

Abstract
Large-scale motions of biomolecules involve linear elastic deformations along low-frequency normal modes, but for function nonlinearity is essential. In addition, unlike macroscopic machines, biological machines can locally break and then reassemble during function. We present a model for global structural transformations, such as allostery, that involve large-scale motion and possible partial unfolding, illustrating the method with the conformational transition of adenylate kinase. Structural deformation between open and closed states occurs via low-frequency modes on separate reactant and product surfaces, switching from one state to the other when energetically favorable. The switching model is the most straightforward anharmonic interpolation, which allows the barrier for a process to be estimated from a linear normal mode calculation, which by itself cannot be used for activated events. Local unfolding, or cracking, occurs in regions where the elastic stress becomes too high during the transition. Cracking leads to a counterintuitive catalytic effect of added denaturant on allosteric enzyme function. It also leads to unusual relationships between equilibrium constant and rate like those seen recently in single-molecule experiments of motor proteins.
The regulation of biological machinery through allostery is a dominant theme in our modern molecular understanding of life. Allostery requires a biomolecule to have at least a pair or, more likely, a multiplicity of conformational states of nearly equal free energy. How can we describe movement between such states? When the pair of states exhibits large-scale structural differences, it is tempting to connect the states by routes using the low-frequency collective elastic vibrations around each structure, the normal modes with the smallest restoring forces. Even in its simplest form, the notion of normal modes is remarkably successful for visualizing and predicting the character of the motions. The motions are in reality overdamped, but their structure often parallels the low-frequency normal modes (1). Yet clearly, a linear normal mode description cannot be complete because the very existence of the two low-lying conformations requires us to acknowledge considerable anharmonicity. The normal mode picture strictly describes the excitations about a single minimum. The limited adequacy of a normal mode description becomes even more apparent when we try to embed our picture of the motion between two dominant conformational states in the complete energy landscape of a biomolecule, which is replete with a myriad of local minima, ranging from the more subtle conformational substrates apparent in kinetic experiments (2) to the still more disordered states that are partially unfolded. Our goal in this article is to describe how allosteric conformational switches function by using a theoretical framework that unites an energy landscape description with the elastic model based on normal modes. To do so we need to go beyond the usual approaches that describe only the geometry of allosteric structural changes to estimate the energetics, i.e., the free energy barriers between different minima. In the simplest interpolation between initial and final structures this barrier depends on the relative stabilities of the two forms and the elastic, geometric properties of the structures, discernible by crystallography. We also argue that allosteric changes need not always go by way of a single transition-state structure but rather sometimes pass through a transition-state ensemble of structures that are partially unfolded.
Our approach will be to see, first, how far the idea of a single distortion path can be pushed by following the normal modes from each of the dominant conformational states of an allosteric protein as determined crystallographically. The strict harmonic picture holds only very close to each minimum. On the other hand, modest adjustments of the small amplitude motions allow the protein to follow adiabatically the large-scale movements quite well for substantial distances (1). This range might be called the regime of nonlinear elasticity. As the amplitude of the displacement grows, however, the elastic limit is reached (3). At this point, one possibility is that a glissile motion much like dislocation flow in a defective solid will occur. This requires special structural elements preexisting in the native structure. Looking for such pivot points is, of course, very worthwhile. When such a pivot exists, the resulting dislocational motion can be energetically modeled by simply switching from the elastic model that starts from one dominant conformation to that from the other structure (4). Another possibility is that the biomolecule will “crack” under the stress of the restoring forces of deforming along these low-frequency modes, just as titin unfolds when externally pulled in single-molecule experiments (5, 6). In this case, the molecule will locally unfold and partially reassemble before following a low-frequency elastic motion downhill to the product. The activated transition then does not occur through a single path but a multiplicity of detailed routes, involving partially unfolded states of the stressed regions. Such cracking would be disastrous for macroscopic machines, but unlike macroscopic machinery, a biological machine can break during its ordinary function and still complete its task because biomolecules can unfold and later refold properly as needed. The possibility of such a mechanism [a “proteinquake” (7)] has recently been invoked in describing myosin conformational change by Terada et al. (8), where they elegantly reconcile several single-molecule experiments that cannot be explained by the rigid lever arm model. Here we explore how the structural details of such cracking can be inferred from models like those already used to describe the free energy profile for the folding of proteins (9–11). Cracking need not occur in all conformational transitions, but when it does occur it leads to a predictable relationship between the thermodynamics of folding and the kinetics of allosteric conformational changes that can be tested in the laboratory. The linkage between partial protein unfolding and functional cooperativity in thermodynamics is well established in the work of Hilser, Freire, and collaborators (12, 13). The focus of the present article is on kinetics and mechanism where unfolding is less expected, because both initial and final structures may be completely folded in the critical regions.
