

Abstract
The HOX11/TLX1 homeobox gene is aberrantly expressed in a subset of T-cell acute lymphoblastic leukemia (T-ALL). Here we employed oligonucleotide microarrays to compare the expression profiles of the K3P and Sil leukemic cell lines originating from patients with HOX11+ T-ALL to that of Jurkat cells which originated from a distinct subtype of T-ALL (TAL1+). To distinguish potential HOX11 target genes from those characteristic of the stage of HOX11 leukemic arrest, we also performed gene expression analysis on Jurkat cells genetically-engineered to express exogenous HOX11. The resulting HOX11 gene expression signature, which was validated for representative signaling pathways by transient transfection of reporter constructs, was characterized by elevated expression of transcriptional programs involved in cell proliferation, including those regulated by E2F, c-Myc and CREB. We subsequently showed that ectopic HOX11 expression resulted in hyperphosphorylation of the retinoblastoma protein (Rb) which correlated with inhibition of the major Rb serine/threonine phosphatase PP1. HOX11 also inhibited PP2A serine/threonine phosphatase activity concomitant with stimulation of the AKT/PKB signaling cascade. These results suggest that transcriptional deregulation of G1/S growth-control genes, mediated in large part through blockade of PP1/PP2A phosphatase activity, plays an important role in HOX11 pathobiology.
Keywords: T-cell acute lymphoblastic leukemia, HOX11 oncogene, oligonucleotide microarrays, G1/S transcriptional networks
Introduction
The HOX11/TLX1 homeobox gene encodes an oncogenic transcription factor that is activated in approximately 5−10% of childhood and 30% of adult T-cell acute lymphoblastic leukemia (T-ALL), frequently due to chromosomal translocations involving the T cell receptor δ or β genes (Owens and Hawley, 2002). Ferrando and colleagues previously reported the gene expression profiles of primary leukemic lymphoblasts from T-ALL patients (Ferrando et al., 2002). In the case of HOX11+ samples, the expression profile was indicative of leukemic arrest at the early cortical thymocyte stage of T cell development. However, it was not possible in that study to distinguish HOX11 target genes from genes that are normally expressed at this stage of T cell differentiation. Thus, while the transforming potential of HOX11 is well established (Hawley et al., 1994a; Hawley et al., 1997; Keller et al., 1998; Owens et al., 2003), the transcriptional programs affected by its aberrant expression in developing T cell precursors remain to be elucidated.
Our group previously performed a comprehensive structure-function analysis of HOX11 and inferred from hematopoietic precursor immortalization assays that specific homeodomain-DNA interactions are required for HOX11-mediated transformation (Owens et al., 2003). In addition, these experiments revealed that the amino terminus of HOX11 (amino acids 1−200) was essential for transforming function. In addition to DNA binding activity (Owens et al., 2003; Dear et al., 1993; Allen et al., 2000), HOX11 was shown by others to interact directly with the catalytic subunits of the protein serine/threonine phosphatases PP2A and PP1 via a region (amino acids 149−190) upstream of the homeodomain (Kawabe et al., 1997), with high affinity interaction requiring amino acids 1−190. PP2A and PP1 are important regulators of multiple intracellular signaling pathways (Janssens and Goris, 2001; Cohen, 2002). In the context of leukemic transformation, HOX11-mediated inhibition of PP2A was proposed to eliminate a G2/M cell-cycle checkpoint based on results obtained with γ-irradiated Jurkat T-ALL cells (Kawabe et al., 1997).
Here we conducted oligonucleotide microarray analyses of two cell lines established from patients with HOX11+ T-ALL [K3P (Dube et al., 1991) and Sil (Hatano et al., 1991)] and compared the gene expression profiles to those of TAL1+ Jurkat T cells (Pulford et al., 1995) before and after introduction of a HOX11 transgene. We next searched for HOX11 “signature” genes conserved between human and murine cells by determining the gene expression profile of a murine HOX11+ tumor cell line that originated from a T-cell leukemia/lymphoma induced in mice transplanted with HOX11-transduced bone marrow cells (Hawley et al., 1997). We confirmed that HOX11 modulated selected gene expression programs identified in this manner in transient transfection experiments with reporter constructs linked to appropriate cis-acting regulatory elements. The results obtained indicate that HOX11 stimulates multiple signaling pathways, most notably those regulating G1/S-phase progression. Moreover, the combined data support a mechanism of HOX11 oncogenicity in which inhibition of PP1/PP2A activity plays an important role.
Results
Expression profiling of HOX11+ T-ALL cell lines
Ferrando and colleagues previously determined the expression profiles of primary leukemic lymphoblast samples from T-ALL patients using oligonucleotide microarrays representing ∼7,300 genes (Affymetrix HU6800 GeneChip) (Ferrando et al., 2002). The expression signature associated with HOX11+ T-ALL reflected leukemic arrest at the early cortical stage of thymocyte differentiation whereas that identified for TAL1+ leukemic lymphoblasts resembled thymocytes transiting the late cortical stage. Building on this framework, we employed Affymetrix HG-U133A GeneChips comprising ∼14,500 genes to compare the expression patterns of the HOX11+ K3P (Dube et al., 1991) and Sil (Hatano et al., 1991) T-ALL cell lines with that of Jurkat T-ALL cells, which are TAL1+ (Pulford et al., 1995).
Analysis of microarray data from hybridization experiments performed in triplicate for genes differentially expressed in HOX11+ K3P and Sil cells versus TAL1+ Jurkat cells identified high levels of expression of HOX11 itself as expected (and, vice versa, TAL1 in Jurkat cells) and increased levels of expression of a number of associated genes (24 probe sets) placed by Ferrando and colleagues into the “Cell proliferation” and “Chemotherapy response and drug targets” functional groups (Figure 1A; see also Supplementary Information file “Filtered data.xls”, Ferrando sheet). Collectively, the mean signal intensities of expression values for these HOX11 signature genes in K3P cells (3,000 ± 90) and Sil cells (2,770 ± 60) were significantly higher than in Jurkat cells (600 ± 100) (P < 0.002; Table 1). We interpreted these results to indicate that the K3P and Sil cell lines were valid in vitro models of HOX11+ T-ALL.
Figure 1.


HOX11+ T-ALL molecular phenotype. (A) Upper graph: Expression levels of representative HOX11 signature genes involved in “Cell proliferation” and “Chemotherapy response and drug targets” identified by Ferrando and colleagues (Ferrando et al., 2002) in K3P and Sil cells (dark bars) and Jurkat cells (white bars) are shown for triplicate experiments. Horizontal lines represent the mean signal intensities for the set of selected HOX11 signature genes in K3P (*; 3000 ± 90) and Jurkat (**; 600 ± 100) cells. Lower graph: Log2 ratios of signal intensities illustrating relative differences in expression levels of the selected HOX11 signature genes in K3P (blue bars) and Sil (pink bars) cells by comparison with Jurkat cells. Gene descriptions: TOP2A, topoisomerase (DNA) II alpha 170kDa; RPA1, replication protein A1, 70kDa; MCM2, minichromosome maintenance deficient 2; TK1, thymidine kinase 1; DHFR, dihydrofolate reductase; TOPB1, topoisomerase (DNA) II binding protein 1; MCM6, minichromosome maintenance deficient 6; POLA, polymerase (DNA directed), alpha; MAD1L1, MAD1 mitotic arrest deficient-like 1; LAP18/STMN1, stathmin 1/oncoprotein 18; POLD2, polymerase (DNA directed), delta 2, regulatory subunit 50kDa; TDT, terminal deoxynucleotidyl transferase; MCM4, minichromosome maintenance deficient 4; UNG, uracil-DNA glycosylase; ERCC1, excision repair cross-complementing rodent repair deficiency; POLG, polymerase (DNA directed), gamma; MCM3, minichromosome maintenance deficient 3; POLR2E, polymerase (RNA) II (DNA directed) polypeptide E, 25kDa; HPRT1, hypoxanthine phosphoribosyltransferase 1; and MCM5, minichromosome maintenance deficient 5. Values of multiple probe sets recognizing DHFR and MCM4 are plotted to highlight extent of variability of the microarray data due to differences in hybridization efficiencies. See Table 1 for P values and Supplementary Information file “Filtered data.xls”, K3P and Sil vs Jurkat >4 sheet. (B) Expression patterns of genes (with unique identifiers) that recapitulated the HOX11+ T-ALL molecular phenotype following transduction of Jurkat cells with the MSCVhyg-HOX11 retroviral vector. Ectopic HOX11 expression resulted in increased expression of E2F and c-Myc transcriptional targets, c-Myc interacting partners, transcripts modulated by AKT and IFN signaling pathways, and genes associated with chromatin remodeling and transcription, whereas expression of WNT-related genes was down-regulated. Graphs were generated using GeneSpring. The colorbar range is from 0−13 for K3P and Sil versus Jurkat, and from 0−1.3 for Jurkat/MSCVhyg-HOX11 versus Jurkat/MSCVhyg.
Table 1.
Relative expression levels of putative HOX11 target gene sets
| Genesa | No. of probe sets |
Mean |
Signal |
Intensities |
P values | P values |
|---|---|---|---|---|---|---|
| K3P | Sil | Jurkat | K3P/Jurkat | Sil/Jurkat | ||
| HOX11 signature | 24 | 3,000 ± 90 | 2,770 ± 60 | 600 ± 100 | 0.002 | 0.001 |
| E2F targets | 50 | 3,100 ± 50 | 3,000 ± 100 | 1,000 ± 200 | 0.006 | 0.002 |
| Myc targets (set I) | 22 | 10,000 ± 500 | 10,500 ± 100 | 4,400 ± 1,000 | 0.02 | 0.02 |
| Myc targets (set II) | 35 | 10,300 ± 500 | 10,200 ± 100 | 6,000 ± 2,000 | 0.04 | 0.06 |
| CREB targets | 17 | 7,300 ± 300 | 7,000 ± 200 | 2,500 ± 700 | 0.03 | 0.02 |
| Wnt-related | 43 | 70 ± 9 | 75 ± 4 | 260 ± 10 | 0.001 | 0.001 |
| IFN-induced | 14 | 4,600 ± 200 | 7,000 ± 200 | 270 ± 40 | 0.02 | 0.001 |
| Mitochondrial | 29 | 7,200 ± 200 | 7,200 ± 200 | 3,500 ± 800 | 0.001 | 0.001 |
Values represent mean signal intensities for each set of genes from three microarray experiments ± SD. P values were calculated using the paired Student's t-test.
