

Abstract
Using a tetrazolium-based assay, a NAD(P)H oxidoreductase was purified from plasma membranes prepared from soybean (Glycine max) hypocotyls. The enzyme, a tetramer of 85 kD, produces O2·− by a reaction that depended on menadione or several other 1,4-naphthoquinones, in apparent agreement with a classification as a one-electron-transferring flavoenzyme producing semiquinone radicals. However, the enzyme displayed catalytic and molecular properties of obligatory two-electron-transferring quinone reductases of the DT-diaphorase type, including insensitivity to inhibition by diphenyleneiodonium. This apparent discrepancy was clarified by investigating the pH-dependent reactivity of menadionehydroquinone toward O2 and identifying the protein by mass spectrometry and immunological techniques. The enzyme turned out to be a classical NAD(P)H:quinone-acceptor oxidoreductase (EC 1.6.5.2, formerly 1.6.99.2) that reduces menadione to menadionehydroquinone and subsequently undergoes autoxidation at pH ≥ 6.5. Autoxidation involves the production of the semiquinone as an intermediate, creating the conditions for one-electron reduction of O2. The possible function of this enzyme in the generation of O2·− and H2O2 at the plasma membrane of plants in vivo is discussed.
The plasma membrane of plant cells displays transmembrane electron transport from cytoplasmic NAD(P)H to various external electron acceptors, which is accompanied by depolarization of the membrane potential and secretion of H+ into the apoplast (Bérczi and Møller, 2000; Vuletić et al., 2005; Lüthje, 2007). Peculiar properties of this redox system, which can be demonstrated in right-side-out plasma membrane vesicles (Menckhoff and Lüthje, 2004), are as follows: (1) use of both NADH and NADPH as electron donors; (2) use of artificial substrates such as ferricyanide (FeCN), dichlorophenoleindophenole (DCPIP), or quinones as electron acceptors; and (3) insensitivity to KCN. The molecular composition and biochemical function of this so-called “standard system” (Bienfait and Lüttge, 1988) found in monocotyledonous and dicotyledonous plants are not well understood at present, despite numerous attempts to isolate and characterize potentially related enzyme activities from solubilized plasma membranes. The artificial electron acceptors generally used for assaying these activities provide no hints to the natural electron transfer reaction catalyzed by this system in the plasma membrane in vivo. The various operationally defined names coined for these enzymes from their ill-characterized catalytic activities, such as NAD(P)H dehydrogenase, NAD(P)H oxidase, NAD(P)H-FeCN reductase, NAD(P)H-duroquinone reductase, and NAD(P)H-superoxide synthase, obscure the fact that there are obviously several enzymes with overlapping substrate specificities that have not yet been properly disentangled.
In this context, it is of particular interest to know whether the plasma membrane redox system is capable of transferring electrons from NAD(P)H to O2 and whether such an activity is involved in the generation of reactive oxygen species at the outer membrane face (Murphy and Auh, 1996; van Gestelen et al., 1997; Vuletić et al., 2003). Based on molecular genetic studies, this capacity is generally attributed to the “respiratory burst oxidase” homologues (RBOHs; i.e. plant equivalents to the NADPH oxidase [NOX] of mammalian phagocytes) that catalyze a transmembrane electron transport from NADPH to O2 via FAD and cytochrome (Cyt) b558, resulting in the formation of superoxide radicals (O2·−; Sumimoto et al., 2005). So far, the expression of RBOH genes in plants has been studied mainly at the level of transcription. Moreover, Arabidopsis (Arabidopsis thaliana) mutants defective in one or another member of the RBOH family have been characterized (Torres and Dangl, 2005; Gapper and Dolan, 2006). Using antibodies raised against RBOH peptides, RBOH proteins have been identified by immunoblotting in plasma membranes from several plant species (Keller et al., 1998; Sagi and Fluhr, 2001; Simon-Plas et al., 2002) and by in situ imaging of GFP-RBOH fusion protein (Kobayashi et al., 2006). However, RBOH proteins have not yet been isolated from plants, and their catalytic properties and cofactor requirements are still largely unknown. The ability of diphenylene iodonium (DPI) to inhibit mammalian NOX at low concentrations (<10 μm; Doussiere and Vignais, 1992) has often been used as a diagnostic feature for NOX activity in plant cells or cell fractions, although DPI also inhibits other flavin- or heme-containing enzymes, especially at higher concentrations (≥10 μm; Frahry and Schopfer, 1998; Doussiere et al., 1999); therefore, it is no suitable tool to unequivocally identify NOX enzymes.
Attempts to isolate proteins with O2·− synthase activity from plasma membranes by biochemical methods have not yet led to homogeneous results. van Gestelen et al. (1997) separated an O2·−-producing, FMN-containing protein fraction from FeCN/DCPIP oxidoreductases in solubilized Phaseolus plasma membranes. No cytochrome was detected. O2·− generation was much higher with NADPH than with NADH and could be inhibited by DPI, although only at high concentrations (60 μm for 50% inhibition). The O2·−-producing activity of plasma membrane vesicles was strongly elevated by 1,4-naphthoquinones (NQs) such as juglone, and the active proteins could be divided by ion exchange chromatography into a NADPH-specific, NQ-independent fraction and three NQ-dependent fractions (van Gestelen et al., 1998). The main NQ-dependent fraction showed high specificity for NADH compared with NADPH. The plasma membrane reduced Cyt c at pH 6 in a juglone-stimulated manner, leading to the conclusion that semiquinones are involved in the catalytic cycle.
Using Rosa cells as a source of plasma membranes, Murphy et al. (2000) partly purified an O2·−-producing enzyme activity that showed apparent specificity for either NADH or NADPH, depending on whether lucigenin luminescence or Cyt c reduction was used to assay O2·− formation. Spectroscopic evidence indicated the presence of pterin rather than flavin in the preparation. The activity was stimulated by NQ (menadione [MD]) and strongly inhibited by DPI (≤0.5 μm for 50% inhibition) and NaN3 but not by KCN.
Taken together, this fragmentary experimental evidence illustrates that the enzymatic mechanism(s) for O2·− generation in the plant plasma membrane may be more complex than suggested by the concept modeled on phagocyte NOX. In an attempt to elucidate this complex picture, we are investigating the molecular and catalytic properties of NAD(P)H-oxidizing enzymes that are potentially involved in O2·− generation by plasma membranes isolated from soybean (Glycine max) hypocotyls. In this article, we describe an enzyme that is responsible for the NQ-stimulated production of O2·− in these membranes. O2·−-producing activity was measured using the tetrazolium compound Na,3′-(1-[phenylamino-carbonyl]-3,4-tetrazolium)-bis(4-methoxy-6-nitro)benzenesulfonic acid hydrate (XTT), which can be reduced by O2·− to a photometrically detectable formazan (Able et al., 1998). To analyze the reaction mechanism of O2·− generation, we developed spectrophotometric methods enabling the measurements of concentration changes of NAD(P)H, quinones, and XTT in the same reaction mixture.
RESULTS AND DISCUSSION
Purification of a NAD(P)H Oxidoreductase Activity from Solubilized Plasma Membranes
A plasma membrane fraction showing NADH-dependent XTT reduction in the presence of MD and detergent was isolated from the upper hypocotyl region of dark-grown soybean seedlings by established techniques and checked for contaminating proteins (see “Materials and Methods”). Extensive washing of the membranes with 1.5 m NaCl or CaCl2 did not affect this activity (data not shown). The proteins solubilized from these membranes by Tween 20 were subjected to affinity chromatography on a Blue Sepharose column. Extensive washing with buffer removed the bulk of the nonbound proteins, including a FeCN reductase, from the bound XTT-reducing fraction that could be eluted from the column with 1 μm NADH (Fig. 1A). The protein concentration of the active enzyme fraction was very low (4–8 μg mL−1). The elution profile obtained with the active enzyme fraction after chromatography on a calibrated Superdex 200 column revealed a single peak of an apparent molecular mass of approximately 85 kD (Fig. 1B). Redox activities of this enzyme fraction in the absence and presence of MD in comparison with the solubilized plasma membrane are summarized in Table I.
Figure 1.

Purification of a MD-dependent NAD(P)H oxidoreductase activity by chromatography on Blue Sepharose (A) and Superdex (B). A, A sample of 5 mL of solubilized plasma membrane extract was applied to a 16- × 1-cm column of Blue Sepharose Fast Flow equilibrated with 10 mm HEPES (pH 8.0) containing 0.2% (w/w) Tween 20. The column was washed (0.5 mL min−1) with 100 mL of the same buffer (fractions 0–107), followed by gradient elution (0–5 μm NADH; fractions 108–167) and 1 m NaCl (fractions 168–200) in the same buffer. B, A sample of 0.7 mL of enzyme purified as shown above (fractions 110–120) together with a small amount of vitamin B12 was applied to a Superdex 200 HR 10/30 column, eluted (6 mL h−1) with 10 mm HEPES (pH 8.0), and collected in 300-μL fractions. The column was calibrated with Cyt c (12.4 kD), myoglobin (17.8 kD), chymotrypsin (25 kD), ovalbumin (43 kD), bovine serum albumin (67 kD), aldolase (160 kD), catalase (230 kD), and ferritin (440 kD) as shown in the inset. The position of the oxidoreductase is indicated by dashed lines.
Table I.
