

Abstract
Human replication factor C (RFC, also called activator 1) is a five-subunit protein complex (p140, p40, p38, p37, and p36) required for proliferating cell nuclear antigen (PCNA)-dependent processive DNA synthesis catalyzed by DNA polymerase δ or ɛ. Here we report the reconstitution of the RFC complex from its five subunits simultaneously overexpressed in baculovirus-infected insect cells. The purified baculovirus-produced RFC appears to contain equimolar levels of each subunit and was shown to be functionally identical to its native counterpart in (i) supporting DNA polymerase δ-catalyzed PCNA-dependent DNA chain elongation; (ii) catalyzing DNA-dependent ATP hydrolysis that was stimulated by PCNA and human single-stranded DNA binding protein; (iii) binding preferentially to DNA primer ends; and (iv) catalytically loading PCNA onto singly nicked circular DNA and catalytically removing PCNA from these DNA molecules.
Keywords: eukaryotic DNA replication, proliferating cell nuclear antigen, DNA polymerase δ, activator I
Replication factor C (RFC; also known as activator 1) functions as an accessory factor for proliferating cell nuclear antigen (PCNA)-dependent DNA synthesis catalyzed by DNA polymerase δ or ɛ (pol δ or ɛ) (1–7). RFC contains multiple activities including its ability to preferentially bind DNA primer ends and catalyze DNA-dependent ATP hydrolysis.
Following its association with DNA at a primer end, RFC recruits PCNA (the clamp) and loads it onto DNA in the presence of ATP (clamp loading) (8, 9). This complex then tethers pol δ to the DNA primer junction in a reaction that requires ATP hydrolysis and results in highly processive DNA chain elongation (1–7, 10). This RFC-dependent PCNA loading mechanism is conserved among three species examined (human, Escherichia coli, and T4 bacteriophage). In E. coli and T4, the functional homologs of RFC are the γ complex and T4 gene products (gp) 44/62, while the counterparts of PCNA are the β subunit of the pol III holoenzyme and gp45, respectively.
In E. coli, the β clamp remains on DNA after completion of DNA synthesis and dissociation of the DNA polymerase. It is likely that a similar mechanism occurs in eukaryotes. In E. coli and HeLa cells, the estimated number of Okazaki fragments formed during one round of replication is >10 and 100 × the amounts of β and PCNA, respectively. As a result, the clamps must be recycled to ensure continuous DNA synthesis. The clamp loaders, the γ complex in E. coli and RFC in eukaryotes, also carry out the unloading of clamps to recycle these proteins. However, in T4 bacteriophage, clamp unloading may not be an active process but results from the spontaneous dissociation of gp45 as a result of its intrinsic instability on DNA (for summary, see ref. 8).
RFC is highly conserved from yeast (sc) to humans (h) in its subunit structure. It contains five subunits ranging between 36–140 kDa as revealed by SDS/PAGE. Genes encoding each of these subunits have been cloned from both mammals (12–17) and Saccharomyces cerevisiae, and each subunit has been shown to be essential following deletion analysis in yeast (18–23). The predicted amino acid sequences of each of the yeast and human RFC subunits reveals significant homology in seven regions referred to as RFC boxes (box II–VIII) (17, 20). The large subunit (p140) contains an additional box (box I) within its N-terminal region that shares homology with prokaryotic DNA ligases (14–16, 20).
The role of individual RFC subunits has been studied using the RFC complex isolated from human cells and individual subunits overexpressed and purified from E. coli. It has been demonstrated that hRFCp140 binds DNA (24), whereas both hRFCp37 and its yeast counterpart (scRFC2 gene product) interact with primed DNA templates with low affinity (22, 25). hRFCp40 directly interacts with both PCNA and ATP (24, 25), while the scRFC3 gene product (equivalent to hRFCp36) was shown to contain DNA-dependent ATPase activity (18).
Overexpression of the five scRFC gene products in S. cerevisiae was shown to increase the scRFC activity in a partially purified fraction (21). Recently, we reported the reconstitution of biologically active hRFC from its five subunits in a coupled in vitro transcription-translation system (26). We also demonstrated that the p40, p37, and p36 subunits of RFC formed a core complex to which hRFCp140 and hRFCp38 bound only when both were present.
To facilitate biochemical characterization, we reconstituted the hRFC from its five subunits expressed in recombinant baculovirus infected insect cells. Characterization of the purified baculovirus-produced RFC (bRFC) is reported here demonstrating that bRFC is functionally identical to its native counterpart.
MATERIALS AND METHODS
Preparation of DNAs and Proteins.
Poly(dA)4500 was obtained from Life Sciences (St. Petersburg, FL) and poly(dA)300 and oligo(dT)12–18 were from Pharmacia LKB. Poly(dA)4500 was annealed to oligo(dT)12–18 at a nucleotide ratio of 20:1; poly(dA)300 was annealed to oligo(dT)12–18 at a molar ratio of 1:0.4, 1:2, and 1:10, as described (3). φX174 single-stranded circular/SSC viral DNA was obtained from New England Biolabs. Singly primed M13 circular ssDNA and singly nicked pBluescript DNA were prepared as described (27, 28). Human ssDNA binding protein (HSSB), pol δ, PCNA, and hRFC were purified from HeLa cytosolic extracts as described (refs. 1, 3, 29, and 30, respectively). 32P-labeled PCNA was prepared using recombinant PCNA containing a cAMP-dependent protein kinase consensus sequence at its N terminus as described (31).