We illustrate the ideas by using adenylate kinase. Kinases are an important family of proteins, constituting ≈1.7% of human genes (14). Their critical role in signal pathways is made possible by an allosteric conformational change, which occurs when they are phosphorylated or bind ATP or other proteins. We chose adenylate kinase because it has rather well resolved crystal structures for both dominant conformational states, here called “open” (15) and “closed” (16). One feature, common to other allosteric proteins, is that even in the x-ray structures some parts of the protein are “disordered,” perhaps partially unfolded. That observation alone makes clear the overlap of the energy landscapes for folding and functional motion.
Methods
Preparation of Structure. Two conformations, open (Protein Data Bank ID code 4AKE) (15) and closed (Protein Data Bank ID code 1AKE) (16), of adenylate kinase are used. In addition to atoms included in the crystallographic coordinates, polar hydrogen atoms are added based on the amber united atom model (17). The hydrogen atoms are relaxed with a relatively short minimization by using the amber6 program (18).
The Protein Elastic Model: Tirion Potential. In this study, we use a coarse-grained model, Tirion potential, which is defined as follows. An interaction between two atoms, a and b, is the Hookean pairwise potential:
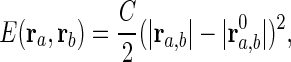 |
where ra denotes the coordinates of atom a, and ra, b = ra – rb. The zero superscript indicates the coordinate at the original conformation. The strength of the potential C is assumed to be the same for all interacting pairs. The total potential is
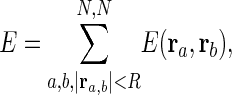 |
where R is the cut-off parameter, so that the interactions are limited to pairs separated by < R. We use R of 8 Å, which has already been shown to be appropriate for normal mode studies of protein (23).
Originally, the Tirion potential was proposed for normal mode analysis. We go beyond normal mode analysis since the Tirion potential is not entirely harmonic as a function of the atomic Cartesian coordinates. The spring constant of the Tirion potential, C, is optimized so that the average of B factors of Cα atoms from x-ray crystallography and the normal mode analysis coincide (19, 20). The normal modes were found by using the rtb approach (21).
This potential is crude but adequate for low-frequency motions. In many respects it is the elastic counterpart of the Go model landscapes used in protein folding simulations (22). Others have used normal modes to model the path of conformational change (23–27). These studies suggest that large-scale conformational transitions are dominated by low-frequency modes. Nevertheless activation barriers and therefore rates are not be obtained from a pure normal mode approach, as pointed out by Mouawad and Perahia (28) in their study of hemoglobin allostery.
Overlap Between Normal Mode and Conformational Change. The overlap, a measure of the relevance of a given normal mode to the conformational change, is defined as cosθn = d·an/|d||an|, where an is the direction of the normal mode n and d is the conformational change (21).
Iteration Procedure for Generating Nonlinear Conformational Change Path. From our initial attempt for describing energy profile with normal modes, we found that a single normal mode does not accurately represent the conformational changes (see Results). Thus we needed to define the conformational change pathway iteratively by the following procedure. We define the initial state S0 = SI and final state SF. For a structure, Sk, we redefine a Tirion potential, whose original conformation is Sk, and perform normal mode analysis, which defines a new normal mode coordinate, 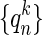 , with vectors,
, with vectors, 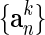 . The conformational difference between Sk and S is recalculated, dk. Structure Sk is displaced along a chosen number of highest overlap normal modes (one or three in this study) toward the final state leading to the Sk+1 structure, which is defined as a point on the current coordinate,
. The conformational difference between Sk and S is recalculated, dk. Structure Sk is displaced along a chosen number of highest overlap normal modes (one or three in this study) toward the final state leading to the Sk+1 structure, which is defined as a point on the current coordinate, 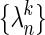 , by
, by 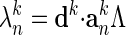 , where Λ is a parameter that determines how far the structure is to be deformed; In our calculation, we use Λ = 0.1. In addition, displacement is limited by an rms deviation (rmsd) of 0.5 Å to the current structure Sk, i.e., Λ has to satisfy the following inequality:
, where Λ is a parameter that determines how far the structure is to be deformed; In our calculation, we use Λ = 0.1. In addition, displacement is limited by an rms deviation (rmsd) of 0.5 Å to the current structure Sk, i.e., Λ has to satisfy the following inequality:
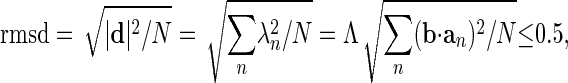 |
where N is the number of atoms. This iterative procedure is continued 100 steps.
Mapping Two Energy Profiles from Open and Closed Structures. When we need to superimpose two energy profiles from the open and closed forms (see Results), since the two profiles are actually defined with different coordinates, i.e., rmsd to open and closed forms, respectively, a mapping between the two coordinates is used. We calculate the pairwise rmsds between structures along the conformational change path from open or closed form, f(Ropen, Rclosed), where Ropen is the rmsd to the open form from each structures on the conformational change path and Rclosed is to the closed form. The mapping between two reaction coordinates, Rclosed = L(Ropen), is defined as the path L between open and closed form that minimizes the function 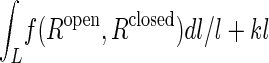 , where l is the length of the path L, and k is a parameter, which enforces that the length of path is short enough, k = 1 was used for our calculation.