HOX11 signature genes were identified by Ferrando and colleagues (Ferrando et al., 2002); E2F target genes were characterized by Ren and colleagues (Ren et al., 2002) and Weinmann and colleagues (Weinmann et al., 2002); set I Myc target genes are c-Myc-inducible genes identified in c-Myc global chromatin immunoprecipitation experiments that contain promoter-associated E-boxes (Fernandez et al., 2003); set II Myc “target” genes were identified in c-Myc global chromatin immunoprecipitation experiments and their expression positively correlates with c-myc mRNA levels in multiple tissues and cell lines (correlation coefficient >0.69) (Li et al., 2003); CREB target genes were extracted from the study by Conkright and colleagues (Conkright et al., 2003); the set of Wnt-related genes was compiled from this work; IFN-induced genes were selected from the study by Krasnoselskaya-Riz and colleagues (Krasnoselskaya-Riz et al., 2002); and mitochondrial genes were chosen from the current data. Gene lists described in the text are provided as Supplementary Information.
Among the previously described HOX11 signature genes, we noted examples known to be involved in cell cycle progression and G1/S transition, including those regulated by E2F and c-Myc (Sears and Nevins, 2002). These findings prompted us to systematically examine the microarray data for E2F and c-Myc target genes. A subgroup of E2F target genes (50 probe sets) was created by in silico screening of genes identified by several groups in experiments that combined E2F global chromatin immunoprecipitation of cross-linked protein-DNA complexes with DNA microarray analyses (Ren et al., 2002; Weinmann et al., 2002; Wells et al., 2002). To complete the list, we also included known direct E2F-induced genes such as dihydrofolate reductase (DHFR) and cyclin D3 (Fry et al., 1999; Ma et al., 2003). In aggregate, the mean signal intensities of expression levels for all extracted E2F targets were significantly increased in K3P (3,100 ± 50) and Sil (3,000 ± 100) cells versus Jurkat cells (1,000 ± 200) (P < 0.006; Table 1), suggesting that the HOX11+ molecular phenotype is associated with elevated E2F activity. To compare gene expression levels of c-Myc target genes in HOX11+ versus HOX11− T-ALL cells (Zeller et al., 2003), we used a similar bioinformatics approach to compile two subgroups: set I consists of c-Myc-inducible genes (22 probe sets) identified in c-Myc global chromatin immunoprecipitation experiments that contain promoter-associated E-boxes (Fernandez et al., 2003) and set II consists of genes (35 probe sets) identified in c-Myc global chromatin immunoprecipitation experiments whose expression positively correlates with c-myc mRNA levels in multiple tissues and cell lines (correlation coefficient >0.69) (Li et al., 2003). As can be seen in Table 1, the mean expression level for the combined set I c-Myc targets was increased ∼2.3-fold in K3P and Sil cells versus Jurkat cells (P < 0.02) whereas the mean expression level for the combined set II c-Myc targets was increased ∼1.7-fold, although the results obtained for the latter comparison did not reach statistical significance for Sil versus Jurkat cells (P < 0.06). (See Supplementary Information file “Filtered data.xls”, E2F and Myc sheets, for a complete listing of E2F and Myc targets.)
Among other genes expressed at significantly higher levels in Sil and K3P cells than in Jurkat cells were targets (17 probe sets) of the cAMP response element binding protein (CREB) (>2.8-fold, P < 0.03; Table 1 and Supplementary Information file “Filtered data.xls”, CREB sheet) (Conkright et al., 2003) and a subset of genes (29 probe sets) involved in mitochondrial function (2-fold, P < 0.001; Table 1 and Supplementary Information file “Filtered data.xls”, Mitochondrial sheet), the latter genes potential downstream targets of the c-Myc/NRF-1 and CREB transcriptional networks (Kelly and Scarpulla, 2004). In addition, significantly elevated expression (17- to 26-fold; P < 0.02 and 0.001, respectively, in K3P and Sil cells) was observed for a subset of genes (14 probe sets) induced by IFN (Table 1 and Supplementary Information file “Filtered data.xls”, IFN sheet) (Krasnoselskaya-Riz et al., 2002).
Lastly, a large subset of genes (43 probe sets) related to Wnt signaling (Varas et al., 2003), including Wnt family members, Wnt receptors and Wnt-induced genes, were found to be expressed at reduced levels compared with the average signal for the entire microarray in all cases. More striking, however, was the observation that their collective expression was down-regulated in the HOX11+ K3P and Sil cell lines compared with HOX11− Jurkat cells (∼3.5-fold lower, P < 0.001; Table 1 and Supplementary Information file “Filtered data.xls”, Wnt sheet).
Gene expression changes induced by ectopic expression of HOX11 in Jurkat cells
To begin to address the question of which features of the molecular portrait of HOX11+ T-ALL could be attributed to HOX11 expression, the HOX11 gene was stably introduced into HOX11− Jurkat cells by retroviral-mediated gene transfer using an amphotropic MSCVhyg-HOX11 retrovirus that also expressed the hygromycin resistance gene as selectable marker (Hawley et al., 1994a; Hawley et al., 1994b). Ectopic expression of exogenous HOX11 in Jurkat/MSCVhyg-HOX11 cells was confirmed by Western blot analysis (see Supplementary Figure 1). Jurkat cells transduced with amphotropic MSCVhyg retrovirus containing only the hygromycin resistance gene served as controls (Hawley et al., 1994b). To avoid clone-specific variations in gene expression patterns, bulk populations of Jurkat/MSCVhyg-HOX11 and Jurkat/MSCVhyg cells were subjected to microarray analysis.
The microarray data was filtered to identify genes whose median values of the signal log2 ratio in triplicate experiments in Jurkat/MSCVhyg-HOX11 cells versus Jurkat/MSCVhyg cells were >1 and all values for the corresponding K3P/Jurkat and Sil/Jurkat signal log2 ratios were >1. A list of 112 genes identified by these criteria was created (see Supplementary Information file “Filtered data.xls”, HOX11 groups sheet) and uploaded into the NetAffx Gene Ontology Data tool (Ashburner et al., 2000) to generate functional groups (see Supplementary Information file “HOX11 groups.jpg”). The most strongly represented functional groups were “Cell cycle”, “DNA replication and chromosome cycle”, “Chromosome organization and biogenesis”, and “Regulation of transcription”. Notably, included within these groups were E2F and c-Myc target genes, genes associated with chromatin remodeling and transcription, and transcripts modulated by the phosphatidylinositol 3-kinase (PI3K)/AKT and IFN signaling pathways, all of which were up-regulated. Selected genes identified by this approach are presented in Figure 1B.
HOX11+ T-ALL molecular phenotype exhibited by murine HOX11+ T-cell tumors
Previously, we reported that enforced retroviral expression of HOX11 in murine bone marrow cells transplanted into syngeneic BALB/c recipients gave rise to T-ALL-like malignancies after long latency (Hawley et al., 1997). To determine whether the murine HOX11+ T-cell tumors shared common molecular features with HOX11+ T-ALL, we carried out microarray hybridizations on the THY137 cell line, which originated from one of the HOX11+ T-cell tumors. THY137 cells resemble CD4+CD8+ double-positive cortical thymocytes that have acquired surface CD3 expression. For comparison, microarray hybridizations were also performed on the HOX11− THY112.2C cell line, which was derived from a T-cell tumor that arose in a BALB/c mouse transplanted with bone marrow cells transduced with a retroviral vector constitutively expressing the v-H-ras gene (Hawley et al., 1995). THY112.2C thymic lymphoma cells express low levels of CD4, CD8 and CD3.
Included among the genes differentially expressed in HOX11+ THY137 cells versus HOX11− THY112.2C cells were E2F- and c-Myc-related genes and CREB targets, all of which were expressed at higher levels in THY137 cells. Selected genes are presented in Figure 2A. A list of all genes (43 probe sets) expressed in human and murine HOX11+ T-ALL at least 2-fold higher than in the corresponding HOX11− control cells was created using RESOURCERER software (see Supplementary Information file “Filtered data.xls”, HOX11 all >2 sheet). The log2 ratios of signal intensities of selected genes are shown in Figure 2B. Highlighted are genes involved in cell proliferation, chromatin remodeling, transcription, signaling pathways and mitochondrial function.
Figure 2.

HOX11+ T-ALL molecular phenotype exhibited by murine HOX11+ T cell tumors. (A) Increased expression of E2F, c-Myc and CREB transcriptional targets (unique identifiers) in murine HOX11+ THY137 cells (HOX11) compared with murine v-Ha-ras-transformed THY112.2C cells (Control). The graph was generated using GeneSpring. The colorbar range is from 0−6. (B) Log2 ratios of signal intensities demonstrating increased expression of selected genes in human and murine HOX11+ cells by comparison with respective HOX11− controls in independent experiments. Human and murine array data were combined based on gene ortholog tables using RESOURCERER software. Gene descriptions: SOCS-1, suppressor of cytokine signaling 1; NDUFB8, NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 8, 19kDa; HTATIP, HIV-1 Tat interacting protein, 60kDa; GTF3A, general transcription factor IIIA; ATP50, ATP synthase H+transporting, mytochondrial F1 complex, O subunit; UQCR, ubiquinol-cytochrome c reductase (6.4kD) subunit; MRPL9, mitochondrial ribosomal protein L9; and GFI1, growth factor independent 1. Log2 ratios of the signal intensities of two probe sets of the NDUFB8 gene are plotted to highlight extent of variability of the microarray data due to differences in hybridization efficiencies. See Supplementary Information for unique identifiers and additional details. Abbreviations: K, K3P cells; S, Sil cells; J, Jurkat cells; H, HOX11+ THY137 thymoma cells; C, control HOX11− THY112.2C thymoma cells.