Replication Assays.
Replication assays using either poly(dA)4500/oligo(dT)12–18 or singly primed M13 DNA as the template were carried out as previously described (3, 26). One unit of RFC activity was defined as the incorporation of 1 nmol of radiolabeled dTMP or dCMP under the reaction conditions used.
ATPase Assay.
ATPase activity was assayed as previously described (3) except that incubation was carried out for 60 min at 37°C. One unit of ATPase activity was defined as the formation of 1 nmol of Pi under the reaction conditions used as quantitated by PhosphorImager (Molecular Probes) analysis.
DNA Binding Assay.
A nitrocellulose filter binding assay was carried out in reaction mixtures (25 μl) containing binding buffer (25 mM Hepes·NaOH, pH 7.5/5 mM MgCl2/1 mM DTT/100 μg/ml BSA/175 mM NaCl), bRFC (as indicated), and 40 fmol of 5′ 32P-labeled poly(dA)300 (700–1000 cpm/fmol), 5′ 32P-labeled oligo(dT)12–18 (4000–5000 cpm/fmol), or 5′ 32P-labeled poly(dA)300 hybridized to unlabeled oligo(dT)12–18 at various molar ratios as indicated. After 30-min incubation at 0°C, the mixture was filtered through an alkaline-washed nitrocellulose filter (Millipore, hemagglutinin 0.45 μm) that was then washed three times with 0.5 ml of binding buffer. The radioactivity adsorbed to the filter was measured by liquid scintillation counting.
PCNA Loading and Unloading Assays.
The loading and unloading of PCNA onto and off singly nicked pBluescript DNA was carried out as described for the loading and unloading of the β subunit of the E. coli pol III holoenzyme (8, 32).
Preparation of Recombinant Viruses.
The cDNAs coding for RFC subunits were subcloned into the following baculovirus transfer vectors: PVL1393 for the p40 cDNA; pBlueBacII for the p140, p36, and p38 cDNAs; and pBlueBacIII for p37. A sequence encoding His-6 as a tag was inserted into the coding sequences of the p38 and p140 cDNAs at the 5′- and 3′-ends, respectively. The 5′-untranslated regions of each cDNA (except for the p40) were modified to match a perfect Kozak sequence (ACCATGg or GCCATGg). The recombinant viruses were produced according to manufacturer’s instructions (Invitrogen).
Large-Scale Infection with hRFC–Recombinant Baculovirus and Preparation of Cell Extracts.
Monolayer high five (HF) cells (Invitrogen) were grown at 27°C to about 80% confluency in Grace’s medium supplemented with 10% fetal bovine serum. HF cells (6 × 108) were infected simultaneously with recombinant viruses that produce each of the five RFC subunits at a multiplicity of infection of 2 for each virus and were maintained at 27°C for 48 hr. The cells were then harvested by centrifugation at 180 × g for 15 min. The cell pellet was washed with ice-cold PBS, resuspended in two volumes of hypotonic buffer (50 mM Tris·HCl, pH 8.0/10 mM KCl/1.5 mM MgCl2/20 mM sodium phosphate, pH 8.0/0.5 mM phenylmethanesulfonyl fluoride/0.2 μg/ml aprotinin/0.2 μg/ml leupeptin/0.1 μg/ml antipain) per volume of packed cells and lysed with 10 strokes of a Dounce homogenizer. After centrifugation at 2400 × g for 30 min at 4°C, the nuclear pellet was resuspended in two vol of extraction buffer (hypotonic buffer without 10 mM KCl) per vol of packed cells, and the mixture was adjusted to a final concentration of 0.42 M NaCl. The suspension was centrifuged immediately at 43,500 × g for 30 min at 4°C and the supernatant (nuclear extract) was used for subsequent purification of bRFC.
Purification of bRFC.
Nuclear extract (10 ml, 80 mg of protein), prepared as described above, was incubated with 0.3 ml of nickel resin (Invitrogen) pre-equilibrated in buffer containing 50 mM Tris·HCl (pH 8.0), 20 mM sodium phosphate (pH 8.0), and 0.5 M NaCl. The mixture was rocked at 4°C overnight. The Ni beads were then washed twice with 10 ml buffer containing 20 mM sodium phosphate (pH 7.4), 0.5 M NaCl, and 10% glycerol and packed in a 0.7 × 4 cm column (Kontes). Bound protein [2.16 mg, 8250 units determined in the poly(dA)4500/oligo(dT)12–18 assay] was eluted from the column using 0.5 M imidazole dissolved in the wash buffer (20 mM sodium phosphate, pH 7.4/0.5 M NaCl/10% glycerol). A portion of the Ni-column eluate (0.4 mg, 1643 units) was subjected to two consecutive rounds of centrifugation (250,000 × g for 24 hr) through 15–35% glycerol gradients (5 ml) in buffer containing 25 mM Tris·HCl (pH 7.5), 1 mM EDTA, 0.01% Nonidet P-40, 0.4 M NaCl, 1 mM DTT, 0.2 μg/ml aprotinin, 0.2 μg/ml leupeptin, and 0.1 μg/ml antipain; 23 fractions of 220 μl were then collected from the bottom of each tube. Approximately 25% of the protein and all of the RFC activity was recovered following the two glycerol gradient centrifugation steps. The specific activity of the isolated bRFC in the poly(dA)4500/oligo(dT)12–18 assay was about 15,000 units/mg protein. Pooled glycerol gradient fractions were stored at −80°C and showed no loss of activity over a period of 3 months with repeated freezing and thawing.