, where l is the length of the path L, and k is a parameter, which enforces that the length of path is short enough, k = 1 was used for our calculation.
Results
The Linear Elastic Model. We first study the deformation of the kinase starting from the open structure. The closed structure was fitted to the open structure, giving a rmsd between the two structures of 7.2 Å. The low-frequency modes were determined by using a coarse-grained model, the Tirion potential (19). Among the normal modes for the initial structure, normal mode 1 (the lowest-frequency mode) is most relevant, having the highest overlap to the conformational change. By generating some deformed structures after mode 1, the energy surface of the open form is evaluated (Fig. 1). If the Tirion potential were perfectly harmonic, this would give a quadratic function of the displacement from the open form with a curvature dependent on the frequency of normal mode frequency. The Tirion energy surface and this ideal harmonic description only agree in the vicinity of the energy minimum structure. The Tirion energies are larger than expected from the standard normal mode analysis, because a single normal mode does not accurately represent the conformational changes during the deformation. Examining the open and closed forms suggests the conformational change resembles the bending of a rod. Obviously, when bending a rod through a finite angle, a single normal mode only indicates the initial direction of the transformation. Bending is kinematically nonlinear in a Cartesian basis. This kinematic nonlinearity, well known in macroscopic elastic theory, is the main shortcoming of the strictly linear model.
Fig. 1.

Comparison of energy surfaces with different models. When the energy surface is perfectly harmonic as a function of Cartesian coordinates and the conformational change path is linear, the energy surface along the conformational change path can be derived from the normal mode frequency of mode 1 at the initial state (solid line). However, the energy surface along the normal mode 1 (×) computed explicitly with the full anharmonic Tirion potential yields a much higher energy than the harmonic approximation owing to nonlinearities. The energy surface along the nonlinear conformational change path generated by using an iterative method with one mode (○) agrees with the harmonic approximation quite well up to 4 Å of rmsd but exceeds the harmonic result beyond this point. The energy using three modes to connect initial and final states (□) is also shown.
Nonlinear Elastic Models. To extend the model to include the bending nonlinearity, we explore a nonlinear path by using combinations of normal modes that adiabatically change as we move from one global structure to the other as a basis. We used the combinations of one or three normal modes that are most relevant to conformational change (high overlap modes). The procedure to find the best combination of modes is iterative (see Movie 1, which is published as supporting information on the PNAS web site, www.pnas.org). For each structure on the pathway, the strain energy is first calculated by using the Tirion potential with a spring network defined from the open form structure. Then the spring network of connectivities is redefined at each step of the calculation before determining a new set of normal modes for the distorted structure. Following the energy along the resulting nonlinear path made by using only one mode gives much lower energy than when the energy was strictly computed along the linear path moving along the first low-frequency normal mode (Fig. 1). The resulting adiabatic approximation surface agrees well with the quadratic approximation. Thus, although the harmonic approximation as a function of Cartesian coordinates is not accurate, the harmonic approximation is quite good when the energy is written as a function of a nonlinear reaction coordinate.
Strain Energy Is Localized. The strain in the molecule when it is deformed along the reaction path is not uniform. We assign a strain energy to each protein residue as the sum of the atomic strain energies. The strain intensity for all residues is determined for each structure along the nonlinear conformational change. Fig. 2a shows the spatial pattern of residue strain as the molecule is deformed. When the rmsd to the open form becomes >3 Å, some residues become particularly highly strained. These must rearrange to allow the transition to come to completion. The residue strain energies as a function of the sequence are shown in Fig. 2b. Fig. 2c shows the 3D structure of the protein with the residues colored according to their strain energies; red corresponds to high strain and blue corresponds to low (see Movie 2, which is published as supporting information on the PNAS web site). The T1 region, residues 10–12, is under high strain. The helix α6, residues 110–120, and helix α7, residues 160–170, are also under high strain. There is a clear correlation between the high-strain energy regions along the nonlinear conformational conversion path and what have been called hinges (15, 29).
Fig. 2.

Residue strain energy of structures along nonlinear conformational change path. (a) The change of the strain energy localized in individual residues as the structure is deformed is shown. The rmsd of each structure from the open structure is indicated on the right. Residues in blue have no strain energy while red residues have high strain energy. The maximum strain (indicated in red) is 0.5 kcal/mol. (b) The residue strain energy of the structure after 15 steps of iteration is shown in a 2D plot. The secondary structure of this protein is indicated on the top of the plot. (c) The structure after 15 steps of iteration is shown along with the residue strain energy. The residues again are colored according to the strain energy; blue corresponding to no strain and red residues corresponding to high strain energy as before. c was prepared with vmd (43) and raster3d (44).
