

Abstract
Despite many scientific advances, human exposure to, and intoxication by, toxic metal species continues to occur. Surprisingly, little is understood about the mechanisms by which certain metals and metal-containing species gain entry into target cells. Since there do not appear to be transporters designed specifically for the entry of most toxic metal species into mammalian cells, it has been postulated that some of these metals gain entry into target cells, through the mechanisms of ionic and/or molecular mimicry, at the site of transporters of essential elements and/or molecules. The primary purpose of this review is to discuss the transport of selective toxic metals in target organs and provide evidence supporting a role of ionic and/or molecular mimicry. In the context of this review, molecular mimicry refers to the ability of a metal ion to bond to an endogenous organic molecule to form an organic metal species that acts as a functional or structural mimic of essential molecules at the sites of transporters of those molecules. Ionic mimicry refers to the ability of a cationic form of a toxic metal to mimic an essential element or cationic species of an element at the site of a transporter of that element. Molecular and ionic mimics can also be sub-classified as structural or functional mimics. This review will present the established and putative roles of molecular and ionic mimicry in the transport of mercury, cadmium, lead, arsenic, selenium, and selected oxyanions in target organs and tissues.
Keywords: Metal, Mercury, Lead, Mimicry, Cadmium, Transport
Introduction
Metals, including essential and nonessential species, make up a significant fraction of all elements. Nutritive (essential) metals, such as copper (Cu), zinc (Zn), and iron (Fe), are required for normal cellular processes in both prokaryotes and eukaryotes, and thus, there are mechanisms in place to regulate their cellular uptake and accumulation. In contrast, toxic (nonessential) metals, such as mercury (Hg), cadmium (Cd), and lead (Pb) have no known nutritive value. Accordingly, no specific, dedicated mechanisms have evolved for their uptake, at least in most animal species. Yet, many studies have proven that these toxic metals do indeed gain entry into various target cells (for reviews, see Clarkson, 1993; Ballatori, 2002; Zalups, 2000a; Zalups and Ahmad, 2003).
A number of different mechanisms for the transport of toxic metal species exist in the animal kingdom. In recent years, the concepts of molecular mimicry and ionic mimicry have been postulated as mechanisms by which certain toxic metal species can gain entry into target cells. The term, “mimic” is defined by Webster's 3rd New International Dictionary of the English Language (2002) as a verb meaning “to copy or imitate very closely.” Molecular mimicry refers to the phenomenon whereby the bonding of metal ions to nucleophilic groups on certain biomolecules results in the formation of organo-metal complexes that can behave or serve as a structural and/or functional homolog of other endogenous biomolecules or the molecule to which the metal ion has bonded (Fig. 1; Ballatori, 2002; Clarkson, 1993; Zalups, 2000a). Ionic mimicry, on the other hand, refers to the ability of an unbound, native, cationic species of a metal to mimic an essential element or cationic form of that element (Clarkson, 1993; Wetterhahn-Jennette, 1981; Zalups and Ahmad, 2003). Molecular and ionic mimics may be classified as structural and/or functional mimics. A structural mimic refers to an elemental or molecular species that is similar in size and shape to another element or molecule. A functional mimic is one that can elicit the same effect (i.e., physiological response) as the native element or molecule. In the following sections, we discuss structural and functional mimicry in relation to molecular and/or ionic mimicry and the specific metal involved in these phenomena.
Fig. 1.
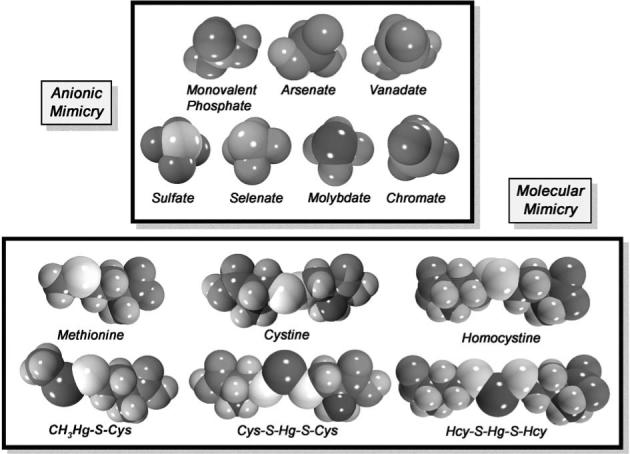
Space-filled models of selected mercuric conjugates and oxyanions implicated in molecular mimicry. Note the similarities in chemical structure between the cysteine (Cys) S-conjugate of methylmercury (CH3Hg-S-Cys) and the amino acid methionine. Also, note the similarities between the Cys S-conjugate of inorganic mercury (Cys-S-Hg-S-Cys) and amino acid cystine, and the homocysteine (Hcy) S-conjugate of inorganic mercury (Hcy-S-Hg-S-Hcy) of homocystine. A significant body of current evidence supports the hypothesis that mercuric conjugates of certain amino acids (such as Cys and Hcy) may act as molecular mimics of naturally occurring amino acids that are similar structurally to the mercuric complexes. Recent experimental findings from renal proximal tubular cells, transfected Madin-Darby canine kidney (MDCK) cells and oocytes from Xenopus laevis have demonstrated that Cys-S-Hg-S-Cys and Hcy-S-Hg-S-Hcy can act as molecular mimics of cystine and homocystine, respectively, at the sites of the luminal amino acid transporter, system b0,+ and the basolateral organic anion transporter, OAT1. There is also evidence from cultured endothelial cells and Xenopus laevis indicating that CH3Hg-S-Cys can serve as a molecular mimic of the amino acid methionine at the site of system L, which can explain the movement of methylmercury across the endothelium of the blood–brain barrier. There are also similarities in structure between monovalent phosphate and the oxyanionic forms of the toxic metals, arsenic (arsenate) or vanadium (vanadate). Both arsenate and vanadate have been shown to mimic phosphate at the site of phosphate transporters. In addition, the structure of sulfate is shown in comparison with the structures of selenate, molybdate, and chromate, which are homologous to sulfate. There is evidence indicating that these oxyanions can mimic sulfate at the site of transporters responsible for its uptake.
Molecular mimicry
It is thought that when metals bind to nucleophilic sites on certain biological molecules, the complexes formed are able to “mimic” structurally and/or functionally endogenous substrates that normally bind to, or occupy, the active sites of carrier proteins, channels, structural proteins, enzymes, and/or transcription factors. In recent years, a number of carrier proteins have been implicated in the transport of some toxic metals. In particular, amino acid transporters (i.e., system b0,+, system L; Bridges and Zalups, 2004; Bridges et al., 2004; Simmons-Willis et al., 2002) and organic anion transporters (i.e., OAT1 and OAT3; Aslamkhan et al., 2003; Zalups and Ahmad, 2004; Zalups et al., 2004) have been implicated in the absorptive transport of inorganic and organic forms of Hg in renal epithelial cells, endothelial cells and glial cells. Molecular mimicry has been implicated as the primary means for the entry of certain metals via these transporters. Interestingly, cationic forms of some metals can apparently mimic certain anionic complexes (i.e., oxyanions), as a form of molecular mimicry.
Ionic mimicry
In principle, ionic mimicry is similar to molecular mimicry. The term ionic mimicry has, for the most part, been used to describe the ability of an unbound, cationic species of a metal to behave or serve as a structural and/or functional homolog or mimic of another (usually an essential) element at the site of a carrier protein, ion channel, enzyme, structural protein, transcription factor and/or metal-binding protein. For example, a great deal of evidence has been accrued showing that the cationic species of certain toxic metals, such as Cd, can use ion channels (in particular calcium (Ca2+) channels) and certain membrane transporters, such as the divalent metal transporter 1 (DMT1/DCT1/Nramp1), to gain access into target cells of mammalian organisms.
A considerable amount of scientific data on molecular and ionic mimicry has been published in recent years. Notwithstanding, numerous questions remain unanswered. This review will focus on known and putative mechanisms by which several toxic metals gain access to the intracellular compartments of target cells affected adversely by these metals. Evidence supporting the phenomena of molecular and/or ionic mimicry for selected toxic metals will be outlined individually by species of metal and the organs, tissues, and cells involved in the process.
Mercury
Hg is a unique toxic metal-pollutant that is found in many environmental and occupational settings. It can exist in elemental (metallic), inorganic, and/or organic forms. Elemental Hg (Hg0) is unique among all metals in that it exists as a liquid at room temperature. Due to its high vapor pressure, Hg0 can be released readily into the atmosphere as Hg vapor. Inorganic forms of Hg, as mercurous (Hg1+) or mercuric (Hg2+) ions, commonly combine with anionic species of chlorine, sulfur, or oxygen to form mercurous or mercuric salts. The primary cation of Hg found in environmental settings is the mercuric ion. Organic mercuric compounds form when mercuric ions bind covalently with carbon atoms of certain small organic functional groups such as methyl, ethyl, or phenyl groups. The most frequently encountered form of organic Hg in the environment is methylmercury (CH3Hg+). It is formed primarily by methylation of inorganic mercuric ions by microorganisms in soil and water (reviewed in ATSDR, 2003a; Zalups, 2000a).
All forms of Hg are toxic to almost all members of the animal kingdom. However, the extent of the adverse effects induced by Hg in an organism depends on the form of Hg at the time of exposure, the duration of exposure, and the route of exposure. Exposure to all forms of Hg, at least to some extent, has been shown to cause deleterious effects in the cardiovascular system (Carmignani et al., 1992; Soni et al., 1992; Warkany and Hubbard, 1953; Wakita, 1987), gastrointestinal system (Afonso and deAlvarez, 1960; Bluhm et al., 1992; Lundgren and Swensson, 1949; Murphy et al., 1979), liver (Jaffe et al., 1983; Murphy et al., 1979; Samuels et al., 1982), kidneys (Murphy et al., 1979; Rowens et al., 1991; Samuels et al., 1982; Yasutake et al., 1991), and neurological system (Jaffe et al., 1983; Kutsuna, 1968; Lin and Lim, 1993; Tsubaki and Takahashi, 1986).
Unfortunately, the risk of humans being exposed to Hg is significant. Humans are not only exposed to Hg in numerous occupational and environmental settings, but they are also exposed to this toxic metal by dental amalgams and/or medicinal and dietary sources (ATSDR, 2003a; Zalups, 2000a). The majority of human exposure results from the ingestion of food and water contaminated with CH3Hg+. Major predatory freshwater and saltwater fish, such as northern pike, salmon, swordfish and shark, can contain high levels of CH3Hg+, which is largely related to the longevity of fish in contaminated waters. Upon ingestion of contaminated muscular tissue of fish, CH3Hg+ is released and absorbed by the gastro-intestinal tract of humans and other mammals (ATSDR, 2003a). After entering systemic circulation, mercuric ions can then be delivered to target organs affected adversely by the metal.
It is important to note that within biological systems, mercurous or mercuric ions do not exist as inorganic salts, or in an unbound, “free” ionic state (Hughes, 1957). Mercuric ions have a very high affinity for thiol-containing biomolecules, such as glutathione (GSH), cysteine (Cys), homocysteine (Hcy), N-acetylcysteine (NAC), metallothionein (MT) and albumin. Thus, in biological systems, mercuric ions are always bound to one or more of these compounds. Several studies have shown that in the presence of excess low molecular weight, thiol-containing molecules, mercuric ions will bind to these molecules in a linear II, coordinate covalent manner (Fig. 1; Canty et al., 1994; Fuhr and Rabenstein, 1973; Rubino et al., 2004). Mercuric-thiol conjugates formed under these conditions appear to be stable in an aqueous environment from pH 1 to 14 (Fuhr and Rabenstein, 1973). Furthermore, the formation constant for the thiol–Hg bond has been estimated to be as much as 10 orders of magnitude greater than the formation constant for the bonding of mercuric ions to other nucleophiles present in the same environment (reviewed by Zalups, 2000a).
Numerous studies over the last 20 years have implicated some form of molecular mimicry in the uptake of thiolconjugates of inorganic and organic mercuric ions in selective target cells. Part of the impetus behind implicating molecular mimicry as a mechanism for the uptake of mercuric ions stems from the fact that mercuric conjugates of some non-protein thiols, such as Cys, Hcy, or GSH, are similar structurally to endogenous compounds such as cystine, homocystine, glutathione disulfide (GSSG), or methionine. Consequently, a number of investigators believe that selective mercuric species gain entry into certain target cells by serving as molecular mimics of these compounds at the sites of carrier proteins involved in the extracellular to intracellular transport of these biological molecules. Some of the most direct evidence implicating molecular mimicry in the transport of a toxic metal in a mammalian epithelial cell comes from studies of the transport of inorganic Hg (Hg2+) in renal tubular epithelia.
Molecular mimicry and the renal transport of Hg2+
The kidney is the primary target organ that takes up and accumulates Hg2+ from the blood (reviewed by Zalups, 2000a). The accumulation of this metal in the kidneys is very rapid, with as much as 50% of a nontoxic dose being present there within a few hours after exposure (Zalups, 1993). Within the kidney, the segments of the proximal tubule are the principal portions of the nephron that take up and accumulate Hg2+. Until recently, the mechanisms by which Hg2+ gains access to the intracellular compartment of renal tubular epithelial cells were largely unknown.
A number of theories regarding the transport of Hg2+ into proximal tubular epithelial cells have been put forth over the years. One hypothesis postulates that some mercuric ions bound to albumin are taken up by endocytosis at the luminal plasma membrane of proximal tubular cells (Madsen, 1980; Zalups and Barfuss, 1993a). Albumin is the most abundant protein in plasma and possesses a free SH group (Brown and Shockley, 1982) to which Hg2+ can bind (reviewed by Zalups, 2000a). As more information on the uptake of Hg2+ along the proximal tubule has become available, it appears that Hg2+-albumin complexes are not the primary species of Hg taken up by proximal tubular epithelial cells.
Recent studies provide strong evidence indicating that there are at least two distinct mechanisms (or sets of mechanisms) responsible for the uptake of Hg in proximal tubular cells: at least one localized in the luminal membrane (Bridges et al., 2004; Zalups, 1995, 1997, 1998a, 1998b; Zalups and Barfuss, 1993b, 1998a; Zalups and Minor, 1995; Zalups and Lash, 1997b; Zalups et al., 1991, 1998; Bridges and Zalups, 2004) and at least another one in the basolateral membrane (Aslamkhan et al., 2003; Zalups, 1995, 1997, 1998a; Zalups and Ahmad, 2004; Zalups and Barfuss, 1993b, 1995, 1998a; Zalups and Lash, 1997b; Zalups and Minor, 1995; Zalups et al., 2004).
A large body of evidence has linked the preponderance of the luminal uptake of Hg2+ in the proximal tubule to the actions of γ-glutamyltransferase and cysteinylglycinase. Numerous in vivo experiments have demonstrated that inhibition of γ-glutamyltransferase with acivicin (an irreversible alkylating agent of γ-glutamyltransferase) reduces significantly the renal (proximal) tubular uptake and accumulation of systemically administered Hg2+ (Bernt et al., 1985; de Ceaurriz et al., 1994; Tanaka et al., 1990; Tanaka-Kagawa et al., 1993; Zalups, 1995). These studies led to the hypothesis that GSH S-conjugates of Hg2+ (G-S-Hg-S-G), entering the luminal compartment of the proximal tubule, are degraded rapidly and efficiently by γ-glutamyltransferase and cysteinylglycinase to yield thiol S-conjugates of Hg2+, including Cys S-conjugates of Hg2+ (Cys-S-Hg-S-Cys). Indeed, studies in brush-border membrane vesicles (isolated from the renal cortex and outer stripe of the outer medulla of rats) indicate that mercuric ions are taken up more readily when they are in the form of Cys-S-Hg-S-Cys than when they are in the form of G-S-Hg-S-G or mercuric conjugates of 2,3-dimercaptopropane-1-sulfonate (DMPS; Zalups and Lash, 1997b). Moreover, studies in suspensions of rabbit proximal tubules (Wei et al., 1999; Zalups et al., 1993) and isolated perfused proximal tubules from rabbits (Cannon et al., 2000, 2001) have provided additional evidence for the luminal uptake of Cys-S-Hg-S-Cys. Since this conjugate is similar structurally to the amino acid cystine, our laboratory hypothesized that Cys-S-Hg-S-Cys can serve as a mimic of cystine at the site of one or more transporters (of this amino acid) located in the luminal plasma membrane of proximal tubular epithelial cells. Subsequent experiments in isolated, perfused proximal tubules from rabbits provide substantive evidence that amino acid transporters are indeed involved in the luminal uptake of Cys-S-Hg-S-Cys (Cannon et al., 2001). Evidence from these experiments indicates that there is at least one Na+-dependent and one Na+-independent carrier involved in the luminal transport of this conjugate along the proximal tubule.