Validation of microarray data and HOX11 transcriptional mechanisms
As shown in Figure 1B, the direct introduction of HOX11 into Jurkat cells caused activation of E2F targets, c-Myc targets and c-Myc cooperating genes such as CDC6, ORC1L, RAD51, PCNA, MYB, MYCBP(AMY), RAF1 and NMI (Hateboer et al., 1998; Zeller et al., 2003; Ren et al., 2002; Weinmann et al., 2002; Wells et al., 2002). Among the genes differentially expressed at higher levels in HOX11+ THY137 cells were c-myc itself and the E2F target cyclin D3 (CCND3; Figure 2A), deregulated expression of which may predispose to malignant transformation (Chen et al., 2003; Sonoki et al., 2001).
E2F- and Myc-dependent transcription is regulated by the retinoblastoma (Rb) family of proteins (“pocket” proteins), which includes Rb and the related proteins p107 and p130 (Sears and Nevins, 2002; Classon and Harlow, 2002; Beijersbergen et al., 1994; Luo et al., 2004). This suggested that the observed activation of G1/S-specific transcription could be due to HOX11-mediated derepression of cell cycle regulatory genes under the control of pocket proteins. Consistent with this notion, Western blot analysis showed increased levels of phosphorylation of Rb in HOX11-expressing Jurkat/MSCVhyg-HOX11 cells compared with control Jurkat/MSCVhyg cells (Figure 3A). To examine this possibility in more detail, we performed transient transfection experiments in NIH3T3 cells with a luciferase reporter plasmid pRb-TA-Luc containing an Rb response element upstream of a minimal promoter from the herpes simplex virus thymidine kinase (HSV tk) gene. Compared with the empty vector pcDNA3, cotransfection with pcDNA3-HOX11 resulted in a 1.8-fold increase in expression from pRb-TA-Luc (Figure 3B). This result was noteworthy as we had previously observed that HOX11 down-regulated transcription from the intact HSV tk promoter, albeit at higher concentrations of input DNA (Owens et al., 2003). Although the transfection conditions were different in that study, deletion of the third helix of the homeodomain (H3d mutant), which is essential for DNA binding, abrogated HOX11-mediated repression. To determine whether the HOX11-mediated increase in Rb response element-controlled transcription involved DNA binding-dependent or -independent mechanisms, the HOX11 H3d mutant (pcDNA3-HOX11 H3d) was cotransfected into NIH3T3 cells together with pRb-TA-Luc. As can be seen in Figure 3B, the DNA binding function of HOX11 was not essential for the induction. In fact, reporter gene transcription was further increased 1.9-fold compared with wild-type HOX11 (3.4-fold compared with the empty vector control), congruent with our previous data indicating a separable repressive function of the HOX11 homeodomain on RNA polymerase II-mediated transcription (Owens et al., 2003).
Figure 3.
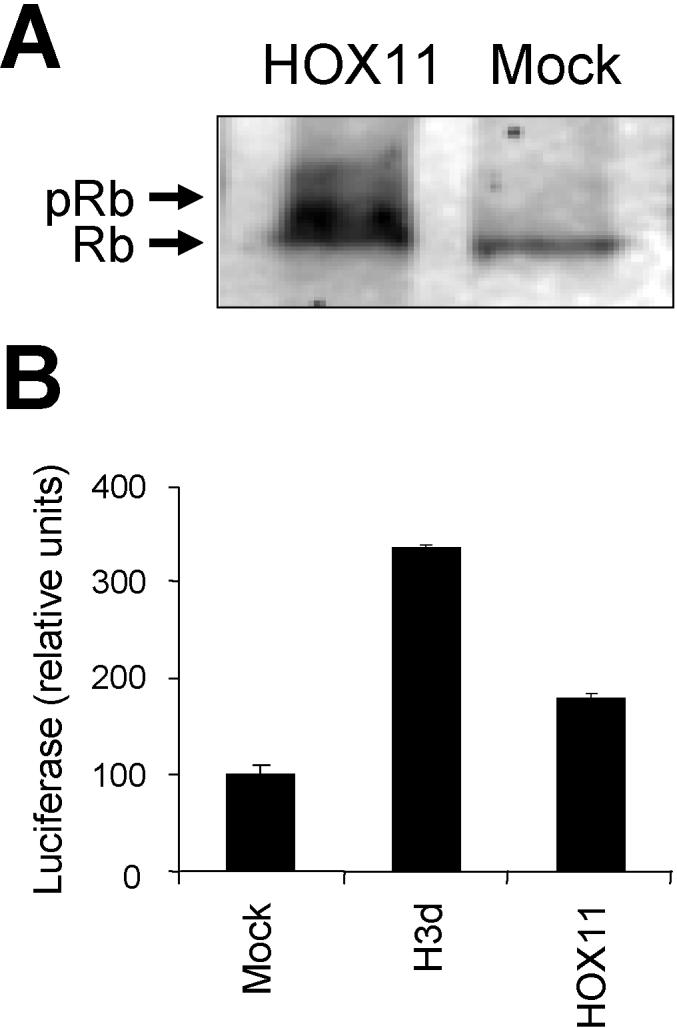
Consequences of ectopic HOX11 expression on Rb phosphorylation state and function. (A) Rb phosphorylation state in HOX11-transduced Jurkat cells (HOX11) versus empty vector-transduced Jurkat cells (Mock). Arrows indicate the migration positions of hyperphosphorylated (upper) or hypophosphorylated (lower) Rb. (B) Rb-mediated repression of gene expression in NIH3T3 cells is partially reversed by coexpression of HOX11. Transient cotransfection and luciferase reporter assays were performed in NIH3T3 cells using pRb-TA-Luc containing an Rb response element plus the empty pcDNA3 plasmid (Mock), pcDNA3-HOX11 expressing wild-type HOX11 (HOX11) or pcDNA3-HOX11 H3d, a DNA-binding deficient mutant of HOX11 that contains a deletion (from amino acid 246 to amino acid 251) of the third helix of the homeodomain (H3d) (Owens et al., 2003). The data represent three independent experiments.
HOX11 was previously reported to physically interact with the catalytic subunit of the protein serine/threonine phosphatase PP2A in T-ALL cells and to disrupt a G2/M cell-cycle checkpoint (Kawabe et al., 1997). In addition to regulating G2/M transition, PP2A has also been suggested to regulate S-phase progression through direct interaction with Rb and p107 (Avni et al., 2003; Voorhoeve et al., 1999; Cicchillitti et al., 2003). We confirmed that HOX11 interacted with PP2A in Jurkat/MSCVhyg-HOX11 cells (Figure 4A). HOX11 was also reported to target the protein serine/threonine phosphatase PP1 in T-ALL cells (Kawabe et al., 1997). By comparison with PP2A, PP1 has been shown to play a role in the G1/S transition by directly dephosphorylating Rb (Ludlow and Nelson, 1995; Berndt et al., 1997; Yan and Mumby, 1999). Therefore, we next examined whether the levels of PP1 and/or PP2A activity were affected by expression of exogenous HOX11 in Jurkat cells. Using specific antibodies against PP1 and PP2A, the respective proteins were immunoprecipitated from Jurkat/MSCVhyg-HOX11 and control Jurkat/MSCVhyg cells and PP1/PP2A phosphatase activities were measured using a commercial serine/threonine phosphatase assay kit. As shown in Figure 4B, HOX11 significantly inhibited the phosphatase activities of both PP1 and PP2A (by ∼40% and ∼69%, respectively), being ∼60% and ∼83% as effective as okadaic acid at suppressing PP1 and PP2A phosphatase activities, respectively. These observations in human T-ALL cells confirm and extend previous findings of an ∼50% relative suppression of total phosphatase activity by HOX11 in comparison with okadaic acid following injection into Xenopus oocytes (Kawabe et al., 1997). Taken together, the combined data indicated that HOX11 indirectly up-regulated expression of E2F and c-Myc transcriptional networks by disrupting PP1/PP2A regulation of Rb (and, by inference, the p107 pocket protein).
Figure 4.

HOX11 inhibits PP1/PP2A phosphatases. (A) HOX11 co-immunoprecipitates with PP2A in HOX11-expressing Jurkat and NIH3T3 cells. Abbreviations: M, Jurkat cells expressing the empty MSCVhyg vector backbone; HOX11, Jurkat cells expressing MSCVhyg-HOX11; WT, wild-type NIH3T3 cells; iHOX11, NIH3T3 cells bearing a doxycycline-inducible HOX11 expression cassette; DOX, doxycycline; IP, antibody used for immunoprecipitation; IB, antibody used for immunoblotting; IgG HC, immunoglobulin heavy chain. (B) Jurkat cells expressing MSCVhyg-HOX11 (HOX11) or the MSCVhyg vector backbone (Mock) were pretreated with (OA) or without 1μM okadaic acid for 1 hr, whole lysates were prepared, immunoprecipitated with anti-PP1 or anti-PP2A antibodies and examined for PP1 or PP2A phosphatase activity, respectively, as described in Materials and methods. The data represent three independent experiments.