RESULTS
Reconstitution of hRFC in Baculovirus-Infected Insect Cells That Supports PCNA-Dependent DNA Synthesis.
We have constructed recombinant baculoviruses that produce each of the hRFC subunits. Extracts prepared from HF cells infected with each of the five baculoviruses were examined for subunit protein expression by Coomassie blue staining (Fig. 1A, lanes 1–6) and immunoblot analyses using antibodies against each subunit (Fig. 1A, lanes 7–11). Expression of the p140 subunit (lanes 2 and 7) was lower than the expression of the other subunits and migration of the p40 and p38 subunits was similar due to the His-6 tag fusion on the p38 subunit.
Figure 1.
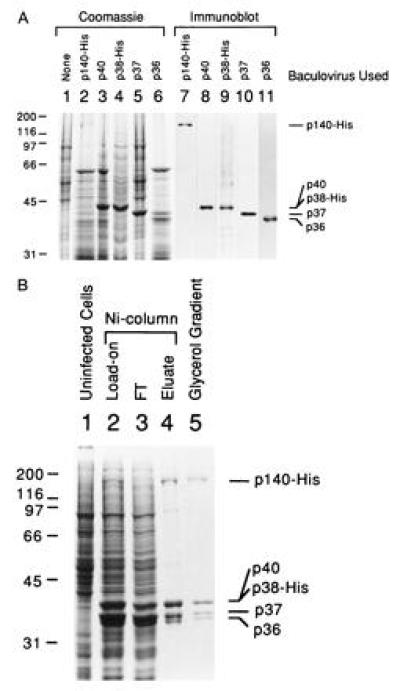
Reconstitution of RFC from its five subunits in baculovirus-infected HF cells. (A) Overexpression of the RFC subunits in baculovirus-infected HF cells. The five subunits of RFC were individually expressed in HF cells and cell lysates were analyzed by SDS/9% PAGE followed by staining with Coomassie brilliant blue (lanes 1–6) or by Western blot analysis (lanes 7–11). The additions to each lane were: lane 1, 30 μg of uninfected HF cell extract; lanes 2–6, 30 μg of extract from cells producing the p140, p40, p38, p37, and p36 subunit, respectively; lanes 7–11, immunoblots of cell lysates as described in lanes 2–6. Each blot was probed with specific antibodies against the corresponding subunit as follows: lane 7, p140; lane 8, p40; lane 9, p38; lane 10, p37; lane 11, p36. Molecular weight markers are indicated at the left of the figure and the position of each of the RFC subunits are indicated at the right. (B) Purification of reconstituted bRFC. The bRFC was reconstituted by coinfecting HF cells with all five recombinant viruses capable of expressing each RFC subunit. The reconstituted RFC was purified from nuclear extracts of the coinfected cells by Ni-affinity chromatography and subsequent glycerol gradient centrifugation as described. Proteins were visualized after SDS/PAGE by Coomassie blue staining. The additions to each lane were: lane 1, 30 μg of uninfected HF cell extract; lane 2, 25 μg of infected cell nuclear extract (Ni column load on); lane 3, 25 μg of protein that flowed through the Ni column; lane 4, 1.4 μg of Ni column eluate; lane 5, 0.9 μg of protein from glycerol gradient fractions 11–13. The markers at the left and right of the figure are as indicated in A.
Reconstitution of hRFC was accomplished following Ni-affinity column purification of nuclear extracts derived from insect cells coinfected with the five recombinant viruses that produce the RFC subunits. As shown in Fig. 1B, the column eluate contained four major protein bands visualized by Coomassie blue staining following SDS/PAGE that corresponded to the five RFC subunits. The p40 and His-tagged p38 subunits comigrated on the gel as a single intensely stained band. The identity of each subunit was confirmed by Western blot analysis (data not shown). All five subunits bound to the Ni column despite only two of the subunits containing a His-tag (the p140 and p38), indicating that a stable complex of the five RFC subunits was formed in the coinfected insect cells. The intensity of Coomassie blue-stained protein bands indicated an equal stoichiometry between the subunits, consistent with the subunit stoichiometry of the native hRFC complex (3).