Likely amino acid transporters involved in the Na+-dependent transport of Cys-S-Hg-S-Cys include systems B0, B0,+, and/or ASC. Direct evidence supporting the involvement of these specific carriers is currently lacking. One transporter that is most likely involved in the Na+-independent transport of Cys-S-Hg-S-Cys is system b0,+. This heterodimeric transporter, which is comprised of the subunits, b0,+AT, and 4F2hc (Palacin et al., 1998, 2001), has a high affinity for cystine as well as neutral and basic amino acids. Recent direct molecular findings from type II Madin-Darby canine kidney (MDCK) cells transfected stably with system b0,+ indicate that this transport system can mediate the luminal uptake of Cys-S-Hg-S-Cys (Fig. 2A; Bridges et al., 2004). Analysis of substrate-specificity indicates that the uptake of Cys-S-Hg-S-Cys and cystine are inhibited by the same amino acids, meaning that these two molecular species are substrates for the same carrier, that is, system b0,+. Additional findings from the transfected cells indicate that this carrier (Bridges et al., 2004) does not readily transport mercuric conjugates of GSH (G-S-Hg-S-G), N-acetylcysteine (NAC-S-Hg-S-NAC), and cysteinylglycine (CysGly; CysGly-S-Hg-S-CysGly). The ability of system b0,+ to transport Hcy S-conjugates of Hg2+ (Hcy-S-Hg-S-Hcy) has also been tested recently in these transfected MDCK cells. Since Cys-S-Hg-S-Cys and Hcy-S-Hg-S-Hcy are structural homologs, we hypothesized that they both are substrates of the same transporter. In a separate study which utilized MDCK cells transfected with system b0,+, it was demonstrated that Hcy-S-Hg-S-Hcy is indeed a transportable substrate of system b0,+ (Bridges and Zalups, 2004). Collectively, these data provide the most concrete lines of evidence supporting the hypothesis that Cys-S-Hg-S-Cys and Hcy-S-Hg-S-Hcy serve as molecular mimics of the amino acids cystine and homocystine, respectively, at the site of system b0,+.
Fig. 2.
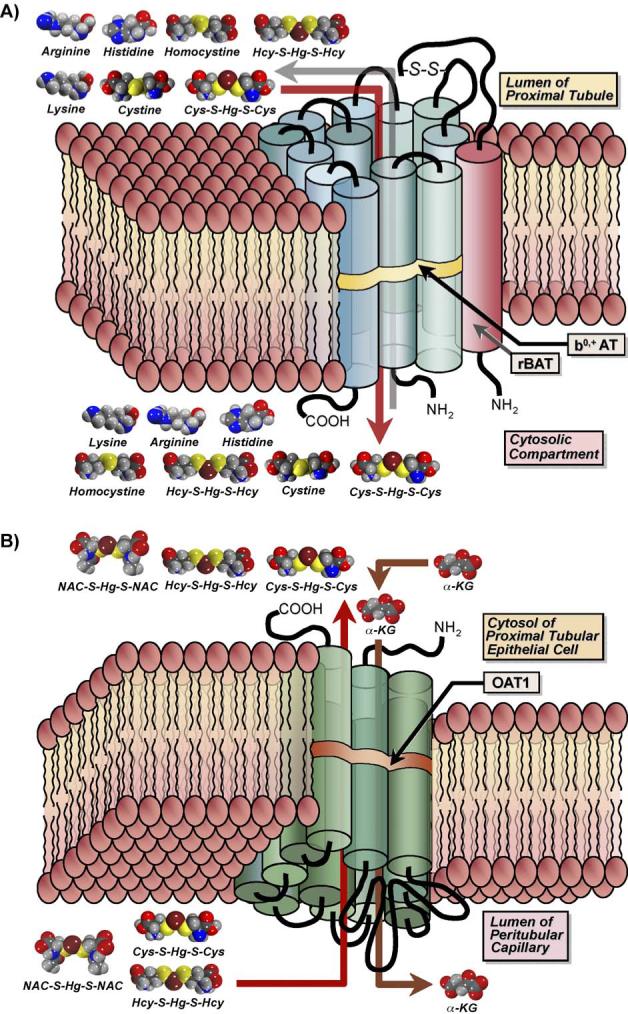
Diagrammatic representation of the transport of amino acids and mercuric conjugates of amino acids by the amino acid transporter, system b0,+ (A) and the organic anion transporter 1 (OAT1; B). (A) System b0,+ is a Na+-independent transporter comprised of a heavy chain and a light chain, which are linked together by a disulfide bond (S–S). The light chain, b0,+ AT (blue cylinders), possesses 12 transmembrane domains, while the heavy chain, rBAT (red cylinder), traverses the plasma membrane only once. This carrier is localized in the luminal plasma membrane of transporting epithelia (such as renal proximal tubular epithelial cells) and functions as an amino acid exchanger that mediates the transport of cystine as well as a variety of neutral and cationic amino acids. Recent studies have identified additional substrates for this carrier, including mercuric conjugates of cysteine (Cys; Cys-S-Hg-S-Cys) and homocysteine (Hcy; Hcy-S-Hg-S-Hcy), which are similar structurally to the amino acids cystine and homocystine, respectively. Experiments carried out in Madin-Darby canine kidney (MDCK) cells transfected stably with both subunits of system b0,+ showed that Cys-S-Hg-S-Cys and Hcy-S-Hg-S-Hcy mimic cystine and homocystine, respectively, at the site of this transporter. (B) The organic anion transporter 1 is a multi-specific carrier that is localized in the basolateral plasma membrane of many types of epithelial cells. Its expression is especially pronounced in renal proximal tubular epithelial cells. This transporter spans the plasma membrane 12 times and has two large intracellular loops, with the first between the first and second transmembrane domains and the second joining the sixth and seventh domains. The inward transport of organic anions is driven by the outward flux of α-ketoglutarate (α-KG). Data from recent studies in which MDCK cells were transfected stably with OAT1 demonstrate that mercuric conjugates of N-acetylcysteine (NAC-S-Hg-S-NAC), Cys-S-Hg-S-Cys, and Hcy-S-Hg-S-Hcy are transportable substrates of this carrier. As hypothesized for system b0,+, these mercuric species likely act as molecular mimics of endogenous substrates of OAT1.
In addition to the uptake of Hg2+ at the luminal plasma membrane, a preponderance of evidence indicates that approximately 40−60% of the proximal tubular uptake of Hg2+ occurs via one or more transporters located at the basolateral membrane (Zalups, 1995, 1997, 1998a, 1998b; Zalups and Barfuss, 1995, 1998a, 1998b; Zalups and Minor, 1995). In experiments where the rates of glomerular filtration in rats were reduced to negligible levels, a 40% decrease in the renal tubular uptake of Hg2+ was observed, indicating that the basolateral uptake of Hg2+ comprises a significant fraction of the total renal uptake of Hg2+ (Zalups and Minor, 1995). Data from these experiments also demonstrated that the addition of para-aminohippurate (PAH), which is a specific inhibitor of the organic anion transporter (OAT) family (Ferrier et al., 1983; Pritchard, 1988; Roch-Ramel et al., 1992; Shimomura et al., 1981; Ullrich et al., 1987a, 1998b), effectively inhibited Hg2+ uptake. These findings indicate that the majority of the basolateral uptake of Hg2+ is likely mediated by one or more OATs, which are multi-specific carriers that mediate the uptake of a wide variety of substrates (Wright and Dantzler, 2003). Current evidence implicates only OAT1 and OAT3 in the uptake of Hg2+. Both of these transporters are localized in the basolateral plasma membrane of proximal tubular epithelial cells (Kojima et al., 2002; Motohashi et al., 2002). The preponderance of current evidence indicates that OAT1 is the major mechanism for the uptake of Hg2+ at the basolateral plasma membrane of proximal tubular cells (Zalups, 1995, 1997, 1998a, 1998b; Zalups and Barfuss, 1995, 1998a, 1998b, 2000; Zalups and Lash, 1994; Zalups et al., 1998).
Recent findings from MDCK cells (which normally do not express OAT1) that were stably transfected with OAT1 show that various mercuric conjugates, including Cys-S-Hg-S-Cys (Zalups et al., 2004), NAC-S-Hg-S-NAC (Aslamkhan et al., 2003), and Hcy-S-Hg-S-Hcy (Zalups and Ahmad, 2004) are substrates of this transporter (Fig. 2B). OAT1 and OAT3 have also been implicated in the transport of Cys-S-Hg-S-Cys in oocytes from Xenopus laevis altered at a molecular level to express these two transporters (Aslamkhan et al., 2003; Zalups et al., 2004). A significant body of recent molecular evidence indicates that the mercuric conjugates of Cys, Hcy, and NAC are taken up via a mechanism involving molecular mimicry.
Molecular mimicry and the intestinal transport of Hg2+
Gastrointestinal absorption of Hg2+, although inefficient, occurs following consumption of food and/or liquids contaminated with inorganic forms of Hg. Thus, understanding the intestinal absorption, accumulation, and excretion of Hg2+ is important. Foulkes (2000) suggested that the uptake of Hg2+ from the lumen of the intestine is dependent upon the composition of the contents in the intestinal lumen. In other words, the mechanism(s) by which Hg2+ is transported is/are dependent upon the ligands to which Hg2+ is bound. Food that is digested in the stomach and small intestine contains a great number of thiol-containing molecules, such as amino acids and peptides, to which Hg2+ may bind. Given the prevalence of amino acid and peptide transporters in enterocytes lining the three segments of the small intestine (Dave et al., 2004; Ganapathy et al., 2001), it is reasonable to hypothesize that Hg2+ may be taken up by one or more of these carriers. Inasmuch as ingested Hg2+ likely forms complexes with thiol-containing molecules in the lumen of the small intestine, these complexes may serve as structural or functional mimics of some of the endogenous molecules, such as amino acids and/or polypeptides, which are absorbed along the small intestine. Surprisingly, even though the intestine appears to be the initial site of Hg2+ absorption, very little is known about the mechanisms involved in the gastrointestinal handling of this metal.
In vivo studies, in which sections of rat duodenum, jejunum, ileum and stomach were perfused with HgCl2 for various time intervals, demonstrated that the duodenum is the primary site of Hg2+ absorption within the gastrointestinal tract of rats (Endo et al., 1984). Interestingly, in rats with ligated bile ducts, the absorption of Hg2+ was decreased significantly. Subsequent co-administration of bile and HgCl2 increased the absorption of Hg2+ in the duodenum to levels similar to those observed in control rats. Furthermore, it was shown that the accumulation of Hg2+ in the cells of the small intestine was greatest when the pH of the perfusion solution was 4.7 (Endo et al., 1984, 1986). In contrast, when the pH of the perfusion solution was 8.0, the accumulation of Hg2+ in the intestine was significantly lower than that at pH 4.7. This difference in accumulation may be due to an increase in the absorptive transport of Hg2+ from the intestinal lumen into the blood. Accordingly, the content of Hg2+ in blood was the highest when the perfusion solution was more alkaline (pH 8.0). These data suggest that alkalinity increases the absorption of Hg2+ across the intestine; however, they do not implicate a specific mechanism in this process.
Foulkes and Bergman (1993) described a potential mechanism for the uptake of Hg2+ in the intestine. Experiments in which HgCl2 was added directly to everted sacs of rat jejunum have shown that Hg2+ absorption is a two-step process in which Hg2+ first binds to the plasma membrane in the form of an anion such as . Secondly, the Hg2+ traverses the plasma membrane in an internalization step. Interestingly, an inhibitor of anion transport, 4,4′-diisothiocyanostilbene-2,2′-disulphonic acid (DIDS) did not affect this uptake.
More recently, studies in blue crabs have provided additional insight into the mechanisms involved in the intestinal uptake of Hg2+. The findings from these studies indicate that there are passive and active mechanisms involved in the uptake of Hg2+ across the plasma membranes of enterocytes (Andres et al., 2002; Laporte et al., 2002). It is likely that amino acids and peptides present in digested food bind to Hg2+ and that the complexes formed are taken up by active and/or facilitated mechanisms involving amino acid and/or peptide transporters. Owing to the fact that amino acid transporters have been implicated in the transport of Cys-S-Hg-S-Cys in renal proximal tubular cells, and the prevalence of amino acid transporters in the luminal plasma membrane of enterocytes, it seems reasonable to postulate that a similar mechanism plays a role in the intestinal absorption of Hg2+. However, direct evidence supporting such a mechanism has yet to be provided.
The intestine also plays an important role in the elimination of Hg2+. Two mechanisms appear to be involved in the fecal elimination of Hg2+: (1) transcellular and/or paracellular secretion of Hg2+ via enterocytes, and (2) delivery of Hg2+ into the intestinal lumen via bile (Zalups and Lash, 1994). Data from in vivo studies in rats with cannulated or ligated (Zalups, 1998c) bile ducts indicate that intestinal secretion of Hg2+ from the blood into the lumen of the intestine accounts for a substantial fraction of the total pool of Hg2+ that is excreted in the feces. Up until these studies, it had been assumed that biliary secretion of Hg2+ was the principal mechanism involved in the fecal elimination of systemic Hg2+. The intestinal secretion of Hg2+ may involve the transport of an Hg2+–thiol complex, which could act as a mimic or a structural homolog of an endogenous molecule normally secreted by enterocytes. Amino acid transporters are potential mechanisms for this secretion. Given that many of these transporters are actually counter-exchangers, they have the potential to transport substrates both into and out of cells. Consequently, Hg2+–thiol complexes, such as Cys-S-Hg-S-Cys, may utilize these carriers to enter and exit enterocytes.
Molecular mimicry and the hepatic transport of Hg2+
The transport of Hg2+ across the sinusoidal membrane into hepatocytes is not well defined. Hepatocytes contain some of the same transporters that have been implicated in the transport of Hg2+ in other organs, including a transporter of GSSG and numerous amino acid carriers. Therefore, it can be postulated that these transporters may be involved in the uptake of Hg2+ across the sinusoidal membrane of the hepatocytes. It has been established that Hg2+ forms complexes with GSH and/or amino acids, such as Cys and Hcy. Although transporters for GSSG (multidrug resistance proteins 1 and 2) have been identified in the canalicular membrane (Akerboom et al., 1984, 1991; Keppler et al., 1998; Leslie et al., 2001), they have not been localized in the sinusoidal membrane. Furthermore, various amino acid carriers have been identified in the liver (Bode, 2001; Wagner et al., 2001); however, it is presently unclear whether these carriers are present on the sinusoidal membrane.