The Cracking Model. The observations that some particular contiguous residues are under high strain leads us to hypothesize that these special regions may crack, i.e., unfold partially, during the conformational change. To include this possibility, a residue will be allowed to unfold if the strain energy of residue is higher than the difference of local folding energy and the structural entropy to be gained by locally unfolding. Under this local approximation, the free energy per residue can be written as
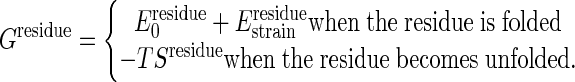 |
For simplicity, we assume that all residues have identical folding energy, 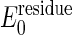 , at the initial state and that their entropy change by unfolding, Sresidue, is also identical. Experimentally determined stabilities are used to set these parameters. It is easy to incorporate residue specific energies for these thresholds as in the work of Munoz and Eason (10). The unfolding free energy of the adenylate kinase from Escherichia coli has been reported to be ≈4 kcal/mol for guanidine hydrochloride denaturation (30) and ≈10 kcal/mol for urea denaturation (31). Heat denaturation of adenylate kinase from Saccharomyces cerevisiae is reported to give a stability of ≈4 kcal/mol (32). Dividing the total unfolding free energy by the number of residues (≈200) yields
, at the initial state and that their entropy change by unfolding, Sresidue, is also identical. Experimentally determined stabilities are used to set these parameters. It is easy to incorporate residue specific energies for these thresholds as in the work of Munoz and Eason (10). The unfolding free energy of the adenylate kinase from Escherichia coli has been reported to be ≈4 kcal/mol for guanidine hydrochloride denaturation (30) and ≈10 kcal/mol for urea denaturation (31). Heat denaturation of adenylate kinase from Saccharomyces cerevisiae is reported to give a stability of ≈4 kcal/mol (32). Dividing the total unfolding free energy by the number of residues (≈200) yields 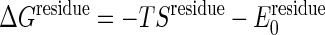 of ≈0.02–0.05 kcal/mol. This simple model of dividing the unfolding energy evenly neglects the fact that a good share of the binding in the folded state comes from nonadditive interactions such as hydrophobicity and side-chain conformations that enhance the cooperative aspect of denaturation. The unfolding energy for a segment should therefore be larger than the equilibrium value spread uniformly throughout the chain. Thus we examine the result by using two values of the threshold, one with
of ≈0.02–0.05 kcal/mol. This simple model of dividing the unfolding energy evenly neglects the fact that a good share of the binding in the folded state comes from nonadditive interactions such as hydrophobicity and side-chain conformations that enhance the cooperative aspect of denaturation. The unfolding energy for a segment should therefore be larger than the equilibrium value spread uniformly throughout the chain. Thus we examine the result by using two values of the threshold, one with 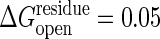 and the other with 0.1 kcal/mol for unfolding. The cooperativity coming from nonadditive interactions also requires that several contiguous residues must be simultaneously under high strain to unfold (33). Thus, as in unfolding calculations (10), we require that at least a number Nmin of contiguous residues should have strain energies exceeding the threshold, where
and the other with 0.1 kcal/mol for unfolding. The cooperativity coming from nonadditive interactions also requires that several contiguous residues must be simultaneously under high strain to unfold (33). Thus, as in unfolding calculations (10), we require that at least a number Nmin of contiguous residues should have strain energies exceeding the threshold, where 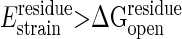 , to be treated as being cracked. Models of folding kinetics fit experiment well by using Nmin = 5 (10).
, to be treated as being cracked. Models of folding kinetics fit experiment well by using Nmin = 5 (10).
Fig. 3 shows the strain energy along the nonlinear conformational change path when cracking is allowed. Obviously partial unfolding gives a lower energy barrier. The extent of cracking strongly depends on the threshold 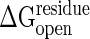 . Cracking leads to a critical yield stress so the energy becomes an approximately linear function of deformation rather than following the quadratic dependence of the elastic regime. Such linear relations have also been invoked to explain the efficiency of motor proteins by Bustamante et al. (34). The cracking model suggests the linear behavior may be much more general. The critical yield stress found here, 140 pN, is comparable to values seen in single-molecule pulling experiments. As it becomes clear in the specific analysis exhibited later in this article, cracking leads to linear behavior of the rate versus driving force over a large range of reaction driving forces, a situation that would not be true if only elastic deformations were allowed. Studies on allosteric motions in hemoglobin by Eaton et al. (35) have also exhibited such a linear free energy dependence supporting the general applicability of the cracking mechanism.
. Cracking leads to a critical yield stress so the energy becomes an approximately linear function of deformation rather than following the quadratic dependence of the elastic regime. Such linear relations have also been invoked to explain the efficiency of motor proteins by Bustamante et al. (34). The cracking model suggests the linear behavior may be much more general. The critical yield stress found here, 140 pN, is comparable to values seen in single-molecule pulling experiments. As it becomes clear in the specific analysis exhibited later in this article, cracking leads to linear behavior of the rate versus driving force over a large range of reaction driving forces, a situation that would not be true if only elastic deformations were allowed. Studies on allosteric motions in hemoglobin by Eaton et al. (35) have also exhibited such a linear free energy dependence supporting the general applicability of the cracking mechanism.