To further ascertain whether the five RFC subunits eluting from the Ni-affinity column were stably associated in a complex capable of supporting PCNA-dependent DNA synthesis, glycerol gradient centrifugation analyses of the Ni-purified bRFC were carried out (Fig. 2). As shown in Fig. 2A, the five RFC subunits cosedimented through the gradient, peaking at fractions 11–13 corresponding to an S value of 7.4, similar to that of native hRFC (3). A minor protein peak appeared in fractions 15–17 and contained only the small subunits of RFC. The glycerol gradient fractions were assayed for their ability to support RFC-dependent DNA synthesis using poly(dA)4500/oligo(dT)12–18 as the template as well as for the DNA-dependent ATPase activity. Quantitation of the results (Fig. 2B) indicated that both of these activities peaked at fraction 12, coincidental with the bRFC protein peak. Fractions 15–17 of the minor protein peak did not support DNA synthesis but were associated with a trailing edge of DNA-dependent ATPase activity. This residual activity may be due to the presence of the three subunit complex, p40·p37·p36, which has been shown to contain DNA-dependent ATPase (data not shown).
Figure 2.
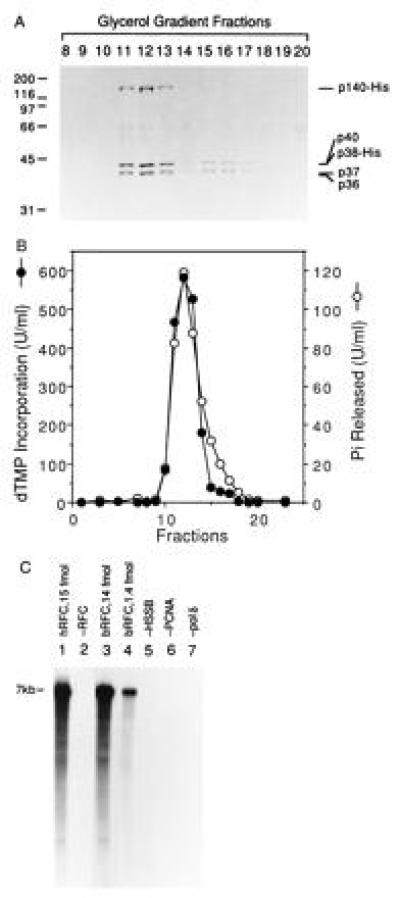
Cosedimentation of RFC-dependent DNA synthesis activity and ATPase activity through a 15–35% glycerol gradient. (A) SDS/PAGE analysis. The bRFC, eluted from a Ni-column (Fig. 1B), was further purified by two consecutive 15–35% glycerol gradient centrifugations. Following acetone precipitation and centrifugation, the pellets were analyzed by SDS/PAGE followed by Coomassie staining. The numbers at the top of the figure represent the fraction analyzed. The bands that migrated between 55–70 kDa in all lanes were artifactual. (B) RFC-dependent DNA synthesis and DNA-dependent ATPase activity. Each glycerol gradient fraction (0.5 μl after 3-fold dilution) was assayed for its ability to support DNA synthesis in the presence of a multiply primed poly(dA)4500/oligo (dT)12–18 template and ATPase activity as described. (C) RFC-dependent nucleotide incorporation using a singly primed M13 DNA template. Reactions were carried out as described using 4.4 fmol of M13 DNA template prior to separation through alkaline agarose gel followed by autoradiography. Reactions shown in lanes 1 and 2 were carried out in the presence and absence of 15 fmol of hRFC, respectively; reactions shown in lanes 3–7 were carried out with bRFC as follows: lane 3, 14 fmol of bRFC; lane 4, 1.4 fmol of bRFC; lanes 5–7, 14 fmol of bRFC in the absence or presence of HSSB, PCNA, or pol δ as indicated. Nucleotide incorporation (pmol), measured following acid precipitation and liquid scintillation counting, was as follows: lane 1, 26; lane 2, 0.4; lane 3, 24.4; lane 4, 8.8; lane 5, 2.8; lane 6, 0.48; lane 7, 0.08.
To further characterize bRFC in supporting PCNA-dependent DNA synthesis by pol δ, singly primed M13 DNA was used as a template for elongation assays. Following the incorporation of 32P-labeled nucleotides, the DNA products were separated by electrophoresis through an alkaline agarose gel (Fig. 2C). Like native hRFC (lane 1), bRFC supported the synthesis of full-length M13 DNA catalyzed by pol δ (lanes 3 and 4), and this reaction required PCNA, HSSB, and pol δ (lanes 5–7, respectively). No DNA synthesis was observed if RFC was omitted (lane 2). The specific activity of bRFC (15 × 103 units/mg), measured in the poly(dA)4500/oligo(dT)12–18 assay, was 2- to 3-fold higher than that of the most highly purified hRFC [6 × 103 units/mg (3)]. These results demonstrate that bRFC is identical to the native hRFC in supporting PCNA-dependent pol δ-catalyzed DNA synthesis.
Characterization of bRFC-Catalyzed ATP Hydrolysis.
Purified hRFC has previously been shown to contain an ATPase activity that is stimulated by the addition of DNA, PCNA, and HSSB. The effects of various DNA effectors on bRFC ATPase activity were examined. Results were quantitated and are shown in Fig. 3A. bRFC possessed weak ATPase activity that could be stimulated maximally (8-fold) by φX174 circular ssDNA, 3-fold by poly(dA)4500, 4-fold by poly(dA)4500/oligo(dT)4500, and only marginally stimulated by oligo(dT)12–18. Each bRFC molecule hydrolyzed ≈20 molecules of ATP/min at 37°C in the presence of φX174 circular ssDNA and 10.3 molecules of ATP per min in the presence of poly(dA)4500/oligo(dT)12–18. These values are three times higher than those observed for hRFC (3).