Redox activities of solubilized plasma membranes and the purified enzyme fraction eluted with NADH from the Blue Sepharose column
Activities were measured in the absence (−MD) or presence (+MD) of 100 μm MD at pH 7.5. Protein concentrations were 1.5 ± 0.5 mg mL−1 (solubilized proteins) and 6.4 ± 0.4 μg mL−1 (purified enzyme). XTT concentration was 500 μm. Ratio = (specific activity + MD)/(specific activity − MD); yield = (specific activity in the purified enzyme fraction/specific activity in the solubilized protein fraction).
| Assay Reaction | Specific Activity
|
Yield
|
||||||
|---|---|---|---|---|---|---|---|---|
| Solubilized Protein Fraction
|
Purified Enzyme Fraction
|
−MD | +MD | |||||
| −MD | +MD | Ratio | −MD | +MD | Ratio | |||
| μmol min−1 mg−1 protein | % | |||||||
| NADPH oxidation | 0.0016 ± 0.0004 | 0.15 ± 0.01 | 94 | <0.1a | 29 ± 1 | >290 | – | 83 |
| NADH oxidation | 0.0018 ± 0.0020 | 0.16 ± 0.01 | 89 | <0.1a | 30 ± 1 | >300 | – | 79 |
| XTT reduction with NADPH | 0.0038 ± 0.0001 | 0.10 ± 0.002 | 26 | <0.1a | 22 ± 0.1 | >220 | – | 92 |
| XTT reduction with NADH | 0.025 ± 0.002 | 0.10 ± 0.005 | 4 | <0.1a | 19 ± 1 | >190 | – | 80 |
| Cyt c reduction with NADPH | 0.009 ± 0.001 | 0.30 ± 0.01 | 33 | 0.16 ± 0.04 | 60 ± 6 | 380 | 7 | 86 |
| Cyt c reduction with NADH | 0.017 ± 0.003 | 0.31 ± 0.01 | 18 | 0.11 ± 0.05 | 63 ± 4 | 570 | 3 | 87 |
| FeCN reduction with NADPH | 0.21 ± 0.01 | 0.38 ± 0.02 | 1.8 | 25 ± 1 | 54 ± 1 | 2.2 | 50 | 61 |
| FeCN reduction with NADH | 0.39 ± 0.01 | 0.53 ± 0.03 | 1.4 | 25 ± 2 | 52 ± 2 | 2.1 | 27 | 42 |
| DCPIP reduction with NADPH | 0.029 ± 0.003 | 0.032 ± 0.001 | 1.1 | 3.0 ± 0.2 | 3.3 ± 0.2 | 1.1 | 43 | 43 |
| DCPIP reduction with NADH | 0.028 ± 0.003 | 0.031 ± 0.003 | 1.1 | 2.2 ± 0.2 | 2.2 ± 0.2 | 1.0 | 33 | 30 |
Below detection limit.
The reaction catalyzed by the purified enzyme in the presence of NAD(P)H and MD consumed O2. The addition of XTT suppressed the O2 consumption, and this suppression could be partially counteracted by adding superoxide dismutase (SOD; Fig. 2). These results are in agreement with a reaction mechanism involving the reduction of O2 to O2·− that subsequently reduces XTT to XTTH2, whereby O2 is regenerated according to:
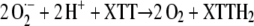 |
This reaction is suppressed by the disproportionation of O2·− with SOD.
Figure 2.

Consumption of O2 by the NAD(P)H oxidoreductase, its suppression by XTT, and the partial reversion of the suppression by SOD (pH 7.5). The addition of NADPH (200 μm), MD (100 μm), enzyme, XTT (100 μm), and SOD (100 μg mL−1) is indicated by arrows. The measurements were performed in a 2-mL total volume containing 1.3 μg mL−1 purified enzyme.
A number of conclusions can be drawn from these data. (1) Utilizing adsorption to Blue Sepharose followed by desorption with NADH resulted in an at least 200-fold increase in specific activity of an 85-kD NAD(P)H oxidoreductase isolated from solubilized plasma membranes in a single purification step. A closely related protocol was used by Luster and Buckhout (1989) to purify an “electron transport protein” with similar properties from maize (Zea mays) plasma membranes. (2) Our purified enzyme consumes O2 and reduces XTT and Cyt c in a MD-dependent reaction. Its ability to reduce FeCN is slightly stimulated by MD, while DCPIP is independent of MD. NADH and NADPH are oxidized at similar rates in the presence of MD. (3) In the absence of an electron acceptor except O2, the NAD(P)H-oxidizing activity is very low in the solubilized membrane fraction and not detectable in the purified enzyme fraction. No NOX-type activity could be detected in the elution profile of the Blue Sepharose column (Fig. 1A), although the solubilized membrane fraction does contain measurable amounts of MD-independent XTT- and Cyt c-reducing activity. (4) The purified enzyme accounts for 80% to 90% of the NAD(P)H oxidoreductase activity detected by the XTT assay in the presence of MD and about 50% of the FeCN/DCPIP reductase activity present in the solubilized membrane fraction.
Peroxidase Activity of the Purified Plasma Membranes
The formation and decomposition of reactive oxygen species is affected by peroxidases. Moreover, hydroquinones can be converted into semiquinones by these enzymes (Ohnishi et al., 1969). Plasma membranes isolated from maize roots were reported to contain at least two tightly bound guaiacol peroxidases (Mika and Lüthje, 2003). We tested aliquots (50 μg of protein) of our solubilized plasma membrane fraction with the assay used by these authors and found no detectable peroxidase activity. Using a tetramethylbenzidine-based assay with increased sensitivity revealed a peroxidase activity of 12 nmol min−1 mL−1 (6 nmol min−1 mg−1 protein), corresponding to 1.7 ng of horseradish peroxidase equivalents per milligram of protein estimated from a standard curve with pure horseradish peroxidase. The peroxidase activity solubilized from the plasma membrane accounted for 0.06% of the soluble peroxidase activity in the tissue homogenate, indicating that significant amounts of peroxidases appear not to be obligatory components of plant plasma membranes.
Effects of Oxidoreductase Inhibitors
We investigated the effect of several diagnostic inhibitors on the NAD(P)H-oxidizing and XTT-reducing activities of the enzyme purified from plasma membranes to compare them with known classes of oxidoreductases. The data summarized in Figure 3 demonstrate that: (1) KCN and NaN3 (≤10 mm) had no effect on XTT reduction; (2) Zn2+ and Cd2+ inhibited XTT reduction only at very high concentrations (50% inhibitory concentration [I50] = 2 mm and 5 mm, respectively); (3) DPI (≤500 μm) had no effect on both assay reactions; (4) Cibacron Blue and dicumarol inhibited both assay reactions (I50 = 3–5 μm and 300 μm, respectively); and (5) Cu2+ and Mn2+ inhibited XTT reduction but not NADPH oxidation (I50 = 1 μm and 100 μm, respectively), indicating that these redox-active ions interfered with the electron transfer from the reduced MD to XTT rather than with the initial catalytic activity of the enzyme.
Figure 3.

Effects of various inhibitors on the NAD(P)H oxidoreductase activity, measured either as oxidation of NADPH (200 μm; white symbols, dashed lines) or reduction of XTT (100 μm; black symbols) in the presence of MD (100 μm) at pH 7.5. NADPH oxidation was measured in the absence of XTT. Reaction mixtures with dicumarol and DPI contained up to 2% dimethyl sulfoxide, which had no effect on enzyme activity (100% = 4 μm min−1 produced by 1.3 μg mL−1 purified enzyme).
The following compounds exhibited no effect on XTT reduction: rotenone (10 μm), piericidin A (100 μm), nitrofurantoin (100 μm), quinacrine (100 μm), CaCl2 (2 mm), MgCl2 (2 mm), EDTA (4 mm), N-ethylmaleimide (10 mm), 4-chloromercuribenzoate (400 μm), FMN (10 μm), and FAD (10 μm; data not shown).
An important result emerging from this list deserves further consideration, namely the insensitivity of the enzyme to DPI, an inhibitor inactivating many flavoenzymes, including NOX-type enzymes at less than 10 μm (Cross, 1987; Doussiere and Vignais, 1992). DPI binds to, and thereby inactivates, the catalytic center of these enzymes only after its reduction to the phenyl radical (O'Donnell et al., 1994); therefore, it attacks specifically one-electron-transferring flavoenzymes such as NOX (Cross, 1987) or mitochondrial NADH:ubiquinone reductase (Ragan and Bloxham, 1977). As a consequence, flavoenzymes transferring two electrons in one step from NAD(P)H to various acceptors are not subject to inhibition by DPI (Cross, 1987). Examples of this class are the NAD(P)H:quinone-acceptor oxidoreductases known as DT-diaphorases in animals (Lind et al., 1990; Ross, 1997). Functionally related flavoenzymes have also been found in plants (Spitsberg and Coscia, 1982; Luster and Buckhout, 1989; Valenti et al., 1990; Serrano et al., 1994; Rescigno et al., 1995; Trost et al., 1995, 1997; Sparla et al., 1996, 1998). These enzymes catalyze the transfer of a hydride atom to various benzoquinones and naphthoquinones, directly forming the corresponding hydroquinones and thus avoiding the intermediate semiquinone radical that can reduce O2 to O2·− (Iyanagi and Yamazaki, 1970; Lind et al., 1990; Sparla et al., 1996; Ross, 1997). A purified quinone oxidoreductase investigated by Trost et al. (1997) was shown to be insensitive to DPI. A further remarkable feature of this group of enzymes is that they can be inhibited by Cibacron Blue (Prestera et al., 1992) and dicumarol (Lüthje et al., 1994; Rescigno et al., 1995).
Evidently, the enzyme isolated from soybean plasma membranes shares some important properties with two-electron-transferring quinone reductases. Therefore, the basic problem is to explain the ability of this enzyme to elicit one-electron transfer from NAD(P)H, presumably via quinone intermediates, to O2, a property that appears to be incompatible with the reaction mechanism of DPI-insensitive NAD(P)H oxidoreductases.
Catalytic Properties of NAD(P)H Oxidoreductase
The following experiments were conducted with the purified enzyme fraction eluted from Blue Sepharose by NADH (Fig. 1A). Table II shows the kinetic parameters for the substrates involved in the O2-reducing reaction measured by the XTT assay. In order to elucidate the role of quinones, we examined the ability of several NQs and 1,4-benzoquinones (BQs) to replace MD in this reaction (Table III). Unsubstituted NQ, 5-hydroxy-NQ, and 5-hydroxy,2-methyl-NQ supported XTT reduction similar to MD. The enzyme was inactive with 2-hydroxy- and 2-methoxy-substituted NQs as well as phylloquinone and menaquinone-4. Only those NQs that can be reduced by NADPH to the respective hydroquinones supported XTT reduction. However, the accumulation of hydroquinones fell short of NADPH consumption to an extent depending on the type of NQ. XTT reduction in the presence of active NQs could only partially be inhibited by removing O2·− with a saturating amount of SOD, and again this depended on the particular NQ. None of the tested BQs supported XTT reduction, although several of them were reduced by NADPH to the corresponding hydroquinones that accumulated in stoichiometric amounts. This result is in conflict with the reaction catalyzed by one-electron-transferring reductases that includes the intermediary formation of semiquinone.