Fig. 3.

The energy profiles for open and closed states with and without cracking. Calculations of free energy profiles without cracking and with cracking for two values of 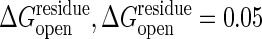 and 0.1 kcal/mol are shown. The threshold of the closed state,
and 0.1 kcal/mol are shown. The threshold of the closed state, 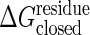 , is set so that the open and the closed states have the same energy at the totally unfolded state. The strain energy computed from the open form is shown in black. The one computed from the closed form is shown in red for free energy change of ΔGeq = 0 kcal/mol and is shown in blue for ΔGeq = –3 kcal/mol. Results computed without allowing cracking are shown as solid lines, and the broken lines correspond to a threshold for cracking of
, is set so that the open and the closed states have the same energy at the totally unfolded state. The strain energy computed from the open form is shown in black. The one computed from the closed form is shown in red for free energy change of ΔGeq = 0 kcal/mol and is shown in blue for ΔGeq = –3 kcal/mol. Results computed without allowing cracking are shown as solid lines, and the broken lines correspond to a threshold for cracking of 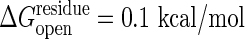 . The dotted lines use a cracking threshold of
. The dotted lines use a cracking threshold of 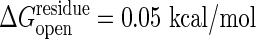 .
.
Our theoretical analysis predicts that for adenylate kinase, the structure that corresponds to the beginning of cracking (at rmsd = 4 Å along the three-mode iterative path from the open form) shows high strain energy in α6. This helix would likely become partially unfolded during conformational change and figure prominently in the transition-state ensemble for allosteric change. Evaluating whether a mere sliding of the helix without losing contacts is energetically competitive with unfolding would require a much more detailed atomistic calculation.
Free Energy Barrier. A full understanding of the allosteric transition requires consideration of the product energy surface to describe the final approach to the closed structure. This is obtained by using the same strategy describe above but starting for the closed structure (16) deforming toward the open structure (15). The crossing of these two surfaces locates approximately the transition state for the motion (Fig. 3). Obviously small-scale rearrangements are needed to complete the motion from one surface to the other. To locate the transition region we must know the relative stability of the closed and open forms. The conformational change in kinase doubtless is affected by the binding of ligand. The x-ray crystal structure of the closed form is a complex with the inhibitor Ap5A, which is a bisubstrate analog inhibitor that connects ATP and AMP by a fifth phosphate, thus mimicking both substrates. Before a ligand binds, the open form is more stable, while upon ligand binding, the closed form becomes energetically favorable.
Sanders et al. (36) have experimentally determined the standard binding free energy, ΔG°, of ATP and AMP, obtaining –6.3 and –4.6 kcal/mol, respectively. If we imagine the ligands are in rapid binding equilibrium, from these data, depending on the concentration of substrate, the effective binding free energy of Ap5A can be varied in the range of a few kcal/mol. Thus, we use the reaction free energy for the allosteric transition accompanied by binding, ΔGeq, of –3 kcal/mol and 0 kcal/mol as reasonable gaps when drawing Fig. 3. As discussed below, however, this topic needs further investigation. The threshold of the ligand bound, the closed state, 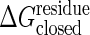 , is related to the threshold of the open state,
, is related to the threshold of the open state, 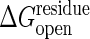 , in such a way that the open and the closed states have the same energy when completely unfolded, i.e.,
, in such a way that the open and the closed states have the same energy when completely unfolded, i.e., 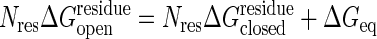 , where Nres is the number of residues.
, where Nres is the number of residues.
To find where the binding event would occur, the energy profile from the closed form is superimposed on the energy profile from the open form (Fig. 3). The two profiles are actually defined with different coordinates, i.e., rmsd to open and closed forms, respectively. Thus, a mapping between the two coordinates is used as described in Methods.
Discussion
In allosteric proteins, the association of ligands to enzymes, a bimolecular step, may precede or follow the unimolecular conformational change or, if binding occurs at states far from equilibrium, the bimolecular step may occur during the unimolecular change. In the first two cases, the unimolecular transition described here will be independent of the bimolecular components of the reaction. The free energy difference between the two surfaces in Fig. 3 can be determined between the ligand-bound conformations in the first case and for the ligand-free conformations in the second one. If ligand binding, however, occurs concurrently with the structural transformation, then the situation becomes substantially more complex. For example, if ligand binding is fast compared with protein structural changes, the free energy drive on the reaction would be ligand-concentration-dependent, with the caveat that the existence of ligand preequilibrium states might destroy this effect. A similarity to the nonadiabatic and adiabatic patterns of electron transfer is immediately apparent (37).