Much more information is available on the transport of Hg2+ across the canalicular plasma membrane of hepatocytes. Several studies have provided important data regarding potential mechanisms involved in the hepatocellular export of Hg2+ across the canalicular membrane. The prevailing theory regarding this export implicates a mechanism by which the transport of Hg2+ is dependent on the cytosolic concentration of GSH (Ballatori and Clarkson, 1983, 1984, 1985a, 1998b; Dutczak and Ballatori, 1992). Subsequent in vivo studies, in which hepatocellular concentrations of GSH were reduced with pretreatment with buthionine sulfoximine (BSO) or diethylmaleate (DEM), offer additional evidence supporting the role of GSH for the transport of Hg2+ into the canalicular compartment (Zalups and Lash, 1997a). It appears that in the absence of adequate levels of cytosolic GSH, mercuric ions are unable to exit the hepatocyte, which leads to increased cytosolic accumulation of Hg2+. Though the actual mechanism(s) responsible for the transport of mercuric ions from within hepatocytes across the canalicular plasma membrane has/have not been demonstrated directly, experimental evidence indicates that Hg2+ bonds GSH to form G-S-Hg-S-G, which appears to be transported into the biliary canalicular compartment. Since G-S-Hg-S-G is similar structurally to GSSG, this mercuric complex may serve as a molecular mimic of GSSG at the site of a GSSG transporter. However, a definitive role for a transporter of GSSG in the transport of Hg2+ has yet to be elucidated. This transporter may be one of the multiple drug resistance proteins (MRPs), which apparently mediate the transport GSH S-conjugates (Leslie et al., 2001; Suzuki and Sugiyama, 1998).
Transport of Hg2+ in placenta
Normally, very little Hg2+ enters and accumulates in the placenta (Ask et al., 2002; Inouye and Kajiwara, 1990; Suzuki et al., 1967), and the mechanism(s) that mediate placental uptake of Hg2+ are poorly understood. Interestingly, experiments in brush-border membrane vesicles from human placenta suggest that an amino acid transporter may be involved in the uptake of Hg2+ in this organ (Iioka et al., 1987). These experiments demonstrated that the Na+-dependent uptake of alanine was inhibited significantly by HgCl2, indicating that one or more amino acid transporters may be involved in this uptake. As these studies used HgCl2 rather than an Hg-thiol complex, such as Cys-S-Hg-S-Cys, the results may not reflect accurately physiological processes that occur in vivo. They do provide, however, valuable preliminary data. Based on these data and the prevalence of amino acid transporters in the placenta (Jansson, 2001; Kudo and Boyd, 2002), we can postulate that Hg2+, as a thiol-conjugate, mimics a structurally similar amino acid and is utilized as a substrate by one or more amino acid transporters. This proposed mechanism of molecular mimicry may be similar to that demonstrated in other tissues (e.g., system b0,+ in proximal tubular cells, system L in endothelial and glial cells of the blood–brain barrier).
Intracellular mimicry of Hg2+
Just as some Hg–thiol complexes can mimic endogenous molecules at the site of proteins present in the plasma membrane, these complexes may also mimic molecules at the binding site(s) of intracellular proteins and enzymes. Owing to the fact that Cys-S-Hg-S-Cys mimics cystine at the site of an amino acid transporter on the plasma membrane (Bridges et al., 2004), it is not surprising to find that this conjugate also acts as a mimic of this amino acid at binding sites of intracellular molecules that utilize cystine as a substrate. An example of this mimicry has been demonstrated by the preliminary findings of Cooper et al. (2004), which indicate that Cys-S-Hg-S-Cys acts as a mimic of cystine at the binding site of the intracellular enzyme, γ-cystathionase. This enzyme is activated normally by the binding of cystine or cystathionine. As the binding of Cys-S-Hg-S-Cys was shown to inactivate the enzyme, rather than activate it, it can be concluded that this conjugate is behaving as a structural, but not a functional, mimic of cystine and cystathionine. These findings suggest that the ability of molecular species of metals to mimic endogenous molecules may have serious, deleterious effects on intra-cellular processes.
Molecular mimicry and the transport of CH3Hg+ in brain
The brain and central nervous system (CNS) are the primary target sites where the adverse affects of CH3Hg+ are manifested (ATSDR, 2003a,b; WHO, 2000). Accordingly, a great number of studies have focused on mechanisms by which this organic form of Hg gains access to the CNS, and more specifically, how it crosses the blood–brain barrier. As with inorganic mercuric ions, the methyl mercuric ion (CH3Hg+) does not exist as a free, unbound cation in biological systems (Hughes, 1957), but rather, is found conjugated to thiol-containing biomolecules, such as GSH, Cys, Hcy, or NAC (reviewed by Clarkson, 1993). In fact, initial studies utilizing homogenates of rat cerebrum demonstrated that the primary non-protein thiol bound to CH3Hg+ is GSH (Thomas and Smith, 1979). Subsequent studies in rats and primary cultures of bovine brain endothelial cells revealed a possible role for Cys in the transport of CH3Hg+ across the blood–brain barrier (Hirayama, 1980; Aschner and Clarkson, 1988, 1989). In particular, co-administration of Cys with CH3Hg+ has been shown to increase the uptake of CH3Hg+ into capillary endothelial cells of the blood–brain barrier. Interestingly, experiments in rats demonstrated that the uptake of CH3Hg+ is inhibited significantly by the neutral amino acid, phenylalanine (Hirayama, 1980, 1985; Thomas and Smith, 1982). The data from these experiments led to the hypothesis that the Cys S-conjugate of CH3Hg+ (CH3Hg-S-Cys) is a transportable substrate of a neutral amino acid transporter in the capillary endothelium of the blood–brain barrier. In vivo studies in rat brain (Aschner and Clarkson, 1988) and in vitro studies in bovine cerebral capillary endothelial cells (Aschner and Clarkson, 1989) demonstrated that the uptake of CH3Hg-S-Cys is inhibitable by neutral amino acids, providing additional support for the theory that this complex is taken up by a neutral amino acid carrier. The investigators in these studies suggested that CH3Hg-S-Cys behaves as a “mimic” of an amino acid in order to cross the blood–brain barrier. Indeed, it is similar structurally to the amino acid methionine (Jernelov, 1973; Landner, 1971), which is a substrate of the neutral amino acid carrier, system L.
System L is present in the basolateral plasma membrane of many types of transporting epithelia. Interestingly, in the endothelial cells lining the blood–brain barrier, this carrier is found in both the apical and basolateral plasma membranes (Betz and Goldstein, 1978). Not surprisingly, system L is considered a major carrier of large neutral amino acids into the substance of the brain. In addition, this transporter has a broad substrate-specificity (Oldendorf, 1973), which may allow it to utilize CH3Hg-S-Cys as a substrate. Indeed, in vivo studies in rats (Kerper et al., 1992) and in vitro studies utilizing primary cultures of rat astrocytes (Aschner et al., 1990, 1991) provided evidence that CH3Hg-S-Cys is a transportable substrate of system L. The involvement of this transporter in the uptake of CH3Hg-S-Cys was also demonstrated in another study utilizing cultured endothelial cells from the brain (Mokrzan et al., 1995). Interestingly, this study measured the uptake of CH3Hg+, as a conjugate of Hcy (CH3Hg-S-Hcy), or as CH3Hg-S-Cys and found that these two complexes were transported similarly. Though the authors of this study suggest that system L is involved in the uptake of CH3Hg-S-Cys, they did not conclude that this carrier also mediates the uptake of CH3Hg-S-Hcy.
Since these initial studies, two isoforms of the system L family have been identified at the molecular level: the L-type, large neutral amino acid transporters, LAT1 (Kanai et al., 1998; Prasad et al., 1999), and LAT2 (Pineda et al., 1999). These transporters are heterodimeric proteins, comprised of a heavy chain, 4F2hc, and a light chain, LAT1 or LAT2, bound together by a disulfide bond (reviewed by Chillaron et al., 2001). With this knowledge, it has become possible to identify and characterize further the specific mechanisms involved in the uptake of CH3Hg-S-Cys. To illustrate this point, Simmons-Willis et al. (2002) utilized oocytes from Xenopus laevis to study directly the involvement of LAT1 and LAT2 in the transport of this conjugate. These investigators provided the first line of direct molecular evidence implicating CH3Hg-S-Cys as a transportable substrate of LAT 1 and 2 (Simmons-Willis et al., 2002). These data also provide substantive evidence for the phenomenon of molecular mimicry, where CH3Hg-S-Cys appears to mimic methionine at the site of system L (Fig. 3).
Fig. 3.
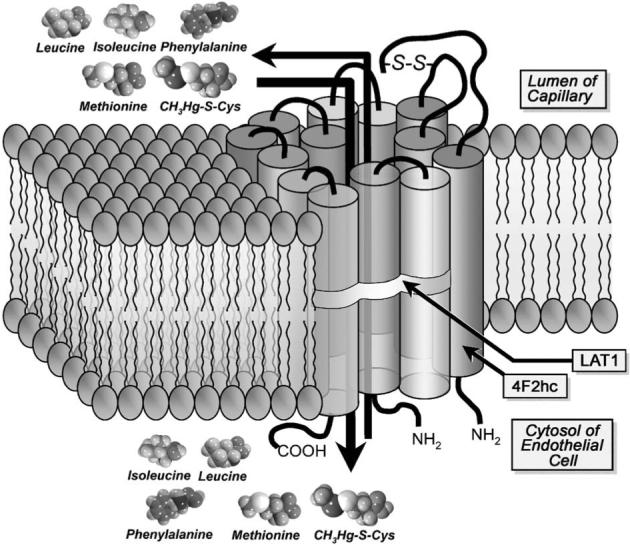
Schematic representation of the transport of methylmercuric conjugates of cysteine (Cys; CH3Hg-S-Cys) by the amino acid transporter system L in the capillary endothelium of the blood–brain barrier. System L is a heterodimeric transporter that has been shown to mediate the Na+-dependent transport of a variety of large, neutral amino acids, including methionine, phenylalanine, leucine, and isoleucine. This transporter is an amino acid exchanger whose activity is dependent on the disulfide linkage (S–S) between the heavy chain 4F2hc and a light chain LAT1 or LAT2. System L has been identified in the basolateral plasma membranes of numerous types of transporting epithelia. Interestingly, it has been localized in the apical and basolateral plasma membranes of the endothelial cells lining the blood–brain barrier. Moreover, system L has been shown recently to take up and transport CH3Hg-S-Cys across this endothelial lining. Inasmuch as CH3Hg-S-Cys is similar structurally to the amino acid methionine, it has been suggested that this conjugate acts as a molecular mimic of methionine at the site of system L.
Molecular mimicry and the renal transport of CH3Hg+
Though the primary target of CH3Hg+ is the central nervous system, it also induces significant detrimental effects in other organs, including the kidneys (Friberg, 1959; Prickett et al., 1950; Norseth and Clarkson, 1970a, 1970b; Magos and Butler, 1976; Magos et al., 1981, 1985; McNeil et al., 1988; Zalups et al., 1992). Until recently, it was unclear as to how this organo-metal complex is taken up by renal tubular epithelial cells. Richardson and Murphy (1975) demonstrated that the renal tubular uptake of CH3Hg+ is dependent upon the cellular concentration of GSH. Moreover, several studies have shown that when CH3Hg+ is co-administrated with GSH, the renal uptake and accumulation of CH3Hg+ increases (Alexander and Aaseth, 1982; Tanaka et al., 1992).
It has been proposed that γ-glutamyltransferase and cysteinylglycinase, which are present in abundance on the luminal (brush-border) plasma membrane of proximal tubular cells, act upon GSH S-conjugates of CH3Hg+ (CH3Hg-S-G) to yield CH3Hg-S-Cys (reviewed by Zalups, 2000a). In vitro evidence indicates that the methyl mercuric ion remains bonded to the sulfur atom of Cys during the catabolism of GSH (Naganuma et al., 1988). Experimental evidence supporting the role of γ-glutamyltransferase in the renal tubular uptake of CH3Hg+ comes from studies in which the activity of this enzyme was inhibited by the alkylating agent acivicin. Following the pretreatment with acivicin, the renal tubular uptake of CH3Hg+ was shown to decrease while the urinary excretion of GSH and CH3Hg+ was shown to increase (Bernt et al., 1985; de Ceaurriz and Ban, 1990; DiSimplicio et al., 1990; Gregus et al., 1987; Mulder and Kostyniak, 1985; Naganuma et al., 1988; Tanaka et al., 1990, 1991, 1992; Yasutake et al., 1989). The observed changes in the renal cellular uptake and excretion of CH3Hg+ indicate that the catabolism of the CH3Hg-S-G complex is a necessary step in the renal proximal tubular absorption of CH3Hg+.
Findings from some studies indicate that a fraction of the CH3Hg+ that enters into systemic circulation is oxidized to Hg2+ either before and/or after it enters the proximal tubular epithelial cells of the kidney (Dunn and Clarkson, 1980; Gage, 1964; Norseth and Clarkson, 1970a, 1970b; Omata et al., 1980; Zalups et al., 1992). These findings lead one to suggest that some or all of the mercuric ions taken up in the kidneys after exposure to CH3Hg+ may be due to the transport of some chemical form of Hg2+ rather than CH3Hg+.
Tanaka et al. (1992) demonstrated in mice the existence of one or more luminal and basolateral mechanisms involved the renal tubular uptake of CH3Hg+. These investigators found that the luminal mechanism(s) are greatly dependent upon the actions of γ-glutamyltransferase. The role of cysteinylglycinase, however, was not studied. Collectively, their data indicate that the species of CH3Hg+ taken up is most likely in the form of a cysteinylglycine S-conjugate of CH3Hg+ (CH3Hg-S-CysGly) or CH3Hg-S-Cys. The mechanism(s) responsible for the uptake of CH3Hg-S-Cys in the proximal tubule has/have not yet been identified. However, we can draw parallels from the information available for the transport of Hg2+, as Cys-S-Hg-S-Cys. Inasmuch as Cys-S-Hg-S-Cys appears to mimic cystine at the site of an amino acid transporter in proximal tubular cells (Bridges et al., 2004), it is possible that CH3Hg-S-Cys behaves in a similar way. Additionally, since CH3Hg-S-Cys has been implicated as a molecular mimic of methionine at the site of system L in endothelial and glial cells, this complex may also mimic methionine at the site of one or more carriers of this amino acid in the kidney.
Uptake of CH3Hg+ at the basolateral membrane may also involve a multi-specific carrier, such as the organic anion transporter 1 (OAT1). As mentioned above, in the kidneys, this transporter is localized exclusively in the basolateral membrane of proximal tubular epithelial cells (Kojima et al., 2002; Motohashi et al., 2002). There is some evidence from studies in Xenopus laevis oocytes implicating this transporter in the cellular uptake of NAC and DMPS S-conjugates of CH3Hg+ (CH3Hg-S-NAC and (CH3Hg-S)2-DMPS, respectively; Koh et al., 2002). Thus, CH3Hg-S-Cys may also be taken up at the basolateral membrane of proximal tubular epithelial cells by OAT1.
The mechanism(s) by which CH3Hg+ is transported out of the proximal tubular cell into the tubular lumen have not been tested directly. However, Koh et al. (2002) proposed that the efflux of CH3Hg+ across the luminal plasma membrane is mediated by MRP2. MRP2 is an ATP-binding cassette (ABC) transport protein that is localized in the luminal membrane of the proximal tubule (Schaub et al., 1997, 1999) and has been shown to be involved in the transport of glutathione-S conjugates of other metals (Leslie et al., 2004). Clearly, a great deal about this potential mechanism remains to be clarified.