Table II.
Km and Vmax values for various steps of the reaction system catalyzed by the NAD(P)H oxidoreductase at pH 7.5
Km and Vmax values were obtained from linear Lineweaver-Burk plots using equal amounts of purified enzyme. NAD(P)H (30–200 μm) oxidation was determined from the decrease in A340 in the presence of 100 μm MD. XTT (12–100 μm) reduction was determined from the increase in A470 in the presence of 200 μm NADPH and 100 μm MD. MD (20–200 μm) reduction was determined from the consumption of NADPH (200 μm) in the absence of XTT.
| Reaction | Km | Vmax |
|---|---|---|
| μm | μm min−1 | |
| NADPH oxidation | 90a | 4.1 |
| NADH oxidation | 200a | 5.6 |
| XTT reduction | 14 | 3.6 |
| MD reduction | 70 | 7.1 |
Equivalent values were obtained by measuring the reduction of XTT (500 μm) under similar conditions.
Table III.
Quinone substrate specificity of the NAD(P)H oxidoreductase determined as NADPH oxidation, hydroquinone accumulation, or XTT reduction with or without SOD
Enzyme activity was measured at pH 7.5 with 200 μm NADH, 100 μm quinone, 100 μm XTT with or without 100 μg mL−1 SOD (in the case of XTT reduction), and 0.13 μg mL−1 purified enzyme. The percentage of the normalized activity is given in parentheses (the value for NADPH oxidation or XTT reduction without SOD is set to 100%).
| Sample | Enzyme Activity
|
|||
|---|---|---|---|---|
| NADPH Oxidation | Hydroquinone Accumulation | XTT Reduction without SOD | XTT Reduction with SOD | |
| μm min−1 | ||||
| NQ | 6.9 ± 0.4 (100) | 1.1 ± 0.1 (16) | 4.9 ± 0.4 (100) | 1.0 ± 0.1 (20) |
| 2-Methyl-NQ (MD, vitamin K3) | 4.5 ± 0.2 (100) | 2.0 ± 0.1 (44) | 4.0 ± 0.1 (100) | 1.9 ± 0.1 (48) |
| 5-Hydroxy-NQ (juglone) | 5.7 ± 0.4 (100) | 0.01 ± 0.05 (<1) | 4.1 ± 0.3 (100) | 0.5 ± 0.1 (12) |
| 5-Hydroxy,2-methyl-NQ (plumbagin) | 3.5 ± 0.1 (100) | 0.4 ± 0.1 (11) | 2.5 ± 0.1 (100) | 1.4 ± 0.2 (56) |
| 2-Hydroxy-NQ (lawsone) | <0.1 | <0.1 | <0.1 | – |
| 2-Methoxy-NQ | <0.1 | <0.1 | <0.1 | – |
| 2,3-Dimethoxy-NQ | 0.1 ± 0.2 | <0.1 | 0.2 ± 0.1 | – |
| 2-Methyl,3-phythyl-NQ (phylloquinone, vitamin K1) | 0.4 ± 0.5 | <0.1 | <0.1 | – |
| Menaquinone-4 (vitamin K2) | <0.1 | 0.4 ± 0.2 | <0.1 | – |
| BQ | 4.3 ± 0.3 (100) | 4.8 ± 1.0 (112) | 0.02 ± 0.02 | – |
| 2-Methyl-BQ (toluquinone) | 6.1 ± 1.0 (100) | 5.9 ± 0.8 (97) | 0.01 ± 0.02 | – |
| 2,5-Dimethyl-BQ (xyloquinone) | 6.3 ± 0.9 (100) | 5.2 ± 0.7 (83) | 0.08 ± 0.02 | – |
| 2,6-Dimethyl-BQ | 6.8 ± 1.0 (100) | 4.8 ± 0.9 (71) | 0.01 ± 0.02 | – |
| 2,3-Dimethoxy,5-methyl-BQ (ubiquinone 0) | 5.0 ± 0.2 (100) | 4.7 ± 0.2 (94) | 0.04 ± 0.02 | – |
| Tetramethyl-BQ (duroquinone) | 0.3 ± 0.1 | 0.4 ± 0.3 | <0.01 | – |
| 2,6-Dimethoxy-BQ | <0.1 | <0.1 | <0.2 | – |
| 2,5-Dihydroxy-BQ | <0.1 | <0.2 | <0.02 | – |
| Tetrahydroxy-BQ | <0.1 | <0.1 | <0.2 | – |
Taken together, these results indicate that the enzyme is capable of utilizing several NQs and BQs as electron acceptors, but only certain NQs can mediate electron transfer to XTT. This situation is highly reminiscent of the findings of Ernster et al. (1962) that DT-diaphorase catalyzes the reduction of Cyt c in the presence of some NQs but not in the presence of BQs. It also appears from our data that the reduction of XTT in the presence of active NQs can only in part be attributed to the formation of O2·−.
In order to clarify the connections between NADPH oxidation and O2·− production, we examined the stoichiometries between NADPH oxidation, O2 consumption, and XTT reduction in the MD-mediated reaction system (Fig. 4). If all electrons delivered by NADPH are utilized for the reduction of O2 to O2·− (NADPH + 2 O2 → NADP+ + H+ + 2 O2·−), the molar ratio of NADPH oxidation to O2 consumption would be 1:2. If the disproportionation of O2·− into H2O2 (2 O2·− + 2 H+ → H2O2 + O2) is taken into account, the ratio will be 1:1. Figure 4, A and B, shows that after a lag of about 20 s, the steady-state rates of these changes show a ratio of 1:0.7, indicating that at the air-saturated O2 concentration (260 μm) about two-thirds of the electrons removed from NADPH are utilized for reducing O2. The O2 consumption could be reduced by half with catalase, indicating that H2O2 is produced that decomposes according to H2O2 → H2O + ½ O2 (Fig. 4A). About one-third of the electrons removed from NADPH are utilized for accumulating menadionehydroquinone (MDH2). This portion can be significantly increased by reducing the O2 concentration in the reaction mixture from 260 μm to about 10 μm (Fig. 4B), suggesting that MDH2 accumulation competes with O2 reduction for electrons. (For technical reasons, it was not possible to establish O2 concentrations lower than 10 μm during the reaction.) If XTT was added to the reaction mixture, it captured the electrons removed from NADPH with a molar ratio that varied between 1:1 and 1:0.7 in different experiments at both air-saturated and reduced O2 concentrations (Fig. 4C). This correlation presumably reflects a variable contribution of MDH2 accumulation that in turn is influenced by the rate of the enzyme reaction. Similar to XTT, Cyt c was able to capture most of the electrons removed from NADPH (molar ratio of 1:1.7, in good agreement with NADPH + 2 Cyt coxidized → NADP+ + H+ + 2 Cyt creduced; Fig. 4D).
Figure 4.

Concentration changes of various substrates involved in the transfer of electrons from NADPH to XTT or Cyt c in the reaction catalyzed by the NAD(P)H oxidoreductase (200 μm NADPH and 100 μm MD, pH 7.5). A, O2 consumption with and without catalase (200 μg mL−1). B, MD reduction and simultaneously measured NADPH oxidation at 260 μm O2 (aerobic) or about 10 μm O2 (anaerobic). C, Reduction of added XTT (100 μm) and simultaneously measured NADPH oxidation under aerobic or anaerobic conditions. D, Reduction of added Cyt c (50 μm) and simultaneously measured NADPH oxidation under aerobic or anaerobic conditions.
It can be concluded from these results that the electron transfer from NADPH can be accounted for by the accumulation of MDH2 and, after a lag phase, the consumption of O2. In the presence of XTT or Cyt c, the electron transfer to these acceptors is essentially complete and can occur via two competing routes, only one of which involves the reduction of O2. This conclusion is further supported by the finding that SOD at saturating concentrations inhibits XTT reduction by 50% without affecting O2 reduction, NADPH oxidation, or MDH2 accumulation (Table IV).
Table IV.
Effects of SOD (100 μg mL−1) on various steps involved in the transfer of electrons from NADPH to XTT in the reaction catalyzed by the NAD(P)H oxidoreductase
Experimental details are as described for Figure 4. Inhibition of the XTT reduction by SOD was saturated at ≥1 μg mL−1. SOD (50 μg mL−1) inhibited XTT reduction by >95% if the enzyme was replaced by a similarly active concentration (50 nm) of methoxyphenazine methosulfate catalyzing the univalent reduction of O2 by NAD(P)H.
| Reaction | Reaction Rate
|
|
|---|---|---|
| −SOD | +SOD | |
| μm min−1 | ||
| O2 consumption | 1.9 ± 0.3 | 1.7 ± 0.3 |
| NADPH consumption | 3.3 ± 0.5 | 3.4 ± 0.1 |
| MDH2 accumulation | 1.2 ± 0.3 | 1.1 ± 0.2 |
| XTT reduction | 2.5 ± 0.3 | 1.3 ± 0.1 |
Another interesting property of the reaction system catalyzed by the oxidoreductase emerged from the investigation of the pH dependence of the NADPH-oxidizing and the XTT-reducing activities (Fig. 5). NADPH oxidation displays a broad asymmetric peak between pH 4.5 and >9.0, with an optimum at pH 5.5. However, XTT reduction covers only the high pH range (pH > 6) of this curve, with an apparent optimum at pH 7.5. Hence, in the range of pH 5 to 6, NADPH oxidation takes place at a high rate, but the electrons cannot be utilized for the reduction of XTT. Figure 6 shows that under these conditions, no O2 consumption can be observed and the NADH oxidation is accompanied by MDH2 accumulation with a molar ratio of 1:1. Obviously, at pH 6, O2·− production is completely shut down in favor of the accumulation of MDH2.
Figure 5.