When unfolding or cracking is allowed, the rate dependence on the stability difference of open and closed forms differs from the purely elastic models. Without unfolding, the energy surfaces are quadratic, so the activation barrier varies quadratically with the free energy difference of the two forms, much as in the Marcus theory for electron transfer (37). On the other hand, when unfolding occurs, the free energy surfaces are locally linear, giving a linear dependence of the activation energy on the reaction free energy change. Using protein engineering to differentially affect the stability of the two forms is the natural way to test this model.
Recent observations of the Kern group (D. Kern, personal communication) suggest that the structural transition may be the rate-determining step for catalysis in adenylate kinase. This result encourages us to make specific quantitative predictions for this system, which we now describe. Fig. 4 shows how the transition-state barrier depends on the reaction driving force. Although a clear curvature characteristic of intersecting parabolas is observed for a fully elastic model, inclusion of cracking makes the dependence linear for a very large range of driving forces. Although the calculations have been performed for adenylate kinase, we note such a large linear range is observed in many systems. In particular, linearity with load has been observed by Bustamante et al. (34) in motor proteins where such linearity has been argued to be essential to the motor's efficiency.
Fig. 4.

The transition-state barrier dependence on the reaction driving force. Calculations of how the transition-state barrier, ΔG*, depends on the reaction driving force, (–ΔGeq), both without cracking (a) and with cracking (c) are shown. The corresponding energy surfaces at the different driving forces (–ΔGeq) without cracking (b) and with cracking (d) are also shown. A quadratic curvature is observed for the fully elastic model without cracking. Including the cracking effect makes the barrier dependence on driving force linear for a very large range of driving forces.
More importantly the cracking model allows residues to become unfolded in the transition state that are folded in both stable states. Thus an unexpected dependence on folding stability change should be seen: simultaneously lowering the stability of both conformations without changing their relative stability will speed the reaction. Using site-directed mutagenesis, one can determine the probability of individual residues to remain structured or to crack during the transition as in folding Φ value analysis (38). Evidence for cracking can also be gleaned by using the global effects of denaturants, such as urea, on conformational change kinetics. Denaturant will enhance the local unfolding and therefore should lower the barrier height. Such anomalous activation of enzymes by adding low concentrations of denaturant has been observed in this and other systems (39, 40). Under the assumption that the conformational change is the rate-determining step, we have calculated the reaction rate dependence on denaturant. This relationship is not monotonic. At higher concentration, as anyone would expect, denaturation reduces the activity. But at low levels of denaturant the activity does indeed increase in accord with our model. The activity increase observed experimentally by adding denaturant (39), corresponds to a ∂ΔG*/∂[urea] in the range of 0.2 and 0.7 kcal/mol per M. Here M is the molar concentration of urea. We have computed the analogous slope for our theoretical model of the conformational change by itself, ∂ΔG*/∂[urea]. Details are presented in Fig. 5 for a reasonable driving force ΔGeq = –3 kcal/mol and 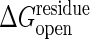 in the range between 0.15 and 0.1 kcal/mol, the model yields results consistent with the experimental speedup, ∂ΔG*/∂[urea] of between 0.22 and 0.65 kcal/mol per M. More careful direct measurement of the conformational change kinetics by itself, not as a composite with other chemical steps, as a function of denaturant, however, is needed to test our model more completely. Although the conformational change step has been kinetically isolated in the related systems of carboxyl-terminal Src kinase and cAMP-dependent protein kinase (41, 42), the denaturant dependence has yet to be studied. Although the present model can be refined, this initial agreement between theory and experiment shows the potential of the present approach for quantitatively understanding the underlying mechanisms governing allosteric motions.
in the range between 0.15 and 0.1 kcal/mol, the model yields results consistent with the experimental speedup, ∂ΔG*/∂[urea] of between 0.22 and 0.65 kcal/mol per M. More careful direct measurement of the conformational change kinetics by itself, not as a composite with other chemical steps, as a function of denaturant, however, is needed to test our model more completely. Although the conformational change step has been kinetically isolated in the related systems of carboxyl-terminal Src kinase and cAMP-dependent protein kinase (41, 42), the denaturant dependence has yet to be studied. Although the present model can be refined, this initial agreement between theory and experiment shows the potential of the present approach for quantitatively understanding the underlying mechanisms governing allosteric motions.
Fig. 5.

