

Abstract
The epidermis and its appendages develop from a single layer of multipotent embryonic progenitor keratinocytes. Embryonic stem cells receive cues from their environment that instruct them to commit to a particular differentiation programme and generate a stratified epidermis, hair follicles or sebaceous glands. Exciting recent developments have focused on how adult skin epithelia maintain populations of stem cells for use in the natural cycles of hair follicle regeneration and for re-epithelialization in response to wounding.
At the surface of our body, the skin epidermis and its appendages provide a protective barrier that keeps microbes out and essential body fluids in. It receives daily assaults, including harmful ultraviolet radiation from the Sun, and scratches and wounds. It confronts these attacks by undergoing continual self-renewal to repair damaged tissue and replace old cells. To do this, it depends on stem cells, which reside in the adult hair follicle, sebaceous gland and epidermis for the purpose of maintaining tissue homeostasis, regenerating hair and repairing the epidermis after injury. Where do these adult stem cells come from and when during embryonic development are they determined? How do they specify on demand one differentiation programme versus another, and what governs these lineages? How do stem cells in these tissues know how many cells to replenish and when? And, finally, how do the skin epithelial cells communicate with the immune system to prevent infection? Here I place particular emphasis on recent studies to review what is known about these fascinating features of mammalian skin epithelium from embryogenesis to adult. I also speculate on how dysregulation of the normal process of wound healing in the skin epidermis can lead to skin disorders, including cancers and chronic inflammation. The review concentrates heavily on mouse skin, given the accelerated pace of scientific discoveries made possible by mouse genetics.
Embryonic origins of skin epithelium
After gastrulation, the embryo surface emerges as a single layer of neuroectoderm, which will ultimately specify the nervous system and skin epithelium. At the crossroads of this decision is Wnt signalling, which blocks the ability of ectoderm to respond to fibroblast growth factors (FGFs). In the absence of FGF signalling the cells express bone morphogenetic proteins (BMPs), and become fated to develop into epidermis. Conversely, the acquisition of neural fate arises when, in the absence of a Wnt signal, the ectoderm is able to receive and translate activating cues by FGFs, which then attenuate BMP signalling through inhibitory cues1 (Fig. 1a). The embryonic epidermis that results from this process consists of a single layer of multipotent epithelial cells. It is covered by a transient protective layer of tightly connected squamous endodermis-like cells, known as a periderm, which are shed once the epidermis has stratified and differentiated2.
Figure 1. Early signalling steps in specification of embryonic skin.

a, In the absence of Wnt signalling, ectodermal progenitors respond to FGFs, downregulate BMP signalling and progress towards neurogenesis1. Wnt signalling blocks the ability of early ectodermal progenitor cells to respond to FGFs, allowing them to respond to BMP signalling and adopt an epidermal fate. As development progresses, single-layered embryonic epidermis expresses Wnts. Some cells fail to respond to Wnts, and these become fated to become epidermal cells through BMP, EGF and Notch signalling. The cells that do respond to Wnt signalling also receive underlying FGF and BMP inhibitory signals from the mesenchyme and, together, these signals instruct the cells to make an appendage4,6-10. Collectively, the inhibition of BMP inhibitory signals and Wnt activating signals produce the hair placode9,12,13. Additional dermal messages from below further instruct the placodes to make the follicle.
Hair formation
Mesenchymal cells from the dorsal backskin dermis are derived from dermomyotome, in which Wnt signalling specifies their fate3. As these mesenchymal cells populate the skin, their interactions with the over-lying epithelium induce the formation of hair placodes, which appear as small epidermal invaginations into the underlying dermis. Pioneering studies on mesenchymal–epithelial tissue recombination in chicks and mice revealed that early cues from the mesenchyme determine the positioning of placodes and specify their commitment4,5 (Fig. 1). Key components of these mesenchymal-bud-promoting signals include FGFs6,7 and, subsequently, BMP-inhibitory factors8,9. In the presence of excess BMPs, or the absence of either the BMP-inhibitor noggin or the FGF10/FGF7 receptor FGFRIIIb, follicle density is reduced7,10,11.
In conjunction with these early dermal signals, ectodermal Wnt signals instruct the epidermal cells to grow downwards to form a hair bud or placode12-14. Among the early genes expressed in the placode are sonic hedgehog (Shh), which has a crucial role in organizing the dermal cells into a condensate, or dermal papilla, which thereafter becomes a permanent associate of the hair follicle15-17. Once formed, the dermal papilla signals to the placode to grow downwards to form the hair follicle, which matures through additional reciprocal mesenchymal–epithelial crosstalk4.
Increasing evidence places Wnt signalling at the helm of placode formation. Although the single layered epithelium uniformly expresses a number of Wnts, including Wnt10b, the placode and dermal condensate seem to respond most robustly, as judged by the presence of nuclear β-catenin, the outcome of a Wnt signal13,14,18,19. Embryonic placodes and dermal papillae also exhibit activation of the Wnt reporter gene TOPGAL. This drives β-galactosidase expression under the control of an enhancer composed of multiple recognition sites for the LEF1/TCF family of DNA-binding proteins, which function concomitantly with Wnt/β-catenin signalling13,19,20. LEF1/TCF proteins can act as transcriptional repressors, but upon association with β-catenin act as a functional bipartite transcriptional activator. On the basis of timing and morphology, Wnt activity in the epithelium seems to precede that in the dermal condensates of developing hair follicles.
The TCF family member TCF3 is expressed early in embryogenesis when the skin epithelium exists as a single layer of unspecified progenitor cells21. Placode initiation and Wnt reporter activity are accompanied by a reduction in TCF3 and upregulation of LEF1. LEF1 concentrates in developing hair placodes and the dermal papilla. TCF3 seems to maintain epithelial progenitor status: when it is sustained transgenically in these progenitors, all three differentiation lineages (epidermis, hair follicle and sebaceous gland) are repressed21. Conversely, LEF1 is important for follicle morphogenesis, and when Lef1 is knocked out a reduction in hair follicles is observed22.
Mice expressing a constitutively stabilized form of β-catenin in the epidermis develop excess hair follicles12,23-25. When β-catenin is conditionally targeted or when the Wnt inhibitor dickkopf-1 (DKK-1) is expressed ectopically, epidermal appendage formation is impaired26,27. On the basis of these findings, Wnt signalling seems to be an integral part of the early epidermal–dermal discourse that specifies hair patterning and morphogenesis.
In apparent contrast to the epidermal/neural fate switch, activation of Wnt signalling and inhibition of BMP signalling seem to be on the same side of the epidermal/hair placode switch. Thus, the β-catenin/LEF1-responsive, TOPGAL-positive hair placodes lack nuclear phosphorylated (activated) SMAD1, the outcome of BMP signalling11. Moreover, in mice that lack noggin, ectodermal LEF1 is no longer expressed and TOPGAL Wnt reporter activity is diminished28. Conversely, LEF1 expression is maintained upon conditional ablation of Bmpr1a, which encodes the key BMP receptor for the epidermis and its appendages29,30. If inhibition of BMP signalling promotes LEF1 accumulation, this could explain in part how BMP inhibitory and Wnt-promoting signals converge in placode formation.
Hair follicle maturation
Once the placode has formed, downstream signalling events drive the downgrowth and maturation of the hair follicle. A myriad of changes take place during follicle morphogenesis, as exemplified by the differences between the transcriptional profile of placode cells and their epidermal counterparts31. The purified placode cells are positive for TOPGAL, and the large number of Wnt pathway and Wnt target genes comprising the placode signature is reflective of the programme of gene expression that occurs as a downstream consequence of Wnt signalling.