Figure 3.
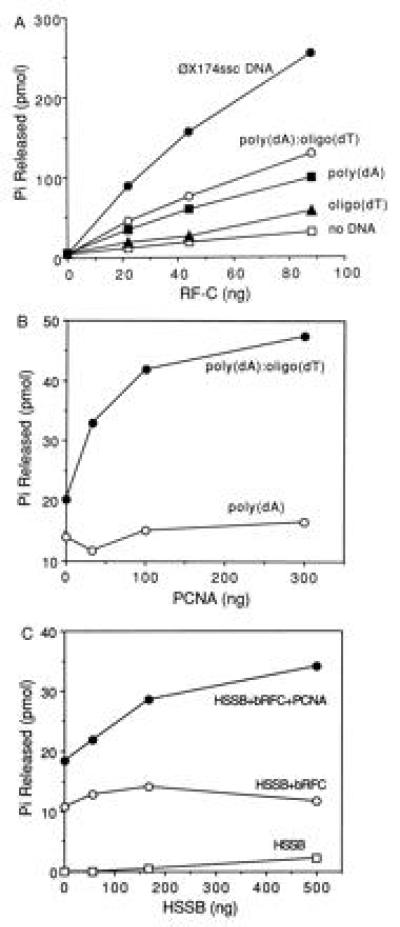
Characterization of bRFC ATPase activity. (A) Stimulation of bRFC ATPase by DNA effectors. ATPase reactions were carried out as described. Reaction mixtures contained bRFC in amounts as indicated, in the absence or presence of 12.5 μM (nucleotide concentration) of the following DNAs: oligo(dT)12–18, poly(dA)4500, poly(dA)4500/oligo(dT)12–18 (20:1 nucleotide ratio), or φX174 circular ssDNA. (B) PCNA stimulation of RFC ATPase activity. PCNA was added in amounts as indicated to reaction mixtures containing 15 ng of bRFC in the presence of 12.5 μM of poly(dA)4500 or poly(dA)4500/oligo(dT)12–18. (C) HSSB stimulation of RFC ATPase activity. HSSB was added in amounts as indicated to reaction mixtures containing poly(dA)4500/oligo(dT)12–18 in the absence or presence of 10 ng of bRFC and/or 40 ng of PCNA.
PCNA addition further stimulated bRFC ATPase activity in the presence of poly(dA)4500/oligo(dT)12–18 but not poly(dA)4500 (Fig. 3B). HSSB stimulation of ATP hydrolysis by bRFC was dependent upon the presence of poly(dA)4500/oligo(dT)12–18 and PCNA (Fig. 3C). These observations indicated that bRFC ATPase activity was maximally activated in the presence of primed DNA termini, PCNA, and HSSB factors required to assemble a PCNA clamp onto a DNA-primer end and recruit pol δ or ɛ prior to DNA synthesis.
bRFC Binds Specifically to DNA Primer Ends.
We examined bRFC for its ability to bind ssDNA and multiply primed DNA templates using a nitrocellulose filter binding assay. As shown in Fig. 4, bRFC bound to poly(dA)300 inefficiently and did not bind to oligo(dT)12–18. However, bRFC bound to poly(dA)300 annealed to oligo(dT)12–18 and the binding efficiency was markedly increased as the molar ratio of oligo(dT)12–18 to poly(dA)300 (1:0.4, 1:2, and 1:10) increased, suggesting that bRFC specifically recognized DNA primer ends. The selective binding of bRFC to primed DNA and not ssDNA required the presence of 175 mM NaCl; at lower salt concentrations, bRFC bound to both DNAs identically (data not shown). Similar observations were made when hRFC was used (data not shown).
Figure 4.
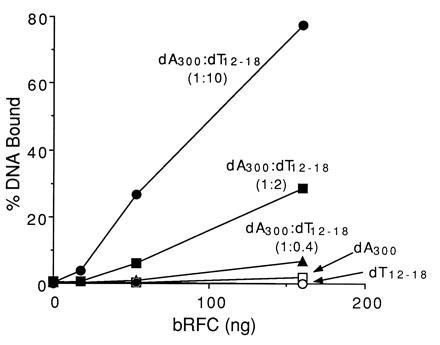
bRFC preferentially binds to DNA primer ends. Binding of bRFC to DNA was examined using a nitrocellulose filter binding assay as described. bRFC, in amounts as indicated, was incubated with poly(dA)300, oligo(dT)12–18, or poly(dA)300/oligo(dT)12–18 at a molar ratio of 1:0.4, 1:2 and 1:10, respectively, as indicated. The 100% value represented 40 fmol of input DNA.
bRFC Efficiently Catalyzes PCNA Loading and Unloading Onto and Off DNA.
bRFC catalyzed loading of 32P-labeled PCNA onto DNA was followed after A15m agarose gel filtration that separated 32P-PCNA on pBluescript DNA from free 32P-PCNA. Increasing amounts of bRFC (0.08, 0.3, and 1.2 pmol) resulted in the loading of proportionately increasing amounts of PCNA trimer (0.34, 1.55, and 5.2 pmol, respectively) onto 0.5 pmol of singly nicked pBluescript DNA in a reaction completely dependent on bRFC and ATP (ref. 8; data not shown) (Fig. 5A).