The transition-state barrier dependence on the cracking threshold. Shown is how the transition-state barrier, ΔG*, depends on the cracking threshold, 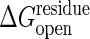 . A driving force of ΔGeq =–3 kcal/mol is used. The data (○) are fitted by a hyperbolic relation,
. A driving force of ΔGeq =–3 kcal/mol is used. The data (○) are fitted by a hyperbolic relation, 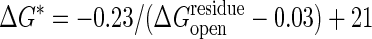 . From the slope of the line, the Tafel coefficient
. From the slope of the line, the Tafel coefficient 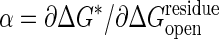 is estimated as 47 at
is estimated as 47 at 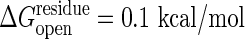 and 16 at
and 16 at 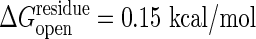 . The experimentally determined urea dependence of the stability, m = ∂ΔGD–N/∂[urea] = 2.9 kcal/mol per M (31), here M is the molar concentration of urea, corresponds to a change of cracking threshold
. The experimentally determined urea dependence of the stability, m = ∂ΔGD–N/∂[urea] = 2.9 kcal/mol per M (31), here M is the molar concentration of urea, corresponds to a change of cracking threshold 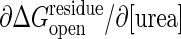 with the value m/Nres ≈0.014 kcal/mol per M, where the total number of residues Nres = 214. Combining these, the urea dependence of the transition barrier ∂ΔG*/∂[urea] is ≈0.65 kcal/mol per M if
with the value m/Nres ≈0.014 kcal/mol per M, where the total number of residues Nres = 214. Combining these, the urea dependence of the transition barrier ∂ΔG*/∂[urea] is ≈0.65 kcal/mol per M if 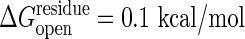 and ≈0.22 kcal/mol per M if
and ≈0.22 kcal/mol per M if 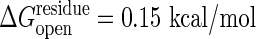 is used.
is used.
Conclusion
Allosteric conformational changes are among the slowest and most potentially complex events in structural biology. While many aspects of such motions may resemble macroscopic machines and be described as the motion of hinges (1), unlike macroscopic machines, the functioning of biological machines may involve catastrophic events such as cracking and subsequent reassembly. These large-scale events would normally be unseen in conventional molecular dynamics studies owing to the limited simulation time used. These events can now be explored by using the present theoretical methods in concert with experimental tests.
Supplementary Material
Acknowledgments
We thank Y. H. Sanejouand and F. Tama for making the program package rtb available to us and F. Tama for helpful discussion for developing the nonlinear approach. This work has been funded by the National Science Foundation-sponsored Center for Theoretical Physics (Grants PHY0216576 and 0225630). O.M. acknowledges the La Jolla Interfaces in Science postdoctoral training program supported by the Burroughs Wellcome Fund. Computations were carried out at the University of California, San Diego Keck II computing facility (partially supported by the National Science Foundation, Division of Molecular and Cellular Biosciences) and the San Diego Supercomputer Center.
Abbreviation: rmsd, rms deviation.
References
- 1.McCammon, J. A., Gelin, B. R., Karplus, M. & Wolynes, P. G. (1976) Nature 262 325–326. [DOI] [PubMed] [Google Scholar]
- 2.Frauenfelder, H., Sligar, S. G. & Wolynes, P. G. (1991) Science 254 1598–1603. [DOI] [PubMed] [Google Scholar]
- 3.Horiuchi, T. & Go, N. (1991) Proteins Struct. Funct. Genet. 10 106–116. [DOI] [PubMed] [Google Scholar]
- 4.Nabarro, F. R. N. (1947) Proc. Phys. Soc. 59 256–272. [Google Scholar]
- 5.Rief, M., Gautel, M., Oesterhelt, F., Fernandez, J. M. & Gaub, H. E. (1997) Science 276 1109–1112. [DOI] [PubMed] [Google Scholar]
- 6.Kellermayer, M. S., Smith, S. B., Granzier, H. L. & Bustamante, C. (1997) Science 276 1112–1116. [DOI] [PubMed] [Google Scholar]
- 7.Ansari, A., Berendzen, J., Bowne, S. F., Frauenfelder, H., Iben, I. E., Sauke, T. B., Shyamsunder, E. & Young, R. D. (1985) Proc. Natl. Acad. Sci. USA 82 5000–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terada, T. P., Sasai, M. & Yomo, T. (2002) Proc. Natl. Acad. Sci. USA 99 9202–9206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onuchic, J. N., Luthey-Schulten, Z. & Wolynes, P. G. (1997) Annu. Rev. Phys. Chem. 48 545–600. [DOI] [PubMed] [Google Scholar]
- 10.Munoz, V. & Eaton, W. A. (1999) Proc. Natl. Acad. Sci. USA 96 11311–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brooks, C. L., III, Onuchic, J. N. & Wales, D. J. (2001) Science 293 612–613. [DOI] [PubMed] [Google Scholar]
- 12.Hilser, V. J. & Freire, E. (1997) Proteins Struct. Funct. Genet. 27 171–183. [DOI] [PubMed] [Google Scholar]
- 13.Luque, I., Leavitt, S. A. & Freire, E. (2002) Annu. Rev. Biophys. Biomol. Struct. 31 235–256. [DOI] [PubMed] [Google Scholar]
- 14.Manning, G., Whyte, D. B., Martinez, R., Hunter, T. & Sudarsanam, S. (2002) Science 298 1912–1934. [DOI] [PubMed] [Google Scholar]
- 15.Muller, C. W., Schlauderer, G. J., Reinstein, J. & Schulz, G. E. (1996) Structure (London) 4 147–156. [DOI] [PubMed] [Google Scholar]
- 16.Muller, C. W. & Schulz, G. E. (1992) J. Mol. Biol. 224 159–177. [DOI] [PubMed] [Google Scholar]
- 17.Weiner, S. J., Kollman, P. A., Case, D. A., Singh, U. C., Ghio, C., Alagona, G., Profeta, S. & Weiner, P. (1984) J. Am. Chem. Soc. 106 765–784. [Google Scholar]
- 18.Case, D. A., Pearlman, D. A., Caldwell, J. W., Cheatham, T. E., III, Ross, W. S., Simmerling, C. L., Darden, T. A., Merz, K. M., Stanton, R. V., Cheng, A. L., et al. (1999) amber6 (University of California, San Francisco).