pH dependence of NADPH oxidation and XTT reduction affected by the NAD(P)H oxidoreductase in the presence of MD. Experimental details are as described for Figure 4, except that the pH was adjusted by mixing 20 mm HEPES and 20 mm MES. NADPH oxidation was measured in the absence of XTT; however, similar results were obtained in the presence of XTT.
Figure 6.

NADPH oxidation, MDH2 accumulation, O2 consumption, and XTT reduction affected by the NAD(P)H oxidoreductase at pH 6. Experimental details are as described for Figure 4, except that the reaction mixture was adjusted to pH 6.0 with MES.
Molecular Properties of the NAD(P)H Oxidoreductase
Two genetically unrelated families of two-electron-transferring NAD(P)H:quinone-acceptor oxidoreductases have been identified in plants. Both contain FMN and are based on subunits of similar size. The enzyme cloned from Arabidopsis, AtNQR [for Arabidopsis NAD(P)H:quinone-acceptor oxidoreductase], by Sparla et al. (1999) belongs to a family of tetrameric flavoproteins composed of 21- to 22-kD subunits (Trost et al., 1995, 1997; Sparla et al., 1998). The NAD(P)H:quinone-acceptor reductase cloned from Arabidopsis, AtFQR1, by Laskowski et al. (2002) belongs to the WrbA family of flavoproteins that show sequence similarities to flavodoxins and other prokaryotic and fungal proteins. These enzymes are dimers or tetramers of 21-kD subunits (Patridge and Ferry, 2006).
To test whether the NAD(P)H oxidoreductase purified from soybean plasma membranes was related to one of these families, we examined its affinity to antibodies raised against recombinant AtNQR and AtFQR1. Native PAGE of cytosolic proteins, solubilized plasma membranes, and purified oxidoreductase revealed similar bands for MD-dependent O2·−-producing activity using nitroblue tetrazolium chloride (NBT) in-gel staining (Fig. 7A). Immunoblots of these gels demonstrated that the activity purified from the plasma membranes can be attributed to NQR (Fig. 7B), while the activity in the cytosolic fraction can be attributed to FQR1 (Fig. 7C). For unknown reasons, the activity in the plasma membrane fraction showed only a very weak signal with the NQR antibody. When similar samples were subjected to SDS-PAGE, no clear protein bands could be detected by silver staining in the purified enzyme preparation (Fig. 8). Only faint bands at 60, 80 to 90, and 110 kD became visible, indicating the presence of impurities. NQRs are characterized by an extraordinarily high specific activity and, as such, the protein associated with the catalytic activity has been reported to be difficult to detect (Trost et al., 1997). Immunoblots of the SDS gels showed a single band at approximately 22 kD, similar to recombinant AtNQR, with the antibody against NQR but not with the antibody against FQR1 in the purified oxidoreductase preparation. On the other hand, the FQR1 antibody gave a positive reaction with a cytosolic antigen of very similar size (Fig. 8).
Figure 7.

Native PAGE and immunoblot analysis of protein fractions containing cytosolic proteins (lane 1), solubilized plasma membrane proteins (lane 2), and purified NAD(P)H oxidoreductase protein (lane 3). Amounts of samples giving the same activity of XTT reduction (10 nmol XTTH2 min−1 for A and 30 nmol XTTH2 min−1 for B and C) were loaded on the gel. A, The gel was stained for enzyme activity using the NBT assay. B and C, Immunoblots were decorated with antibodies directed against NQR and FQR1.
Figure 8.

Silver-stained SDS-PAGE (8%) and immunoblot analysis of protein fractions containing cytosolic proteins (lane 1), solubilized plasma membrane proteins (lane 2), purified NAD(P)H oxidoreductase protein (lane 3), and recombinant AtNQR (1 μg; lane 4). Samples with the following activities were loaded onto the gel for silver staining: 8 nmol XTTH2 min−1 (15 μg; lanes 1 and 2) and 140 nmol XTTH2 min−1 (7.5 μg; lane 3); samples with the following activities were loaded onto the gel for blotting: 30 nmol XTTH2 min−1 (lanes 1 and 2) and 140 nmol XTTH2 min−1 (lane 3). Marker positions (kilodaltons) are given on the left side. Immunoblots were decorated with antibodies directed against NQR and FQR1. The amount of NQR protein in the plasma membrane protein fraction (lane 2) was insufficient for detectable NQR antibody binding.
Given the molecular mass of approximately 85 kD for the native oxidoreductase purified from plasma membranes (Fig. 1B), these data are in agreement with the finding that plant NQRs are homotetramers of 21.5-kD subunits (Sparla et al., 1996, 1999). Beyond that, the enzyme demonstrates an immunological relationship to AtNQR identified by Sparla et al. (1999) rather than to AtFQR1 identified by Laskowski et al. (2002). FQR1 appears to be at least in part responsible for the quinone reductase activity in the cytosolic fraction.
Mass spectrometry (MS) analysis performed with the proteins extracted from the NBT-stained band obtained with the purified plasma membrane enzyme (Fig. 7A, lane 3) confirmed the homology with the NAD(P)H:quinone-acceptor oxidoreductase NQR (EC 1.6.5.2, formerly EC 1.6.99.2). Table V summarizes the peptides that identify a translation of the assembled EST Glycine_max-35276 being homologous with AtNQR (see also the sequence alignment provided as Supplemental Table S1). Interestingly, the NQR contains a FMN-binding site that was first identified in a FMN reductase of Pseudomonas aeruginosa by x-ray crystallography and that is clearly distinguishable from the flavodoxin key fingerprint motif (Agarwal et al., 2006). This FMN reductase shows high sequence homology with NQR. The largest quantity of the NQR peptides coincided with the center of the NBT-stained gel bands. Most of the peptides identified in the stained region of the gel were from lipoxygenase-4 (swissprot identifier P38417), which was not responsible for the NBT reduction. Other peptides detected were mainly from soybean ESTs, which were homologous with the fasciclin-like arabinogalactan proteins 1 and 2 from Arabidopsis (swissprot identifiers Q9FM65 and Q9SU13). However, the profiles of these signals did not match the profile of the NBT-staining signal (data not shown). Two weak signals in the MS spectra identify peptides of a FQR1 homolog (EST Glycine_max-394114201), but their integrated intensity was more than 30-fold smaller than that of peptides from NQR. If significant, these signals are likely due to a minor contamination with soluble proteins.
Table V.
Mass spectrometric identification of soybean NQR following in-gel digestion by trypsin of the NBT-stained band from native gel electrophoresis (Fig. 7A, lane 3)
| Peptidea | Retention Timeb | Mr(obs)c | zd | Mr(cal) | Difference | E Valuee | Sequence (Modification) |
|---|---|---|---|---|---|---|---|
| 16–27 | 38.4 | 1,101.6039 | 2 | 1,101.6029 | 0.0010 | 1.7E-10 | M.AAVAGASSSVIK.V (acetylated N terminus) |
| 28–36 | 33.5 | 872.5078 | 2 | 872.5079 | −0.0001 | 2.7E-04 | K.VAALSGSLR.K |
| 47–55 | 30.9 | 959.5034 | 2 | 959.5036 | −0.0002 | 1.4E-03 | R.SAIELSQGR.V |
| 56–90 | 56.7 | 3,928.0185 | 3 | 3,928.0145 | 0.0040 | 5.9E-10 | R.VEGLQIEYVDISPLPLLNTDLEVN GTYPPQVEAFR.Q |
| 93–115 | 55.2 | 2,468.2830 | 2 | 2,468.2838 | −0.0008 | 1.4E-11 | K.ILAADSILFASPEYNYSVASPLK.N |
| 93–115 | 55.2 | 2,468.2842 | 3 | 2,468.2838 | 0.0004 | 4.7E-15 | K.ILAADSILFASPEYNYSVASPLK.N |
| 132–146 | 36.1 | 1,272.6573 | 2 | 1,272.6574 | −0.0001 | 3.2E-12 | K.PAAIVSAGGGFGGGR.S |
Sequence numbering is for PlantGDB-assembled EST PUT-161a-Glycine_max-35276.
Retention time (minutes) from reverse-phase HPLC.
Relative molecular mass determined from mass/charge ratio and charge.
Charge determined from isotope distribution observed.
Expected number of random hits having an equal or better score (probability scoring by the OMSSA program).
From the sequence data (Supplemental Table S1), it is clear that NQR lacks all diagnostic features of an integral membrane protein with membrane-spanning helices. However, in agreement with previous investigators (Luster and Buckhout, 1989; Valenti et al., 1990; Serrano et al., 1994; Trost et al., 1997; van Gestelen et al., 1997, 1998), we conclude that NQR can be present as a peripheral membrane protein tightly bound to the plasma membrane based on the following criteria: (1) the enzyme can be liberated from the plasma membrane by detergent but not by high salt (1.5 m CaCl2 or NaCl), and (2) soluble proteins present in the homogenate, such as peroxidase, Glc-6-P dehydrogenase, and FQR, were undetectable, or detectable only in traces, in the solubilized membrane fraction. The other proteins, identified as impurities in our NQR preparation by mass spectrometry, show membrane association domains. The lipoxygenase contains a polycystin/lipoxygenase/α-toxin domain that may be involved in protein-lipid interaction (Bateman and Sandford, 1999) and the fasciclin-like arabinogalactan-protein A glycosylphosphatidylinositol anchor. The presence of membrane-associated proteins in the purified plasma membrane protein preparation may be taken as a further indication of the membrane-associating nature of NQR. Further studies are needed to clarify whether NQR is attached to the inner or outer surface of the plasma membrane (Bérczi and Møller, 1998).