One of the key regulators downstream of Wnt/noggin is SHH. Embryonic skin deficient in SHH still expresses TOPGAL and LEF1 and shows placode formation15,16,31. However, in its absence the overall number of placodes is diminished and they do not progress past the hair germ stage, although a small number of terminally differentiated hair cells can still be detected. Microarray analyses of developing wild-type hair placodes have confirmed the elevated expression of SHH effectors such as GLI-Kruppel family member 1 (GLI1) and patched homologue 2 (PTC2)31. In addition, transcriptional profiling has uncovered genes encoding a possible SHH-regulatory circuit involving the transcription factors FOXC1, TBX1, DACH1, SIX1 and LHX2, whose family members have been shown to mediate eye development31. Loss-of-function mutations in at least one of these downstream effectors, Lhx2, result in a sparsity of follicles and a failure to maintain follicle stem-cell behaviour in mice. Thus, the functions of SHH signalling seem to extend beyond merely governing proliferation and follicle downgrowth.
It is curious that with the exception of β-catenin, whose loss affects all follicle formation, the loss of other crucial inductive factors, such as SHH, noggin, LEF1 and LHX2, diminishes rather than abolishes hair germ and/or follicle formation. One clue to understanding these differences is provided by the fact that mice defective in ectodysplasin-A receptor (EDAR)/EDA signalling lack guard hair follicles, whereas mice lacking LEF1 have guard hairs but lack other types of hair follicle11,32,33. Underlying these variations seems to be the different ways in which BMP inhibition can be achieved in order to permit transmission of the follicle Wnt signal. Thus, guard hairs rely on EDAR/EDA/nuclear factor-κB (NF-κB) signalling, which leads to activation of the BMP inhibitor CTGF (connective tissue growth factor)11; the other follicle types seem to be more dependent on noggin for BMP inhibition and LEF1 activation22.
Whereas different modes of BMP inhibition affect the types of follicle that are specified, active BMP signalling seems to affect follicle density once specification has occurred. Soon after the placode forms, BMP4 levels are increased, even though it is the ectoderm, and not the placode, that expresses nuclear phospho-SMAD1 — that is, recognition that a cell is responding to a BMP — at this time31. The importance of this molecular idiosyncrasy seems to be rooted in negative feedback regulation, as suggested by the fact that nascent follicles always emerge at the apexes of a hexagon at the centre of which is an existing follicle from the prior wave of follicle morphogenesis. Such spacing places follicles at the maximum distance from one another, and is suggestive of an inhibitory signal transmitted by the placode to restrict its neighbours from staking their claims too closely. Placodes also express Wnt inhibitory factor 1 (WIF1), suggesting that the negative feedback circuitry might be complex. The dermis also influences follicle size and density, and the recent molecular differences found in dermal cells from different body sites seem likely to expedite progress in deciphering these anatomical distinctions in epithelial–mesenchymal interactions34.
The epidermis
At first glance, it might seem as though the epidermis is simply a default option in the crossfire of signalling pathways directed at making an appendage. However, it is important to consider that BMP signalling is active in the interfollicular epidermis (IFE), and, as such, is as much an epidermis-promoting signal as it is a follicle-inhibitory signal11,29,30 (Fig. 1). In addition, studies on chicks indicate that epidermal growth factor receptor (EGFR) signalling occurs in the epidermal cells at the boundary of each feather placode. Moreover, elevated EGF promotes the proliferation and expansion of interbud epidermal cells, whereas inhibition of EGFR signalling results in increased acquisition of feather bud fates35. Whether EGFR signalling has a similar role in inhibiting the formation of hair follicles is less clear, although elevated EGFR signalling in mammalian skin epithelium is associated with enhanced epidermal proliferation and hair loss36. Thus, when taken together, follicle density seems to be governed by competition between placode-stimulating (epidermis-restricting) and placode-restricting (epidermis-stimulating) signals.
Hair follicle maturation and lineage differentiation
As an embryonic hair follicle grows downwards, its leading front (matrix) is highly proliferative and, through its sustained contact with the dermal papilla, adopts a programme of gene expression that is distinct from that at the root. The outer root sheath (ORS) maintains contact with the basement membrane while the inner layers begin to differentiate to form the inner root sheath (IRS), which will serve as the channel for the protruding hair (Fig. 2a). At about embyonic day (E) 18.5 in mice, the earliest vestiges of the central core of hair shaft cells emerge coincident with the appearance of the sebaceous gland precursor cells in the upper part of the hair follicle. Maturation continues for another 7–8 days, as a companion layer separates the ORS and IRS, and the IRS and hair shaft each develop further into three concentric cell layers (Fig. 2a). The discrete stages of hair follicle development are readily distinguished by their morphological and biochemical differences, and have been described elsewhere37.
Figure 2. Rapidly proliferating matrix-cell progenitors are spatially organized in the hair bulb to respond to distinct differentiation-specific cues.

a, Schematic of the hair bulb. Rapidly proliferating matrix cells at the base (blue) give rise to seven differentiation-specific lineages. The various genetic markers for these lineages are shown. Matrix cells express the proliferative marker Ki67. As these cells differentiate, they express keratins differentially. Ch, hair shaft cuticle; Co, cortex; Cp, companion layer; DP, dermal papilla; Me, medulla29,46. b, Ultrathin section of hair bulb. The single vertical layer of core cells corresponds to the dermal papilla. Note the onion-skin-like organization of matrix cells and their differentiating progeny at the base of the follicle. (Image courtesy of H. A. Pasolli, The Rockefeller University, New York.)
Within each mature hair follicle the seven concentric rings of terminally differentiating cells are derived from matrix cells. Each ring has a distinctive ultrastructure (Fig. 2b). Closer inspection of the hair bulb provides some insight into how these rings might form, and what driving forces might orchestrate their differentiation. At the height of the growth phase (full anagen) of the follicle the dermal papilla exists as a thin strand of cells, separated from the hair bulb surrounding it by a basement membrane. Some mitotic matrix cells seem to orient their spindle plane at an angle relative to the basement membrane, suggesting that distinct differentiation lineages might be generated in an onion-skin-like fashion. Lineage tracing experiments provide support for this model17,38,39, which is similar to that postulated for the epidermis40 (discussed below).
Hair follicle lineages
During the years, researchers have uncovered molecular differences that are diagnostic for the lineages in the hair follicle. The most prominent structural proteins of these lineages are keratins (Fig. 2a), which are differentially expressed in specific patterns that distinguish most, if not all, of the concentric layers within the hair follicle41. Efforts to understand how these keratin genes are differentially regulated led to the discovery that the hair shaft Ha/Hb keratin genes are genuine Wnt target genes, and that Wnt signalling has a crucial role in the differentiation of matrix cells along a hair shaft lineage19. Constitutively active β-catenin in humans and mice leads to pilomatricomas — hair tumours composed exclusively of the hair lineage12,42. Additional hair shaft transcriptional regulatory proteins of importance are HOXC13 and FOXN1, both of which cause hair defects when mutated43,44.
By contrast, mutations in the genes encoding GATA-binding factor (Gata-3) and CCAAT displacement protein (Cdp) yield primary IRS defects, which lead to alterations in the shaft45,46 (Fig. 2a). GATA-3 and CDP are both able to recruit histone deacetylases (HDACs) and transcriptionally repress their target genes. On the basis of studies in other tissues, candidate genes for CDP-mediated repression in the IRS include c-myc, N-cam and p21. GATA-3 represses its own expression in the IRS, but its other targets remain unknown. The transcriptional repressor BLIMP1 also concentrates in differentiating IRS cells, and like CDP, one of BLIMP's target genes is c-myc47-49. If BLIMP1 associates with a histone arginine methyl transferase as it does in germ cells47,48 then it could add an epigenetic mark to the chromatin it represses in the skin. Together, these observations suggest that epigenetic remodelling of chromatin might have a crucial role in the IRS lineage. Such hints merit further investigation in the future.
Although at first glance it might seem that IRS differentiation is governed by repression and relief of repression, positive-acting signalling pathways are also involved. One of these is Notch, a transmembrane receptor that, when activated by its ligand, can be cleaved by γ-secretase to generate a nuclear intracellular cofactor (NICD) for the transcriptional repressor protein RBP-J50. Although Notch signalling is not required for follicle specification, IRS cells of γ-secretase-deficient follicles fail to maintain their fates; perhaps as a consequence, ORS cells aberrantly activate epidermal differentiation, hair differentiation is compromised and follicles develop into epidermal cysts51,52.