Recent studies have demonstrated that c-Myc expression is controlled by PP2A via mechanisms other than those involving p107, including through the AKT (protein kinase B) signaling pathway (Yeh et al., 2004; Ivaska et al., 2002; Dominguez-Caceres et al., 2004). Moreover, the AKT-c-Myc axis represses CDKN1B (KIP1, p27) (Baudino et al., 2003). We noted that CDKNIB/KIP1 expression was reduced in HOX11+ T-ALL cells and down-regulated following introduction of HOX11 into Jurkat cells (Figure 1B). On the other hand, the MCL1 gene, which is a target of an AKT-activated protein complex containing CREB, was up-regulated (Figure 1B) (Wang et al., 1999). These data suggested that HOX11 impacted the AKT signaling pathway via PP2A. AKT is a downstream effector of PI3K that is involved in an variety of processes, including playing a role in controlling the function of E2F family members in T cells (Franke et al., 1997; Brennan et al., 1997). Because Jurkat cells exhibited high basal levels of AKT phosphorylation due to defects in the lipid phosphatases Src homology 2 domain containing inositol polyphosphate phosphatase (SHIP) and phosphatase and tensin homolog deleted on chromosome ten (PTEN) (Shan et al., 2000; Freeburn et al., 2002), we explored possible AKT involvement using NIH3T3 cells in which the exogenous HOX11 gene had been introduced under the control of a doxycycline-regulatable promoter. We first verified that human HOX11 interacted with the mouse PP2A protein in NIH3T3 cells (Figure 4A). As shown in Figure 5, five to fifteen minutes following serum stimulation, NIH3T3 cells expressing HOX11 in the presence of doxycycline exhibited higher levels of AKT phosphorylation than control NIH3T3 cells. By comparison, no differences were observed in the levels of phosphorylation of Erk kinases between HOX11-expressing and HOX11-nonexpressing NIH3T3 cells under these conditions, indicating specificity for the AKT signaling pathway and implicating PP2A in the process.
Figure 5.

Increased AKT phosphorylation in HOX11-expressing cells. NIH3T3 cells or NIH3T3 cells bearing a doxycycline-inducible HOX11 expression cassette (iHOX11) were starved in serum-free medium for 6 hrs and then stimulated with 10% FBS in the presence or absence of 1μg/ml doxycycline (DOX). Whole cell lysates were prepared and examined by Western blotting with anti-phospho-AKT (pAKT) and anti-phosphospho-Erk (pErk1, pErk2) antibodies. The blot was stripped and reprobed with anti-AKT (not shown) and anti-Erk (lower panel) to demonstrate that equal amounts of the respective proteins were loaded. Representative results of three independent experiments are shown.
Given that PP1 and PP2A were reported to be the primary phosphatases involved in dephosphorylating activated CREB (Wadzinski et al., 1993; Alberts et al., 1994; Canettieri et al., 2003), and the implied role of CREB in regulating a number of the genes whose expression patterns were modulated by HOX11 (Table 1), we next performed luciferase reporter assays in NIH3T3 cells using the pCRE-Luc plasmid which contains multiple copies of a CRE binding site. Compared with the empty vector pcDNA3, cotransfection with pcDNA3-HOX11 resulted in a 2-fold increase in expression from pCRE-Luc (Figure 6A). Cotransfection with the HOX11 H3d mutant (pcDNA3-HOX11 H3d) gave essentially identical results (1.8-fold induction), consistent with an indirect (DNA binding-independent) mechanism of CREB activation involving PP1/PP2A.
Figure 6.

HOX11 modulates multiple transcriptional networks. (A) Relative luciferase activity from cotransfection of the reporter plasmids pCRE-Luc containing multiple copies of the CRE-binding sequence (CRE), pISRE-Luc containing five copies of the ISRE-binding sequence (ISRE), and pAP1(PMA)-TA-Luc containing six tandem copies of the sequence TAATGAC resembling a HOX11-binding site [TAAT (Owens et al., 2003)] into NIH3T3 cells with HOX11 expression vectors: pcDNA3-HOX11 expressing wild-type HOX11 (HOX11); pcDNA3-HOX11 H3d, a DNA-binding deficient mutant of HOX11 that contains a deletion (from amino acid 246 to amino acid 251) of the third helix of the homeodomain (H3d); and pcDNA3-HOX11 Lys55Gln, a mutant of HOX11 in which lysine 55 in the third helix of the homeodomain, which forms a salt bridge between the protein and the phosphate groups of DNA and which may be involved in interactions with CBP/p300 coregulators, has been changed to glutamine (K55Q) (Owens et al., 2003; Shen et al., 2001). Controls included the empty pcDNA3 plasmid (Mock) as well as the pTAL-Luc luciferase reporter plasmid containing a minimum promoter (not shown). The data presented are average of at least 3 independent transfections. (B) CPB protein levels are reduced in Jurkat cells expressing MSCVhyg-HOX11 (HOX11) compared with Jurkat cells expressing the empty MSCVhyg vector (Mock). CPB was immunoprecipitated from whole cell extracts with a rabbit anti-CBP polyclonal antibody (anti-CBP NT) followed by immunoblotting with a mouse anti-CBP monoclonal antibody (anti-CBP C1). (C) Repression of transcription mediated by TCF/LEF family members was examined by transiently cotransfecting a plasmid expressing a stabilized β-catenin [ΔN87βcat (Gat et al., 1998)] from the human elongation factor 1 promoter (A. Ramezani and R.G.H., unpublished data) and the TOPflash luciferase reporter plasmid containing TCF/LEF binding sites (TCF) into 293T cells together with the HOX11 expression plasmids described in (A). The FOPflash luciferase reporter plasmid containing mutated TCF/LEF binding sites (mTCF) served as a negative control.
Rb/E2F, c-Myc and CREB all interact with proteins – histone deacetylases and histone acetyltransferases – that regulate chromatin structure (“chromatin remodeling factors”) (Luo et al., 1998; Zhang et al., 2000; Blobel, 2000; Goodman and Smolik, 2000; Frank et al., 2003; Taubert et al., 2004). In particular, HDAC1 and HDAC2 are histone deacetylases that are dephosphorylated by PP1, and inhibition of PP1 phosphatase activity disrupts their interactions with transcriptional corepressors (Galasinski et al., 2002). Included in this HDAC-PP1 complex is CREB, which is dephosphorylated by PP1 at Ser133 (Canettieri et al., 2003). Phosphorylation of Ser133 promotes recruitment of the histone acetyltransferases CREB-binding protein (CBP)/p300 (Blobel, 2000; Goodman and Smolik, 2000), which in turn stimulate acetylation of promoter-bound histones. HDAC inhibitors seem to potentiate CREB activity by prolonging Ser133 phosphorylation in response to a cAMP stimulus (Canettieri et al., 2003). It was notable, therefore, that CBP RNA levels were reduced in HOX11+ K3P and Sil cells compared with Jurkat cells (mean signal intensities, 550 ± 60 for K3P cells, 380 ± 20 for Sil cells, and 1200 ± 200 for Jurkat cells; see below), and also in HOX11+ THY137 cells versus HOX11− THY112.2C cells (Figure 2A). On the other hand, the histone acetyltransferase Htatip (Tip60) was found to be induced in all human and murine HOX11+ T cells (Figure 2B). Tip60 is recruited to c-Myc target genes and postulated to contribute to histone acetylation in response to mitogenic signals (Frank et al., 2003). Recently, E2F1 was shown to bind Tip60 and recruit it to chromatin in late G1 (Taubert et al., 2004); E2F1 had previously been shown to interact with CBP/p300 (Blobel, 2000; Goodman and Smolik, 2000). Another gene involved in the regulation of chromatin structure that was more highly expressed in HOX11+ T-ALL cells and up-regulated in Jurkat/MSCVhyg-HOX11 was the SET gene (Figure 1B), which encodes one of two essential subunits of the inhibitor of acetyltransferases complex (INHAT) that functions as a histone chaperone (Schneider et al., 2004; Kutney et al., 2004). SET was originally discovered as a leukemia-associated protein encoded by the SET-CAN translocation (von Lindern et al., 1992), and subsequently demonstrated to be an inhibitor of PP2A (Li et al., 1996).
We also detected a group of IFN-inducible genes that were expressed at higher levels in K3P and Sil cells than in Jurkat cells, some of which were confirmed to be up-regulated following introduction of HOX11 into Jurkat cells (Figure 1B). To analyze the ability of HOX11 to activate luciferase reporter gene expression via the IFN-stimulated response element ISRE, we cotransfected pcDNA3-HOX11 into NIH3T3 cells together with the pISRE-Luc plasmid. Ectopic HOX11 expression increased ISRE-driven reporter gene expression by 1.5-fold and required an intact homeodomain (Figure 6A), ruling out a mechanism involving PP1/PP2A. In parallel cotransfection experiments with the pAP1(PMA)-TA-Luc plasmid containing the HOX11 target sequence TAAT (Dear et al., 1993), no stimulation of luciferase production was detected (Figure 6A). Because the sequence of the ISRE and the promoter region of pISRE-Luc does not contain this or any other known HOX11 binding motif (Owens et al., 2003; Allen et al., 2000; Dear et al., 1993), stimulation of transcription by HOX11 through the ISRE may be mediated in a manner analogous to Aldh1 (Greene et al., 1998), either via protein-protein interactions or indirectly by an as yet unidentified intermediate HOX11 target gene. Interestingly, cotransfection of pcDNA3-HOX11 Lys55Gln (Owens et al., 2003), a mutant of HOX11 in which lysine 55 in the third helix of the homeodomain was changed to glutamine, stimulated ISRE-driven reporter gene expression at levels 2.3-fold higher than wild-type HOX11 (3.5-fold greater than empty vector) (Figure 6A). The mutated residue coincides with a conserved lysine that has been shown in other homeodomain proteins to be part of the binding surface involved in interactions with CBP/p300 which prevent their binding to DNA (Shen et al., 2001). These findings implicated a role for CBP/p300 in HOX11-mediated modulation of ISRE-dependent transcriptional induction (Weaver et al., 1998). In this context, it was previously shown that homeodomain protein interactions with CBP/p300 were mutually repressive, inhibiting CBP/p300 histone acetyltransferase activity (Shen et al., 2001). As noted above, CBP mRNA levels were reduced in both HOX11+ human and murine cells, and we confirmed that Jurkat/MSCVhyg-HOX11 cells synthesized lower levels of CBP protein than control Jurkat/MSCVhyg cells (Figure 6B). Thus, HOX11 appears to inhibit CBP/p300 at the transcriptional as well as posttranslational levels.