Redox Properties of MDH2
The results reported to date indicate that the purified NQR shares many features with the two-electron-transferring DT-diaphorases of mammals (Märki and Martius, 1960; Ernster et al., 1962; Lind et al., 1990; Ross, 1997) and certain quinone reductases of plants (Luster and Buckhout, 1989; Trost et al., 1995; Sparla et al., 1999), with the notable exception of the ability to reduce O2 to O2·− at pH ≥ 6.5. This reaction implies the formation of a semiquinone intermediate and the capacity for one-electron-transferring reaction (van Gestelen et al., 1998). A key to explain this seemingly disparate property emerged when we investigated the stability of MDH2 as a function of pH. When added to buffered water containing 260 μm O2, MDH2 was stable at acidic pH. However, autoxidation increased steeply when the pH was raised to ≥6.5 (Fig. 9A). XTT reduction was inhibited by SOD by only 50% (Fig. 9B). The molar ratios (1:1 in the case of O2 and XTT, 1:2 in the case of Cyt c) showed that the electron transfer from MDH2 to these acceptors was complete (Fig. 10). Net O2 consumption was prevented when XTT was present (Fig. 10A), similar to the enzyme-catalyzed reaction (Fig. 2). Moreover, in agreement with the results shown in Table IV, SOD reduced the molar ratio of MDH2 oxidation/XTT reduction from 1:1 to 1:0.5, indicating that 50% of the electrons provided by MDH2 could be diverted to XTT without the participation of O2·− (Fig. 10B). In the case of Cyt c, this fraction increased to 70% (Fig. 10C).
Figure 9.

MDH2 autoxidation (A) and accompanying XTT reduction in the absence and presence of SOD (B) as a function of pH. A, MDH2 (100 μm) from a freshly prepared 5 mm stock solution in ethanol was added to HEPES (20 mm) adjusted to pH 5.0 to 7.7. The decrease of A290 due to the conversion of MDH2 to MD was recorded for ≤1 min. At pH > 7.5, the rates were too rapid to be followed accurately. B, Similar measurements recording the reduction of XTT (100 μm) with or without SOD (100 μg mL−1) in the presence of MDH2 (100 μm).
Figure 10.

Molar relations in the electron transfer from MDH2 to O2 (A), XTT (B), or Cyt c (C) at pH 7.5. A, Various amounts of MDH2 dissolved in ethanol were added to HEPES in the absence or presence of XTT (100 μm). The amounts of O2 consumed were measured when the reaction was completed (after 6–10 min). B and C, Reduction of XTT (100 μm; B) or Cyt c (100 μm; C) in the absence or presence of SOD (100 μg mL−1) measured after completion of the reaction (after 1–2 min).
The kinetics of MDH2 autoxidation, concomitant O2 uptake, and reduction of added XTT or Cyt c at pH 7.5 revealed that XTT and Cyt c are more potent acceptors than O2 for electrons provided by MDH2 (Fig. 11). Reducing the O2 concentration to about 10 μm strongly reduced MDH2 autoxidation (Fig. 11A) but affected XTT reduction only slightly (Fig. 11C) and Cyt c reduction not at all (Fig. 11D). Hence, electron transfer from MDH2 to XTT or Cyt c also can take place without the intermediary formation of O2·−. Moreover, aerobic MDH2 oxidation was slowed by SOD, indicating that this reaction depends in part on the formation of O2·− (Fig. 11A).
Figure 11.

Kinetics of the autoxidation of MDH2 (A), concomitant uptake of O2 (B), and reduction of added XTT (C) or Cyt c (D) at pH 7.5. A, Oxidation of MDH2 (100 μm) was followed (A290) in 260 μm O2 (aerobic) and about 10 μm O2 (anaerobic) in the absence or presence of SOD (100 μg mL−1). B, Decrease of O2 concentration during the oxidation of MDH2. C and D, Reduction of XTT (200 μm; C) or Cyt c (200 μm; D) added to 100 μm MDH2 aerobically or anaerobically.
In summary, basic features observed in the redox reaction catalyzed by the isolated oxidoreductase are also found in the nonenzymatic reactions of MDH2 in neutral or alkaline solutions. The enzyme functions as a bona fide NQR that catalyzes an obligate two-electron reduction of certain quinones and is consequently insensitive to inhibition by DPI. However, some naphthohydroquinones produced by the enzyme, such as MDH2, can act as converters from two-electron- to one-electron-transferring reactions, depending on their propensity to undergo autoxidation after activation at high pH (compare with Table III). During the reaction, intermediates are produced that can react either with O2-forming O2·− or with artificial one-electron acceptors such as XTT or Cyt c. In retrospect, it is interesting that an NQR assay has been developed based on this principle (Prochaska and Santamaria, 1988). The differing susceptibility to autoxidation of various naphthohydroquinones produced by DT-diaphorase has been described by Buffinton et al. (1989). Lind et al. (1982) noted that MDH2 was stable at pH ≤ 6.5 but underwent autoxidation at pH 7.5.
Reaction Mechanisms Involved in the Oxidation of MDH2 by O2 or XTT
The ability of redox-labile hydroquinones to form semiquinones in alkaline solution (i.e. in their anionic form) is well known in chemistry (Brunmark and Cadenas, 1989). A general scheme illustrating the reactions involved in MDH2 autoxidation via the semiquinone (MD·−) intermediate state is depicted in Figure 12. MDH2 produced by NQR (reaction 1) and activated by deprotonation at pH ≥ 6.5 (reaction 2) can undergo conversion to MD via two routes (A and B) involving three one-electron-transferring reactions (Cadenas, 1995):
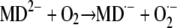 |
(3) |
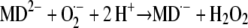 |
(4) |
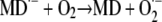 |
(5) |
Figure 12.

Redox chemistry involved in the MD-mediated univalent reduction of O2 by route A and route B. Reaction 1, Divalent enzymatic reduction of MD to MDH2 by NQR; reaction 2, activation of MDH2 by deprotonation; reaction 3, univalent oxidation of MD2−, producing MD·− (semiquinone) by reducing O2 to O2·−; reaction 4, univalent oxidation of MD2−, producing MD·− by reducing O2·− to H2O2; reaction 5, oxidation of MD·− by reducing O2 to O2·−.
After initiation by reaction 3, autoxidation can be propagated through the redox cycle of route B, reaching a steady-state rate after overcoming a lag phase (compare with. Figs. 4A and 11, A and B). If present, XTT reacts with O2·− according to:
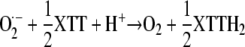 |
(6) |
Moreover, the data from Figures 10 and 11 demonstrate that XTT can replace O2 by reacting either with MD2− or with MD·−:
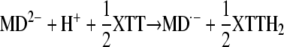 |
(7) |
or
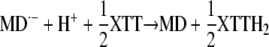 |
(8) |
Because of the negative standard redox potential of the MD·−/MD couple (−203 mV) compared with the MD2−/MD·− couple (+193 mV; Öllinger et al., 1990), reaction 8 will be thermodynamically strongly favored over reaction 7. However, in either case, only 50% of the XTT will be reduced via O2·− (reaction 6) and route B will be shut off due to the lack of O2·−. This explains the finding that the overall reaction can be inhibited by 50% with SOD (compare with Table IV; Figs. 9B and 10B). The lower inhibitory effect of SOD in the case of Cyt c (Fig. 10C) could be due to the ability of this acceptor to react to some extent with both MD2− and MD·−. Evidently, XTT and Cyt c reduction specifically detect O2·− production only to the extent that can be inhibited by SOD.
The autoxidation of the hydroquinones is affected by SOD in various ways (Öllinger et al., 1990). As shown in Figure 11A, SOD causes a partial inhibition of the O2-limited MDH2 oxidation. The removal of O2·− eliminated electron flow through route B. No effect of SOD on NADPH oxidation, MDH2 accumulation, and O2 consumption was detected in the much slower autoxidation reaction limited by the enzymatic production of MDH2 (Table IV), suggesting that O2·− had no major influence on autoxidation under these conditions.
Similar to SOD, electron acceptors such as XTT or Cyt c will decrease the concentration of O2·− in the reaction system, shutting down route B. Nevertheless, electron flow is strongly enhanced even at very low O2 concentrations (Fig. 11, C and D). These results show that route A is sufficient to mediate rapid electron flow provided that suitable sinks are available to remove O2·− and MD·−.
Potential Function of NQR in the Plasma Membrane of Intact Cells
Based on their biochemical properties, quinones and their reduced forms can exhibit both antioxidant and prooxidant activities. For example, linoleic acid autoxidation can be inhibited by phylloquinone and enhanced by phyllohydroquinone (Canfield et al., 1985). With respect to their biological functions in animal cells, cytosolic two-electron-transferring quinone reductases are often considered to act as antioxidant enzymes, protecting the cell against the harmful accumulation of quinones that can produce toxic radicals upon reduction by one-electron-transferring reductases (Lind et al., 1982; Cadenas, 1995; Ross, 1997). In plants, an antioxidant function of two-electron-transferring quinone reductases was recently supported by the observation that the expression of these enzymes can be upregulated in vivo under stress conditions that involve the formation of toxic oxidants (Matvienko et al., 2001; Greenshields et al., 2005). On the other hand, the supposition that these enzymes generally serve as antioxidant tools to protect against quinone toxicity has been challenged on the ground that redox-labile hydroquinones can autoxidize in vivo with the formation of reactive oxygen species, including hydroxyl radicals (Nutter et al., 1992; Dicker and Cederbaum, 1993; Cadenas, 1995). The plasma membrane plays an important role as a site of O2·− and H2O2 generation, serving functions such as cell wall expansion or pathogen defense. Although this performance is often ascribed to NOX-type enzymes (Gapper and Dolan, 2006), alternative sources of reactive oxygen species cannot be excluded. This is especially true in those cases in which O2·− production cannot be inhibited by even high concentrations of DPI and in which a significant part of XTT or Cyt c reduction is resistant to inhibition by SOD. Examples of striking DPI insensitivity of O2·− production or transmembrane electron transport by plasma membrane vesicles have been reported (Menckhoff and Lüthje, 2004; Mojović et al., 2004). As shown here, the NQR purified from plasma membranes has a potential to elicit O2·− production, unaffected by DPI, if supplied with a quinone substrate such as MD. Autoxidation of the hydroquinone produced in this way can lead directly to O2·− and H2O2 formation. Alternatively, coupling of O2·− generation to hydroquinone oxidation could be accomplished by peroxidase at the apoplastic side of the plasma membrane. Ohnishi et al. (1969) have shown that horseradish peroxidase catalyzes the conversion of hydroquinones to semiquinone radicals in the presence of H2O2, even at acidic pH not permitting autoxidation. In both cases, a low-potential cytochrome as a one-electron donor for the reduction of O2 would be unnecessary. A low-potential b-type cytochrome is an essential component of NOX enzymes but in fact has never been detected in plasma membranes of plants (Bérczi and Møller, 2000). Quinones such as ubiquinone or plastoquinone function as lipid-soluble, mobile electron carriers in mitochondrial or thylakoid membranes, suggesting a similar condition in the plasma membrane (Barr et al., 1992). Specifically, this property could be utilized to mediate an electron transport from NAD(P)H oxidation by NQR at the cytoplasmic face to O2 reduction at the apoplastic face of the plasma membrane. In this case, only catalytic amounts of quinone would be required, which undergo redox cycling and thereby transport electrons (and protons) across the membrane, in agreement with the net equation describing the reaction catalyzed by NOX:
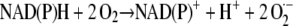 |
Basic questions arising in this context concern the access to substrates and the environmental conditions for the activity of NQR in or at the plasma membrane in vivo. These questions are still wide open. Nevertheless, several findings have been reported hinting at a redox-mediating function of quinones also in the plasma membrane (e.g. the inhibition of electron transport processes by “vitamin K antagonists” such as dicumarol; Döring et al., 1992; Lüthje et al., 1994). Although MD (vitamin K3) is not a normal constituent of the plasma membrane, a substance with the chromatographic and spectroscopic properties of phylloquinone (menadione with a phytyl side chain, vitamin K1) has been isolated from plasma membrane preparations of maize roots (Lüthje et al., 1998; Lochner et al., 2003). Phylloquinone serves as an electron transfer component in PSI of photosynthesis (Brettel, 1997). Unfortunately, the solubility of phylloquinone in water is extremely low, preventing a direct test of its activity as a substrate for NQR in aqueous solution (compare with Table III). The reactivity of the prenyl-menadione derivatives (vitamin K series) with DT-diaphorase in aqueous solution decreases with the length of their prenyl side chain and parallel to their insolubility in water (Märki and Martius, 1960). This suggests that in lipophilic surroundings, phylloquinone can act as a substrate for NQR. It has indeed been demonstrated that phylloquinone is easily reduced by the DT-diaphorase if it is incorporated into the membrane of liposomes and thus can transport electrons from the outside to the inside of this vesicle (Martius et al., 1975).