Molecular mimicry and the transport of CH3Hg+ in placenta
One of the most publicized and serious toxicological consequences of CH3Hg+ exposure is the deleterious neurological effects observed in fetuses whose mothers were exposed to this metal during pregnancy (Amin-Zaki et al., 1974; Harada, 1978, 1995; Inouye and Kajiwara, 1988; Kajiwara and Inouye, 1986, 1992; Matsumoto et al., 1965). CH3Hg+ crosses the placenta readily and accumulates in the fetus (Inouye and Kajiwara, 1988; Inouye et al., 1985; Suzuki et al., 1967) and placenta (Ask et al., 2002) at levels higher than that in maternal tissues and blood. Yet, little is known about the mechanism(s) by which this metal is taken up and transported across this organ. Kajiwara et al. (1996) showed that CH3Hg+ is transported across the rat placenta by a neutral amino acid carrier in a time- and dose-dependent manner. These investigators demonstrated that co-injection with methionine increased the uptake of CH3Hg+. In addition, they proposed that this increase might be the result of the intracellular conversion of methionine to Cys, which may subsequently combine with CH3Hg+ to form the readily transportable conjugate, CH3Hg-S-Cys. This conjugate may then mimic methionine at the site of system L to gain access to the placenta. Accordingly, the authors concluded that the neutral amino acid carrier, system L (Kajiwara et al., 1996), mediated the uptake of CH3Hg+ in placenta. The exact species of CH3Hg+ that was transported was not determined in this study, nor was there direct evidence supporting the conclusion that system L was involved in this transport. However, since system L has been shown to mediate the transport of CH3Hg-S-Cys across the epithelial cells of and the astrocytes associated with the blood–brain barrier (Aschner et al., 1990; Kerper et al., 1992; Mokrzan et al., 1995), it is feasible that this same carrier is also responsible for the uptake of CH3Hg-S-Cys in placenta. System L has been identified in the placenta (Kanai et al., 1998; Pineda et al., 1999; Prasad et al., 1999) and is an important participant in the transfer of nutrients from the maternal to the fetal circulation.
It is important to note that a number of other protein carriers have been identified in the placenta. These include MRPs, organic anion-transporting polypeptides (OATPs), OATs, organic cation transporters (OCTs), and zinc transporters (Leazer and Klaassen, 2003). The localization of most of these transporters in the placental membrane has not been determined. However, one can suggest that one or more of them may play a role in the uptake and/or efflux of CH3Hg+-complexes.
Interestingly, MRP1, 2, and 3 have been identified in the apical membrane of the syncytiotrophoblast. MRP1 and MRP3 were also present in the endothelial cells of placental blood vessels (St. Pierre et al., 2000). Given the role of MRPs in detoxification (Leslie et al., 2001), it is logical to hypothesize that these carriers mediate the efflux of unwanted substances from the fetal circulation. Thus, in cases of CH3Hg exposure, some of the CH3Hg may be transported back across the placenta into the maternal circulation.
Molecular mimicry and the intestinal transport of CH3Hg+
Ingestion of food and water contaminated with CH3Hg+ is the primary route of human exposure to this compound. Thus, a thorough understanding of the mechanisms involved in the intestinal absorption of CH3Hg+ is an important determinant of understanding the transport and toxicity of CH3Hg+ in the body. Studies in ligated rat intestinal segments demonstrated that the uptake of CH3Hg-S-Cys and CH3Hg-S-CysGly was 1.5 times greater than the uptake of CH3Hg-S-G (Urano et al., 1990). Interestingly, when the activity of γ-glutamyltransferase in the striated border of the intestine was inhibited by acivicin, the uptake of CH3Hg+ (when presented as CH3Hg-S-G) in the enterocytes was reduced by 50%. These data indicate that CH3Hg-S-Cys and/or CH3Hg-S-CysGly is/are the most likely species of CH3Hg+ taken up at the luminal plasma membrane of enterocytes. Since the luminal plasma membrane of enterocytes contains dehydropeptidases, a fraction of CH3Hg-S-CysGly formed in the lumen is likely degraded to yield CH3Hg-S-Cys. Any CH3Hg-S-CysGly that escapes degradation, however, may be transported by a peptide transporter present in the luminal membrane. In the intestine, di- and tripeptide transport is the primary means for the uptake of amino acids. Given this, and the similarity between CH3Hg-S-CysGly and a small peptide, it is possible that this complex mimics an endogenous di- or tripeptide to gain access to enterocytes. In addition, CH3Hg-S-Cys has been identified as the primary species of CH3Hg+ that is delivered into lumen of the intestine by bile. Once in the lumen, CH3Hg-S-Cys is reabsorbed rapidly by the enterocytes possibly by acting as a mimic of methionine at the site of an amino acid carrier (Ballatori et al., 1998; Dutczak and Ballatori, 1992; Norseth and Clarkson, 1971; Wang et al., 2000) present in the luminal plasma membrane. Indeed, the findings from a recent in vitro study of isolated, perfused catfish intestines, indicate that system L mediates at least some of the luminal transport of CH3Hg-S-Cys in enterocytes (Leaner and Mason, 2002). The investigators in this study suggested that there is/are one or more active-transport carrier proteins involved in this uptake. Competitive inhibition experiments provided indirect evidence that a neutral amino acid transporter, possibly system L, mediates (at least some of) the uptake of CH3Hg-S-Cys in intestinal enterocytes.
Additional data from experiments in ligated segments of rat intestine have demonstrated that treatment with probenecid (an inhibitor of OAT1), in addition to acivicin, significantly reduced the luminal uptake of CH3Hg+ (as CH3Hg-S-G; Urano et al., 1990). It was concluded that there are two independent transport systems for the uptake of CH3Hg+ across the luminal plasma membrane of enterocytes. One of these mechanisms is dependent upon the activity of γ-glutamyltransferase, while the other appears to be inhibited by probenecid. These findings lead one to postulate that one or more OATs may also be involved in the intestinal uptake of CH3Hg+. It is important to note, however, that probenecid is not a specific inhibitor of OAT, and may act upon other transporters. Furthermore, there is currently no direct evidence to suggest that OATs are present in the intestine.
The efflux of CH3Hg+ across the basolateral membrane of the enterocyte into the extracellular compartment is less clear than the luminal transport of this molecule. Foulkes (1993) suggested that the intracellular concentrations of GSH play a role in regulating the efflux of CH3Hg+ out of the intestine, although data supporting this notion are lacking. The apparent similarities between the structures of CH3Hg-S-G and GSH may lead one to postulate that this conjugate is a transportable substrate of a GSH transporter present in the basolateral plasma membrane of enterocytes, which would facilitate its transport into blood.
Molecular mimicry and the hepatic transport of CH3Hg+
After CH3Hg+ is absorbed by the intestine, it is delivered to the liver via portal blood. Although, little is known about the mechanisms involved in the hepatocellular uptake of CH3Hg+ from sinusoidal blood, the data from an in vivo study in rodents showed that the uptake and accumulation of CH3Hg+ are enhanced after co-administration of or subsequent administration with Cys or GSH (Thomas and Smith, 1982). These data also indicate that CH3Hg-Cys is the most likely species of CH3Hg+ taken up at the sinusoidal membrane of hepatocytes (Thomas and Smith, 1982). Overall, these findings are consistent with the notion that an amino acid transporter, such as system L is involved in this uptake.
The current evidence indicates that the transport of CH3Hg+ from the hepatocytes into the biliary canaliculus is dependent on GSH (Ballatori and Clarkson, 1982, 1983, 1985a, 1998b; Refsvik, 1982; Refsvik and Norseth, 1975). Indeed, early studies in hepatic tissues indicate that the preponderance of CH3Hg+ within hepatocytes is bound to GSH (Omata et al., 1978). Magos et al. (1978) demonstrated that increasing hepatic levels of GSH enhanced the biliary excretion of GSH and CH3Hg+. In contrast, Refsvik (1978) showed that compounds that reduce significantly the hepatic and biliary levels of GSH cause the accumulation of CH3Hg+ in the liver to decrease. It appears that the intracellular concentration of GSH has a significant effect on the transport of CH3Hg+. Indeed, studies in mice deficient in γ-glutamyltransferase have demonstrated that the distribution and accumulation of CH3Hg+ in liver is affected by the actions of γ-glutamyltransferase and cysteinylglycinase (Ballatori et al., 1998). Furthermore, experiments in cultured human hepatocytes (HepG2 cells) in which γ-glutamyltransferase was inhibited indicate that the transport of CH3Hg+ in these cells is dependent upon the intracellular concentration of GSH (Wang et al., 2000). One can hypothesize that CH3Hg-S-G is formed within the hepatocytes and is subsequently transported into the bile at the canalicular membrane. It is reasonable to hypothesize that CH3Hg-S-G may act as a mimic of GSH at the site of a GSH transporter in the canalicular membrane of hepatocytes. Accordingly, Dutczak et al. (1993) have suggested that a GSH transport system on the canalicular membrane serves a primary role in the biliary secretion of CH3Hg-S-G. A GSH-transporter has since been identified on the canalicular membrane of hepatocytes (Ballatori and Dutczak, 1994; Ballatori and Truong, 1995; Fernandez-Checa et al., 1992, 1993; Garcia-Ruiz et al., 1992) and it most likely plays a crucial role in the export of CH3Hg+.
After being secreted into the bile, CH3Hg+ may be reabsorbed along the biliary tree as a conjugate of GSH or one of its metabolites, CysGly, and/or Cys (reviewed by Ballatori, 1994). Experimental evidence indicates CH3Hg+ is absorbed more readily by ductal epithelial cells when it is administered as a complex of GSH or Cys (Dutczak et al., 1991). Once in the biliary tree, CH3Hg-S-G appears to be catabolized to yield CH3Hg-S-Cys, which can be reabsorbed by cells lining the bile ducts and the enterocytes in the intestine (Dutczak and Ballatori, 1992). Though the actual mechanism(s) involved in this uptake have yet to be determined, it is reasonable to hypothesize that CH3Hg-Cys acts as a mimic of an amino acid at the site of an amino acid transporter, such as system L. A number of amino acid transporters, including system L (LAT3; Babu et al., 2003), have been identified in the liver (Bode, 2001; Wagner et al., 2001); however, the exact localization of any one of them has not yet been determined.
Molecular mimicry and the transport of CH3Hg+ in erythrocytes
Studies of the uptake of CH3Hg+, as CH3Hg-S-G, in erythrocytes have provided additional information regarding the cellular handling of this conjugate. Experiments carried out at 5 °C, indicated that the following systems were involved in this transport: (1) one or more OATs, which appear to be the primary mechanism of uptake, (2) a d-glucose diffusive transporter, (3) a cysteine facilitated transporter, and (4) a Cl− transporter (Wu, 1995). Based on these findings, it appears that CH3Hg-S-G mimics an endogenous substrate of each of these transporters in order to gain access into the erythrocyte. In later studies, Wu (1996) measured the uptake of CH3Hg-S-G at 5 °C and 20 °C and concluded that an OAT is the primary mechanism of CH3Hg-S-G uptake at both temperatures. Additional studies in which probenecid inhibited the uptake of CH3Hg-S-G confirmed previous data indicating that this conjugate is a transportable substrate of OAT (Wu, 1997). These data support the hypothesis that molecular mimicry at the site of one or more transporters plays a role in the transport of CH3Hg-S-G in erythrocytes.
Cadmium
Cd is a naturally occurring group IIB element found in the earth's crust. The ionic form of Cd (Cd2+) is usually combined with ionic forms of oxygen (cadmium oxide, CdO), chlorine (cadmium chloride, CdCl2), or sulfur (cadmium sulfate, CdSO4). Approximately 30,000 tons of Cd are released into the atmosphere each year, with an estimated 4000 to 13,000 tons coming from human activities. Since Cd2+ does not break down in the environment, the risk of human exposure is increasing constantly (ATSDR, 2003b).
Humans are exposed to Cd2+ primarily through the ingestion of contaminated food or water and the inhalation of cigarette smoke. Major sources of dietary Cd2+ are fish, liver, grains, leafy vegetables, potatoes, and other root vegetables. On average, a person in the United States will consume approximately 30 μg of Cd2+ per day, with 1−3 μg of that absorbed by the gastrointestinal tract. Of a more serious nature is the inhaled Cd2+ from cigarette smoke, which is primarily in the form of CdO, (ATSDR, 2003b; Oberdorster, 1992). Each cigarette contains approximately 1−2 μg of Cd2+, with 40−60% of that being absorbed through the lungs directly into the systemic circulation (ATSDR, 2003b; Elinder et al., 1976; Lewis et al., 1972). Exposure to Cd2+ on a chronic basis can cause adverse affects in the kidneys, liver, lung, pancreas, testis, placenta, and bone (ATSDR, 2003b; Bhattacharyya et al., 2000; Diamond and Zalups, 1998; Friberg et al., 1986; Goyer et al., 1984; Habeebu et al., 1998; Jarup et al., 1998; Kamiyama et al., 1995; Kazantzis, 1978; Liu et al., 1998a, 1998b, 2000; Min et al., 1986, 1996; Nordberg and Nordberg, 2000; Nordberg et al., 1985; Oteiza et al., 1999; Sarkar et al., 1998; Zalups and Ahmad, 2003; Zalups et al., 1992).
Experimental evidence indicates that Cd2+ may interact with membrane transporters involved in the uptake of nutritive metals, such as Ca2+, Fe, and Zn, as a means to gain entry into target cells of organs affected adversely by this metal. This uptake has been proposed recently to occur through a mechanism of ionic mimicry (Zalups and Ahmad, 2003), whereby Cd2+ mimics the divalent cationic species one or more of these nutritive metals at the binding site of one or more carrier proteins and/or channels that transport these metals.
There is also experimental evidence supporting the hypothesis that Cd2+ can form linear II coordinate covalent complexes with certain sulfhydryl-containing biomolecules, such as GSH, Cys, or Hcy in certain compartments of the body (Rabenstein, 1989; Rabenstein et al., 1983). Much like mercuric conjugates of these molecules, the Cd-containing complexes may serve or behave as molecular mimics of endogenous amino acids, oligopeptides, organic anions, or organic cations at the site of membrane transporters of these substrates.
Receptor-mediated endocytosis of a Cd2+–protein complex, such as CdMT or Cd-albumin, also appears to be an important mechanism by which Cd2+ is taken up by some epithelial cells. All of these potential mechanisms of uptake will be discussed in relation to individual organs in the following sections.
Mimicry and the hepatic transport of Cd2+
Following oral exposure, Cd2+ is absorbed by the intestines and subsequently delivered to the liver by portal blood. In the liver, Cd2+ is taken up avidly from sinusoidal blood by hepatocytes. Cd2+ is also taken up preferentially by the liver following parenteral exposure (ATSDR, 2003b; Liu et al., 2001; Zalups, 2000b). For example, as much as 60% of a nontoxic dose of Cd2+ (5 μmol/kg) has been shown to accumulate in the liver of rats within 1 h after intravenous administration (Zalups, 2000b). It has also been shown in rats injected intravenously with a low dose CdCl2, G-S-Cd-S-G, or Cys-S-Cd-S-Cys, that less than 1% of the dose of Cd2+ remained in the blood after 1 h. This indicates that Cd2+ is extracted from the blood very rapidly by the liver and other organs and tissues (Zalups, 2000b). Of the Cd2+ remaining in the blood, approximately 50% is distributed among the cellular components of blood, with the majority being present in erythrocytes. It has been suggested that the absorption of Cd2+ by erythrocytes may be mediated by an anion exchanger (Dawson and Ballatori, 1995).
Although the mechanisms by which Cd2+ is taken up across the sinusoidal membrane of hepatocytes remain unclear, several hypotheses to explain this transport have been proposed. One possibility is that Cd2+ may mimic other elements or metals at the site of membrane transporters or channels (via some form of ligand exchange reaction) in the sinusoidal membrane of hepatocytes. It is also thought that Cd2+ may bind to proteins that are taken up into hepatocytes via receptor-mediated endocytosis.