The diminished expression of certain Wnt-induced and Wnt-related genes in HOX11+ T-ALL and Jurkat/MSCVhyg-HOX11 cells that was observed was in accordance with the documented ability of HOX11 to also act as a negative regulator of gene expression (Owens et al., 2003; Greene et al., 1998). To further investigate this observation, we next performed luciferase reporter assays in 293T cells using the TOPflash luciferase reporter plasmid. Transcription of the luciferase gene in the TOPflash plasmid is under the control of a transcription factor complex consisting of members of the T cell factor (TCF)/lymphoid enhancer factor (LEF) family of DNA-binding molecules associated with stabilized β-catenin (Verbeek et al., 1995; Ioannidis et al., 2001; Gounari et al., 2001; Varas et al., 2003). The TOPflash plasmid was cotransfected together with an expression plasmid encoding a constitutively stabilized form of β-catenin [ΔN87βcat (Gat et al., 1998)] and either pcDNA3-HOX11, pcDNA3-HOX11 H3d, pcDNA3-HOX11 Lys55Gln, or the empty pcDNA3 vector. Parallel experiments were carried out with a negative control luciferase reporter plasmid, FOPflash, which contains mutated TCF/LEF binding sites. We detected a potent repressive effect (86% inhibition) of wild-type HOX11 on β-catenin-TCF/LEF-driven TOPflash expression compared with that observed in the presence of pcDNA3 (Figure 6B). Background levels of expression from the FOPflash plasmid with mutated TCF/LEF binding sites increased substantially (5.1-fold) in the context of the HOX11 H3d mutant due to a generalized derepression of RNA polymerase II-mediated transcription (Owens et al., 2003). Nonetheless, significant repression of β-catenin-TCF/LEF-directed transcription was still observed upon deletion of helix 3 of the HOX11 homeodomain (48% of empty vector). Collectively, these results supported a DNA binding-independent mechanism for HOX11-mediated repression of Wnt target genes. It is known that TCF/LEFs are negatively regulated by phosphorylation on serine/threonine residues (Ishitani et al., 2003); considering that a role for PP2A in Wnt signaling downstream of stabilized β-catenin has been reported (Ratcliffe et al., 2000), it seems reasonable that TCF/LEF function may be positively regulated by PP2A(PP1).
Discussion
Genome-wide expression profiling provides a powerful tool for studying how complex signaling cascades are linked to downstream transcriptional programs (Riz et al., 2003). Toward a better understanding of how aberrant HOX11 expression during normal thymocyte differentiation leads to T-ALL, we conducted microarray profiling experiments involving HOX11+ T-ALL cell lines and T-ALL experimental models engineered to express exogenous HOX11. Extracting biologically meaningful information from the large amount of expression data generated by microarray profiling can be challenging and is fraught with potential artifacts (Ebert and Golub, 2004; Tan et al., 2003). Therefore, rather than initially focusing on differences in the expression levels of individual genes in HOX11+ and HOX11− cells, a key feature of our experimental design was to employ a hypothesis-driven approach to first construct sets of functionally-related genes whose collective expression levels significantly differed between sample groups. We then confirmed that HOX11 regulated the implicated pathways and processes by independent biochemical and functional analyses. These investigations revealed that HOX11 modulated multiple G1/S transcriptional networks (Sears and Nevins, 2002). On the basis of results obtained with a large series of mutant HOX11 proteins, we previously speculated that a significant component of the leukemogenic properties of HOX11 are most likely due to direct interactions between the homeodomain and the regulatory regions of specific target genes (Owens et al., 2003). The novel finding of the current study is that a major complementary mode of oncogenic transcriptional deregulation by HOX11 is via homeodomain-independent mechanisms. Moreover, the data presented herein identified the protein serine/threonine phosphatases PP1 and PP2A as being central effectors of this facet of HOX11 transcriptional activity.
We noticed that target genes of the E2F and c-Myc transcription factors were prominent among the HOX11 signature genes previously reported by Ferrando and colleagues (Ferrando et al., 2002) which were expressed at higher levels in HOX11+ K3P and Sil cells. These observations raised the possibility of a generalized deregulation of the Rb/E2F and p107/c-Myc pathways in HOX11+ T-ALL (Sears and Nevins, 2002; Classon and Harlow, 2002; Beijersbergen et al., 1994; Luo et al., 2004). Characterization of the collective behavior of test sets of E2F and c-Myc targets identified in chromatin immunoprecipitation studies (Ren et al., 2002; Weinmann et al., 2002; Wells et al., 2002; Fernandez et al., 2003; Fry et al., 1999; Ma et al., 2003; Zeller et al., 2003) corroborated this notion: certain E2F and c-Myc targets had increased expression in K3P and Sil cells versus Jurkat cells, were up-regulated following introduction of HOX11 into Jurkat cells, and displayed elevated transcript levels in a murine HOX11+ T-cell tumor compared with a HOX11− T-cell tumor. Additionally, we showed that stable expression of exogenous HOX11 in Jurkat cells resulted in hyperphosphorylation of Rb, while ectopic HOX11 expression in NIH3T3 cells alleviated repression mediated through an Rb-response element. Data from others has indicated a central role for PP1 in the dephosphorylation of Rb during G1 progression (Ludlow and Nelson, 1995; Berndt et al., 1997; Yan and Mumby, 1999). We demonstrated that HOX11 was ∼60% as effective as okadaic acid at inhibiting PP1 phosphatase activity in stably transduced Jurkat/MSCVhyg-HOX11 cells. In view of an earlier report that HOX11 physically associates with PP1 in Jurkat cells (Kawabe et al., 1997), these findings imply one aspect of HOX11-mediated growth deregulation that involves inhibition of PP1-dependent dephosphorylation of Rb. We further showed that HOX11 was ∼83% as effective as okadaic acid at inhibiting PP2A phosphatase activity in stably transduced Jurkat/MSCVhyg-HOX11 cells. PP2A has been implicated in the regulation of S-phase progression through direct interaction with Rb and p107 (Avni et al., 2003; Voorhoeve et al., 1999; Cicchillitti et al., 2003), and to control c-Myc and E2F function via other mechanisms including that involving the AKT signaling pathway (Yeh et al., 2004; Ivaska et al., 2002; Dominguez-Caceres et al., 2004; Brennan et al., 1997). Considered together with our observation of prolonged AKT phosphorylation following serum stimulation of HOX11-expressing NIH3T3 cells, these results point to an expanded role of this phosphatase in T-cell oncogenesis beyond disruption of the G2/M cell-cycle checkpoint (Kawabe et al., 1997). Along these lines, CREB is another transcription factor whose activity has been reported to be negatively modulated by PP1 and PP2A (Wadzinski et al., 1993; Alberts et al., 1994; Canettieri et al., 2003). We found that a number of CREB target genes were more highly expressed in HOX11+ human and murine T-cell tumors. The demonstration that HOX11 transactivated transcription of a reporter gene linked to a CRE-binding sequence in a homeodomain-independent manner is also consistent with a PP1/PP2-coupled mechanism. On the other hand, we observed down-regulation of a set of Wnt-regulated target genes. In this instance as well, a DNA-binding-independent mechanism of HOX11 regulation potentially involving PP1/PP2A (Ishitani et al., 2003; Ratcliffe et al., 2000) was suggested from the reporter assays with stabilized β-catenin.
These latter findings suggest additional ways in which HOX11 mediates its transforming function besides promoting G1/S (this report) and G2/M (Kawabe et al., 1997) transition. Signaling through the pre-T cell receptor (pre-TCR), which controls the differentiation of immature CD4−CD8−CD25+CD44− double-negative thymocytes into CD4+CD8+ double-positive cells, stimulates CRE-dependent transcription (Grady et al., 2004). Wnt family members are expressed during various stages of T-cell development (Ioannidis et al., 2001; Gounari et al., 2001; Ishitani et al., 2003; Verbeek et al., 1995; Varas et al., 2003); the complex of β-catenin with TCF or LEF activates transcription of T-cell specific genes such as CD3ε, CD4, TCRα, TCRβ and TCRδ (Varas et al., 2003), and is required for progression to the CD4+CD8+ double-positive T cell stage (Verbeek et al., 1995). However, Wnt signaling must be transiently switched off to ensure the rearrangement and expression of the TCRβ chain before CD4 and CD8 expression (Ioannidis et al., 2001; Gounari et al., 2001). We posit that concomitant activation of CREB-mediated signaling and down-regulation of Wnt signaling would be the outcome of inappropriate activation of HOX11 during transit of the CD25+ double-negative thymocyte stage by juxtaposition to TCRδ and TCRβ regulatory elements as a result of the t(10;14)(q24;q11) and t(7;10)(q35;q24) chromosomal translocations. Moreover, there is an increasing body of evidence showing a role of histone modification in T cell differentiation, especially during the CD4+CD8+ double-positive to CD4+/CD8+ single-positive transition (Mathieu et al., 2000; Agata et al., 2001). By down-regulating CBP protein levels, HOX11 could prevent de novo histone acetylation, contributing to the CD4+CD8+ block. In accord with this supposition, the Lys55Gln mutant of HOX11 does not immortalize mouse bone marrow precursors (Owens et al., 2003) nor does it arrest development of hematopoietic progenitors into mature T cells in fetal thymic organ cultures (Owens et al., 2004) (B. Owens and R.G.H., unpublished data). In other homeodomain proteins, the residue mutated was shown to be involved in direct inhibitory protein-protein interactions with CBP (Shen et al., 2001). Loss of CBP expression is associated with lymphomagenesis in mice (Kang-Decker et al., 2004). Moreover, hyperphosphorylation of CBP reduces interaction with transcription factors such as NF-κB, a process that can be mimicked by blocking PP2A activity (Yalcin et al., 2003). In addition, there is a critical developmentally-regulated interplay between pre-TCR signaling through NF-κB and Notch signaling during the transition of CD4−CD8− double-negative thymocytes to the CD4+CD8+ double-positive stage of T cell development (Ciofani et al., 2004). The very recent finding that >50% of human T-ALL cases harbor activating mutations in the NOTCH1 gene, including HOX11+ Sil T-ALL cells, provides direct proof of the importance of synergistic aberrations involving these signaling pathways in T-ALL pathogenesis (Weng et al., 2004).