MATERIALS AND METHODS
Materials
Seedlings of soybean (Glycine max ‘Jutro’) were grown on wet vermiculite in darkness for 4.5 d at 25°C. The top 2 cm of the hypocotyl was excised and used for plasma membrane preparation.
Chemicals were obtained from Merck (polyethylene glycol 6000), Sigma or Aldrich (quinones, horseradish peroxidase, Cibacron 3G-A, dicumarol, DPI, nitrofurantoin, rotenone, piericidin A, N-ethylmaleimide, 4-chloromercuribenzoate, quinacrine, and bicinchoninic acid), Amersham Bioscience (Blue Sepharose 6 Fast Flow), Pharmacia (Superdex 200 HR 10/30 column), Roche Biochemicals (Cyt c, Cu/Zn-SOD, and catalase), and Polyscience (XTT).
MDH2 was synthesized after Fieser et al. (1939) and purified by silica gel chromatography.
Quinone stock solutions (100 mm) were prepared in dimethyl sulfoxide or ethanol (vitamins K1 and K2), diluted with assay buffer to 1 mm, and used within the next 2 h. Juglone solutions had to be kept in darkness to prevent disintegration. MDH2 was dissolved in water-free ethanol and added directly to the reaction medium.
Preparation and Solubilization of Plasma Membranes
The procedure devised by Thein and Michalke (1988) was adopted with some modifications. Batches (100 g) of hypocotyl segments were homogenized with a blender in 400 mL of cold medium containing 10 mm Tris-HCl (pH 8.0), 20 mm Na2EDTA, 300 mm NaCl, 0.5 mm phenylmethylsulfonyl fluoride, and 1 g L−1 bovine serum albumin. After filtering the slurry through nylon cloth, a membrane fraction was isolated by differential centrifugation (30 min at 1,500g, 45 min at 100,000g, 4°C) and resuspended in 180 mL of medium (without phenylmethylsulfonyl fluoride and albumin). Intracellular membranes were precipitated by adding 0.043 g mL−1 polyethylene glycol 6000 and stirring for 30 min on ice. After removing the precipitate by centrifugation (30 min at 1,500g, 4°C), the plasma membranes contained in the supernatant were pelleted (45 min at 100,000g, 4°C), resuspended in 3 mL of 20 mm HEPES (pH 7.8) containing 20 mm Na2EDTA and 250 mm Suc, and stored at −70°C.
The purity of the isolated membrane fraction was ascertained by separating the proteins of the different fractions by SDS-PAGE followed by semidry blotting on polyvinylidene difluoride membranes and decoration of the membranes with specific polyclonal antibodies directed against the Rieske protein (mitochondria) and the plasma membrane P-ATPase. The plasma membrane fraction showed the strongest signal with the anti-P-ATPase antibodies and no signal with the anti-Rieske antibodies, indicating that there was no detectable contamination by mitochondrial membranes (Supplemental Fig. S1). Conventional two-phase partitioning (Larsson et al., 1994) of microsomes resulted in a plasma membrane fraction with very similar properties if tested according to these criteria.
The cytosolic protein fraction was prepared from the homogenate by removing insoluble material by centrifugation (45 min at 100,000g, 4°C).
Membrane localization of the oxidoreductase activity was tested by incubating plasma membranes in 1.5 m CaCl2 or 1.5 m NaCl for 1 h at 4°C in 20 mm HEPES (pH 7.8), followed by washing three times with the same buffer.
Plasma membrane suspensions were solubilized by adding an equal volume of 4% (w/v) Tween 20 and incubating for 30 min at 30°C. After clearing by centrifugation (15 min at 12,000g), the enzyme solution could be stored at −70°C without loss of activity for several months. The contamination of the solubilized plasma membrane fraction with soluble proteins was estimated by measuring the activity of the cytosolic marker enzyme Glc-6-P dehydrogenase. The concentration of the Glc-6-P dehydrogenase in the membrane fraction was about 5% of the concentration expected if the membrane vesicles contained the same concentration as the cytosolic tissue fraction.
Protein Determination
The protein contents of enzyme extracts were estimated with the bicinchoninic acid procedure as described by Kaushal and Barnes (1986) using bovine serum albumin as a standard. This method provides reliable results in the presence of detergents such as Tween 20 at protein concentrations of ≥1 μg mL−1.
Enzyme Assays
Photometric measurements of reaction rates at specific wavelengths allowed the determination of XTT reduction, NAD(P)H oxidation, and hydroquinone accumulation in mixtures of these components. The standard assay mixture contained 200 μm NADPH, 100 μm MD, 100 μm XTT, and enzyme extract (start of reaction) in 20 mm HEPES (pH 7.5) in a total volume of 500 μL (25°C). XTT reduction was measured at 470 nm (ɛ470 = 24.2 mm−1 cm−1) or 550 nm (ɛ550 = 9.8 mm−1 cm−1) for 1 to 2 min. NADPH oxidation was measured at 340 nm (ɛ340 = 6.21 mm−1 cm−1) and corrected for absorbance changes caused by XTT reduction (ɛ340 = 5.4 mm−1 cm−1). Absorbance changes due to MD reduction at 340 nm (ɛ340 = −0.09 mm−1 cm−1) are negligible. Because of overlapping absorbance spectra, the determination of coupled NADPH oxidation and MDH2 accumulation (in the absence of XTT) required measurements at two wavelengths (290 and 340 nm) and mathematical separation of the two partial reactions using the following formulas (1-cm cuvette):
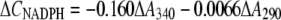 |
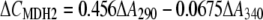 |
Suitable wavelengths pairs and extinction coefficients for corresponding measurements with other quinones were determined from difference spectra (oxidized → reduced) as outlined in Supplemental Appendix S1. Modifications of the standard assays are described in the figure legends.
FeCN (500 μm) reduction was measured at 420 nm (ɛ420 = 1.02 mm−1 cm−1). DCPIP (30 μm) reduction was measured at 600 nm (ɛ600 = 22.0 mm−1 cm−1). Cyt c (50 μm) reduction was measured at 550 nm (ɛ550 = 21.0 mm−1 cm−1).
Anaerobic conditions during the enzyme reaction were obtained by evacuating the reaction mixture followed by gassing with argon. This lowered the O2 concentration by about 96% (10 μm).
Peroxidase activity was measured at 25°C either with 8.3 mm guaiacol and 8.8 mm H2O2 in 50 mm citrate-NaOH at pH 5.0 (470 nm, ɛ470 = 26.6 mm−1 cm−1; Mika and Lüthje, 2003) or with 100 μm 3,3′,5,5′-tetramethylbenzidine and 5 mm H2O2 in the same buffer at pH 4.5 (654 nm, ɛ654 = 39 mm−1 cm−1; Imberty et al., 1984). Tested with horseradish peroxidase (Sigma type VI; 300 purpurogallin units mg−1 protein), the detection limits of these assays were 1 ng and 10 pg, respectively, in 1 mL of reaction mixture.
Oxygen consumption was measured with a Clark-type oxygen electrode using 2 mL of standard assay mixture (25°C).
PAGE and Western Blotting
SDS-PAGE was carried out in 8% polyacrylamide. Proteins were visualized by silver staining. Native PAGE was carried out using 0.1% CHAPS instead of SDS. The sample buffer contained 40 mm Tris-HCl (pH 7.8), 0.1% CHAPS, 10% glycerol, and 0.002% bromphenol blue. Samples were mixed with sample buffer in a 2:1 ratio. The running buffer contained 25 mm Tris, 1.44% Gly, and no detergent. Gels were incubated for 20 min in 50 mm Tris-HCl (pH 7.4), 0.2 mm NBT, 0.1 mm MgCl2, and 1 mm CaCl2 in the dark. The reaction was started after 20 min of preincubation by adding 0.2 mm NADH and 0.1 mm MD and continued for approximately 15 min.
Proteins were transferred onto nitrocellulose (SDS-PAGE) or polyvinylidene difluoride (native PAGE) membranes by semidry blotting using a Multiphor II Novablot unit (Amersham Bioscience). For detection, the ECL system (Amersham Bioscience) was used according to the manufacturer's protocol.