One membrane transporter that is likely involved in the sinusoidal uptake of the cationic form of Cd2+ is DMT1. This transporter has been cloned and it has been characterized as a proton-coupled Fe transporter (Gruenheid et al., 1995; Gunshin et al., 1997). In hepatocytes, DMT1 is localized in the sinusoidal membrane (Trinder et al., 2000). Although DMT1 has not been implicated in the uptake of Cd2+ in hepatocytes, it has been shown to participate in Cd2+ transport in enterocytes (Elisma and Jumarie, 2001; Park et al., 2002; Tallkvist et al., 2001) and distal tubular cells (Olivi et al., 2001; Friedman and Gesek, 1994). Inasmuch as DMT1 has been shown to play a role in the uptake of Cd2+ in other organs, it is logical to hypothesize that it may also mediate the uptake of Cd2+ in hepatocytes. If DMT1 does participate in the hepatic uptake of Cd2+, it likely involves a mechanism of ionic mimicry, whereby Cd2+ mimics the ferrous form of Fe (Fe2+) to access the cytosolic compartment of hepatocytes.
Hepatocellular uptake of Cd2+ may also involve Ca2+ channels. As the ionic radius of Cd2+ (0.95 Å) is similar to that of Ca2+ (1.00 Å; Jacobson and Turner, 1980), it seems possible that Cd2+ can mimic Ca2+ at and in Ca2+ channels in order to gain entry into the hepatocytes. Indeed, in vitro studies utilizing primary cultures of rat hepatocytes (Blazka and Shaikh, 1991, 1992) and cultured immortalized hepatocytes (WRL-68; Souza et al., 1997) indicate that Cd2+ may be transported through Ca2+ channels. In these studies, it was demonstrated that Ca2+ channel antagonists, diltiazem and verapamil, blocked significantly the uptake of Cd2+ into hepatocytes. Blazka and Shaikh (1991) concluded that Cd2+ gained entry into these cells via voltage-gated L-type Ca2+ channels. Additional studies in which hepatocytes were exposed to Ca2+ channel antagonists demonstrated that approximately one third of the Cd2+ entering the cells did so through Ca2+ channels (Souza et al., 1997).
Receptor-mediated and fluid-phase endocytosis account for a large amount of membrane turnover and fluid absorption in hepatocytes (Oka et al., 1989). The endocytosis of transferrin and ferritin is one of the best-characterized forms of receptor-mediated endocytosis in hepatocytes and represents a major pathway in the hepatic uptake of Fe (Mack et al., 1983; Morgan and Baker, 1986; Osterloh and Aisen, 1989). It has been shown that Cd2+ may substitute for Fe2+ at the site of DMT1, thus one would not be surprised to find that this substitution may also occur at the site of one or more Fe-binding proteins, such as ferritin. Indeed, ferritin has been shown to bind Cd2+ (Huebers et al., 1987; Price and Joshi, 1983). Therefore, it has been hypothesized that Cd–ferritin complexes may be endocytosed by hepatocytes in a means similar to that characterized for Fe–ferritin (Zalups and Ahmad, 2003). It is also important to note that albumin, which is the most abundant plasma protein, is a potential carrier of Cd2+ in blood, and thus endocytosis of Cd–albumin complexes may serve as a route for the hepatic entry of Cd2+. Furthermore, the intracellular metal binding protein, MT, binds Cd2+ readily (Cherian and Goyer, 1978; Kägi and Vallee, 1960; Scheuhammer and Cherian, 1986) and CdMT complexes may be, under certain circumstances, transported into cells via receptor-mediated endocytosis.
It is important to note that within hepatocytes, a significant amount of Cd2+ is bound to MT. When hepatocellular necrosis and/or apoptosis is/are induced, it has been hypothesized that complexes of CdMT are released into systemic circulation (Dudley et al., 1985). Some of this Cd2+ is delivered to the kidneys, where it is filtered by the glomeruli and is then reabsorbed by the epithelial cells of the proximal tubule (Dudley et al., 1985; Foulkes, 1978; Webb, 1986). Importantly, CdMT has been implicated as the primary form of Cd2+ that induces renal tubular injury and death (Cherian and Nordberg, 1983; Cherian et al., 1976; Dorian et al., 1992; Felley-Bosco and Diezi, 1987, 1989; Murakami et al., 1983; Nordberg et al., 1975; Zalups et al., 1992).
Cd2+ may also be transported out of hepatocytes back into the sinusoidal blood. Several transport proteins that may be involved in this transport include the organic anion transporting polypeptides, OATP1, OATP2, and OATP4 (Cattori et al., 2001; Jacquemin et al., 1994; Kullak-Ublick et al., 1994), the organic cation transporter, OCT1 (Grundemann et al., 1994), the metal transport protein, MTP1 (Abboud and Haile, 2000) and amino acid or peptide transporters that are localized in the sinusoidal plasma membrane. Although these carriers have not been implicated in the transport of metals out of hepatocytes, most of them are multi-specific transporters and may be able to carry metal ions or conjugated forms of metals.
Various lines of evidence indicate that a fraction of the Cd2+ that enters into hepatocytes is secreted into the bile, and is subsequently delivered to the duodenum for excretion in the feces (Cherian and Vostal, 1977; Leslie et al., 2001). Evidence from thin-layer chromatography of bile from CdCl2-treated animals indicates that Cd2+ is transported from the hepatocyte into bile as a GSH S-conjugate (presumably as G-S-Cd-S-G) by a carrier-mediated process (Cherian and Vostal, 1977). Though a specific carrier has not been identified for this transport, MRP2 is a possible candidate. This transporter is localized in the canalicular membrane and has been shown to mediate the transport of GSH, GSSG, and GSH S-conjugates out of hepatocytes (Heijn et al., 1997; Keppler et al., 1998; Leier et al., 1996, 2001). The structural similarities between G-S-Cd-S-G and GSSG provide a strong rationale for the hypothesis G-S-Cd-S-G may act as a mimic of GSSG at the site of this transporter.
Molecular mimicry and the renal transport of Cd2+
The kidney is one of the primary organs affected adversely in humans following chronic oral or inhalation exposure to Cd2+ (ATSDR, 2003b; Friberg, 1950). The deleterious effects of Cd2+ on the kidneys was realized when factory workers producing nickel-cadmium batteries were exposed to cadmium oxide dust and cadmium fumes. The renal function of these workers was altered significantly, resulting in proteinuria and a lowered rate of glomerular filtration (Friberg, 1950).
Mechanisms of proximal tubular uptake of Cd2+
The majority of Cd2+ found in the kidney is localized in the epithelial cells lining the proximal tubule (Felley-Bosco and Diezi, 1989). Indeed, studies in isolated perfused proximal tubules from the rabbit (Robinson et al., 1993) and cultured proximal tubular cells (Endo, 2002; Endo et al., 1998a, 1998b, 1998c, 1998d, 1999) exposed to CdCl2 provided direct evidence that Cd2+ is taken up in a cationic form by proximal tubular epithelial cells. As these experiments utilized CdCl2, the data obtained may not represent accurately physiological processes that occur in vivo. Owing to the high affinity that Cd2+ has for thiol-containing biomolecules, such as GSH and Cys, likely species of Cd2+ that are presented to the luminal membrane of proximal tubular cells are Cd complexes of these molecules, for example, G-S-Cd-S-G or Cys-S-Cd-S-Cys. In addition, since γ-glutamyltransferase and cysteinylglycinase are present in abundance in the luminal plasma membrane of proximal tubular cells, it is likely that G-S-Cd-S-G is not taken up as an intact complex. As Cys-S-Cd-S-Cys is the primary product formed by the actions of these enzymes on G-S-Cd-S-G, it is more likely that this Cys-complex is a transportable form of Cd2+ at the luminal plasma membrane of proximal tubular epithelial cells. Indeed, in vivo micro-perfusion of renal proximal tubules in the rat has demonstrated that co-perfusion of Cys and Cd2+ increased the tubular absorption of Cd2+ by 82% (Felley-Bosco and Diezi, 1987). Additional in vivo studies in rats have also shown that when CdCl2 is administered subcutaneously with excess Cys, greater levels of Cd accumulate in the epithelial cells lining the proximal tubule (Murakami and Webb, 1981; Murakami et al., 1987). Studies in rats show that when GSH or Cys are simultaneously injected (intravenously) with Cd2+, the renal uptake of Cd2+ increases significantly (Zalups, 2000b). These studies also demonstrated that there is at least one luminal and one basolateral mechanism involved in the renal uptake of Cd2+ (Zalups, 2000b). Unfortunately, no studies to date have identified a specific luminal mechanism for the uptake of a Cd2+-Cys conjugate by proximal tubular or, for that matter, any other epithelial cells. As discussed previously, Cys-S-Hg-S-Cys is a structural mimic of cystine, and is transported across the luminal plasma membrane of proximal tubular epithelial cells by the cystine transporter, system b0,+ (Bridges et al., 2004). Given that Cd2+ forms complexes with Cys, which are structurally similar to Cys-S-Hg-S-Cys, we can hypothesize Cys-S-Cd-S-Cys may be taken up by the same or one or more similar mechanism(s) that mediate(s) the uptake of Cys-S-Hg-S-Cys.
It is also possible that Cd2+ is taken up from the proximal tubular lumen through a mechanism involving ionic mimicry following ligand exchange. There is unpublished support for this coming from our laboratory and from studies in which Cd2+ has been shown to dissociate from MT under certain conditions (Scheuhammer and Cherian, 1986). The process of ligand exchange would permit Cd2+ to exchange from a protein or non-protein thiol to the binding site of a cation transporter, such as DMT1.
With respect to DMT1, there is some controversy regarding its localization in proximal tubular epithelial cells (Canonne-Hergaux and Gros, 2002; Ferguson et al., 2001). In one study by Ferguson et al. (2001), immunolocalization of DMT1 with a polyclonal antibody revealed that DMT1 was present in the cytoplasm of rat proximal tubular cells and did not localize in the brush border membrane. In contrast, Canonne-Hergaux and Gros (2002), also using a polyclonal antibody, showed that DMT1 was localized in the apical membrane of mouse proximal tubular cells. If DMT1 is indeed present in the luminal membrane of proximal tubular cells, it may play a role in the transport of Cd2+ into these cells. It is important to note that the observed discrepancies in the localization of DMT1 may be due to differences between the species used for these experiments and/or variations in the experimental protocols.
In addition to other mechanisms, Cd2+ may gain entry into proximal tubular cells through Ca2+ channels, yet there is no definitive evidence supporting this theory. However, Cd2+ has been shown to utilize Ca2+ channels in isolated cells from other organs, including liver and intestine (Blazka and Shaikh, 1991, 1992; Friedman and Gesek, 1994; Hinkle et al., 1987; Souza et al., 1997). Given these findings, it is reasonable to hypothesize that Cd2+ also utilizes Ca2+ channels for its transport along the nephron.
Endocytosis has also been postulated as an important mechanism for the transport of Cd2+ into proximal tubular cells (Erfurt et al., 2003; Murakami et al., 1983; Zalups and Ahmad, 2003). It appears that CdMT is released from necrotic/apoptotic hepatocytes into hepatic circulation following chronic exposure to Cd2+ (Dudley et al., 1985). CdMT is small enough to be filtered freely at the glomerulus and then be taken up by the epithelial cells of the proximal tubule via an endocytotic mechanism (Dudley et al., 1985; Foulkes, 1978; Webb, 1986). Indeed, the cells of the proximal convoluted tubule are the primary sites affected adversely by CdMT (Cherian and Nordberg, 1983; Dorian et al., 1992; Felley-Bosco and Diezi, 1987, 1989; Murakami et al., 1983; Nordberg et al., 1975; Zalups et al., 1992).
The basolateral entry of Cd2+ into the proximal tubule has important implications in Cd2+ nephrotoxicology. It is thought that the Cd2+ present in the blood may be transported across the basolateral membrane of the proximal tubular epithelial cells as a conjugate of Cys or other non-protein thiols. However, no specific mechanisms for this uptake have yet been identified. One potential candidate for this transport is OAT1. It has been shown to mediate the inward transport of Hg2+ in the form of Cys-S-Hg-S-Cys, Hcy-S-Hg-S-Hcy, and NAC-S-Hg-S-NAC across the basolateral membrane of proximal tubular cells through a mechanism of molecular mimicry (Aslamkhan et al., 2003; Zalups and Ahmad, 2004; Zalups et al., 2004). It is believed that at least some of the Cd2+ in blood, especially after acute exposures, is in the form of G-S-Cd-S-G, Cys-S-Cd-S-Cys, and/or NAC-S-Cd-S-NAC. As these Cd2+ conjugates are similar structurally to those of Hg2+, we can hypothesize that OAT1 not only acts as a basolateral point of entry for conjugates of Hg2+, but may also mediate the uptake of similar conjugates of Cd2+.
Mimicry and transport of Cd2+ in the distal nephron
There is substantial evidence indicating that Cd2+ may be taken up by distal segments of the nephron (Dorian et al., 1992; Felley-Bosco and Diezi, 1987; Ferguson et al., 2001; Friedman and Gesek, 1994; Olivi et al., 2001). It has been hypothesized that some of the Cd2+ delivered to the luminal compartment of the distal nephron and collecting duct is taken up in an absorptive manner by DMT1, likely through some form of ligand exchange reaction. This hypothesis is supported in part by immunolocalization experiments demonstrating that DMT1 is present in the luminal plasma membrane of the epithelial cells lining the ascending thick limb of the loop of Henle, the distal convoluted tubule and the principal cells of the cortical collecting duct (Ferguson et al., 2001). Additional support for this hypothesis comes in part from experiments demonstrating that Cd2+ is a potent inhibitor of Fe2+ uptake in cells derived from the distal nephron (Friedman and Gesek, 1994; Olivi et al., 2001). In one particular study, Friedman and Gesek (1994) demonstrated that Fe2+ is able to inhibit significantly the uptake of Cd2+ in an immortalized line of mouse distal convoluted tubular cells. Olivi et al. (2001) provided data from experiments in MDCK cells showing that Cd2+ and Fe2+ are able to inhibit competitively the uptake of the other. In addition, these investigators showed that the uptake of Cd2+ was greater in renal fibroblasts (HEK-293) that overexpress DMT1 than in corresponding wild-type cells. Together, these findings provide indirect evidence suggesting that Cd2+ may act as an ionic mimic of Fe2+ at the site of the luminal transporter, DMT1, in epithelial cells of the distal nephron and collecting duct.
Ca2+ channels may also provide a route for the absorption of luminal Cd2+ in cells lining the distal nephron. In vitro studies in an immortalized line of mouse distal tubular cells have implicated Ca2+ channels in the transport of Cd2+ (Friedman and Gesek, 1994). Exposure of these cells to parathyroid hormone, which promotes Ca2+ absorption in the distal nephron (Borke et al., 1990; Friedman, 1988), markedly increased the cellular uptake of Cd2+. Furthermore, the observed Cd2+ uptake was inhibited by the Ca2+ channel antagonist, nifedipine, and was enhanced by the Ca2+ channel agonist BAY K 8644 (Friedman and Gesek, 1994). These data suggest that Cd2+ may enter distal tubular epithelial cells via mechanisms of ionic mimicry whereby Cd2+ mimics Ca2+ at the site of Ca2+ channels.
Mimicry and intestinal transport of Cd2+
The duodenum and the proximal jejunum are responsible for the majority of the absorption of ingested Cd2+ (Andersen et al., 1994). Interestingly, the duodenum is also a major site of Fe2+ absorption (Bothwell et al., 1979; Conrad and Umbreit, 2000, 2002; Crichton, 1991). These facts, as well as the similarity in ionic radius between Cd2+ (0.95 Å) and Fe2+ (0.55 Å), suggest that these two cations may utilize some of the same transport mechanisms. Indeed, in vivo experiments in rats indicate that Cd2+ interferes with the intestinal absorption of Fe2+, suggesting that these two metal ions may utilize the same transport pathways (Bunn and Matrone, 1966; Hamilton and Valberg, 1974; Hill et al., 1963; Leon and Johnson, 1985; Schafer and Forth, 1984). DMT1 is a likely candidate for this transport.