BMP receptor signalling is also involved and is essential for both IRS and hair shaft differentiation. In the absence of BMPR1A, masses of undifferentiated, highly proliferative matrix-like cells accumulate that rapidly progress to tumours53. The fact that epidermal cysts arise from Notch signalling defects whereas matrix-like cysts result from BMP signalling defects underscores distinct differences in the ways these pathways affect hair follicle lineages. Sifting through the underlying mechanisms involved and the myriad of factors that affect fate specification and differentiation of follicle matrix cells is one of the main challenges currently facing skin biologists.
The hair cycle
The hair follicle is one of the few organs of the body that undergoes cyclic bouts of degeneration and regeneration throughout life (Fig. 3). Matrix cells are sometimes referred to as transit-amplifying cells because they only survive through the growth (anagen) phase of the cycle. After the first 2 weeks of postnatal mouse life, the initial supply of matrix cells declines, hair shaft and IRS differentiation slow, and the follicle enters a destructive phase known as catagen54. During this phase, which in mice lasts for 3–4 days, apoptosis reduces the follicle to an epithelial strand. This drags the dermal papilla upward to rest just below the permanent, non-cycling upper follicle.
Figure 3. The hair cycle.

The stages of the hair cycle are depicted, starting from the first postnatal anagen, when the hair shaft is growing and protruding through the skin surface. Follicles progress synchronously to the destructive (catagen) phase, during which the lower two-thirds of the follicle undergo apoptosis and regress. The dermal papilla is brought to rest below the bulge-stem-cell compartment, and after the resting (telogen) phase, a critical threshold of activating factors is reached and the stem cells become activated to regrow the hair100.
A number of molecular regulators of the anagen–catagen transition have been identified, although how they work together to promote catagen or terminate anagen and how they spare progenitor cells needed for anagen re-entry is not yet understood. Recently, keratin 17 was found to be a regulator of this process. It seems to regulate anagen by interacting with and sequestering a death adaptor protein required for tumour-necrosis factor receptor 1 (TNFR1)-dependent signalling and induction of apoptosis to initiate the catagen process55. In addition, genetic evidence has implicated a transcription complex involving the vitamin D-retinoid X receptor-α transcription factor and an interacting repressor protein, hairless. When defective, this complex impairs the catagen-related defects in the epithelial strand, leaving the dermal papilla behind and blocking new hair growth56. Hairless, which is expressed in the ORS and matrix, is a transcriptional repressor that shares sequence similarities with lysine histone demethylase, an enzyme that removes a positive epigenetic mark from chromatin57.
After catagen, follicles lie dormant in a resting phase (telogen). In most mice, the first telogen is short, lasting only 1 or 2 days, from about postnatal day (P)19 to P21 in the mid back. The second telogen, however, lasts more than 2 weeks and begins at about P42 (ref. 37).
Hair follicle stem cells
At the onset of anagen, the cycling portion of the follicle regenerates and undergoes a new round of hair growth. The process necessitates a reservoir of stem cells, which reside in the lowest permanent portion of the follicle. The new downward growth occurs adjacent to the existing club hair, which will eventually be shed. This growth adds a layer to the stem-cell reservoir and causes it to ‘bulge’, thereby giving this region its name58-61 (Fig. 4). Bulge cells divide less frequently than other epithelial cells in the skin, but they are activated at anagen to generate a new hair follicle58. The process of bulge-cell activation has been likened to that of embryonic hair follicle morphogenesis. Despite many parallels, there are also differences, and the relationships between the embryonic hair bud and the adult bulge and hair germ are still unfolding.
Figure 4. Diagram of the follicle stem-cell niche.

The bulge contains infrequently cycling, label-retaining cells, which include multipotent stem cells (green) that can generate the new hair follicle during cycling and repair the epidermis on injury. The bulge is in a specialized niche, surrounded by other cell types, which together provide cues that maintain these cells in an undifferentiated, quiescent state. For stem cells to be activated, the niche environment must change.
To understand the special properties underlying bulge stem cells, researchers have turned to microarray profiling, which has led to the identification of about 150 genes that are preferentially expressed in the bulge relative to the proliferating basal cells of the epidermis61-63. Many of these genes — for example, those in the BMP and transforming growth factor-β pathways — are likely to reflect the special quiescent nature of resting bulge cells, which, in turn, show nuclear localization of phospho-SMAD1 and phospho-SMAD2, respectively11,62. In addition, although bulge cells are Wnt-responsive, as is evident from their expression of TCF3, TCF4 and several Wnt receptor proteins (FZDs), they express proteins typically associated with Wnt inhibition21,62. In this regard, TCF3 can function as a repressor when β-catenin is absent or underrepresented19,21. This could explain how bulge cells maintain an undifferentiated state and why β-catenin stabilization and BMP inhibition are required to activate hair follicle morphogenesis12,23-25. Some of these features have also recently been noted for human bulge stem cells, which could be important for future clinical applications64. These parallels are intriguing, because in human skin epidermal stem cells are likely to have the main role in homeostasis — that is, to supply the thick epidermal barrier of the skin surface — whereas in mice, maintaining the protective hair coat is more important.
Several other stimuli function at or slightly downstream of follicle stem-cell activation. One of these is the drug minoxidil. More commonly known as Rogaine, minoxidil was first identified as a vasodilator, and among the cellular responses that have been attributed to its action is prostaglandin synthesis54. Cyclosporin A is another hair growth stimulus, which seems to act in promoting anagen or inhibiting catagen41,54,56. Given that the stabilization of β-catenin also promotes anagen, it is interesting to speculate that the pharmacological targets of minoxidil and cyclosporin A might be downstream of β-catenin stabilization. In this regard, it should also be noted that Wnt signalling is only one of several pathways that can lead to β-catenin stabilization, and ‘Wnt reporter’ activity reflects merely the activation of β-catenin/LEF/TCF-regulated genes.
A number of questions remain. Although grafting and lineage tracing experiments have shown that some bulge cells are stem cells61,63,65, it is still not known whether this is the case for most bulge cells. During the transition to anagen, most bulge cells remain quiescent, and only a small number residing at the base of this segment seem to be activated62. Moreover, in the whisker cycle of rodents, the dermal papilla never reaches the bulge before the start of a new anagen. It has been proposed that stem cells from the bulge migrate along the ORS to the base of the follicle, where they become activated60. Whether this happens in all hair follicles is not known.
Despite the many questions, it is helpful to place the data at hand in a conceptual model for how stem-cell activation in the hair cycle might be achieved. Upon stem-cell activation, β-catenin/LEF1/TCF becomes activated in the early hair germ and persists at the follicle base throughout anagen. Association with the dermal papilla seems to be critical in accumulating a sufficient level of ‘activating factor’ to trigger proliferation and/or lineage commitment of these cells. When the dermal papilla moves away from the bulge, the epidermal stem cells return to quiescence. The cells at the base of the follicle that receive sustained dermal papilla stimulation are highly proliferative. Through local differences in their microenvironment they progress to a point of no return, executing terminal lineage programmes to produce the hair and IRS. In this way, the levels of β-catenin/TCF/LEF signalling determine the outcome of the stem cells and their lineages.
Follicle stem-cell maintenance
Once follicle stem cells have been expended, niche vacancies must be replenished. The process seems to be one of self renewal, although stem-cell maintenance is still poorly understood. Balancing c-myc expression seems to be crucial, as its overexpression in transgenic mice causes follicle stem cells to proliferate and terminally differentiate, and, conversely, conditional ablation of c-myc results in alopecia owing to a long-term failure to sustain follicle stem cells66-68. LHX2 also seems to function in follicle stem-cell maintenance, as exemplified by the fact that LHX2-deficient bulge stem cells are unable to retain label and use stem cells more rapidly than their wild-type counterparts27. Thus, whereas LHX2 seems to function as a molecular brake in regulating the switch between hair follicle stem-cell maintenance and activation, c-Myc acts as the accelerator.