Protein Identification by HPLC-Electrospray Mass Spectrometry
The NBT-stained region of a native PAGE gel derived from a sample of purified plasma membrane oxidoreductase (Fig. 7A, lane 3) was cut into 10 horizontal 2-mm strips that were processed individually to establish abundance profiles of the identified peptides. After destaining the gel slices and reduction of proteins by 10 mm 1,4-dithiothreitol, Cys residues were modified by iodoacetamide at a final concentration of 55 mm for 30 min at room temperature. After washing in 5 mm NH4HCO3 and dehydration by ethanol, 0.25 μg of trypsin (Promega) in 5 mm NH4HCO3 was added to each sample and incubated on ice for 20 min. The buffer was exchanged to 5 mm NH4HCO3, and digestion was done overnight at 37°C. Peptides were extracted successively with 0.1% formic acid and acetonitrile, dried, and resuspended in 3% acetonitrile and 0.1% formic acid.
Peptide mixtures were separated for nano-LC-electrospray ionization-MS/MS using a FAMOS autosampler (Dionex), an Ultimate inert HPLC system (Dionex), and an Agilent HPLC 1100 pump connected to the nano-electrospray ionization source of a Finnigan LTQ-FT (Thermo Electron Corporation) for online mass detection. Peptides were first collected on a trap column (0.1 × 15 mm, Zorbax Eclipse XDB-C18, 5 μm; Agilent Technology) for desalting and concentrating followed by separation on an analytical column made up by fused silica emitters (0.075 × 105 mm, 6 μm; Proxeon Biosystems) filled with Hydrosphere C18, 3 μm (YMC). Peptides were eluted during a 60-min gradient using 97% water, 3% acetonitrile, and 0.1% formic acid as solvent A and 80% acetonitrile, 20% water, and 0.1% formic acid as solvent B at a flow rate of 0.15 μL min−1.
Mass spectrometric detection consisted of full scans at a resolution of 25,000 followed by data-dependent selected ion scans at a resolution of 50,000 and low-resolution MS/MS scans using a dynamic exclusion of parent ion masses for 60 s.
The MS and MS/MS spectra were searched against the soybean EST assembly from PlantGDB (Dong et al., 2005; release October 2007, based on GenBank release 161) and soybean and Arabidopsis (Arabidopsis thaliana) proteins from the uniprot database (release 12.4, October 2007) using an in-house installation of the program OMSSA (Geer et al., 2004; version 2.1). Search results were filtered and sorted using in-house-written software (F. Drepper, unpublished data). Peptide hits were considered significant if the precursor and product ion masses matched within 2 ppm and 0.5 relative mass units, respectively, and if the E-value was below 0.01.
Statistics
Data represent means or representative examples from measurements repeated three to six times. Typical se values are shown in Figure 5 and Tables I, III, and IV but omitted in other figures for the sake of clarity.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Comparison of two methods of plasma membrane preparation.
Supplemental Table S1. NQR sequence alignment.
Supplemental Appendix S1. Formulas and extinction coefficients used for the determination of NAD(P)H oxidation and quinone reduction in NADPH/quinone mixtures.
Supplementary Material
Acknowledgments
We are grateful to K. Kienzler and B. Knapp (both Universität Freiburg) for excellent technical assistance; Dr. W. Seiche (Universität Freiburg) for synthesizing menadionehydroquinone; Drs. F. Sparla and P. Trost (Università di Bologna) for stimulating discussions and provision of NQR antibodies and AtNQR recombinant protein; and Dr. M. Laskowski (Oberlin College), Dr. W. Michalke (Universität Freiburg), and Dr. U. Schulte (Universität Düsseldorf) for providing antibodies directed against FQR1, P-ATPase, and Rieske protein, respectively. Special thanks are due to Dr. W. Haehnel (Universität Freiburg) for placing his mass spectrometry facilities at our disposal.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Anja Krieger-Liszkay (anja.krieger-liszkay@cea.fr).
The online version of this article contains Web-only data.
References
- Able AJ, Guest DI, Sutherland MW (1998) Use of a new tetrazolium-based assay to study the production of superoxide radicals by tobacco cell cultures challenged with avirulent zoospores of Phytophthora parasitica var nicotinae. Plant Physiol 117 491–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal R, Bonanno JB, Burley SK, Swaminathan S (2006) Structure determination of an FMN reductase from Pseudomonas aeruginosa PA01 using sulphur anomalous signal. Acta Crystallogr D Biol Crystallogr 62 383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr R, Pan RS, Crane FL, Brightman AO, Morré DJ (1992) Destruction of vitamin K1 of cultured carrot cells by ultraviolet radiation and its effect on plasma membrane electron transport reactions. Biochem Int 27 449–456 [PubMed] [Google Scholar]
- Bateman A, Sandford R (1999) The PLAT domain: a new piece in the PKD1 puzzle. Curr Biol 9 588–590 [DOI] [PubMed] [Google Scholar]
- Bérczi A, Møller IM (1998) NADH-monodehydroascorbate oxidoreductase is one of the redox enzymes in spinach leaf plasma membranes. Plant Physiol 116 1029–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bérczi A, Møller IM (2000) Redox enzymes in the plant plasma membrane and their possible roles. Plant Cell Environ 23 1287–1302 [Google Scholar]
- Bienfait F, Lüttge U (1988) On the function of two systems that can transfer electrons across the plasma membrane. Plant Physiol Biochem 26 665–671 [Google Scholar]
- Brettel K (1997) Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim Biophys Acta 1318 322–373 [Google Scholar]
- Brunmark A, Cadenas E (1989) Redox and addition chemistry of quinoid compounds and its biological implications. Free Radic Biol Med 7 435–477 [DOI] [PubMed] [Google Scholar]
- Buffinton GD, Öllinger K, Brunmark A, Cadenas E (1989) DT-diaphorase-catalysed reduction of 1,4-naphthoquinone derivatives and glutathionyl-quinone conjugates: effect of substituents on autoxidation rates. Biochem J 257 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E (1995) Autoxidant and prooxidant functions of DT-diaphorase in quinone metabolism. Biochem Pharmacol 49 127–140 [DOI] [PubMed] [Google Scholar]
- Canfield LM, Davy LA, Thomas GL (1985) Anti-oxidant/pro-oxidant reactions of vitamin K. Biochem Biophys Res Commun 128 211–219 [DOI] [PubMed] [Google Scholar]
- Cross AR (1987) The inhibitory effects of some iodonium compounds on the superoxide generating system of neutrophils and their failure to inhibit diaphorase activity. Biochem Pharmacol 36 489–493 [DOI] [PubMed] [Google Scholar]
- Dicker E, Cederbaum AI (1993) Requirement for iron for the production of hydroxyl radicals by rat liver quinone reductase. J Pharmacol Exp Ther 266 1282–1290 [PubMed] [Google Scholar]
- Döring O, Lüthje S, Böttger M (1992) Inhibitors of the plasma membrane redox system of Zea mays L. roots: the vitamin K antagonists dicumarol and warfarin. Biochim Biophys Acta 1110 235–238 [DOI] [PubMed] [Google Scholar]
- Dong Q, Lawrence CJ, Schlueter SD, Wilkerson MD, Kurtz S, Lushbough C, Brendel V (2005) Comparative plant genomics resources at PlantGDB. Plant Physiol 139 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doussiere J, Gaillard J, Vignais PV (1999) The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry 38 3694–3703 [DOI] [PubMed] [Google Scholar]
- Doussiere J, Vignais PV (1992) Diphenylene iodonium as an inhibitor of the NADPH oxidase complex of bovine neutrophils. Factors controlling the inhibitory potency of diphenylene iodonium in a cell-free system of oxidase activation. Eur J Biochem 208 61–71 [DOI] [PubMed] [Google Scholar]
- Ernster L, Danielson L, Ljunggren M (1962) DT diaphorase. I. Purification from the soluble fraction of rat liver cytoplasm, and properties. Biochim Biophys Acta 58 171–188 [DOI] [PubMed] [Google Scholar]
- Fieser LF, Campbell WP, Fry EM, Gates MD (1939) Naphthoquinones of the vitamin K1 type of structure. J Am Chem Soc 61 3216–3223 [Google Scholar]
- Frahry G, Schopfer P (1998) Inhibition of O2-reducing activity of horseradish peroxidase by diphenyleneiodonium. Phytochemistry 48 223–227 [DOI] [PubMed] [Google Scholar]
- Gapper C, Dolan L (2006) Control of plant development by reactive oxygen species. Plant Physiol 141 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH (2004) Open mass spectrometry search algorithm. J Proteome Res 3 958–964 [DOI] [PubMed] [Google Scholar]
- Greenshields DL, Liu G, Selvaray GLG, Wei Y (2005) Differential regulation of wheat quinone reductases in response to powdery mildew infection. Planta 222 867–875 [DOI] [PubMed] [Google Scholar]
- Imberty A, Goldberg R, Catesson AM (1984) Tetramethylbenzidine and p-phenylenediamine-pyrocatechol for peroxidase histochemistry and biochemistry: two new, non-carcinogenic chromogens for investigating lignification processes. Plant Sci Lett 35 103–108 [Google Scholar]
- Iyanagi T, Yamazaki I (1970) One-electron-transfer reactions in biological systems. V. Difference in the mechanism of quinone reduction by the NADH dehydrogenase and the NAD(P)H dehydrogenase (DT-diaphorase). Biochim Biophys Acta 216 282–294 [DOI] [PubMed] [Google Scholar]
- Kaushal V, Barnes LD (1986) Effect of zwitterionic buffers on measurements of small masses of protein with bicinchoninic acid. Anal Biochem 157 291–294 [DOI] [PubMed] [Google Scholar]