DMT1 is localized in the luminal plasma membrane of enterocytes and it appears to play a major role in the intestinal absorption of Fe2+ (Canonne-Hergaux et al., 1999; Griffiths et al., 2000; Gunshin et al., 1997; Tandy et al., 2000; Trinder et al., 2000). Moreover, it has been hypothesized that DMT1 may participate in the uptake of Cd2+ (Elisma and Jumarie, 2001; Park et al., 2002; Zalups and Ahmad, 2003). As with Hg2+, Cd2+ is not normally found as an unbound cation anywhere in the body. In the lumen of the intestines, Cd2+ is usually bound to nucleophilic binding sites on amino acids and protein-ligands. Thus, given that DMT1 has been characterized as a cation transporter (Gunshin et al., 1997), it may seem unlikely that this carrier can utilize Cd2+ as a substrate when it is conjugated to an amino acid, oligopeptide, or protein. Yet, it is important to realize that Cd2+ can dissociate from its bound ligands under certain conditions (Scheuhammer and Cherian, 1986). Considering this, we can hypothesize that when Cd2+ is presented to a stronger nucleophilic ligand, it is released from the carrier ligand to bind to DMT1 or other transporters at the plasma membrane of the enterocyte, which allows it to be transported across the luminal plasma membrane as a cationic species. Indeed, numerous studies have provided evidence for the transport of Cd2+, by DMT1, as a cationic species (Elisma and Jumarie, 2001; Leazer et al., 2002; Lecoeur et al., 2002; Okubo et al., 2003; Park et al., 2002; Picard et al., 2000; Tallkvist et al., 2001). Findings from Xenopus laevis oocytes have provided some of the most direct evidence for the transport of Cd2+ by DMT1 (Gunshin et al., 1997; Okubo et al., 2003). In oocytes microinjected with mRNA encoding DMT1, and then analyzed using two-microelectrode voltage-clamp techniques, the presence of Cd2+ in the extracellular medium was shown to invoke a large inward current, indicating that Cd2+ is a substrate of DMT1 (Gunshin et al., 1997). Additionally, findings of Okubo et al. (2003) indicate that when oocytes are microinjected with mRNA encoding DMT1, and then subsequently exposed to CdCl2, the rates of inward Cd2+ flux are much greater than those in control oocytes. Furthermore, this transport appears to be saturable, with a Michaelis–Menten constant (Km) of approximately 1.0 μM (Okubo et al., 2003).
Other investigators have also studied the expression of DMT1 at a molecular level in relation to the uptake of Cd2+ (Park et al., 2002; Tallkvist et al., 2001). Tallkvist et al. (2001) carried out experiments in an immortalized line of colonic (Caco-2) cells in which the expression of the gene for DMT1 was reduced following exposure to Fe2+. Interestingly, the uptake of Cd2+ was also reduced in these cells. The decrease in the uptake Cd2+ correlates well with the reduction in DMT1 gene expression, suggesting that the uptake of Cd2+ is linked to the expression of DMT1. These data were confirmed recently by in vivo studies in rats (Park et al., 2002). Rats were fed a diet either deficient in Fe or supplemented with Fe for 4 weeks, following which animals were given an oral dose of CdCl2. The mRNA levels of DMT1 in the small intestine of the animals fed the Fe-deficient diet were 15-fold greater than those in animals fed the supplemented diet. In addition, the content of Cd2+ in the small intestines of animals with depleted Fe stores was significantly greater than that in rats fed a supplemented diet. The Cd2+ content in the duodenum was approximately 10-fold greater than that in other organs tested. These data provide strong evidence supporting the hypothesis that DMT1 mediates the intestinal uptake of Cd2+. Additional studies in Caco-2 cells corroborated the aforementioned findings (Bannon et al., 2003). Stable transfection of Caco-2 cells with an antisense construct of DMT1 successfully “knocked down” the activity of DMT1 in these cells. The uptake of Fe2+, Pb2+, and Cd2+ was decreased in these cells, indicating that DMT1 plays a mechanistic role in the uptake of these metals by enterocytes. Collectively, these molecular studies make an important contribution to understanding the intestinal disposition and transport of Fe2+ and Cd2+.
It is clear that dietary Fe status has a significant effect on the expression of DMT1 (Anderson et al., 2002; Martini et al., 2002; Rolfs et al., 2002; Wareing et al., 2003; Zoller et al., 2001, 2002) and the uptake of Cd2+ in the intestine (Berglund et al., 1994; Choudhury et al., 2001; Flanagan et al., 1978; Fox et al., 1980; Groten et al., 1992). This is particularly evident in pregnant rats (Leazer et al., 2002). The requirement for Fe increases during pregnancy (Svanberg, 1975), and thus the expression of DMT1 is elevated to compensate for the increased need for Fe. An elevation of DMT1 expression has also been observed in anemic (Choudhury et al., 2001) women. During pregnancy and other conditions where Fe stores are lowered, DMT1 expression increases through an Fe-response element. This in turn, appears to promote the enteric absorption of ingested Cd2+.
Another potential mechanism involved in the uptake of Cd2+ in enterocytes is a Zn transporter. Data from apical membrane vesicles from the small intestine of the pig indicate that the cationic form of Zn (Zn2+) and Cd2+ share a binding site on a transporter that does not take up Ca2+ (Tacnet et al., 1990, 1991). It has also been shown that Zn2+ and Cd2+ compete for a transporter that is independent of DMT1 (Elisma and Jumarie, 2001). It has been hypothesized that Cd2+ mimics Zn2+ and is transported into enterocytes via a Zn2+ transporter, such as the human, zinc-regulated zinc transporter 1 (hZTL1). This transporter, which is present in the luminal plasma membrane of enterocytes, is responsible for the inward transport of Zn2+ in these cells (Cragg et al., 2001). Given this, it is possible that Cd2+ can mimic Zn2+ at the site of hZTL1 to gain access to the intracellular compartment of enterocytes.
Cd2+ may also gain access to the cytosolic compartment of enterocytes via Ca2+ channels present on the luminal plasma membrane of these cells. However, there is little data from studies in the intestine that support this hypothesis. On the other hand, the ability of Cd2+ to compete with Ca2+ at the site of membrane Ca2+ channels has been well established in numerous in vitro models (Blazka and Shaikh, 1991, 1992; Friedman and Gesek, 1994; Hinkle et al., 1987; Souza et al., 1997). Data directly supporting this phenomenon in animal models are lacking. In vivo studies in rats have demonstrated that in conditions where dietary Ca2+ is restricted chronically, the intestinal uptake of orally administered Cd2+ was increased greatly (Felley-Bosco and Diezi, 1992). From these studies, we can postulate that Cd2+ may serve or behave as a mimic of Ca2+ at the sites of one or more Ca2+ transporters to gain entry into enterocytes.
Foulkes (1985, 1988, 2000) and Foulkes and McMullen (1987) have proposed a two-step mechanism of Cd2+ transport across the luminal plasma membrane of enterocytes. The first step was postulated to involve binding of Cd2+ to the plasma membrane. This binding was susceptible to chelators, such as EDTA, but was insensitive to changes in temperature. The second step was shown to be temperature-sensitive and chelator-insensitive and likely represented the actual flux of Cd2+ across the luminal plasma membrane into the intracellular compartment of the enterocytes. This second step likely involves one or more of the transport mechanisms alluded to above.
Uptake of Cd2+ across the plasma membrane of enterocytes may also occur in a manner where Cd2+ is bonded to amino acids or peptides. It has been established that Cd2+ forms linear II coordinate covalent complexes with thiol-containing biomolecules such as Cys (Cys-S-Cd-S-Cys) or GSH (G-S-Cd-S-G) (Rabenstein, 1989). As mentioned above, inasmuch as ingested food contains an abundance of such molecules, it is likely that Cd2+ is presented to the plasma membrane of enterocytes as a complex of one or more of these molecules. Although a certain amount of ligand exchange occurs with Cd2+ ions (allowing the transport of the cationic species of Cd2+), it is highly probable that a substantial fraction of the Cd2+ taken up at the luminal plasma membrane of enterocytes is, at least initially, bound to an amino acid or peptide ligand. Though the transport of S-conjugates of Cd2+ has not been studied directly in the intestine, we can form an hypothesis regarding this transport based on the information available for the transport of S-conjugates of Hg2+. Like Cys-S-Hg-S-Cys and G-S-Hg-S-G, the molecules Cys-S-Cd-S-Cys and G-S-Cd-S-G are similar structurally to cystine and GSSG. These similarities in molecular structure lead us to hypothesize that such complexes may serve as mimics of endogenous molecules at the site of membrane transporters of these mimicked molecules. It should be noted that numerous studies have shown that G-S-Hg-S-G is transported out of the liver into the biliary tree, where it is broken down to form Cys-S-Hg-S-Cys before entering the duodenum (Ballatori et al., 1998; Dutczak and Ballatori, 1992; Norseth and Clarkson, 1971; Wang et al., 2000). Experimental evidence suggests that Cd conjugates of GSH are likely handled in the same manner. Indeed, the findings of Cherian and Vostal (1977) indicate that Cd2+ is transported out of bile as a conjugate of GSH. Based on these findings and the fact that the duodenal contents are rich in amino acid and peptides, it is reasonable to suggest that a significant fraction of absorbed Cd2+ is taken up as a conjugate of these ligands by one or more amino acid and/or peptide transporters. To our knowledge, the involvement of these amino acid carriers in the transport of Cd2+–thiol complexes has not been investigated.
The possibility of endocytosis of Cd–peptide complexes in the intestine cannot be ruled out. As endocytosis of CdMT has been implicated as a means of Cd2+ uptake in other organs, it is possible that Cd2+ is absorbed via endocytosis along with various polypeptides.
The similarities between the uptake of Fe2+ and Cd2+ at the luminal plasma membrane of enterocytes leads us to postulate that these two metals may utilize a common mechanism for efflux at the basolateral membrane of enterocytes. The basolateral metal protein transporter 1, (MTP1/Ferroportin 1/Ireg1), has been identified in enterocytes, and is thought to participate in the absorption of dietary Fe by mediating the export of this metal across the basolateral plasma membrane into portal circulation (Abboud and Haile, 2000; Donovan et al., 2000; McKie et al., 2002). Although there is no evidence to support a role for MTP1 in the intracellular to extracellular transport of Cd2+, this transporter may be nonetheless involved. A second carrier protein thought to be involved in the export of Fe into the circulation is hephaestin (Vulpe et al., 1999). Immunolocalization experiments in IEC-6 cells isolated from the small intestine have identified hephaestin in the perinuclear region of the cells. Although this protein has also been detected on the plasma membrane, the labeling was weak and the exact region of membrane to which it is localized was not determined (Frazer et al., 2001; Simovich et al., 2002). It has been suggested that this protein may mediate the intracellular transport of Fe, and possibly Cd2+, to MTP1. Moreover, if hephaestin is present on the basolateral plasma membrane, it may function in conjunction with MTP1 to transport Fe2+, and maybe Cd2+, out of enterocytes (Chung and Wessling-Resnick, 2003).
In addition, Cd2+ may exit the basolateral membrane of the enterocyte via a Zn2+ transporter. An interaction between Zn2+ and Cd2+ has been demonstrated indirectly in several in vivo and in vitro models (Cotzias et al., 1961, 1962; Elisma and Jumarie, 2001; Jacobs et al., 1983). A potential candidate for this efflux is the Zn2+ transporter ZnT-1 (McMahon and Cousins, 1998; Palmiter and Findley, 1995). ZnT-1 was first identified in rat kidney, and it is thought to play an important role in the absorption of Zn2+ (Palmiter and Findley, 1995). Present in the basolateral membrane of rat enterocytes (McMahon and Cousins, 1998), ZnT-1 likely mediates the inward to outward transport of Zn2+ (Palmiter and Findley, 1995). The basolateral localization of this transporter and its postulated role in the export of Zn2+ supports the hypothesis that this carrier may also participate in the outward flux of Cd2+ across the basolateral membrane of enterocytes.
There is evidence indicating that Cd2+ may be a competitive inhibitor of Ca2+ at the site of the intestinal Ca-ATPase present on the basolateral plasma membrane of enterocytes (Verbost et al., 1987; Schoenmakers et al., 1992). It has been suggested that Cd2+ utilizes this carrier for its transport out of the enterocyte into portal and systemic circulation. Inasmuch as Cd2+ has been shown to act as an ionic mimic of Ca2+ in other organs and cellular models (Blazka and Shaikh, 1991, 1992; Friedman and Gesek, 1994; Hinkle et al., 1987; Souza et al., 1997), it is reasonable to postulate that it may also do so at the site of the intestinal Ca-ATPase.
Mimicry of Cd2+ in testis
The testes are another target affected adversely by Cd2+ (Gunn et al., 1963a, 1963b; Kar and Das, 1960; Parizek, 1957; Parizek and Zahor, 1956). Acute testicular necrosis and the destruction of the seminiferous tubules have been shown to occur in rats and mice after a single injection of Cd2+. The mechanism by which Cd2+ gains access to the intracellular compartments of the cells of the testes is unclear. However, several studies have shown that Cd2+ may gain access to these cells by acting as a mimic of Zn2+ at the site of Zn2+ transporters (King et al., 1999; Waalkes and Perantoni, 1988; Waalkes and Poirier, 1985). Experiments using interstitial cells isolated from the testes of rats demonstrated that the Cd2+ uptake into these cells was inhibitable by Zn2+ and N-ethylmaleimide, a sulfhydryl-alkylating agent (Waalkes and Poirier, 1985). These findings suggest that testicular uptake of Cd2+ may be due to the activity of a Zn2+-transporter, as well as a transporter that carries thiol-conjugates of Cd2+ (possibly as G-S-Cd-S-G or Cys-S-Cd-S-Cs). The latter may be an amino acid transporter, of which many have been identified in the testes (Wagner et al., 2001). Subsequent studies of the interaction between Zn2+ and Cd2+ provided additional evidence for the participation of a Zn2+ carrier protein in the transport of Cd2+ (Waalkes and Perantoni, 1988). By using isolated interstitial cells from rat testes, it was shown that there are passive and active mechanisms involved in the uptake of Cd2+. Although the passive uptake of Cd2+ was not affected by Zn2+, the addition of Zn2+ was able to inhibit significantly the active component of Cd2+ uptake. In vivo experiments in mice demonstrated that the uptake of Cd2+ in the testes was a saturable process that was competitively inhibited by Zn2+, but not by Ca2+ (King et al., 1999). The results of these experiments would suggest that a Zn2+ transporter most likely mediates the testicular uptake of Cd2+. A possible candidate for this transport is hZTL1 or a similar Zn2+ transporter (Cragg et al., 2001). The expression of this hZTL1 has not been characterized in testis. If present, it may be involved in the inward transport of Cd2+ via a mechanism of ionic mimicry.
The Fe2+ transporter DMT1 also represents a route of Cd2+ entry in the testes. There are no data implicating DMT1 in the testicular transport of Cd2+, yet this transporter has been identified in the Sertoli cells of the testis. Thus, it may be responsible for the inward transport of Fe2+, and possibly Cd2+ in these cells.