Several other factors have been identified as candidates in follicle stem-cell maintenance, including telomerase, cell-division cycle 42 (Cdc42) and Rac1 (reviewed in ref. 36). It has been postulated that Rac1 might help to maintain stem cells by negatively regulating c-Myc and by enhancing integrin levels69, whereas Cdc42 might control progenitor differentiation through β-catenin stabilization70.
Sebaceous gland development
Sebaceous glands are an appendage of the hair follicle, located above the bulge and arrector pili muscle and just below the hair shaft orifice at the skin surface. Sebaceous gland progenitor cells emerge near the conclusion of embryogenesis, but the gland does not mature until just after birth. The main role of the gland is to generate terminally differentiated sebocytes, which produce lipids and sebum. When sebocytes distintegrate, they release these oils into the hair canal for lubrication and protection against bacterial infections. Sebaceous gland homeostasis necessitates a progenitor population of cells that gives rise to a continual flux of proliferating, differentiating and, finally, dead cells that are lost through the hair canal.
The basement membrane that demarcates the mesenchymal–epithelial border also surrounds the gland. Not surprisingly, the sebaceous gland cells that are attached to the inner surface of this membrane share many of the features of epidermal keratinocytes, including proliferative capacity and expression of c-myc. Overexpression of c-myc induces sebaceous gland hyperplasia at the expense of hair follicle differentiation, which is suggestive of a special role for this transcription factor in the gland71,72. Sebocyte differentiation involves the adipogenic transcription factor peroxisome proliferator-activated receptor-γ (PPAR-γ). PPAR-γ-null mice are not viable, but PPAR-γ-null embryonic stem cells have been shown to contribute poorly to the sebaceous glands of chimaeric mice47. In addition, alterations in Wnt, hedgehog and Notch signalling proteins and their effectors result in perturbations in sebaceous gland development, although it is not yet clear how these factors contribute to the sebaceous gland lineage.
Until recently, little was known about how the sebaceous gland is formed and how it is able to sustain its dynamic balance of growth and terminal differentiation. Stem cells have been implicated in the process of sebaceous gland homeostasis, and support for a resident progenitor-cell population comes from fate mapping by retroviral labelling of epithelial cells in vivo73. BLIMP1 was recently identified as a marker, and genetic lineage studies revealed the ability of BLIMP1-positive cells to generate sebaceous glands47. In culture, BLIMP1-positive cells yield sebocyte colonies and undergo self-renewal. The results of loss-of-function and molecular studies suggest that BLIMP1 functions by repressing c-myc, so that as sebocyte progenitors switch off BLIMP1 they become proliferative and initiate sebocyte differentiation47.
When Blimp1 is mutated, or when the skin is injured, the homeostasis of the sebaceous gland is perturbed. Interestingly, under these conditions, bulge cells can be mobilized to maintain the sebaceous gland. Such a precursor–product relationship has been documented by engraftment experiments with isolated bulge stem cells61,63,65, and by analysing the rudimentary follicle structures of either hairless mutant mice74 or Blimp1 conditionally null mice47.
Epidermal development
Towards the end of embryonic development, the interfollicular epidermis (IFE) reaches maturity. Epidermis is composed of layers, the outermost of which is at the skin surface. After birth, its role is to guard against infection, to prevent dehydration, and to undergo re-epithelialization after wound injuries. To accomplish these feats, the epidermis constantly replenishes itself by a process of homeostasis. During this process, dividing cells in the innermost (basal) layer continually execute a programme of terminal differentiation, move outwards and are sloughed from the skin surface.
When exposed to physical trauma and chemical assaults the epidermis must also protect itself, which it does by producing copious amounts of cytoplasmic heteropolymers known as intermediate filaments that are composed of keratin proteins. As cells exit from the basal layer and begin their journey towards the skin surface, they switch from the expression of keratins K14 and K5 to K1 and K10 (ref. 75). This switch was discovered more than 25 years ago, and remains the most reliable indication that an epidermal cell has undergone a commitment to terminally differentiate. The first suprabasal cells are known as spinous cells, reflecting their cytoskeleton of K1/K10 filament bundles connected to robust cell–cell junctions known as desmosomes. These connections provide a cohesive, integrated mechanical framework across and within stacks of epithelial sheets. K6, K16 and K17 are also expressed suprabasally, but only in hyperproliferative situations such as wound healing. This keratin network not only remodels the cytoskeleton for migration but also regulates cell growth through binding to the adaptor protein 14-3-3σ and stimulating Akt/mTOR (mammalian target of rapamycin) signalling76,77.
As spinous cells progress to the granular layer, they produce electron-dense keratohyalin granules packed with the protein profilaggrin, which, when processed, bundles keratin intermediate filaments even more to generate large macrofibrillar cables. In addition, cornified envelope proteins, which are rich in glutamine and lysine residues, are synthesized and deposited under the plasma membrane of the granular cells. When the cells become permeabilized to calcium, they activate transglutaminase, generating γ-glutamyl ε-lysine crosslinks to create an indestructible proteinaceous sac to hold the keratin macrofibrils. The final steps of terminal differentiation involve the destruction of cellular organelles including the nucleus, and the extrusion of lipid bilayers, packaged in lamellar granules, onto the scaffold of the cornified envelope. The dead stratum corneum cells create an inpenetrable seal that is continually replenished as inner layer cells move outwards and are sloughed from the skin surface.
Controlling basal cell behaviour
Epidermal growth and proliferation must be carefully balanced: too little proliferation results in thinning of the skin and loss of protection, and too much is a characteristic of hyperproliferative disorders, including psoriasis (see page 866) and cancers. During normal homeostasis the epidermis must be able to sense and replace basal vacancies, and when wounds occur cells must migrate and proliferate but also sense when to stop after wound closure. Finally, the epidermis must have an SOS system to recruit immune cells to fight infection and aid in wound repair, and to turn off the response once the wound has been closed. How does the epidermis achieve all this?
Basal cell proliferation relies on an underlying basement membrane that is rich in extracellular matrix (ECM) proteins and growth factors. Basal cells attach to this structure through two types of cell-junction adhesion complexes, composed of integrins. Hemidesmosomes contain a transmembrane core of α6β4 integrins that connect intracellularly to the keratin intermediate filament network and provide mechanical strength. Focal adhesions contain α3β1 integrins, which connect to the actin and microtubule networks. Both types of junction adhere extracellularly to laminin 5, the main ECM ligand for the epidermal basement membrane. αβ1 integrins seem to be especially important in assembling and organizing this membrane78,79.
Integrins also function in growth control and migration. Although these growth regulatory circuitries are many and complex, they involve physical interactions with regulatory kinases. To permit migration in response to wounding, cell–substratum junctions must be dynamically turned over. Integrins differ in their relative roles in wound-repair: whereas cells lacking αβ1 integrins are less migratory, those lacking α6β4 integrins show increased migratory behaviour79-81. The positive role of β1 integrin in migration seems to result in part from its ability to directly bind and control the activity of focal adhesion kinase (FAK). This is a non-receptor tyrosine kinase that, in turn, functions as a master switch in negatively regulating cellular tension imposed by an elaborate actin–myosin network associated with focal adhesions80,81.
To function as a tissue, basal cells must also adhere to one another. They do this not only by means of desmosomes but also through adherens junctions (Fig. 5). Adherens junctions are composed of a transmembrane core of E-cadherin, which binds two related proteins, β-catenin and p120-catenin82. Just as desmosomes and hemidesmosomes form an integral network with intermediate filaments, adherens junctions and focal adhesions orchestrate actin–myosin dynamics throughout the cells of the tissue. Adherens junctions do so by associating with an array of actin regulatory proteins, which include p120-catenin's interactions with Rho-GTPase proteins and β-catenin's association with α-catenin, an unrelated protein that can bind vasodilator-stimulated phosphoprotein (also known as ENA), formins, ajuba and α-actinin proteins82. Exactly how adherens junctions and focal adhesions regulate the actin–myosin network to coordinate adhesion and migration is not yet clear but, typically, whenever cell–cell junctions are reinforced, integrin dynamics are diminished. This inverse regulation is likely to be essential during wound repair, in which epidermal outgrowth must occur in a polarized and orchestrated manner.
