 1988;332:644–646. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 47.Whitman M, Kaplan D, Roberts T, Cantley L. Biochem J. 1987;247:165–174. doi: 10.1042/bj2470165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaufmann S, Ewing C, Shaper J. Anal Biochem. 1987;161:89–95. doi: 10.1016/0003-2697(87)90656-7. [DOI] [PubMed] [Google Scholar]
- 49.Cochet C, Filhol O, Payrastre B, Hunter T, Gill G. J Biol Chem. 1991;266:637–644. [PubMed] [Google Scholar]
- 50.Helin K, Beguinot L. J Biol Chem. 1991;273:8363–8368. [PubMed] [Google Scholar]
- 51.Keating M, Williams L. J Biol Chem. 1987;262:7932–7937. [PubMed] [Google Scholar]
- 52.Chen S, Tsao Y, Yang C, Lin Y, Huang C, Kuo S. J Gen Virol. 1995;76:2653–2659. doi: 10.1099/0022-1317-76-11-2653. [DOI] [PubMed] [Google Scholar]
- 53.Tsao Y, Huang C, Lin Y, Chen S. Cancer Lett. 1995;95:201–205. doi: 10.1016/0304-3835(95)03894-3. [DOI] [PubMed] [Google Scholar]
- 54.Gu Z, Matlashewski G. J Virol. 1995;12:8051–8056. doi: 10.1128/jvi.69.12.8051-8056.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Margolis B, Rhee S, Felder S, Mervic M, Lyall R, Levitzki A, Ullrich A, Zilberstein A, Schlessinger J. Cell. 1989;57:1101–1107. doi: 10.1016/0092-8674(89)90047-0. [DOI] [PubMed] [Google Scholar]
- 56.Meisenhelder J, Suh P, Rhee S, Hunter T. Cell. 1989;57:1109–1122. doi: 10.1016/0092-8674(89)90048-2. [DOI] [PubMed] [Google Scholar]
- 57.Nishibe S, Wahl M, Rhee S, Carpenter G. J Biol Chem. 1989;264:10335–10338. [PubMed] [Google Scholar]
- 58.Kaplan D, Whitman M, Schaffhausen B, Pallas D, White M, Cantley L, Roberts T. Cell. 1987;50:1021–1029. doi: 10.1016/0092-8674(87)90168-1. [DOI] [PubMed] [Google Scholar]
- 59.Molloy J, Bottaro D, Fleming T, Marshall M, Gibbs J, Aaronson S. Nature (London) 1989;342:711–714. doi: 10.1038/342711a0. [DOI] [PubMed] [Google Scholar]
- 60.Ellis C, Moran M, McCormick R, Pawson T. Nature (London) 1990;343:377–381. doi: 10.1038/343377a0. [DOI] [PubMed] [Google Scholar]
- 61.Kaplan D, Morrison D, Wong G, McCormick F, Williams L. Cell. 1990;61:125–133. doi: 10.1016/0092-8674(90)90220-9. [DOI] [PubMed] [Google Scholar]
- 62.Morrison D, Kaplan D, Escobedo J, Rapp U, Roberts T, Williams L. Cell. 1989;58:649–657. doi: 10.1016/0092-8674(89)90100-1. [DOI] [PubMed] [Google Scholar]
- 63.Gulbins E, Coggeshall K, Baier G, Katzav S, Burn P, Altman A. Science. 1993;260:822–825. doi: 10.1126/science.8484124. [DOI] [PubMed] [Google Scholar]
- 64.Gale N, Kaplan S, Lowenstein E, Schlessinger J, Bar-Sagi D. Nature (London) 1993;363:88–92. doi: 10.1038/363088a0. [DOI] [PubMed] [Google Scholar]
- 65.Feig L. Science. 1993;260:767–768. doi: 10.1126/science.8484117. [DOI] [PubMed] [Google Scholar]
- 66.Green M, Lowenstein P. Cell. 1987;51:795–802. doi: 10.1016/0092-8674(87)90102-4. [DOI] [PubMed] [Google Scholar]
- 67.Settleman J, Fazeli A, Malicki J, Horowitz B, DiMaio D. Mol Cell Biol. 1989;9:5563–5572. doi: 10.1128/mcb.9.12.5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B, Subleski M, Shalloway D, Kung H, Kamata T. Proc Natl Acad Sci USA. 1993;90:8504–8508. doi: 10.1073/pnas.90.18.8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li A, Nishimura R, Kashishian A, Batzer A, Kim W, Cooper J, Schlessinger J. Mol Cell Biol. 1994;14:509–517. doi: 10.1128/mcb.14.1.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bennett A, Tang T, Sugimoto S, Walsh C, Neel B. Proc Natl Acad Sci USA. 1994;91:7335–7339. doi: 10.1073/pnas.91.15.7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen S, Tsao Y, Lee J, Liu H, Yang C, Tsao L. J Gen Virol. 1994;75:1953–1960. doi: 10.1099/0022-1317-75-8-1953. [DOI] [PubMed] [Google Scholar]