- 19.Tirion, M. M. (1996) Phys. Rev. Lett. 77 1905–1908. [DOI] [PubMed] [Google Scholar]
- 20.Bahar, I., Atilgan, A. R. & Erman, B. (1997) Folding Des. 2 173–181. [DOI] [PubMed] [Google Scholar]
- 21.Tama, F., Gadea, F. X., Marques, O. & Sanejouand, Y. H. (2000) Proteins Struct. Funct. Genet. 41 1–7. [DOI] [PubMed] [Google Scholar]
- 22.Go, N. (1983) Annu. Rev. Biophys. Bioeng. 12 183–210. [DOI] [PubMed] [Google Scholar]
- 23.Tama, F. & Sanejouand, Y. H. (2001) Protein Eng. 14 1–6. [DOI] [PubMed] [Google Scholar]
- 24.Atilgan, A. R., Durell, S. R., Jernigan, R. L., Demirel, M. C., Keskin, O. & Bahar, I. (2001) Biophys. J. 80 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tama, F. & Brooks, C. L., III (2002) J. Mol. Biol. 318 733–747. [DOI] [PubMed] [Google Scholar]
- 26.Doruker, P., Jernigan, R. L. & Bahar, I. (2002) J. Comput. Chem. 23 119–127. [DOI] [PubMed] [Google Scholar]
- 27.Tama, F., Valle, M., Frank, J. & Brooks, C. L., III (2003) Proc. Natl. Acad. Sci. USA 100 9319–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mouawad, L. & Perahia, D. (1996) J. Mol. Biol. 258 393–410. [DOI] [PubMed] [Google Scholar]
- 29.Kumar, S., Ma, B., Tsai, C. J., Wolfson, H. & Nussinov, R. (1999) Cell Biochem. Biophys. 31 141–164. [DOI] [PubMed] [Google Scholar]
- 30.Tian, G. C., Sanders, C. R., II, Kishi, F., Nakazawa, A. & Tsai, M. D. (1988) Biochemistry 27 5544–5552. [DOI] [PubMed] [Google Scholar]
- 31.Burlacu-Miron, S., Perrier, V., Gilles, A. M., Pistotnik, E. & Craescu, C. T. (1998) J. Biol. Chem. 273 19102–19107. [DOI] [PubMed] [Google Scholar]
- 32.Spuergin, P., Abele, U. & Schulz, G. E. (1995) Eur. J. Biochem. 231 405–413. [DOI] [PubMed] [Google Scholar]
- 33.Plotkin, S. S., Wang, J. & Wolynes, P. G. (1996) Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 53 6271–6296. [DOI] [PubMed] [Google Scholar]
- 34.Bustamante, C., Keller, D. & Oster, G. (2001) Acc. Chem. Res. 34 412–420. [DOI] [PubMed] [Google Scholar]
- 35.Eaton, W. A., Henry, E. R. & Hofrichter, J. (1991) Proc. Natl. Acad. Sci. USA 88 4472–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanders, C. R., II, Tian, G. C. & Tsai, M. D. (1989) Biochemistry 28 9028–9043. [DOI] [PubMed] [Google Scholar]
- 37.Marcus, R. A. & Sutin, N. (1985) Biochim. Biophys. Acta 811 265–322. [Google Scholar]
- 38.Fersht, A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Freeman, New York).
- 39.Zhang, H. J., Sheng, X. R., Pan, X. M. & Zhou, J. M. (1997) Biochem. Biophys. Res. Commun. 238 382–386. [DOI] [PubMed] [Google Scholar]
- 40.Fan, Y. X., Ju, M., Zhou, J. M. & Tsou, C. L. (1995) Biochim. Biophys. Acta 1252 151–157. [DOI] [PubMed] [Google Scholar]
- 41.Adams, J. A. (2001) Chem. Rev. 101 2271–2290. [DOI] [PubMed] [Google Scholar]
- 42.Hamuro, Y., Wong, L., Shaffer, J., Kim, J. S., Stranz, D. D., Jennings, P. A., Woods, V. L., Jr. & Adams, J. A. (2002) J. Mol. Biol. 323 871–881. [DOI] [PubMed] [Google Scholar]
- 43.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14 33–38. [DOI] [PubMed] [Google Scholar]
- 44.Merritt, E. A. & Bacon, D. J. (1997) Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}