Comparison between human and murine T-ALL models revealed other genes that are up-regulated by HOX11 that were not studied further but which would be presumed to contribute to leukemic transformation of T cell precursors (Figure 2B). Among these are Socs-1, which functions at the transition from the CD25+CD44− double-negative thymocyte stage to the CD4+CD8+ double-positive stage stage (Fujimoto et al., 2000; Trop et al., 2001), and the oncogenic T-cell transcription factor Gfi-1 (Schmidt et al., 1998a), which is similarly required at this stage (Schmidt et al., 1998b) and which has been reported to predispose to T cell tumorigenesis by promoting S phase entry concomitant with hyperphosphorylation of Rb (Karsunky et al., 2002).
Finally, although not a focus of this paper, it is worth mentioning that the centrosomal kinase, Aurora-A/STK15, which has been implicated in genome instability and malignant transformation, is modulated through direct interaction with PP1 (Du and Hannon, 2002). Considered together with previous data (Kawabe et al., 1997; Shen et al., 2001), perhaps what is most notable about the current findings is that the oncogenic action of HOX11 is remarkably analogous to that of transforming viral proteins. For example, SV40 large T antigen and adenovirus E1A oncoprotein form complexes with Rb and CBP/p300 (Eckner et al., 1996; Ait-Si-Ali et al., 1998), whereas SV40 small t antigen and polyoma virus small and middle T antigens form complexes with PP2A (Pallas et al., 1990; Sontag et al., 1997). In this regard, a critical role for PP2A in human cell transformation has recently come to light (Chen et al., 2004). We note that leukemic fusion proteins encoded by translocations involving the Drosophila trithorax-related chromatin-modifying proto-oncogene, MLL, form complexes with SET and PP2A (Li et al., 1996; Adler et al., 1997). In Drosophila, the thrithorax protein physically interacts with the catalytic subunit of PP1 (Rudenko et al., 2003), suggesting that mammalian PP1 may also be a physiologically relevant partner of MLL. These observations argue that the leukemogenic activities of MLL fusion proteins share overlapping features with that of HOX11.
The above results notwithstanding, it is necessary to underscore the fact that HOX11 regulation of ISRE-controlled transcription was dependent on an intact homeodomain as is the expression of the HOX11 target gene Aldh1 (Greene et al., 1998; Owens et al., 2003). Using hematopoietic precursor cell immortalization as a surrogate transformation assay (Hawley et al., 1994a; Hawley et al., 1997), our laboratory previously reported that deletion of the third helix of the homeodomain (HOX11 H3d mutant) or the introduction of point mutations within helix 3 (e.g., HOX11Asn51Ala mutant) abrogated HOX11 immortalizing function (Owens et al., 2003). Thus, we appreciate that cooperating HOX11 homeodomain-dependent transcriptional mechanisms still remain to be elaborated and pivotal direct targets responsible for full oncogenic activity of this master regulator remain to be identified.
Materials and methods
Cell lines and gene transfer experiments
The human T-ALL cell lines K3P (Dube et al., 1991), Sil (Hatano et al., 1991), and Jurkat (TIB-152; American Type Culture Collection, Manassas, VA) were cultured in Iscove's Modified Dulbecco's medium (IMDM; Invitrogen Corp., Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen Corp.). The HOX11 gene was introduced into Jurkat cells by retroviral-mediated gene transfer using amphotropic vector stocks of the MSCVhyg-HOX11 retroviral vector containing the hygromycin resistance gene (Hawley et al., 1994a; Hawley et al., 1994b). Control cells were Jurkat cells transduced with amphotropic vector stocks of the empty MSCVhyg retroviral vector backbone (Hawley et al., 1994b). After drug selection (200 μg/ml hygromcyin B; Invitrogen Corp.), HOX11 protein levels were determined by Western blotting using an anti-HOX11 antibody (described below).
The murine THY137 cell line was derived from a T-cell tumor that arose in a BALB/c mouse transplanted with MSCV-HOX11-transduced bone marrow cells (Hawley et al., 1997). The murine THY112.2C cell line, used as a control, was derived from a T-cell tumor that arose in a BALB/c mouse transplanted with bone marrow cells transduced with the MSCV-v-H-ras retroviral vector constitutively expressing the v-H-ras gene (Hawley et al., 1995). Both cell lines were maintained in IMDM supplemented with 10% heat-inactivated FBS.
Murine NIH3T3 cells (CRL-1658, ATCC) and human embryonic kidney 293T cells (obtained from M. Eiden, National Institute of Mental Health, National Institutes of Health, Bethesda, MD) were cultured in Dulbecco's modified Eagle medium with high glucose (Invitrogen Corp.) plus 10% FBS. NIH3T3 cells stably expressing the HOX11 gene were derived by retroviral transduction with MSCV-HOX11 as described previously (Allen et al., 2000). NIH3T3/iHOX11 cells with doxycyline-inducible HOX11 expression were generated by cotransduction with a retroviral vector in which the HOX11 gene was placed under the control of the doxycycline-regulatable promoter P(tetO)7-CMVmin and a retroviral vector driving the expression of an improved version of a reverse tetracycline-controlled transactivator rtTAS-M2 (Urlinger et al., 2000) (R.G.H., J. Molete, and T.S. Hawley, unpublished data). HOX11 expression constructs based on the pcDNA3 backbone were described previously (Owens et al., 2003) and used in transient transfection experiments with luciferase reporter constructs containing cis-acting regulatory elements. The luciferase reporter plasmids included pRb-TA-Luc containing an Rb-response element to measure transcriptional regulation by Rb, pCRE-Luc containing multiple copies of the cAMP response element (CRE) to monitor the activation of cAMP response element binding protein (CREB) and cAMP-mediated signal transduction pathways, and pISRE-Luc containing five copies of the interferon (IFN)-stimulated response element (ISRE) (BD Mercury Pathway Profiling Luciferase Systems; BD Biosciences Clontech, Palo Alto, CA). Modulation of T cell factor (TCF)/lymphoid enhancer factor (LEF)-mediated transcription was investigated by cotransfection of a plasmid expressing a stabilized β-catenin [ΔN87βcat (Gat et al., 1998)] from the human elongation factor 1 promoter (Ramezani et al., 2003) (A. Ramezani and R.G.H., unpublished data) and the TOPflash luciferase reporter plasmid (Upstate USA, Inc., Chicago, IL) containing TCF/LEF binding sites, with the FOPflash luciferase reporter plasmid (Upstate USA, Inc.) containing mutated TCF/LEF binding sites used as a negative control. NIH3T3 or 293T cells were seeded in 6 well dishes 16 hrs prior to transfection. Duplicate wells for each construct were set up in order to measure luciferase activity and HOX11 protein expression levels. Transient transfection was performed using a lipid-mediated gene transfer method and the FuGENE 6 transfection reagent (Roche Diagnostics Corp. Indianapolis, IN). Cells were transfected with 0.75 μg pcDNA3-HOX11 construct and 0.5 μg luciferase reporter plasmid DNA. Thirty six hours posttransfection, cells were harvested and lysed in Reporter Lysis Buffer (Promega Corp., Madison, WI). Luciferase activity was determined using a commercial assay kit (Bright-Glo Luciferase Assay System; Promega Corp.) according to the manufacturer's instructions. A plasmid (0.05 μg) containing the lacZ gene driven by the simian virus 40 (SV40) promoter was used as an internal control to monitor efficiency of transfection. The levels of β-galactosidase activity were measured using the FluoReporter lacZ/Galactosidase Quantitation Kit (Molecular Probes, Inc., Eugene, OR) and varied less than 10% between samples.
Microarray gene expression analysis
Total RNA was isolated by TRIzol Reagent (Invitrogen Corp.) and further purified using RNeasy mini spin columns (QIAGEN Inc., Valencia, CA). Complementary DNA synthesis was performed with an HPLC purified T7-(dT)24 primer (Genset Oligos/Proligo LLC, Boulder, CO). Biotin-labeled complementary RNA (cRNA) synthesis, hybridization, washing, staining and scanning were performed according to protocols provided by Affymetrix Inc. (Santa Clara, CA) as previously described (Krasnoselskaya-Riz et al., 2002). Target cRNA from three independent cultures of the human T-ALL cell lines was hybridized to three Affymetrix HG-U133A GeneChip oligonucleotide arrays (three biological replicates); target cRNA from two independent cultures of the murine T-cell tumor lines was hybridized to two Affymetrix MG-U74Av2 GeneChip oligonucleotide arrays (two biological replicates). The detailed procedures are available on the Affymetrix website (http://www.affymetrix.com/support/technical/manuals.affx).
Quality control and low-level analysis of expression data were performed using Microarray Suite Version 5.0 software (MAS 5.0; Affymetrix). Subsequent data analysis was performed in Microsoft Excel or with GeneSpring software (Silicon Genetics, Redwood City, CA). Gene annotation queries were conducted through the Affymetrix NetAffx Analysis Center (https://www.affymetrix.com/analysis/query/go_analysis.affx) and gene sets categorized based on Gene Ontology (GO) (Ashburner et al., 2000) information from the Locus Link website of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/LocusLink/) and information obtained from the Myc Target Gene Database (Zeller et al., 2003) (http://www.myc-cancer-gene.org/index.asp) maintained by Johns Hopkins University School of Medicine (Baltimore, MD). RESOURCERER software (The Institute for Genomic Research, Rockville, MD) was used to compare human and murine data (http://pga.tigr.org/tigr-scripts/magic/r1.pl).