- Keller T, Damude HG, Werner D, Doerner P, Dixon RA, Lamb C (1998) A plant homolog of the neutrophil NADPH oxidase gp91phox subunit gene encodes a plasma membrane protein with Ca2+ binding motifs. Plant Cell 10 255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Kawakita K, Maeshima M, Doke N, Yoshioka H (2006) Subcellular localization of Strboh proteins and NADPH-dependent O2−-generating activity in potato tissues. J Exp Bot 57 1373–1379 [DOI] [PubMed] [Google Scholar]
- Larsson C, Sommarin M, Widell S (1994) Isolation of highly purified plant plasma membranes and separation of inside-out and right-side-out vesicles. Methods Enzymol 228 451–469 [Google Scholar]
- Laskowski MJ, Dreher KA, Gehring MA, Abel S, Gensler AL, Sussex IM (2002) FQR1, a novel primary auxin-response gene, encodes a flavin mononucleotide-binding quinone reductase. Plant Physiol 128 578–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind C, Cadenas E, Hochstein P, Ernster L (1990) DT-diaphorase: purification, properties, and function. Methods Enzymol 186 287–301 [DOI] [PubMed] [Google Scholar]
- Lind C, Hochstein P, Ernster L (1982) DT-diaphorase as a quinone reductase: a cellular device against semiquinone and superoxide radical formation. Arch Biochem Biophys 216 178–185 [DOI] [PubMed] [Google Scholar]
- Lochner K, Döring O, Böttger M (2003) Phylloquinone, what can we learn from plants? Biofactors 18 73–78 [DOI] [PubMed] [Google Scholar]
- Luster DG, Buckhout TJ (1989) Purification and identification of a plasma membrane associated electron transport protein from maize (Zea mays L.) roots. Plant Physiol 91 1014–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüthje S (2007) Plasma membrane redox systems: lipid rafts and protein assemblies. Prog Bot 69 169–200 [Google Scholar]
- Lüthje S, González-Reyes JA, Navas P, Döring O, Böttger M (1994) Inhibition of maize (Zea mays L.) root plasma membrane-bound redox activities by coumarins. Z Naturforsch 49c 447–452 [Google Scholar]
- Lüthje S, van Gestelen P, Córdoba-Pedregosa MC, González-Reyes JA, Asard H, Villalba JM, Böttger M (1998) Quinones in plant plasma membranes: a missing link? Protoplasma 205 43–51 [Google Scholar]
- Märki F, Martius C (1960) Vitamin K-Reduktase, Darstellung und Eigenschaften. Biochem Z 333 111–135 [PubMed] [Google Scholar]
- Martius C, Ganser R, Viviani A (1975) The enzymatic reduction of K-vitamins incorporated in the membrane of liposomes. FEBS Lett 59 13–14 [DOI] [PubMed] [Google Scholar]
- Matvienko M, Wojtowicz A, Wrobel R, Jamison D, Goldwasser Y, Yoder JI (2001) Quinone oxidoreductase message levels are differentially regulated in parasitic and non-parasitic plants exposed to allelopathic quinones. Plant J 25 375–387 [DOI] [PubMed] [Google Scholar]
- Menckhoff M, Lüthje S (2004) Transmembrane electron transport in sealed and NAD(P)H-loaded right-side-out plasma membrane vesicles isolated form maize (Zea mays L.) roots. J Exp Bot 55 1343–1349 [DOI] [PubMed] [Google Scholar]
- Mika A, Lüthje S (2003) Properties of guaiacol peroxidase activities isolated from corn root plasma membranes. Plant Physiol 132 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojović M, Vuletić M, Bačić GG, Vučinić Z (2004) Oxygen radicals produced by plant plasma membranes: an EPR spin-trap study. J Exp Bot 55 2523–2531 [DOI] [PubMed] [Google Scholar]
- Murphy TM, Auh CK (1996) The superoxide synthases of plasma membrane preparations from cultured rose cells. Plant Physiol 110 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TM, Vu H, Nguyen T, Woo CH (2000) Diphenylene iodonium sensitivity of a solubilized membrane enzyme from rose cells. Protoplasma 213 228–234 [Google Scholar]
- Nutter LM, Ngo EO, Fisher GR, Gutierrez PL (1992) DNA strand scission and free radical production in menadione-treated cells. Correlation with cytotoxicity and role of NADPH quinone acceptor oxidoreductase. J Biol Chem 267 2474–2479 [PubMed] [Google Scholar]
- O'Donnell VB, Smith GCM, Jones OTG (1994) Involvement of phenyl radicals in iodonium compound inhibition of flavoenzymes. Mol Pharmacol 46 778–785 [PubMed] [Google Scholar]
- Ohnishi T, Yamazaki H, Iyanagi T, Nakamura T, Yamazaki I (1969) One-electron-transfer reactions in biochemical systems. II. The reaction of free radicals formed in the enzymic oxidation. Biochim Biophys Acta 172 357–369 [DOI] [PubMed] [Google Scholar]
- Öllinger K, Buffinton GD, Ernster L, Cadenas E (1990) Effect of superoxide dismutase on the autoxidation of substituted hydro- and semi-naphthoquinones. Chem Biol Interact 73 53–76 [DOI] [PubMed] [Google Scholar]
- Patridge EV, Ferry JG (2006) WrbA from Escherichia coli and Archaeoglobus fulgidus is an NAD(P)H:quinone oxidoreductase. J Bacteriol 188 3498–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestera T, Prochaska HJ, Talalay P (1992) Inhibition of NAD(P)H:(quinone-acceptor) oxidoreductase by Cibacron blue and related anthraquinone dyes: a structure-activity study. Biochemistry 31 824–833 [DOI] [PubMed] [Google Scholar]
- Prochaska HJ, Santamaria AB (1988) Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: a screening assay for anticarcinogenic enzyme inducers. Anal Biochem 169 328–336 [DOI] [PubMed] [Google Scholar]
- Ragan CI, Bloxham DP (1977) Specific labelling of a constituent polypeptide of bovine heart mitochondrial reduced nicotinamide-adenine-dinucleotide-ubiquinone reductase by the inhibitor diphenyleneiodonium. Biochem J 163 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescigno A, Sollai F, Masala S, Porcu MC, Sanjust E, Rinaldi AC, Curreli N, Grifi D, Rinaldi A (1995) Purification and characterization of an NAD(P)H:quinone oxidoreductase from Glycine max seedlings. Prep Biochem 25 57–67 [DOI] [PubMed] [Google Scholar]
- Ross D (1997) Quinone reductases. In FP Guengrich, ed, Comprehensive Toxicology, Vol 3. Pergamon Press, New York, pp 179–197
- Sagi M, Fluhr R (2001) Superoxide production by plant homologues of the gp91phox NADPH oxidase: modulation of activity by calcium and by tobacco mosaic virus infection. Plant Physiol 126 1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Córdoba F, González-Reyes JA, Navas P, Villalba JM (1994) Purification and characterization of two distinct NAD(P)H dehydrogenases from onion (Allium cepa L.) root plasma membrane. Plant Physiol 106 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Plas F, Elmayan T, Blein J-P (2002) The plasma membrane oxidase NtrbohD is responsible for AOS production in elicited tobacco cells. Plant J 31 137–147 [DOI] [PubMed] [Google Scholar]
- Sparla F, Tedeschi G, Pupillo P, Trost P (1998) Molecular characterization of NAD(P)H:quinone oxidoreductase of tobacco leaves. Protoplasma 205 52–58 [Google Scholar]
- Sparla F, Tedeschi G, Pupillo P, Trost P (1999) Cloning and heterologous expression of NAD(P)H:quinone reductase of Arabidopsis thaliana, a functional homologue of animal DT-diaphorase. FEBS Lett 463 382–386 [DOI] [PubMed] [Google Scholar]
- Sparla F, Tedeschi G, Trost P (1996) NAD(P)H:(quinone acceptor) oxidoreductase of tobacco leaves is a flavin mononucleotide-containing flavoenzyme. Plant Physiol 112 249–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitsberg VL, Coscia CJ (1982) Quinone reductases of higher plants. Eur J Biochem 127 67–70 [DOI] [PubMed] [Google Scholar]
- Strain HH, Svec WA (1966) Extraction, separation, estimation and isolation of the chlorophylls. In LP Vernon, GR Seely, eds, The Chlorophylls. Academic Press, New York, pp 21–66
- Sumimoto H, Miyano K, Takeya R (2005) Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun 338 677–686 [DOI] [PubMed] [Google Scholar]
- Thein M, Michalke W (1988) Bisulfite interacts with binding sites of the auxin-transport inhibitor N-1-napthylphtalamic acid. Planta 176 343–350 [DOI] [PubMed] [Google Scholar]
- Torres MA, Dangl JL (2005) Functions of the respiratory burst oxidase in biotic interactions, abiotic stress and development. Curr Opin Plant Biol 8 397–403 [DOI] [PubMed] [Google Scholar]
- Trost P, Bonora P, Scagliarini S, Pupillo P (1995) Purification and properties of NAD(P)H:(quinone acceptor) oxidoreductase of sugarbeet cells. Eur J Biochem 234 452–458 [DOI] [PubMed] [Google Scholar]
- Trost P, Foscarini S, Preger V, Bonora P, Vitale L, Pupillo P (1997) Dissecting the diphenylene iodonium-sensitive NAD(P)H:quinone oxidoreductase of zucchini plasma membrane. Plant Physiol 114 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti V, Guerrini F, Pupillo P (1990) NAD(P)H-duroquinone reductase in the plant plasma membrane. J Exp Bot 41 183–192 [Google Scholar]
- van Gestelen P, Asard H, Caubergs RJ (1997) Solubilization and separation of a plant plasma membrane NADPH-O2− synthase from other NAD(P)H oxidoreductases. Plant Physiol 115 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gestelen P, Asard H, Horemans N, Caubergs RJ (1998) Superoxide-producing NAD(P)H oxidases in plasma membrane vesicles from elicitor responsive bean plants. Physiol Plant 104 653–660 [Google Scholar]
- Vuletić M, Šukalović VHT, Vučinić Ž (2003) Superoxide synthase and dismutase activity of plasma membranes from maize roots. Protoplasma 221 73–77 [DOI] [PubMed] [Google Scholar]
- Vuletić M, Šukalović VHT, Vučinić Ž (2005) The coexistence of the oxidative and reductive systems in roots: the role of plasma membranes. Ann N Y Acad Sci 1048 244–258 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.