Figure 5. Model for control of epidermal proliferation.
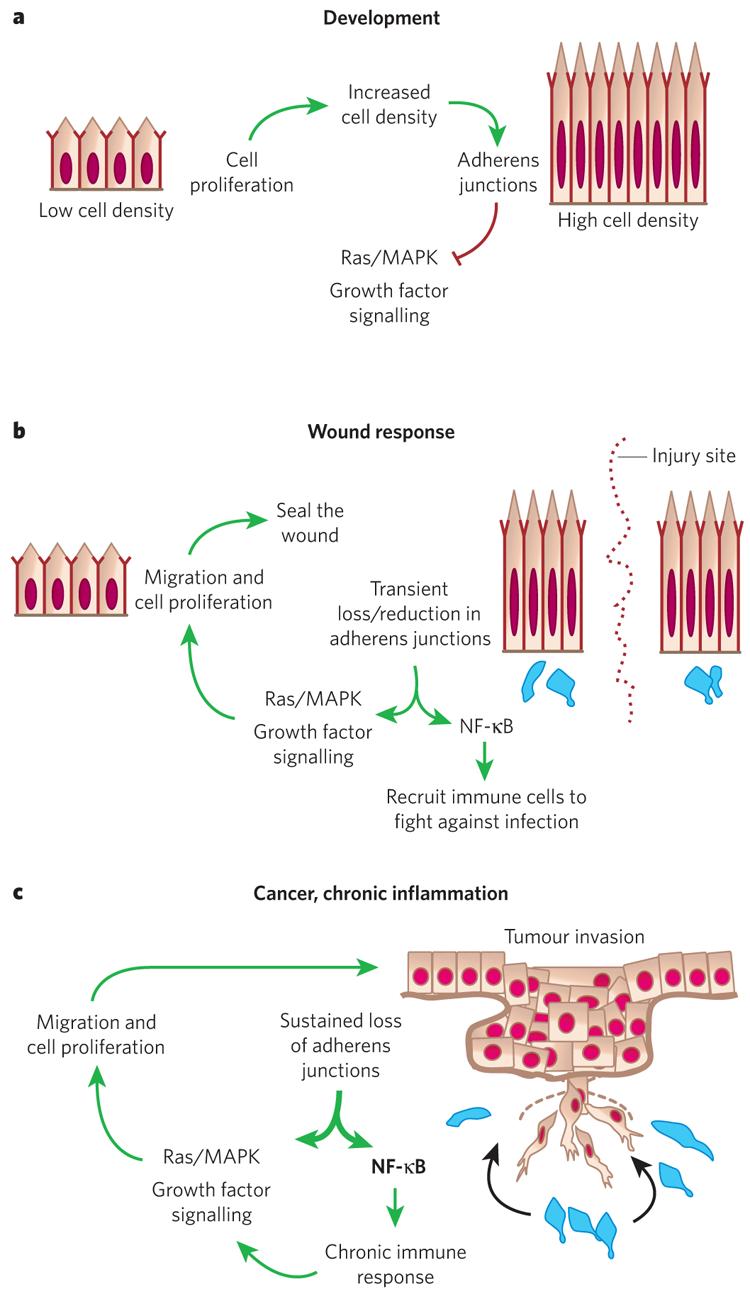
a, During development, basal epidermal cell density is low and cells proliferate. As cells increase in density the number of adherens junctions at cell–cell borders increases. This provides a regulatory feedback loop to control Ras–MAPK activity and cell proliferation. b, In a wound response, adherens junctions are severed at the wound site, stimulating the Ras–MAPK pathway and triggering a hyperproliferative response and cell migration. Adherens junction protein levels also affect NF-κB, which, when active, results in the recruitment of immune cells (blue) to protect against infection. After wound repair, regulatory feedback pathways cause these processes to be dampened and the system returns to normal. c, Cancer and chronic inflammatory disorders can lead to mutations and/or permanent changes in adherens junction proteins. This results in a break in the regulatory circuitry, and the proliferative and NF-κB responses become constitutively activated.
Recently, researchers have begun to realize that cellular junctions act as signalling centres and have functions that extend beyond their classical roles in cell adhesion and cytoskeletal dynamics. The longest standing example is β-catenin, which is now well known for its dual role in adhesion and transcription. p120-Catenin also functions not only in regulating Rho GTPases and adherens junction assembly, but also as a transcriptional cofactor83. In addition, through a mechanism that is not fully understood but seems to involve the small Rho GTPases, p120-catenin and α-catenin can affect the transcriptional status of NF-κB, which, in turn, governs the production and secretion of cytokines and chemokines and the recruitment of immune cells84,85. The abrogation of JunB, a downstream inhibitor of EGF signalling, also triggers chemokine/cytokine expression86.
When sustained activation of NF-κB arises — for example, through dysfunction of p120-catenin, α-catenin and/or JunB — signalling from the epidermis to immune cells goes awry. This triggers a molecular war between the epidermis, which spews out cytokines and chemokines, and the recruited immune cells, which respond in a similar way and send proliferative signals to the epidermis. Further studies will be needed to ascertain the controls on these normal pathways for the repair of the epidermis in response to injury, stress and infection, and to elucidate the extent to which these various pathways intersect in recruiting immune cells to the skin. NF-κB regulation is particularly important because it also mediates a TNFR1-dependent hypoproliferative influence on healthy epidermis87.
The consequences of α-catenin loss are particularly severe, and the epidermis progresses to a condition resembling invasive squamous-cell carcinoma29. Although the mechanisms are not yet fully understood, they are likely to involve an upregulation in integrins and tyrosine kinase activity85,88,89. An emerging view of adherens junctions is that they serve as molecular sensors for ‘crowd control’ and wound repair. In this model, when basal cell density is low or when the epidermis is severed, as in a wound, a reduction in adherens junctions triggers cell migration and proliferation, and immune cells are recruited to protect against possible infection. Once proper cell density has been achieved and adherens junction formation becomes optimal, the system returns to normal (Fig. 5). When this circuitry is defective — for example, when α-catenin or p120 is mutated — the system becomes imbalanced and chronic inflammation and hyperproliferative disorders, including cancer, can result.
Maintaining and controlling the epidermis
Interfollicular skin epithelium is organized into columns of hexagonally packed cells. This organization has been revealed by genetic lineage tracing involving either retrovirally infected, β-galactosidase-expressing cultured keratinocytes grafted onto immunodeficient mice or direct infection of dermabraded skin with a β-galactosidase-expressing virus73,90,91. Initial discrete stacks of β-galactose-positive cells expand to about 10 adjacent stacks over time, which suggests that epidermal proliferative units (EPUs) may be composed of one basal stem cell that occasionally divides laterally to produce cells with a more transient proliferative capacity92,93. Irrespective of the relative proliferative capacity of individual basal cells, their ability to fuel a constant flux of terminally differentiating cells places the epidermis in a constant state of dynamic equilibrium. In humans, the epidermis replenishes itself every 4 weeks throughout life.
The ability of epidermal domains to be maintained individually can be explained by one of two different mechanisms, which are not necessarily mutually exclusive (Fig. 6). In vitro studies support a delamination model, whereby epidermal cells with the highest integrin levels also have the highest proliferative and attachment potential, whereas those with low integrin levels detach and terminally differentiate92. Studies on embryonic skin development in mice have revealed that concomitantly with stratification, basal cells can also orient their spindle poles asymmetrically relative to the basement membrane40,94. Such divisions seem to be asymmetric, leading to one proliferative basal daughter and one detached, suprabasal daughter. Postnatally, cellular divisions in black skin are markedly reduced, but in other areas or other adult tissues in which the epithelium is thicker, asymmetrically oriented spindles are apparent.
Figure 6. Models of how epidermal homeostasis might be achieved.