Figure 5.
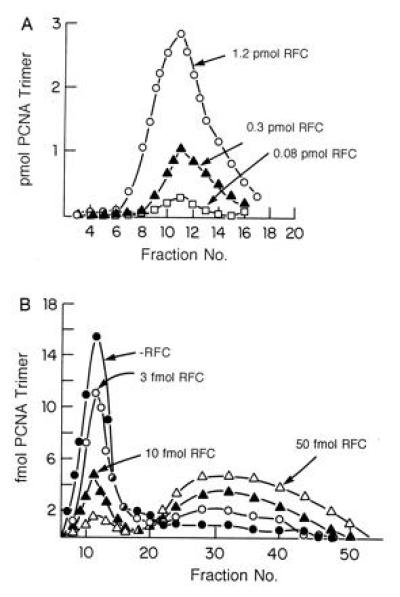
bRFC loads and unloads PCNA onto and off DNA. (A) bRFC catalyzed loading of PCNA onto singly nicked pBluescript DNA. Reaction mixtures (100 μl) containing 40 mM Tris·HCl (pH 7.5), 0.5 mM DTT, 20 μg/ml BSA, 7 mM Mg (OAc)2, 2 mM ATP, 0.5 pmol of singly nicked pBluescript plasmid DNA, 5.2 pmol of 32P-labeled PCNA trimer (574 cpm/fmol) and bRFC (as indicated), were incubated for 10 min at 37°C. Each reaction mixture was then filtered through a 6 ml Bio-Gel A15m column equilibrated with buffer containing 0.02 M Tris·HCl (pH 7.5), 0.1 mM EDTA, 40 μg/ml bovine serum albumin, 8 mM MgCl2, 4% glycerol, 5 mM DTT, and 0.1 M NaCl at 4°C. Fractions (180 μl) were collected and subjected to Cerenkov counting to detect the elution of the labeled PCNA. The peak shown in the graph represented PCNA that eluted in the excluded volume, i.e., PCNA complexed with DNA. (B) bRFC unloads 32P-PCNA catalytically from singly nicked DNA. 32P-PCNA was assembled onto DNA as described in A and the product isolated by BioGel A15m separation. The isolated PCNA–DNA complexes were then incubated in the presence or absence of bRFC (amounts as indicated) in reaction mixtures as described in A prior to gel filtration to resolve free 32P-PCNA from the 32P-PCNA–DNA complex. Both included and excluded material were subjected to Cerenkov counting as described above.
To examine the unloading of PCNA from DNA, bRFC was incubated with the isolated 32P-PCNA–DNA complex. Release of 32P-PCNA from DNA was measured following A15m agarose gel filtration that separated the 32P-PCNA–DNA complex from free 32P-PCNA. As shown in Fig. 5B, incubation of the labeled complex (containing 55.1 fmol of PCNA trimers) in the presence of increasing amounts of bRFC (3, 10, and 50 fmol) resulted in the elution of decreasing amounts of 32P-PCNA in the excluded region (decreases of 12.6, 37.7, and 47.9 fmol, respectively) and the concomitant detection of increasing levels of 32P-PCNA in the included volume (peaking between fractions 28–40). These results indicate that bRFC catalytically unloads PCNA off DNA and are identical to those observed with hRFC (data not shown).
DISCUSSION
The data presented in this study demonstrate that the five-subunit human RFC complex can be reconstituted in baculovirus-infected insect cells. The purified bRFC was functionally identical to its native counterpart in (i) supporting PCNA-dependent DNA synthesis catalyzed by pol δ; (ii) catalyzing DNA-dependent ATP hydrolysis; (iii) specifically binding to DNA primers ends; and (iv) functioning catalytically both in loading PCNA onto singly nicked DNA as well as removing PCNA from the PCNA–DNA complex. The specific activity of bRFC, measured by DNA synthesis and DNA-dependent ATPase assays, was higher (2- to 3-fold) than biologically isolated hRFC. It is likely that the large number of purification steps required to isolate this labile complex from HeLa cells (3) leads to partial inactivation of hRFC, in contrast to the few steps used to isolate bRFC.
All five subunits of RFC are essential for viability in S. cerevisiae and recently a mutation in the gene scrfc5 (equivalent to hRFCp38) exhibited defects in both DNA replication and S phase checkpoint function (33). Overexpression of PCNA in this mutant suppressed the replication defect of the rfc5 mutant but had no effect on the checkpoint defect. These results suggest that scRFC may play a role in linking replication with cell cycle regulation. Pol ɛ has also been implicated as an S phase checkpoint in S. cerevisiae (34), raising the possibility that active replication complexes may link the completion of DNA replication with the onset of mitosis.