Immunoprecipitation, Western blotting and PP1/PP2A phosphatase assay
Immunoprecipitations and Western blotting were carried out as described (Owens et al., 2003; Yu et al., 2003). In brief, cells were lysed on ice in RIPA buffer (50 mM Tris-HCl pH 7.4, 1% NP40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM NaV3O4, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin) for 30 min. Insoluble materials were removed by centrifugation at 14,000 rpm at 4 °C for 10 min. Whole cell lysates (500 μg) were incubated at 4 °C with 1 μg of antibody overnight and then with protein A or protein G beads for an additional 2−3 hrs. Immunoprecipitates were washed three times with HNTG buffer (20 mM Hepes pH 7.5, 150 mM NaCl, 1% glycerol, 0.1% triton X-100 and 1 mM Na3VO4) and then resolved by 10% SDS-PAGE for immunoblotting. Antibodies used included: anti-HOX11 (C-18), anti-PP1 (E-9), anti-ERK1 (K-23), anti-phospho-ERK1 (E-4) and anti-CBP (C-1) (Santa Cruz Biotechnology, Santa Cruz, CA); anti-CBP NT (Upstate USA, Inc.); anti-PP2A-Catalytic α and anti-Rb (BD Biosciences Pharmingen, San Diego, CA); and anti-AKT and anti-phospho-AKT (Thr308) (Cell Signaling Technology Inc. Beverly, MA).
PP1/PP2A phosphatase activities in the immunoprecipitates were measured using a colorimetric serine/threonine phosphatase assay kit (Upstate USA, Inc.). Jurkat cells were pretreated with or without okadaic acid (1 μM) for 1 hr and then lysed on ice in cell lysis buffer containing 50 mM Hepes pH 7.5, 150 mM NaCl, 1 mM EGTA, 10% glycerol, 1.5 mM MgCl2, 1% NP40, 2 μg/ml leupeptin, 2 μg/ml aprotinin and 1 mM PMSF for 30 min. Cell lysates (500 μg) were incubated with 2 μg of anti-PP1(E-9) or anti-PP2A for 1 hr and then with protein A or protein G beads for an additional 2 hrs. Immunoprecipitates were washed twice in TBS buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl) and once in phosphatase assay buffer (50 mM Hepes pH 7.5, 100 mM NaCl, 0.1 mg/ml BSA, 0.1 mM EDTA, 0.1% β-mercaptoethanol). PP1/PP2A phosphatase activities in the immunoprecipitates were measured in 50 μl phosphatase assay buffer containing 200 μM phosphopeptide (KRpTIRR) at 30°C for 30 min (Ambach et al., 2000). The reactions were terminated by the addition of malachite green solution and were examined at 620 nm after 15 min. The amount of phosphate released was calculated based on a standard curve.
Acknowledgements
We gratefully acknowledge Vladimir Kuznetsov for statistical analysis support and Timothy McCaffrey for advice on GeneSpring software. We also thank Wen-mei Yu for technical assistance during part of this work and help with data presentation, Joseph Molete and Teresa Hawley for providing NIH3T3/iHOX11 cells, and Ali Ramezani for the gift of the β-catenin lentiviral vector. This work was supported in part by National Institutes of Health grants R01HL65519, R01HL66305 and R24RR16209.
Footnotes
Supplementary Information is available at Oncogene's website: http://www.nature.com/onc/journal/v24/n36/suppinfo/1208727s1.html?url=/onc/journal/v24/n36/full/1208727a.html
Supplementary Material
{kind=link}
References
- Adler HT, Nallaseth FS, Walter G, Tkachuk DC. J Biol Chem. 1997;272:28407–28414. doi: 10.1074/jbc.272.45.28407. [DOI] [PubMed] [Google Scholar]
- Agata Y, Katakai T, Ye SK, Sugai M, Gonda H, Honjo T, Ikuta K, Shimizu A. J Exp Med. 2001;193:873–880. doi: 10.1084/jem.193.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Si-Ali S, Ramirez S, Barre FX, Dkhissi F, Magnaghi-Jaulin L, Girault JA, Robin P, Knibiehler M, Pritchard LL, Ducommun B, Trouche D, Harel-Bellan A. Nature. 1998;396:184–186. doi: 10.1038/24190. [DOI] [PubMed] [Google Scholar]
- Alberts AS, Montminy M, Shenolikar S, Feramisco JR. Mol Cell Biol. 1994;14:4398–4407. doi: 10.1128/mcb.14.7.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TD, Zhu Y-X, Hawley TS, Hawley RG. Leuk Lymphoma. 2000;39:241–256. doi: 10.3109/10428190009065824. [DOI] [PubMed] [Google Scholar]
- Ambach A, Saunus J, Konstandin M, Wesselborg S, Meuer SC, Samstag Y. Eur J Immunol. 2000;30:3422–3431. doi: 10.1002/1521-4141(2000012)30:12<3422::AID-IMMU3422>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avni D, Yang H, Martelli F, Hofmann F, ElShamy WM, Ganesan S, Scully R, Livingston DM. Mol Cell. 2003;12:735–746. doi: 10.1016/s1097-2765(03)00355-1. [DOI] [PubMed] [Google Scholar]
- Baudino TA, Maclean KH, Brennan J, Parganas E, Yang C, Aslanian A, Lees JA, Sherr CJ, Roussel MF, Cleveland JL. Mol Cell. 2003;11:905–914. doi: 10.1016/s1097-2765(03)00102-3. [DOI] [PubMed] [Google Scholar]
- Beijersbergen RL, Hijmans EM, Zhu L, Bernards R. EMBO J. 1994;13:4080–4086. doi: 10.1002/j.1460-2075.1994.tb06725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt N, Dohadwala M, Liu CW. Curr Biol. 1997;7:375–386. doi: 10.1016/s0960-9822(06)00185-0. [DOI] [PubMed] [Google Scholar]
- Blobel GA. Blood. 2000;95:745–755. [PubMed] [Google Scholar]
- Brennan P, Babbage JW, Burgering BM, Groner B, Reif K, Cantrell DA. Immunity. 1997;7:679–689. doi: 10.1016/s1074-7613(00)80388-x. [DOI] [PubMed] [Google Scholar]
- Canettieri G, Morantte I, Guzman E, Asahara H, Herzig S, Anderson SD, Yates JR, III, Montminy M. Nat Struct Biol. 2003;10:175–181. doi: 10.1038/nsb895. [DOI] [PubMed] [Google Scholar]
- Chen Q, Lin J, Jinno S, Okayama H. Oncogene. 2003;22:992–1001. doi: 10.1038/sj.onc.1206193. [DOI] [PubMed] [Google Scholar]
- Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Cancer Cell. 2004;5:127–136. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- Cicchillitti L, Fasanaro P, Biglioli P, Capogrossi MC, Martelli F. J Biol Chem. 2003;278:19509–19517. doi: 10.1074/jbc.M300511200. [DOI] [PubMed] [Google Scholar]
- Ciofani M, Schmitt TM, Ciofani A, Michie AM, Cuburu N, Aublin A, Maryanski JL, Zuniga-Pflucker JC. J Immunol. 2004;172:5230–5239. doi: 10.4049/jimmunol.172.9.5230. [DOI] [PubMed] [Google Scholar]
- Classon M, Harlow E. Nat Rev Cancer. 2002;2:910–917. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- Cohen PT. J Cell Sci. 2002;115:241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Guzman E, Flechner L, Su AI, Hogenesch JB, Montminy M. Mol Cell. 2003;11:1101–1108. doi: 10.1016/s1097-2765(03)00134-5. [DOI] [PubMed] [Google Scholar]
- Dear TN, Sanchez-Garcia I, Rabbitts TH. Proc Natl Acad Sci USA. 1993;90:4431–4435. doi: 10.1073/pnas.90.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Caceres MA, Garcia-Martinez JM, Calcabrini A, Gonzalez L, Porque PG, Leon J, Martin-Perez J. Oncogene. 2004;23:7378–7390. doi: 10.1038/sj.onc.1208002. [DOI] [PubMed] [Google Scholar]
- Du J, Hannon GJ. Nucleic Acids Res. 2002;30:5465–5475. doi: 10.1093/nar/gkf678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube ID, Kamel-Reid S, Yuan CC, Lu M, Wu X, Corpus G, Raimondi SC, Crist WM, Carroll AJ, Minowada J, Baker JB. Blood. 1991;78:2996–3003. [PubMed] [Google Scholar]
- Ebert BL, Golub TR. Blood. 2004;104:923–932. doi: 10.1182/blood-2004-01-0274. [DOI] [PubMed] [Google Scholar]
- Eckner R, Ludlow JW, Lill NL, Oldread E, Arany Z, Modjtahedi N, DeCaprio JA, Livingston DM, Morgan JA. Mol Cell Biol. 1996;16:3454–3464. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, Lander ES, Golub TR, Look AT. Cancer Cell. 2002;1:75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B. EMBO Rep. 2003;4:575–580. doi: 10.1038/sj.embor.embor861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Freeburn RW, Wright KL, Burgess SJ, Astoul E, Cantrell DA, Ward SG. J Immunol. 2002;169:5441–5450. doi: 10.4049/jimmunol.169.10.5441. [DOI] [PubMed] [Google Scholar]
- Fry CJ, Pearson A, Malinowski E, Bartley SM, Greenblatt J, Farnham PJ. J Biol Chem. 1999;274:15883–15891. doi: 10.1074/jbc.274.22.15883. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Naka T, Nakagawa R, Kawazoe Y, Morita Y, Tateishi A, Okumura K, Narazaki M, Kishimoto T. J Immunol. 2000;165:1799–1806. doi: 10.4049/jimmunol.165.4.1799. [DOI] [PubMed] [Google Scholar]
- Galasinski SC, Resing KA, Goodrich JA, Ahn NG. J Biol Chem. 2002;277:19618–19626. doi: 10.1074/jbc.M201174200. [DOI] [PubMed] [Google Scholar]
- Gat U, DasGupta R, Degenstein L, Fuchs E. Cell. 1998;95:605–614. doi: 10.1016/s0092-8674(00)81631-1. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Gounari F, Aifantis I, Khazaie K, Hoeflinger S, Harada N, Taketo MM, von BH. Nat Immunol. 2001;2:863–869. doi: 10.1038/ni0901-863. [DOI] [PubMed] [Google Scholar]