Symmetric and asymmetric cell division have been observed in the epidermis, and both are likely to contribute to epidermal homeostasis. Models of how they might do so are shown. Both are predicated on the basis of cellular attachment to an underlying basement membrane providing the main means by which basal epidermal progenitor cells are able to preserve their mitotic capability. a, Symmetric division is thought to supply new cells through stratification (the formation of layers) and differentiation by delamination (the detachment of a cell from the basement membrane). b, Asymmetric division is believed to contribute through stratification and differentiation by asymmetric partitioning of cellular contents into the two daughter cells.
Interestingly, mitotic basal epidermal cells with perpendicularly aligned spindles seem to use some of the same proteins involved in an ancient mechanism for asymmetrically partitioning the cellular contents and fates of two daughter cells in Drosophila neuroblasts40. It is tempting to speculate that the attached basal daughter might preferentially receive integrins, growth factor receptors and other key growth promoting machinery at the base of the cell. An additional factor involved is the basal transcription factor p63, which not only promotes proliferation in the epidermis but also seems to be required for epidermal stratification95,96. In the absence of p63, basal cells seem to divide only symmetrically40.
In the fly neuroblast, the asymmetric division machinery preferentially partitions Notch signalling. Although it is not yet clear whether this feature is used by the epidermis, it is intriguing that suprabasal epidermal cells use NICD, the transcriptional effector of Notch signalling, to regulate their differentiation51,52. Additional transcriptional regulators of epidermal differentiation include p21, AP2, C/EBP (CCAAT/enhancer binding protein), Kruppel-like factor and PPAR proteins44. Another important factor in epidermal stratification and differentiation is IKK-α (inhibitor of NF-κB kinase-α), which exerts its action on the epidermis in an NF-κB-independent manner97. By a mechanism not yet determined, the polarity established by the basement membrane, cell substratum and cell–cell junctions generates the epidermal architecture, which, in turn, governs the transcriptional balance between proliferation and differentiation.
The future of skin stem cells in regenerative medicine
Through understanding the basics of its biology, we now know the skin to be a rich source of readily accessible stem cells. Although the main purpose of the stem cells in the bulge is to maintain homeostasis of the hair follicle during cycling, bulge cells can be mobilized to replenish the cells of the sebaceous gland and the epidermis when necessary17,47,59,61,63,65,91. Whether resident progenitor cells in the basal layer of the epidermis, the sebaceous gland and the follicle matrix can also select alternative lineages when required remains unknown. Although in normal skin the molecular steps in these lineages are becoming increasingly well defined both temporally and spatially, their potential for re-routing their lineage when exposed to a new microenvironment, for example in wounding, engraftment or human disease, is less clear.
The level of plasticity afforded these stem cells is becoming increasingly important as the potential of stem cells in regenerative medicine continues to be explored. Developmental biologists have long been aware that embryonic epithelial cells are sensitive to the mesenchyme to which they are exposed. When confronted with mesenchyme from chick wing, epidermis from the leg produces feathers, and, similarly, mesenchyme from leg can prompt wing epidermis to make scales. These special features seem to be lost in most cells as development proceeds, but do multipotent stem cells of the skin retain this potential? If bulge stem cells exposed to corneal mesenchyme produce hair follicles, this would not be helpful, but if they produce cornea, they could be used to treat certain types of blindness. Other possible uses for bulge stem cells might be in treating chronic ulcers or hair disorders. The potential of cultured basal epidermal keratinocytes has already been realized for treating burns patients98 (see page 874).
On the basis of studies on haematopoietic stem cells, the promise of bulge cells seems slim for generating non-epithelial tissues. That said, the results of recent studies suggest that bulge-cell nuclei from adult mice can be reprogrammed when placed in an enucleated, unfertilized oocyte, and cloned mice have been generated with such technology99. If current ethical and technical hurdles can be overcome to permit cloning of human hybrid embryonic stem cells from oocytes and adult skin nuclei, then it might be possible to harness their potential for broader uses in regenerative medicine in the future. A final note in closing is that the accessibility of the skin as a potential source of stem cells has not gone unnoticed in the broader research community. Given that there are many different cell types in the skin, the recent identification of melano-blast stem cells, mesenchymal stem cells and neural-like stem cells seem likely to be the tip of a promising iceberg for future explorations.
Acknowledgments
I am grateful to my many colleagues who helped to establish the groundwork for this review. I also thank J.-F. Nicolas (Pasteur Institute), H. A. Pasolli, H. Rhee and T. Lechler for their helpful suggestions about figures for this review. I am especially indebted to my former mentor, H. Green, his former postdoctoral researchers — T.-T. Sun, J. Rheinwald, F. Watt and Y. Barrandon — and to the members of my laboratory, past and present, all of whom have contributed so heavily to the field of skin biology and who have served as an enormous source of inspiration to my own contributions. E.F. is an Investigator of the Howard Hughes Medical Institute. This work was supported in part by the National Institutes of Health and the Stem Cell Research Foundation.
Footnotes
Reprints and permissions information is available at npg.nature.com/reprintsandpermissions.
The author declares no competing financial interests.
References
- 1.Stern CD. Neural induction: old problem, new findings, yet more questions. Development. 2005;132:2007–2021. doi: 10.1242/dev.01794. [DOI] [PubMed] [Google Scholar]
- 2.M'Boneko V, Merker HJ. Development and morphology of the periderm of mouse embryos (days 9–12 of gestation) Acta Anat. (Basel) 1988;133:325–336. doi: 10.1159/000146662. [DOI] [PubMed] [Google Scholar]
- 3.Atit R, et al. β-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev. Biol. 2006;296:164–176. doi: 10.1016/j.ydbio.2006.04.449. [DOI] [PubMed] [Google Scholar]
- 4.Hardy MH. The secret life of the hair follicle. Trends Genet. 1992;8:55–61. doi: 10.1016/0168-9525(92)90350-d. [DOI] [PubMed] [Google Scholar]
- 5.Olivera-Martinez I, Thelu J, Dhouailly D. Molecular mechanisms controlling dorsal dermis generation from the somitic dermomyotome. Int. J. Dev. Biol. 2004;48:93–101. doi: 10.1387/ijdb.15272374. [DOI] [PubMed] [Google Scholar]
- 6.Davidson D. The mechanism of feather pattern development in the chick. II. Control of the sequence of pattern formation. J. Embryol. Exp. Morphol. 1983;74:261–273. [PubMed] [Google Scholar]
- 7.Petiot A, et al. A crucial role for Fgfr2-IIIb signalling in epidermal development and hair follicle patterning. Development. 2003;130:5493–5501. doi: 10.1242/dev.00788. [DOI] [PubMed] [Google Scholar]
- 8.Jung HS, et al. Local inhibitory action of BMPs and their relationships with activators in feather formation: implications for periodic patterning. Dev. Biol. 1998;196:11–23. doi: 10.1006/dbio.1998.8850. [DOI] [PubMed] [Google Scholar]
- 9.Noramly S, Morgan BA. BMPs mediate lateral inhibition at successive stages in feather tract development. Development. 1998;125:3775–3787. doi: 10.1242/dev.125.19.3775. [DOI] [PubMed] [Google Scholar]
- 10.Botchkarev VA, et al. Noggin is a mesenchymally derived stimulator of hair-follicle induction. Nature Cell Biol. 1999;1:158–164. doi: 10.1038/11078. [DOI] [PubMed] [Google Scholar]