The precise role of each subunit of RFC remains to be elucidated. In human cells, the facile genetic approach available in yeast is considerably more limited. Each of the five human genes encoding the RFC subunits has been mapped to distinct chromosomes (35, 36) and RFC2 (p40) has been localized to chromosomal region 7q11.23, mutations of which cause William syndrome (37). Interestingly, 17 of 17 William syndrome patients examined were found to be deleted for the RFC2 gene but it is not clear whether this contributed to the multiple phenotypes associated with this syndrome since this genetic defect spans a large region.
In addition to their role in DNA replication, clamps and clamp loaders are involved in a number of DNA transactions including repair, Okazaki fragment joining and transcription. In eukaryotes, DNase IV (38–40) (also called flap endonuclease and 5′ → 3′ exonuclease) cleaves branched DNA structures and catalyzes the excision of oligoribonucleotides formed by DNA primase prior to deoxynucleotide incorporation during lagging strand maturation. PCNA was shown to bind to DNase IV and stimulate its activity (40). Following RFC-mediated DNA loading, PCNA through its interaction with DNase IV may target the nuclease to its site of action where it can cleave RNA primers. RFC may also be involved in nucleotide excision repair following removal of damaged DNA, a reaction dependent on PCNA, HSSB and pol δ or ɛ (41, 42). In T4 phage infected E. coli, the T4 clamp (gp45) interacts with E. coli RNA polymerase contributing to the switch from early to late transcription (43). This may require continuous loading of T4 gp45 by the T4 gp44/62 clamp loader complex as the T4 gp45-DNA complex is highly labile.
The development of the baculovirus-based RFC reconstitution system reported here should also enable us to examine how the RFC subunits assemble a functional complex capable of finding DNA primer ends, hydrolyzing ATP and loading PCNA onto DNA. By coinfecting baculoviruses expressing the RFCp40, p37, and p36 subunits, we have isolated a stable heterotrimeric complex consistent with our previous report using coupled in vitro transcription-translation (26). The isolated bRFC p40·p37·p36 complex hydrolyzed ATP in a DNA-dependent manner (data not shown). The biological activity of this three subunit complex and its role in the assembly of the five subunit RFC complex are currently under investigation.
Acknowledgments
We are indebted to Dr. Z.-Q. Pan for helpful discussions. F.U. is a student enrolled in the graduate program at the Physiologisch-Chemisches Institut, Universität Tübingen, and is supported by the German Academic Exchange Service through funds of the Zweites Hochschulsonderprogramm. These studies were supported by National Institutes of Health Grants GM 38839 (M.O.D.) and GM 38559 (J.H.). J.H. is a Professor of the American Cancer Society.
Footnotes
Abbreviations: pol, DNA polymerase; RFC, replication factor C; scRFC, Saccharomyces cerevisiae RFC; hRFC, human RFC; bRFC, baculovirus-produced RFC; PCNA, proliferating cell nuclear antigen; ssDNA, single-stranded DNA; HSSB, human ssDNA binding protein, also called RPA; HF; high five insect cells; gp, gene product.
References
- 1.Lee S-H, Eki T, Hurwitz J. Proc Natl Acad Sci USA. 1989;86:7361–7365. doi: 10.1073/pnas.86.19.7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsurimoto T, Stillman B. Mol Cell Biol. 1989;9:609–619. doi: 10.1128/mcb.9.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee S-H, Kwong A D, Pan Z-Q, Hurwitz J. J Biol Chem. 1991;266:594–602. [PubMed] [Google Scholar]
- 4.Tsurimoto T, Stillman B. Proc Natl Acad Sci USA. 1990;87:1023–1027. doi: 10.1073/pnas.87.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoder B L, Burgers P M. J Biol Chem. 1991;266:22689–22697. [PubMed] [Google Scholar]
- 6.Fien K, Stillman B. Mol Cell Biol. 1992;12:155–163. doi: 10.1128/mcb.12.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Podust V N, Georgaki A, Strack B, Hübscher U. Nucleic Acids Res. 1992;20:4159–4165. doi: 10.1093/nar/20.16.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao N, Turner J, Kelman Z, Stukenberg P T, Pan Z-Q, Hurwitz J, O’Donnell M. Genes Cells. 1996;1:101–113. doi: 10.1046/j.1365-2443.1996.07007.x. [DOI] [PubMed] [Google Scholar]
- 9.Podust L M, Podust V N, Sogo J M, Hübscher U. Mol Cell Biol. 1995;15:3072–3081. doi: 10.1128/mcb.15.6.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgers P M J. J Biol Chem. 1991;266:22698–22706. [PubMed] [Google Scholar]
- 11.Kuriyan J, O’Donnell M. J Mol Biol. 1993;234:915–925. doi: 10.1006/jmbi.1993.1644. [DOI] [PubMed] [Google Scholar]