Mimicry and transport of Cd2+ in breast
As discussed in the previous sections, ionic Cd2+ or molecules containing Cd2+ may act as ionic or molecular mimics, respectively, of endogenous cations or molecules. Additionally, Cd2+ may serve as a functional mimic of certain endogenous molecules. In this instance, Cd2+ would have the ability to elicit the normal physiological response induced by an endogenous molecule. Indeed, several studies have demonstrated that Cd2+ may be a mimic of estrogen (estradiol) at the site of the estrogen receptor (Garcia-Morales et al., 1994; Martin et al., 2003; Stoica et al., 2000a). Garcia-Morales et al. (1994) demonstrated in a breast-cancer cell line (MCF-7) that a 24-h exposure to CdCl2 elicits the same physiological effect that is observed following treatment with estradiol, indicating that these two chemicals can independently activate the estrogen receptor. Furthermore, exposure of the cells to CdCl2 increased the transcription of mRNA encoding the progesterone receptor and increased the rate of growth of the exposed cells. Additional studies from these investigators provided data regarding the mechanism by which Cd2+ is able to activate the estrogen receptor (Martin et al., 2003; Stoica et al., 2000a). These data indicate that Cd2+, like estradiol, activates the estrogen receptor by binding to the hormone-binding domain. Through its interactions with specific amino acids, Cd2+ changes the conformation of the receptor to that created by the binding of estradiol (Martin et al., 2003; Stoica et al., 2000a). It is important to note that Cd2+ does not normally exist in an unbound state in physiological solutions (Zalups and Ahmad, 2003), thus data obtained from studies in which cells were treated with CdCl2 may not accurately represent the physiological events that occur in vivo. However, subsequent in vivo studies in rats provided substantive support for the previous in vitro studies by demonstrating that Cd2+ behaves as a functional mimic of estrogen in the uterus and mammary gland (Johnson et al., 2003). In rats injected intraperitoneally with CdCl2, a 1.9-fold increase in uterine weight and an increase in milk protein synthesis in the mammary gland were observed. The addition of antiestrogen blocked the effects of Cd2+ on these tissues, suggesting that Cd2+ and estradiol are utilizing the same pathway (Johnson et al., 2003). Collectively, these data indicate that Cd2+ may function as a functional mimic of estradiol at the site of the estrogen receptor. Furthermore, these data provide important information relevant to the development of hormone-related diseases such as breast cancer.
Lead
Lead (Pb) is a bluish-gray metal that occurs naturally in the earth's crust. It can exist as a Pb salt or as metallic Pb. Humans continue to be exposed to Pb through many different means. During the years when leaded gasoline was the primary source of fuel for automobiles, large quantities of Pb were added to the environment, especially in proximity to the major highways and thoroughfares. Additionally, up until 1978, much of the paint used in homes contained as much as 40% Pb (ATSDR, 2003c).
Children are particularly sensitive to the effects of the inorganic form of Pb (Pb2+), and are still being exposed to this toxic metal by playing with and ingesting soil contaminated with Pb2+. Eating, inhaling, and coming in dermal contact with older paints containing Pb2+ are also common routes of exposure. Occupational exposure to Pb2+ can occur in work places utilizing welding and in the manufacture of Pb-containing batteries, lead smelting and refining, and the production of pottery. It is estimated that, between 0.5 and 1.5 million, workers are exposed to Pb2+ each year in his/her workplaces (ATSDR, 2003c).
Pb2+ has been shown to have serious effects in the nervous, circulatory, skeletal, renal, hematopoietic, and endocrine systems (ATSDR, 2003c; Goyer, 1993; National Research Council, 1993). Pb2+ poisoning is more common in children than adults and is characterized by neurological symptoms such as headache, convulsions, ataxia, learning disorders, and hyperactive behavior (Blackman, 1937; Goyer, 1993; Needleman et al., 1990). The effects of Pb2+ on the cardiovascular system are contradictory. While a number of clinical studies have suggested that there is a relationship between the exposure to Pb2+ and elevated blood pressure, an equal number of studies have found no correlation between the two (ATSDR, 2003c; Goyer, 1993). The Environmental Protection Agency (EPA), however, has identified elevated blood pressure as a symptom of Pb2+ poisoning (EPA, 1989). The skeletal system, specifically bone, is a major site for the accumulation of Pb2+. This accumulation has been shown to compromise some ability of bone cells to respond to hormones and also may result in alterations in the plasma levels of 1,25-dihydroxy-vitamin D3 (Goyer, 1993; Pounds et al., 1991). Long-term exposure to Pb2+ may also result in a nephropathy or renal adenocarcinoma (Baker et al., 1980; Buchet et al., 1980; Cooper et al., 1985; Fowler, 1993; Lilis, 1981; Goyer, 1982, 1993; Selevan et al., 1985).
Despite the severe clinical consequences of Pb2+ exposure, the mechanisms by which Pb2+ enters target cells are not well understood. To date, there are several putative mechanisms to explain the uptake of Pb2+ in target cells, and some of these may involve ionic mimicry. Early studies of Pb2+ transport in rats have shown a relationship between the toxicity of Pb2+ and Fe deficiencies (Six and Goyer, 1972). Specifically, the toxicological signs of Pb2+ exposure, such as the urinary excretion of δ-aminolevulinic acid, were greater in rats fed an Fe-deficient diet than in rats fed a normal diet (Six and Goyer, 1972). Epidemiological studies of children have demonstrated a relationship between Fe status and blood levels of Pb2+ (Brandman et al., 2001; Schell et al., 2003). Children who were Fe-deficient and were living in environments containing high levels of Pb2+ had higher blood Pb2+ concentrations than children whose iron levels were normal (Brandman et al., 2001). Furthermore, Schell et al. (2003) found that the blood levels of Pb2+ in newborns were related inversely to the Fe status of the mothers. Owing to the observed interaction between Fe2+ and Pb2+, it can be hypothesized that these two metals utilize one or more of the same transport mechanisms, perhaps the Fe2+ transporter, DMT1.
Data implicating DMT1 in the transport of Pb2+ come in part from studies in which the overexpression of DMT1 in yeast and human fibroblasts was shown to increase the transport of Pb2+ in these cells (Bannon et al., 2002). This increased level of Pb2+ transport was inhibited by Fe2+, which is a known substrate of DMT1 (Gruenheid et al., 1995; Gunshin et al., 1997). Contrary to these findings are data from studies carried out in Caco-2 cells in which the expression of DMT1 had been “knocked down” (Bannon et al., 2002). These data suggest that when the expression of DMT1 has been reduced, the uptake of Fe2+ decreases, but the uptake of Pb2+ is unchanged. Since the uptake of Fe2+ and Pb2+ was similar in the wild-type Caco-2 cells, it was suggested that DMT1 mediates some of the uptake of Pb2+ in these cells, but that this uptake likely does not represent the main mechanism involved in the uptake of Pb2+ (Bannon et al., 2002). Considering all of these findings, it does appear that Pb2+ can be transported by DMT1, probably via a mechanism of molecular mimicry where Pb2+ mimics Fe2+ to gain access to the intracellular compartment of cells at the site of this transporter.
An interaction between Pb2+ and Ca2+ has been identified in numerous in vivo and in vitro models (Barton, 1984; Blake and Mann, 1983; Bogden et al., 1992; Goldstein, 1977; Goyer, 1995, 1997; Kapoor and van Rossum, 1984; Mahaffey et al., 1986; Simons, 1986a; Six and Goyer, 1970; Ziegler et al., 1978). Studies in rats (Bogden et al., 1992; Six and Goyer, 1970), humans (Ziegler et al., 1978), ligated intestinal loops of the chicken (Mykkänen and Wasserman, 1981), and perfused intestinal loops of the mouse (Flanagan et al., 1979) have shown that Pb2+ absorption is inversely related to dietary Ca2+. An examination of approximately 3000 children that had been exposed to Pb2+ revealed a relationship between dietary Ca2+ and blood levels of Pb2+ (Mahaffey et al., 1986). It appears that low dietary intake of Ca2+ can lead to higher levels of Pb2+ in blood. The reverse of this relationship was also shown (Barton, 1984; Blake and Mann, 1983; Bogden et al., 1992). High doses of Ca2+ were associated with lowered blood levels of Pb2+ in rats (Barton, 1984; Bogden et al., 1992) and humans (Blake and Mann, 1983). The interaction between Pb2+ and Ca2+ leads to the hypothesis that Pb2+ may gain entry into cells through one or more of the different types of Ca2+ channels expressed in various cells in the body. Moreover, since the ionic radius of Pb2+ (1.19 Å) is similar to that of Ca2+ (1.00 Å), it is possible that this Pb2+ may mimic Ca2+ at the site of Ca2+ transporters.
Indeed, it has been shown that Pb2+ can be carried into cells via Ca2+ channels (Kerper and Hinkle, 1997a, 1997b; Reuter, 1983; Simons, 1993; Simons and Pocock, 1987). Simons and Pocock (1987) used cells isolated from bovine adrenal medulla to demonstrate that Ca2+ channels can mediate the uptake of Pb2+. Their data show that compounds, which depolarize the cell and subsequently open Ca2+ channels, promote the uptake of Pb2+. Furthermore, they showed that the flux of Pb2+ through Ca2+ channels is at least tenfold greater than that observed for Ca2+. Kerper and Hinkle (1997a, 1997b) also suggested that Pb2+ is transported into cells via Ca2+ channels. Experiments in cultured rat pituitary GH3 cells, rat C6 glioma cells, 301 cells (a subclone of Human Embryonic Kidney (HEK) 293 cells; Kerper and Hinkle, 1997a) and bovine brain capillary endothelial cells (Kerper and Hinkle, 1997b) indicate that Pb2+ enters these cells via channels that are activated by the reduction of intracellular stores of Ca2+. These experiments also showed that the uptake of Pb2+ was a time- and concentration-dependent process and was inhibited significantly by the addition of Ca2+.
Additional mechanisms that may be involved in the uptake of Pb2+ include Ca2+ pumps, such as the Ca2+-ATPase. Experiments in human erythrocytes indicate that Pb2+ may substitute for Ca2+ as a ligand for the Ca2+-ATPase (Pfleger and Wolf, 1975). More recently, Simons (1986a) suggested that Pb2+ replaces Ca2+ at the site of an active transporter of Ca2+. Furthermore, studies in human red blood cell ghosts provided direct evidence indicating that Pb2+ is actively transported by a Ca2+-ATPase (Simons, 1988). The maximum velocity (Vmax) for this transport was estimated to be 11 mmol × cell−1 × h−1 while the Km was calculated to be approximately 0.5 μM.
Anion exchangers have also been implicated in the transport of Pb2+ into erythrocytes (Bannon et al., 2000; Simons, 1986b, 1993). Using resealed erythrocyte ghosts, Simons (1986b) demonstrated that over 90% of the uptake of Pb2+ could be attributed to transport via an anion exchanger. He showed that inhibitors of anion exchangers (DIDS, 4-acetamido-4′-isothiocyanostilbene-2,2′-disulphonic acid (SITS), phloretin, furosemide, and bumetanide) blocked nearly all of the Pb2+ uptake in erythrocyte ghosts. The rate of uptake of Pb2+ was shown to be proportional to the external concentrations of Pb2+ and , suggesting that the transport of Pb2+ is dependent upon the formation of one or more complexes. In addition, this transport was dependent upon anion exchange, much like the exchanger. It was concluded, therefore, that the uptake of Pb2+ into erythrocytes is most likely mediated by an anion exchanger (Simons, 1986b, 1993). Subsequent studies in human erythrocytes have provided further support for the findings of Simons (1986b, 1993), confirming that that the transport of Pb2+ into erythrocytes is mediated by an anion exchanger (Bannon et al., 2000). In addition, the uptake of Pb2+ by immortalized renal distal tubular (MDCK) cells was found to be dependent on time of exposure and temperature. This transport, however, was not affected by DIDS, indicating that it is not mediated by an anion exchanger (Bannon et al., 2000).
Interestingly, experiments in rats with cannulated bile ducts indicate that Pb2+ can be transported as a GSH S-conjugate (Alexander et al., 1986). After an injection of DEM, cyclohexene oxide, or methyl iodide, which reduced intracellular concentrations of GSH, the biliary secretion of Pb2+ was reduced significantly. Though the transport of a GSH–Pb2+ complex was not demonstrated directly, these data suggest that such a complex may be a transportable form of Pb2+, possibly at the site of a GSSG transporter.
Endocytosis of Pb2+–protein complexes may also serve as a route for the entry of this metal into cells. Oskarsson et al. (1982) and DuVal and Fowler (1990) identified a 63,000-dalton protein in a cytosolic fraction of rat kidneys that binds Pb2+. This protein was later identified as alpha-2-microglobulin (Fowler and DuVal, 1991). In contrast, Pb2+-binding proteins (PbBPs) in the human kidney have been identified as diazapine-binding inhibitor (DBI) and thymosin beta-4 (Smith et al., 1994, 1998). Fowler and DuVal (1991) proposed that Pb2+, bound to PbBPs, enters cells, such as the proximal tubular epithelial cells, by some form of endocytosis. Once inside the cell, PbBPs containing Pb2+ are directed to lysosomes, and subsequently to the nucleus. Whether or not mimicry is involved in the internalization of PbBPs is not clear. It would appear, however, that Pb2+ bonds with alpha-2-microglobulin in order to gain access to the intracellular compartment of cells.
Pb2+, like the cationic species of some of the other toxic metals, is also capable of acting as a functional mimic of endogenous ions at intracellular binding sites. It has been shown to be a functional substitute for Ca2+ at the site of calmodulin (Goldstein and Ar, 1983; Habermann et al., 1983), a protein that plays a role in the regulation of intracellular Ca2+ (Stoclet et al., 1987). In addition, the activity of protein kinase C may be affected by the binding of Pb2+. Normally, Ca2+ is responsible for the activation of protein kinase C, which mediates cell division, cell-to-cell communication, and cytoskeletal organization. According to Goyer (1997), Pb2+ is a better activator of protein kinase C than Ca2+. It is important to note that the intracellular concentrations of Ca2+ and its actions, unlike Pb2+, are highly regulated. Therefore, the binding of Pb2+ to an enzyme such as protein kinase C may activate the enzyme for longer than necessary and result in deleterious effects. Of significant importance is the interaction between Ca2+ and Pb2+ at the sites of cellular junctions. Many junctional complexes require Ca2+ in order to maintain their integrity. At these sites, Pb2+ may act as a structural and/or a functional mimic of Ca2+. If acting as a structural mimic only, one would expect the integrity of cellular junctional complexes to be compromised. Interestingly, exposure to high levels of Pb2+ appears to result in the disruption of the blood–brain barrier (Goldstein, 1984), leading to edema, and possibly brain damage. Importantly, the endothelial cells in the brain accumulate Pb2+ preferentially (Toews et al., 1978), which can account, in part, for the severe neurological symptoms observed with Pb2+ poisoning.
Selenium
Selenium (Se) is an essential element that is commonly found in rock formations and soil. It is rarely found in its elemental form in the environment, but is usually present as sodium selenite and sodium selenate. Se is essential for the proper function of intracellular antioxidant enzymes and has a recommended daily allowance of 55 μg/day (ATSDR, 2003d). This metal is not classified as a toxic metal, but rather, is considered to be essential for human health. Yet, it is unique in that it has been shown to act as a functional mimic of several endogenous molecules. For this reason, a discussion of Se is included in this review.
Se can be found in paint, certain types of glass, vitamin supplements, plastics, and fungicides (ATSDR, 2003d). In addition, the ash released from the burning of coal is a major source of environmental contamination of Se. Inhalation or consumption of excess quantities of this metal may be harmful to humans (ATSDR, 2003d; Combs and Gray, 1998). Indeed, inhalation of Se compounds, such as hydrogen selenide and selenium dioxide, results in serious injury to the respiratory tract (Dudley and Miller, 1941; Hall et al., 1951), cardiovascular system (Wilson, 1962), gastrointestinal tract (Holness et al., 1989; Glover, 1967; Wilson, 1962), liver (Dudley and Miller, 1941; Hall et al., 1951), and nervous system (Clinton, 1947; Glover, 1967). Ingestion of excess Se has been shown to cause vision impairment, paralysis, and respiratory failure in livestock (ATSDR, 2003d).