- 11.Mou C, Jackson B, Schneider P, Overbeek PA, Headon DJ. Generation of the primary hair follicle pattern. Proc. Natl Acad. Sci. USA. 2006;103:9075–9080. doi: 10.1073/pnas.0600825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gat U, DasGupta R, Degenstein L, Fuchs E. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated β-catenin in skin. Cell. 1998;95:605–614. doi: 10.1016/s0092-8674(00)81631-1. [DOI] [PubMed] [Google Scholar]
- 13.DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- 14.Noramly S, Freeman A, Morgan BA. β-catenin signaling can initiate feather bud development. Development. 1999;126:3509–3521. doi: 10.1242/dev.126.16.3509. [DOI] [PubMed] [Google Scholar]
- 15.St-Jacques B, et al. Sonic hedgehog signaling is essential for hair development. Curr. Biol. 1998;8:1058–1068. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- 16.Oro AE, Higgins K. Hair cycle regulation of Hedgehog signal reception. Dev. Biol. 2003;255:238–248. doi: 10.1016/s0012-1606(02)00042-8. [DOI] [PubMed] [Google Scholar]
- 17.Levy V, Lindon C, Harfe BD, Morgan BA. Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev. Cell. 2005;9:855–861. doi: 10.1016/j.devcel.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Reddy S, et al. Characterization of Wnt gene expression in developing and postnatal hair follicles and identification of Wnt5a as a target of Sonic hedgehog in hair follicle morphogenesis. Mech. Dev. 2001;107:69–82. doi: 10.1016/s0925-4773(01)00452-x. [DOI] [PubMed] [Google Scholar]
- 19.Merrill BJ, Gat U, DasGupta R, Fuchs E. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 2001;15:1688–1705. doi: 10.1101/gad.891401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hurlstone A, Clevers H. T-cell factors: turn-ons and turn-offs. EMBO J. 2002;21:2303–2311. doi: 10.1093/emboj/21.10.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen H, Rendl M, Fuchs E. Tcf3 maintains stem cells and represses cell fate determination in skin. Cell. 2006;127:171–183. doi: 10.1016/j.cell.2006.07.036. [DOI] [PubMed] [Google Scholar]
- 22.van Genderen C, et al. Development of several organs that require inductive epithelial–mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994;8:2691–2703. doi: 10.1101/gad.8.22.2691. [DOI] [PubMed] [Google Scholar]
- 23.Van Mater D, Kolligs FT, Dlugosz AA, Fearon ER. Transient activation of β-catenin signaling in cutaneous keratinocytes is sufficient to trigger the active growth phase of the hair cycle in mice. Genes Dev. 2003;17:1219–1224. doi: 10.1101/gad.1076103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo Celso C, Prowse DM, Watt FM. Transient activation of β-catenin signalling in adult mouse epidermis is sufficient to induce new hair follicles but continuous activation is required to maintain hair follicle tumours. Development. 2004;131:1787–1799. doi: 10.1242/dev.01052. [DOI] [PubMed] [Google Scholar]
- 25.Lowry WE, et al. Defining the impact of β-catenin/Tcf transactivation on epithelial stem cells. Genes Dev. 2005;19:1596–1611. doi: 10.1101/gad.1324905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. β-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 27.Andl T, Reddy ST, Gaddapara T, Millar SE. WNT signals are required for the initiation of hair follicle development. Dev. Cell. 2002;2:643–653. doi: 10.1016/s1534-5807(02)00167-3. [DOI] [PubMed] [Google Scholar]
- 28.Jamora C, DasGupta R, Kocieniewski P, Fuchs E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003;422:317–322. doi: 10.1038/nature01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobielak K, Pasolli HA, Alonso L, Polak L, Fuchs E. Defining BMP functions in the hair follicle by conditional ablation of BMP receptor IA. J. Cell Biol. 2003;163:609–623. doi: 10.1083/jcb.200309042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andl T, et al. Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development. 2004;131:2257–2268. doi: 10.1242/dev.01125. [DOI] [PubMed] [Google Scholar]
- 31.Rhee H, Polak L, Fuchs E. Lhx2 maintains stem cells character in hair follicles. Science. 2006;312:1946–1949. doi: 10.1126/science.1128004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Headon DJ, et al. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature. 2001;414:913–916. doi: 10.1038/414913a. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt-Ullrich R, et al. NF-κB transmits Eda A1/EdaR signalling to activate Shh and cyclin D1 expression, and controls post-initiation hair placode down growth. Development. 2006;133:1045–1057. doi: 10.1242/dev.02278. [DOI] [PubMed] [Google Scholar]
- 34.Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006;2:e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atit R, Conlon RA, Niswander L. EGF signaling patterns the feather array by promoting the interbud fate. Dev. Cell. 2003;4:231–240. doi: 10.1016/s1534-5807(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 36.Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu. Rev. Cell Dev. Biol. 2006;22:339–373. doi: 10.1146/annurev.cellbio.22.010305.104357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller-Rover S, et al. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Invest. Dermatol. 2001;117:3–15. doi: 10.1046/j.0022-202x.2001.01377.x. [DOI] [PubMed] [Google Scholar]
- 38.Kopan R, et al. Genetic mosaic analysis indicates that the bulb region of coat hair follicles contains a resident population of several active multipotent epithelial lineage progenitors. Dev. Biol. 2002;242:44–57. doi: 10.1006/dbio.2001.0516. [DOI] [PubMed] [Google Scholar]
- 39.Legue E, Nicolas JF. Hair follicle renewal: organization of stem cells in the matrix and the role of stereotyped lineages and behaviors. Development. 2005;132:4143–4154. doi: 10.1242/dev.01975. [DOI] [PubMed] [Google Scholar]
- 40.Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437:275–280. doi: 10.1038/nature03922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langbein L, Schweizer J. Keratins of the human hair follicle. Int. Rev. Cytol. 2005;243:1–78. doi: 10.1016/S0074-7696(05)43001-6. [DOI] [PubMed] [Google Scholar]
- 42.Chan EF, Gat U, McNiff JM, Fuchs E. A common human skin tumour is caused by activating mutations in β-catenin. Nature Genet. 1999;21:410–413. doi: 10.1038/7747. [DOI] [PubMed] [Google Scholar]
- 43.Godwin AR, Capecchi MR. Hoxc13 mutant mice lack external hair. Genes Dev. 1998;12:11–20. doi: 10.1101/gad.12.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai X, Segre JA. Transcriptional control of epidermal specification and differentiation. Curr. Opin. Genet. Dev. 2004;14:485–491. doi: 10.1016/j.gde.2004.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellis T, et al. The transcriptional repressor CDP (Cutl1) is essential for epithelial cell differentiation of the lung and the hair follicle. Genes Dev. 2001;15:2307–2319. doi: 10.1101/gad.200101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaufman CK, et al. GATA-3: an unexpected regulator of cell lineage determination in skin. Genes Dev. 2003;17:2108–2122. doi: 10.1101/gad.1115203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horsley V, et al. Blimp1 defines a progenitor population that governs cellular input to the sebaceous gland. Cell. 2006;126:597–609. doi: 10.1016/j.cell.2006.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ancelin K, et al. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nature Cell Biol. 2006;8:623–630. doi: 10.1038/ncb1413. [DOI] [PubMed] [Google Scholar]
- 49.Chang DH, Angelin-Duclos C, Calame K. BLIMP-1: trigger for differentiation of myeloid lineage. Nature Immunol. 2000;1:169–176. doi: 10.1038/77861. [DOI] [PubMed] [Google Scholar]
- 50.De Strooper B, et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 51.Pan Y, et al. γ-Secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev. Cell. 2004;7:731–743. doi: 10.1016/j.devcel.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 52.Blanpain C, Lowry WE, Pasolli HA, Fuchs E. Canonical Notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 2006;20:3022–3035. doi: 10.1101/gad.1477606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ming Kwan K, Li AG, Wang XJ, Wurst W, Behringer RR. Essential roles of BMPR-IA signaling in differentiation and growth of hair follicles and in skin tumorigenesis. Genesis. 2004;39:10–25. doi: 10.1002/gene.20021. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt-Ullrich R, Paus R. Molecular principles of hair follicle induction and morphogenesis. BioEssays. 2005;27:247–261. doi: 10.1002/bies.20184. [DOI] [PubMed] [Google Scholar]