- 12.Chen M, Pan Z-Q, Hurwitz J. Proc Natl Acad Sci USA. 1992;89:2516–2520. doi: 10.1073/pnas.89.7.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, Pan Z-Q, Hurwitz J. Proc Natl Acad Sci USA. 1992;89:5211–5215. doi: 10.1073/pnas.89.12.5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burbelo P B, Utani A, Pan Z Q, Yamada Y. Proc Natl Acad Sci USA. 1993;90:11543–11547. doi: 10.1073/pnas.90.24.11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bunz F, Kobayashi R, Stillman B. Proc Natl Acad Sci USA. 1993;90:11014–11018. doi: 10.1073/pnas.90.23.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu Y, Zeft A S, Riegel A T. Biochem Biophys Res Commun. 1993;193:779–786. doi: 10.1006/bbrc.1993.1693. [DOI] [PubMed] [Google Scholar]
- 17.O’Donnell M, Onrust R, Dean F B, Chen M, Hurwitz J. Nucleic Acids Res. 1993;21:1–3. doi: 10.1093/nar/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Burgers P M. Proc Natl Acad Sci USA. 1994;91:868–872. doi: 10.1073/pnas.91.3.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Burgers P M. J Biol Chem. 1994;269:21880–21884. [PubMed] [Google Scholar]
- 20.Cullmann G, Fien K, Kobayashi R, Stillman B. Mol Cell Biol. 1995;15:4661–4671. doi: 10.1128/mcb.15.9.4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gary M, Burgers P M. Nucleic Acids Res. 1995;23:4986–4991. doi: 10.1093/nar/23.24.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noskov V, Maki S, Kawasaki Y, Leem S-H, Ono I, Araki H, Pavlov Y, Sugino A. Nucleic Acids Res. 1994;22:1527–1535. doi: 10.1093/nar/22.9.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howell E A, McAlear M A, Rose D, Holm C. Mol Cell Biol. 1994;14:255–267. doi: 10.1128/mcb.14.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsurimoto T, Stillman B. J Biol Chem. 1991;266:1950–1960. [PubMed] [Google Scholar]
- 25.Pan Z-Q, Chen M, Hurwitz J. Proc Natl Acad Sci USA. 1993;90:6–10. doi: 10.1073/pnas.90.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uhlmann F, Cai J, Flores-Rozas H, Dean F B, Finkelstein J, O’Donnell M, Hurwitz J. Proc Natl Acad Sci USA. 1996;93:6521–6526. doi: 10.1073/pnas.93.13.6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flores-Rozas H, Kelman Z, Dean F B, Pan Z-Q, Harper J W, Elledge S J, O’Donnell M, Hurwitz J. Proc Natl Acad Sci USA. 1994;91:8655–8659. doi: 10.1073/pnas.91.18.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stukenberg P T, Studwell-Vaughan P S, O’Donnell M. J Biol Chem. 1991;266:11328–11334. [PubMed] [Google Scholar]
- 29.Kenny M K, Lee S-H, Hurwitz J. Proc Natl Acad Sci USA. 1989;86:9757–9761. doi: 10.1073/pnas.86.24.9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S-H, Ishimi Y, Kenny M K, Bullock P, Dean F B, Hurwitz J. Proc Natl Acad Sci USA. 1988;85:9469–9473. doi: 10.1073/pnas.85.24.9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelman Z, Yao N, O’Donnell M. Gene. 1995;166:177–178. doi: 10.1016/0378-1119(95)00556-7. [DOI] [PubMed] [Google Scholar]
- 32.Naktinis V, Turner J, O’Donnell M. Cell. 1996;84:137–145. doi: 10.1016/s0092-8674(00)81000-4. [DOI] [PubMed] [Google Scholar]
- 33.Sugimoto K, Shimomura T, Hashimoto K, Araki H, Sugino K, Matsumoto K. Proc Natl Acad Sci USA. 1996;93:7048–7052. doi: 10.1073/pnas.93.14.7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navas T A, Zhou Z, Elledge S J. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- 35.Okumura K, Nogami M, Taguchi H, Dean F B, Chen M, Pan Z-Q, Hurwitz J, Shiratori A, Murakami Y, Ozawa K, Eki T. Genomics. 1995;25:274–278. doi: 10.1016/0888-7543(95)80135-9. [DOI] [PubMed] [Google Scholar]
- 36.Luckow B, Bunz F, Stillman B, Lichter P, Schutz G. Mol Cell Biol. 1994;14:1626–1634. doi: 10.1128/mcb.14.3.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peoples R, Perez-Jurado L, Nung Y-K, Kaplan P, Franke U. Am J Hum Genet. 1996;58:1370–1373. [PMC free article] [PubMed] [Google Scholar]
- 38.Robbins P, Parrin D, Wood R D, Lindahl T. J Biol Chem. 1994;269:28535–28538. [PubMed] [Google Scholar]
- 39.Murante R S, Rust L, Bambara R A. J Biol Chem. 1995;270:30377–30383. doi: 10.1074/jbc.270.51.30377. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Li J, Harrington J, Lieber M R, Burgers P M J. J Biol Chem. 1995;270:22109–22112. doi: 10.1074/jbc.270.38.22109. [DOI] [PubMed] [Google Scholar]
- 41.Aboussekhra A, Biggerstaff M, Shirji M K K, Vilpo J A, Moncollin V, Podust V N, Protic M, Hübscher U, Egly J-M, Wood R D. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 42.Sancar A. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 43.Herendeen D R, Kassavetis G A, Geiduschek E P. Science. 1992;256:1298–1303. doi: 10.1126/science.1598572. [DOI] [PubMed] [Google Scholar]