- Grady GC, Mason SM, Stephen J, Zuniga-Pflucker JC, Michie AM. J Immunol. 2004;173:1802–1810. doi: 10.4049/jimmunol.173.3.1802. [DOI] [PubMed] [Google Scholar]
- Greene WK, Bahn S, Masson N, Rabbitts TH. Mol Cell Biol. 1998;18:7030–7037. doi: 10.1128/mcb.18.12.7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano M, Roberts CW, Minden M, Crist WM, Korsmeyer SJ. Science. 1991;253:79–82. doi: 10.1126/science.1676542. [DOI] [PubMed] [Google Scholar]
- Hateboer G, Wobst A, Petersen BO, Le CL, Vigo E, Sardet C, Helin K. Mol Cell Biol. 1998;18:6679–6697. doi: 10.1128/mcb.18.11.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley RG, Fong AZC, Lu M, Hawley TS. Oncogene. 1994a;9:1–12. [PubMed] [Google Scholar]
- Hawley RG, Fong AZC, Ngan B-Y, Hawley TS. Oncogene. 1995;11:1113–1123. [PubMed] [Google Scholar]
- Hawley RG, Fong AZC, Reis MD, Zhang N, Lu M, Hawley TS. Cancer Res. 1997;57:337–345. [PubMed] [Google Scholar]
- Hawley RG, Lieu FHL, Fong AZC, Hawley TS. Gene Ther. 1994b;1:136–138. [PubMed] [Google Scholar]
- Ioannidis V, Beermann F, Clevers H, Held W. Nat Immunol. 2001;2:691–697. doi: 10.1038/90623. [DOI] [PubMed] [Google Scholar]
- Ishitani T, Ninomiya-Tsuji J, Matsumoto K. Mol Cell Biol. 2003;23:1379–1389. doi: 10.1128/MCB.23.4.1379-1389.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kahari VM, Heino J. Mol Cell Biol. 2002;22:1352–1359. doi: 10.1128/mcb.22.5.1352-1359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Goris J. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA, Taylor WR, Brindle PK, van Deursen JM. Cancer Cell. 2004;5:177–189. doi: 10.1016/s1535-6108(04)00022-4. [DOI] [PubMed] [Google Scholar]
- Karsunky H, Mende I, Schmidt T, Moroy T. Oncogene. 2002;21:1571–1579. doi: 10.1038/sj.onc.1205216. [DOI] [PubMed] [Google Scholar]
- Kawabe T, Muslin AJ, Korsmeyer SJ. Nature. 1997;385:454–458. doi: 10.1038/385454a0. [DOI] [PubMed] [Google Scholar]
- Keller G, Wall C, Fong AZC, Hawley TS, Hawley RG. Blood. 1998;92:877–887. [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- Krasnoselskaya-Riz I, Spruill A, Chen YW, Schuster D, Teslovich T, Baker C, Kumar A, Stephan DA. AIDS Res Hum Retroviruses. 2002;18:591–604. doi: 10.1089/088922202753747941. [DOI] [PubMed] [Google Scholar]
- Kutney SN, Hong R, Macfarlan T, Chakravarti D. J Biol Chem. 2004;279:30850–30855. doi: 10.1074/jbc.M404969200. [DOI] [PubMed] [Google Scholar]
- Li M, Makkinje A, Damuni Z. J Biol Chem. 1996;271:11059–11062. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. Proc Natl Acad Sci U S A. 2003;100:8164–8169. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow JW, Nelson DA. Semin Cancer Biol. 1995;6:195–202. doi: 10.1006/scbi.1995.0026. [DOI] [PubMed] [Google Scholar]
- Luo Q, Li J, Cenkci B, Kretzner L. Oncogene. 2004;23:1088–1097. doi: 10.1038/sj.onc.1207225. [DOI] [PubMed] [Google Scholar]
- Luo RX, Postigo AA, Dean DC. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- Ma Y, Yuan J, Huang M, Jove R, Cress WD. J Biol Chem. 2003;278:16770–16776. doi: 10.1074/jbc.M212702200. [DOI] [PubMed] [Google Scholar]
- Mathieu N, Hempel WM, Spicuglia S, Verthuy C, Ferrier P. J Exp Med. 2000;192:625–636. doi: 10.1084/jem.192.5.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens BM, Hawley RG. Stem Cells. 2002;20:364–379. doi: 10.1634/stemcells.20-5-364. [DOI] [PubMed] [Google Scholar]
- Owens BM, Hawley RG, Spain LM. Methods Mol Med. 2004;105:311–322. doi: 10.1385/1-59259-826-9:311. [DOI] [PubMed] [Google Scholar]
- Owens BM, Zhu YX, Suen TC, Wang PX, Greenblatt JF, Goss PE, Hawley RG. Blood. 2003;101:4966–4974. doi: 10.1182/blood-2002-09-2857. [DOI] [PubMed] [Google Scholar]
- Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM. Cell. 1990;60:167–176. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- Pulford K, Lecointe N, Leroy-Viard K, Jones M, Mathieu-Mahul D, Mason DY. Blood. 1995;85:675–684. [PubMed] [Google Scholar]
- Ramezani A, Hawley TS, Hawley RG. Blood. 2003;101:4717–4724. doi: 10.1182/blood-2002-09-2991. [DOI] [PubMed] [Google Scholar]
- Ratcliffe MJ, Itoh K, Sokol SY. J Biol Chem. 2000;275:35680–35683. doi: 10.1074/jbc.C000639200. [DOI] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. Genes Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riz I, Eaker S, Hawley RG. Applied Genomics and Proteomics. 2003;1:95–108. [Google Scholar]
- Rudenko A, Bennett D, Alphey L. EMBO Rep. 2003;4:59–63. doi: 10.1038/sj.embor.embor712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Karsunky H, Gau E, Zevnik B, Elsasser HP, Moroy T. Oncogene. 1998a;17:2661–2667. doi: 10.1038/sj.onc.1202191. [DOI] [PubMed] [Google Scholar]
- Schmidt T, Karsunky H, Rodel B, Zevnik B, Elsasser HP, Moroy T. EMBO J. 1998b;17:5349–5359. doi: 10.1093/emboj/17.18.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Weise C, Kouzarides T. J Biol Chem. 2004;279:23859–23862. doi: 10.1074/jbc.C400151200. [DOI] [PubMed] [Google Scholar]
- Sears RC, Nevins JR. J Biol Chem. 2002;277:11617–11620. doi: 10.1074/jbc.R100063200. [DOI] [PubMed] [Google Scholar]
- Shan X, Czar MJ, Bunnell SC, Liu P, Liu Y, Schwartzberg PL, Wange RL. Mol Cell Biol. 2000;20:6945–6957. doi: 10.1128/mcb.20.18.6945-6957.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen WF, Krishnan K, Lawrence HJ, Largman C. Mol Cell Biol. 2001;21:7509–7522. doi: 10.1128/MCB.21.21.7509-7522.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoki T, Harder L, Horsman DE, Karran L, Taniguchi I, Willis TG, Gesk S, Steinemann D, Zucca E, Schlegelberger B, Sole F, Mungall AJ, Gascoyne RD, Siebert R, Dyer MJ. Blood. 2001;98:2837–2844. doi: 10.1182/blood.v98.9.2837. [DOI] [PubMed] [Google Scholar]
- Sontag E, Sontag JM, Garcia A. EMBO J. 1997;16:5662–5671. doi: 10.1093/emboj/16.18.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PK, Downey TJ, Spitznagel EL, Jr., Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC. Nucleic Acids Res. 2003;31:5676–5684. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, Chan HM, Livingston DM, Amati B. Mol Cell Biol. 2004;24:4546–4556. doi: 10.1128/MCB.24.10.4546-4556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trop S, De SP, Zuniga-Pflucker JC, Rottapel R. Blood. 2001;97:2269–2277. doi: 10.1182/blood.v97.8.2269. [DOI] [PubMed] [Google Scholar]
- Urlinger S, Baron U, Thellmann M, Hasan MT, Bujard H, Hillen W. Proc Natl Acad Sci USA. 2000;97:7963–7968. doi: 10.1073/pnas.130192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varas A, Hager-Theodorides AL, Sacedon R, Vicente A, Zapata AG, Crompton T. Trends Immunol. 2003;24:197–206. doi: 10.1016/s1471-4906(03)00033-4. [DOI] [PubMed] [Google Scholar]
- Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te RH, van de WM, Oosterwegel M, Wilson A, MacDonald HR, Clevers H. Nature. 1995;374:70–74. doi: 10.1038/374070a0. [DOI] [PubMed] [Google Scholar]
- von Lindern M, van BS, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Mol Cell Biol. 1992;12:3346–3355. doi: 10.1128/mcb.12.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhoeve PM, Hijmans EM, Bernards R. Oncogene. 1999;18:515–524. doi: 10.1038/sj.onc.1202316. [DOI] [PubMed] [Google Scholar]
- Wadzinski BE, Wheat WH, Jaspers S, Peruski LF, Jr., Lickteig RL, Johnson GL, Klemm DJ. Mol Cell Biol. 1993;13:2822–2834. doi: 10.1128/mcb.13.5.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF. Mol Cell Biol. 1999;19:6195–6206. doi: 10.1128/mcb.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BK, Kumar KP, Reich NC. Mol Cell Biol. 1998;18:1359–1368. doi: 10.1128/mcb.18.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ. Genes Dev. 2002;16:235–244. doi: 10.1101/gad.943102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J, Graveel CR, Bartley SM, Madore SJ, Farnham PJ. Proc Natl Acad Sci U S A. 2002;99:3890–3895. doi: 10.1073/pnas.062047499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Yalcin A, Koulich E, Mohamed S, Liu L, D'Mello SR. J Neurochem. 2003;84:397–408. doi: 10.1046/j.1471-4159.2003.01540.x. [DOI] [PubMed] [Google Scholar]
- Yan Y, Mumby MC. J Biol Chem. 1999;274:31917–31924. doi: 10.1074/jbc.274.45.31917. [DOI] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- Yu WM, Hawley TS, Hawley RG, Qu CK. Oncogene. 2003;22:5995–6004. doi: 10.1038/sj.onc.1206846. [DOI] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.