- 55.Tong X, Coulombe PA. Keratin 17 modulates hair follicle cycling in a TNFα-dependent fashion. Genes Dev. 2006;20:1353–1364. doi: 10.1101/gad.1387406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Potter GB, et al. The hairless gene mutated in congenital hair loss disorders encodes a novel nuclear receptor corepressor. Genes Dev. 2001;15:2687–2701. doi: 10.1101/gad.916701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klose RJ, et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 58.Cotsarelis G, Sun TT, Lavker RM. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61:1329–1337. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- 59.Taylor G, Lehrer MS, Jensen PJ, Sun TT, Lavker RM. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell. 2000;102:451–461. doi: 10.1016/s0092-8674(00)00050-7. [DOI] [PubMed] [Google Scholar]
- 60.Oshima H, Rochat A, Kedzia C, Kobayashi K, Barrandon Y. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell. 2001;104:233–245. doi: 10.1016/s0092-8674(01)00208-2. [DOI] [PubMed] [Google Scholar]
- 61.Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118:635–648. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 62.Tumbar T, et al. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morris RJ, et al. Capturing and profiling adult hair follicle stem cells. Nature Biotechnol. 2004;22:411–417. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- 64.Ohyama M, et al. Characterization and isolation of stem cell-enriched human hair follicle bulge cells. J. Clin. Invest. 2006;116:249–260. doi: 10.1172/JCI26043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Claudinot S, Nicolas M, Oshima H, Rochat A, Barrandon Y. Long-term renewal of hair follicles from clonogenic multipotent stem cells. Proc. Natl Acad. Sci. USA. 2005;102:14677–14682. doi: 10.1073/pnas.0507250102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arnold I, Watt FM. c-Myc activation in transgenic mouse epidermis results in mobilization of stem cells and differentiation of their progeny. Curr. Biol. 2001;11:558–568. doi: 10.1016/s0960-9822(01)00154-3. [DOI] [PubMed] [Google Scholar]
- 67.Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nature Genet. 2001;28:165–168. doi: 10.1038/88889. [DOI] [PubMed] [Google Scholar]
- 68.Zanet J, et al. Endogenous Myc controls mammalian epidermal cell size, hyperproliferation, endoreplication and stem cell amplification. J. Cell Sci. 2005;118:1693–1704. doi: 10.1242/jcs.02298. [DOI] [PubMed] [Google Scholar]
- 69.Benitah SA, Frye M, Glogauer M, Watt FM. Stem cell depletion through epidermal deletion of Rac1. Science. 2005;309:933–935. doi: 10.1126/science.1113579. [DOI] [PubMed] [Google Scholar]
- 70.Wu X, et al. Cdc42 controls progenitor cell differentiation and β-catenin turnover in skin. Genes Dev. 2006;20:571–585. doi: 10.1101/gad.361406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niemann C, Owens DM, Hulsken J, Birchmeier W, Watt FM. Expression of ΔNLef1 in mouse epidermis results in differentiation of hair follicles into squamous epidermal cysts and formation of skin tumours. Development. 2002;129:95–109. doi: 10.1242/dev.129.1.95. [DOI] [PubMed] [Google Scholar]
- 72.Takeda H, et al. Human sebaceous tumors harbor inactivating mutations in LEF1. Nature Med. 2006;12:395–397. doi: 10.1038/nm1386. [DOI] [PubMed] [Google Scholar]
- 73.Ghazizadeh S, Taichman LB. Multiple classes of stem cells in cutaneous epithelium: a lineage analysis of adult mouse skin. EMBO J. 2001;20:1215–1222. doi: 10.1093/emboj/20.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Panteleyev AA, Paus R, Christiano AM. Patterns of hairless (hr) gene expression in mouse hair follicle morphogenesis and cycling. Am. J. Pathol. 2000;157:1071–1079. doi: 10.1016/S0002-9440(10)64621-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fuchs E, Green H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell. 1980;19:1033–1042. doi: 10.1016/0092-8674(80)90094-x. [DOI] [PubMed] [Google Scholar]
- 76.Coulombe PA, Wong P. Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nature Cell Biol. 2004;6:699–706. doi: 10.1038/ncb0804-699. [DOI] [PubMed] [Google Scholar]
- 77.Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441:362–365. doi: 10.1038/nature04659. [DOI] [PubMed] [Google Scholar]
- 78.Watt FM. Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 2002;21:3919–3926. doi: 10.1093/emboj/cdf399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wilhelmsen K, Litjens SH, Sonnenberg A. Multiple functions of the integrin α6β4 in epidermal homeostasis and tumorigenesis. Mol. Cell Biol. 2006;26:2877–2886. doi: 10.1128/MCB.26.8.2877-2886.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raghavan S, Vaezi A, Fuchs E. A role for αβ1 integrins in focal adhesion function and polarized cytoskeletal dynamics. Dev. Cell. 2003;5:415–427. doi: 10.1016/s1534-5807(03)00261-2. [DOI] [PubMed] [Google Scholar]
- 81.Brakebusch C, Fassler R. β1 integrin function in vivo: adhesion, migration and more. Cancer Metastasis Rev. 2005;24:403–411. doi: 10.1007/s10555-005-5132-5. [DOI] [PubMed] [Google Scholar]
- 82.Perez-Moreno M, Fuchs E. Catenins: keeping cells from getting their signals crossed. Dev. Cell. 2006;11:601–612. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van Roy FM, McCrea PD. A role for Kaiso-p120ctn complexes in cancer? Nature Rev. Cancer. 2005;5:956–964. doi: 10.1038/nrc1752. [DOI] [PubMed] [Google Scholar]
- 84.Perez-Moreno M, et al. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124:631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kobielak A, Fuchs E. Links between α-catenin, NF-κB, and squamous cell carcinoma in skin. Proc. Natl Acad. Sci. USA. 2006;103:2322–2327. doi: 10.1073/pnas.0510422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zenz R, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]
- 87.Zhang JY, Green CL, Tao S, Khavari PA. NF-κB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev. 2004;18:17–22. doi: 10.1101/gad.1160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E. Hyperproliferation and defects in epithelial polarity upon conditional ablation of α-catenin in skin. Cell. 2001;104:605–617. doi: 10.1016/s0092-8674(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 89.Zhang W, et al. E-cadherin loss promotes the initiation of squamous cell carcinoma invasion through modulation of integrin-mediated adhesion. J. Cell Sci. 2006;119:283–291. doi: 10.1242/jcs.02738. [DOI] [PubMed] [Google Scholar]
- 90.Kolodka TM, Garlick JA, Taichman LB. Evidence for keratinocyte stem cells in vitro: long term engraftment and persistence of transgene expression from retrovirus-transduced keratinocytes. Proc. Natl Acad. Sci. USA. 1998;95:4356–4361. doi: 10.1073/pnas.95.8.4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ito M, et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nature Med. 2005;11:1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 92.Mackenzie IC. Relationship between mitosis and the ordered structure of the stratum corneum in mouse epidermis. Nature. 1970;226:653–655. doi: 10.1038/226653a0. [DOI] [PubMed] [Google Scholar]
- 93.Potten CS. Cell replacement in epidermis (keratopoiesis) via discrete units of proliferation. Int. Rev. Cytol. 1981;69:271–318. doi: 10.1016/s0074-7696(08)62326-8. [DOI] [PubMed] [Google Scholar]
- 94.Smart IH. Variation in the plane of cell cleavage during the process of stratification in the mouse epidermis. Br. J. Dermatol. 1970;82:276–282. doi: 10.1111/j.1365-2133.1970.tb12437.x. [DOI] [PubMed] [Google Scholar]
- 95.Mills AA, et al. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 96.Yang A, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 97.Hu Y, et al. IKKα controls formation of the epidermis independently of NF-κB. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 98.Green H. Cultured cells for the treatment of disease. Sci. Am. 1991;265:96–102. doi: 10.1038/scientificamerican1191-96. [DOI] [PubMed] [Google Scholar]
- 99.Li J, Greco V, Guasch G, Fuchs E, Mombaerts P. Mice cloned from adult skin cells. Proc. Natl Acad. Sci. USA. doi: 10.1073/pnas.0611358104. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Paus R, Cotsarelis G. The biology of hair follicles. N. Engl. J. Med. 1999;341:491–497. doi: 10.1056/NEJM199908123410706. [DOI] [PubMed] [Google Scholar]