Little is known about the manner in which Se enters mammalian cells. Data from everted sacs of rat ileum indicate that the uptake of selenate across the luminal plasma membrane of enterocytes may be driven by a Na+ gradient (Arduser et al., 1985). Other experiments in isolated rat enterocytes showed that intracellular and extracellular GSH stimulates the uptake of Se (Anundi et al., 1984). The addition of GSH to the extracellular solution of cultured enterocytes increased the cellular accumulation of Se. Interestingly, when the catabolism of intracellular GSH was inhibited, by inhibition of γ-glutamyltransferase with serine-borate, the uptake of Se was reduced. These studies suggest that the transport of Se is dependent upon the products of GSH metabolism (Anundi et al., 1984). In support of this theory, additional studies demonstrated that Cys, in addition to GSH, stimulates the uptake of Se in isolated perfused segments of distal jejunum (Senn et al., 1992) and ligated intestinal loops (Vendeland et al., 1992). Interestingly, in the isolated segments of jejunum, GSH inhibited the uptake of Se while Cys stimulated its uptake. These data suggest that the transportable species of Se are not GSH S-conjugates, but are likely Cys S-conjugates of Se (Senn et al., 1992). Indeed, subsequent studies in primary cultures of enterocytes isolated from sheep showed that Se-Cys complexes are transported readily across the luminal plasma membrane of these cells (Wurmli et al., 1989). This transport was inhibited by the presence of various amino acids, indicating that, an amino acid transporter is involved in the uptake of this metal, perhaps by a mechanism involving molecular mimicry, similar to that utilized in the proximal tubular uptake of Cys-S-Hg-S-Cys.
Interestingly, Se (like Cd) appears to behave as a functional mimic of estrogen at the site of the estrogen receptor. Studies in MCF-7 cells have shown that Se, in the form of sodium selenite, binds to the hormone-binding domain of the estrogen receptor resulting in its subsequent activation (Stoica et al., 2000b). Exposure to Se has also been shown to increase the transcription of the progesterone receptor. These findings have many important implications for the treatment and prevention of hormone-related diseases.
Another important function of Se is its role as an insulin mimic. According to Stapleton (2000), an insulin mimic is any agent that can effectively elicit the effect of insulin on cells. The first apparent evidence of selenium's ability to mimic insulin comes from experiments in isolated rat hepatocytes (Ezaki, 1990). The findings from these experiments demonstrate that selenate stimulates the transport of glucose in hepatocytes in a dose-dependent manner. This transport was associated with the insertion of the glucose transporters, GLUT-1 and GLUT-2, into the plasma membrane of the cells, a response similar to that induced by insulin.
The effect of selenate on the uptake of glucose was also studied in rat soleus muscle (Furnsinn et al., 1996). Furnsinn et al. (1996) showed that selenate stimulated the uptake of glucose and increased the rate of aerobic and anaerobic glycolysis. Subsequent in vivo studies in streptozotocin-induced diabetic rats (Berg et al., 1995; McNeill et al., 1991) and mice (Ghosh et al., 1994) indicate that treatment with selenate effectively lowers plasma levels of glucose. Insulin is involved in the regulation of cellular metabolic processes, such as carbohydrate and fatty acid metabolism (O'Brien and Granner, 1996), glycogen synthesis (Shepherd et al., 1995), glycolysis (Magnuson et al., 1989), gluconeogenesis (O'Brien et al., 1990; Short et al., 1986), fatty acid biosynthesis (Moustaid et al., 1994; Stapleton et al., 1990), and the pentose phosphate pathway (Wagle et al., 1998). Multiple studies have shown that selenate is able to mimic insulin in the above metabolic processes (Battell et al., 1998; Becker et al., 1996; Ghosh et al., 1994; Magnuson et al., 1989). An example of this effect was demonstrated in diabetic rats and mice exposed to selenate (Becker et al., 1996; Ghosh et al., 1994; Magnuson et al., 1989). Oral administration of selenate to diabetic rats restored the expression of enzymes involved in glycogen synthesis and gluconeogenesis (Becker et al., 1996) to normal levels. Similarly, treatment of diabetic rats with selenate also restored the expression of enzymes involved in fatty acid biosynthesis and the pentose phosphate pathway (Ghosh et al., 1994; Magnuson et al., 1989) to normal levels. A characteristic effect of diabetes is a high plasma level of lipids. Interestingly, selenate reduced the plasma levels of lipids, triglycerides, cholesterol and free fatty acids in diabetic rats (Battell et al., 1998). Collectively, the above data provide indirect evidence that selenate may act as a mimic of insulin in a variety of cellular processes. The clinical implications of these findings have yet to be determined.
Arsenic
Arsenic (As) is a highly toxic element found naturally in the earth's crust. When combined with anionic species of oxygen, chlorine, or sulfur, it is referred to as inorganic As (ATSDR, 2003e). Organic As is formed when As ions combine with carbon and hydrogen. Data from studies of animals have shown that organic forms of As are less toxic than the same dose of inorganic As (ATSDR, 2003e). Interestingly, high doses of organic As can produce the same toxicological effects as a lower dose of inorganic As (ATSDR, 2003e).
Inorganic As has long been recognized as a human poison and carcinogen (ATSDR, 2003e; EPA, 1999). Numerous studies have shown that this metal has deleterious effects on the respiratory (Civantos et al., 1995; Dunlap, 1921; Lundgren, 1954; Morton and Caron, 1989; Pinto and McGill, 1953; Sandstrom et al., 1989), neurological (Armstrong et al., 1984; Blom et al., 1985; Civantos et al., 1995; Feldman et al., 1979; Lagerkvist and Zetterlund, 1994), and cardiovascular (Cullen et al., 1995; Jensen and Hansen, 1998; Lagerkvist et al., 1986, 1988) systems. The most common and well-characterized sign of As poisoning is the development of skin lesions and warts (Bickley and Papa, 1989; Dunlap, 1921; Holmqvist, 1951; Lagerkvist et al., 1986; Mohamed, 1998; Perry et al., 1948; Pinto and McGill, 1953).
Humans are exposed to As through environmental, dietary and occupational sources. Water contaminated with inorganic As is a major environmental issue, especially in areas having a rocky terrain. The importance of monitoring drinking water for As contamination was emphasized after millions of Bangladesh residents were poisoned by consumption of arsenic-containing well water (Smith et al., 2000). Soil contamination of inorganic As is also a concern in North America. Since children often play in (and may unintentionally eat) dirt, the ingestion of As-contaminated soil may be a primary source for childhood exposures. Humans may also be exposed to inorganic As upon ingestion of certain fish and various types of seafood, which can contain high levels of this metal. With regard to occupational sources of As in the United States, approximately 90% of all As used in industry is utilized in the “pressure-treatment” of wood. Workers involved in the treatment process or exposed to dust produced when the wood is cut are at higher risk for As poisoning. Inorganic As is also a component of pesticides used for treatment of cotton crops (ATSDR, 2003e).
The mechanisms of As uptake and elimination in target cells have been studied extensively, but unfortunately, are not completely understood. Like the transport of a number of other metals, the transport of As may be dependent on the co-transport of a GSH S-conjugate of As. In vivo studies in rats indicate that the biliary excretion of As is dependent upon the transport of GSH across the canalicular membrane of hepatocytes (Gyurasics et al., 1991). Some studies have implicated the MRPs in the biliary export of As (Cole et al., 1994; Kala et al., 2000; Zaman et al., 1995). Cole et al. (1994) transfected a line of cultured human hepatocytes (HeLa cells) with MRP1 and showed that this transfection confers a phenotype of As resistance upon these cells. Since the control cells remained susceptible to the effects of As, it can be postulated that MRP1 may be responsible for the transport of arsenic out of cells (Cole et al., 1994). In support of this theory, studies in the soil nematode, Caenorhabditis elegans indicate that inactivation of the mrp1 gene increases the sensitivity of these organisms to As (Broeks et al., 1996). Additional experiments in a line of lung cancer (SW-1573/S1) cells that had been transfected stably with MRP1 provided conclusive evidence that As and GSH are co-transported by this carrier (Zaman et al., 1995). Subsequent experiments in rats with cannulated bile ducts demonstrated that MRP2 is involved in the transport of As–GSH complexes across the biliary canaliculus of hepatocytes (Kala et al., 2000). Unfortunately, these experiments did not determine the actual structure of the As–GSH complex involved in this transport. It has been shown, however, that As and GSH form a complex consisting of three GSH molecules bonded to one As atom (As(GS)3) (Scott et al., 1993; Delnomdedieu et al., 1994). This complex is likely the species of As that is transported by MRP1 and MRP2.
Additional mechanisms involved in the uptake of As–GSH complexes were identified in a study utilizing micro-array techniques (Liu et al., 2001). The findings from this study indicate that rat hepatocytes cultured in As-containing media for 18 weeks had become tolerant to As and that this tolerance was due to an increased expression of glutathione S-transferase II, MRP1, MRP2, and P-glycoproteins. The authors of this study postulated that these mechanisms work in consort to reduce effectively the intracellular content of As in hepatocytes. A more short-term study was carried out in primary cultures of rat and human hepatocytes (Vernhet et al., 2001). Cells exposed to sodium arsenite and sodium arsenate for 8−48 h demonstrated a time- and dose-dependent increase in the expression of MRP2, providing additional evidence that this carrier may be involved in the transport of As. Additional studies in HeLa cells overexpressing MRP1 showed that As, as As(GS)3, is transported by this protein carrier. Furthermore, it was demonstrated that the formation of a tri-glutathione conjugate of As is required for its transport (Leslie et al., 2004). These studies are supported by experiments in mice lacking γ-glutamyltransferase, which identified As(GS)3 and methyl arsenic di-glutathione (CH3As(GS)2) in the urine of these mice (Kala et al., 2004). MRP1 has been localized in the basolateral membrane of a line of cultured porcine renal proximal tubular epithelial (LLC-PK1) cells (Evers et al., 1996), and thus may mediate the transport of As(GS)3 and CH3As(GS)2 from the blood into the intracellular compartment of these cells. Additional experiments using mice deficient in MRP1, MRP1a/1b, or MRP2 have demonstrated that administration of MK571, and inhibitor of MRPs, reduced the urinary excretion of As by 50% (Kala et al., 2004). These experiments suggest that there is an additional transporter, other than MRP1, MRP1a/1b, or MRP2, which is involved in the transport of As complexes (Kala et al., 2004).
MRP1 and MRP2 transport a growing number of substrates. Therefore, the role of molecular mimicry in the MRP-mediated transport of As is unclear. There are two possible explanations for the transport of As by the MRPs. Arsenic complexes may act as mimics of one or more substrate(s) that is/are normally transported by the MRPs. Alternatively, as As contamination in the environment has become more prevalent, MRPs have adapted to the changing needs of the cells in which they are localized, consequently broadening their substrate specificities.
Interestingly, As, in the form of arsenite has the apparent ability to act as a functional mimic of estrogen (estradiol) at the site of the estrogen receptor. Experiments in MCF-7 cells demonstrated that arsenite interacts with the hormone-binding domain of the estrogen receptor to activate this protein (Stoica et al., 2000c). Similar to data for Cd2+ and Se, arsenite was shown to increase the transcription of the progesterone receptor gene and increase the rate of cell growth. Furthermore, arsenite blocked the binding of estradiol to the estrogen receptor, suggesting that these two compounds utilize the same pathway. These data suggest that As may be a significant risk factor for the development of breast cancer and other hormone-related diseases.
Oxyanions of toxic metals
It has been noted that endogenous oxyanions, such as monovalent phosphate and sulfate, are similar structurally to oxyanions of several toxic metals (Clarkson, 1993; Wetterhahn-Jennette, 1981). The molecular structure of arsenate and vanadate are very similar to that of monovalent phosphate, while the molecules, chromate, molybdate and selenate are similar in shape and size to sulfate (Fig. 1). Therefore, it is not surprising that the oxyanionic forms of toxic metals have been found to mimic monovalent phosphate or sulfate at the site of some membrane carrier proteins. For example, vanadate seems to interfere with the transport of monobasic phosphate at the sites of several different carriers. According to Clarkson (1993), studies of vanadate in erythrocytes indicate that the inward flux is inhibitable by phosphate and by agents that inhibit the exchanger. More recent findings from studies of vanadate in HEK-293 cells demonstrated that vanadate can inhibit the uptake of phosphate at the site of the NaPi-3 co-transporter (Timmer and Gunn, 1998). In addition, vanadate may act as a mimic of a phosphate molecule on the cytoplasmic side of the Na+-K+ ATPase (Cantley et al., 1978a, 1978b; Karlish et al., 1979). The binding of a phosphate molecule, derived from ATP, is essential for the activity of this exchanger. Substitution of this phosphate with vanadate results in the inhibition of the Na+-K+ ATPase (Cantley et al., 1978a; Karlish et al., 1979).
Arsenate has also been shown to interact with monovalent phosphate at the sites of several different carriers. Experiments in osteosarcoma cells (ROS 17/2.8; Caverzasio et al., 1988), bovine and rabbit renal brush border membrane vesicles (Schali et al., 1986; Vachon et al., 1991), vesicles from the matrix of chicken cartilage (Montessuit et al., 1991), and the luminal plasma membrane of rat intestine (Ishizawa et al., 1990) show that arsenate will compete with phosphate at the site of the sodium-dependent phosphate co-transporter. Additionally, arsenate has been shown to inhibit competitively phosphate transport at the site of an uncharacterized sodium-independent transporter (Azzarolo et al., 1991; Quamme et al., 1989). Interestingly, arsenate does not inhibit the transport of phosphate across the sodium-dependent phosphate exchanger in human erythrocytes (Shoemaker et al., 1988), indicating that there may be some specificity involved in the mimicry of phosphate by arsenate. Arsenate may act as a mimic of phosphate at intracellular sites as well. For example, a phosphate molecule is donated normally to 3-phosphoglyceradehyde to form 1,3-diphosphoglycerate, which in turn, donates a phosphate molecule to ADP to form ATP. Arsenate may substitute for the molecule of phosphate that is donated to 3-phosphoglyceraldehyde and results in the disruption of ATP synthesis (De Master and Mitchell, 1973).
Sulfate has a tetrahedral structure similar to that of oxyanions of several metals, including selenate, molybdate, and chromate (Fig. 1B). Early studies of sulfate transport in rat ileum indicate that molybdate competitively inhibits the uptake of sulfate, suggesting that these two compounds are substrates for the same transporter (Cardin and Mason, 1975; Mason and Cardin, 1977). Subsequent studies in membrane vesicles from human placenta showed that molybdate and chromate are competitive inhibitors of sulfate (Shennan et al., 1988). Furthermore, data from proximal tubular brush-border membrane vesicles from sheep demonstrate that sulfate and molybdate utilize the same transport system (Ryan et al., 1987). Additional support for these findings come from in vivo studies in renal proximal tubules of rats showing that metals, such as selenate and molybdate, are transported by luminal and basolateral sulfate transporters (David and Ullrich, 1992). Collectively, these findings indicate that metal compounds have the ability to act as functional mimics of the oxyanion sulfate at the sites of several different extracellular protein carriers.
Chromium has been shown to inhibit competitively the uptake of sulfate at the site of the anion exchanger in human erythrocytes (Ormos and Manyai, 1978) and placenta (Shennan, 1988). Moreover, it has been shown that sulfate and chromate can inhibit the transport of molybdate into the enterocytes of the small intestine (Huisingh and Matrone, 1976), while molybdate was able to inhibit the transport of sulfate (Shennan, 1988). These data provide further evidence supporting the notion of molecular mimicry.
Summary
As humans are exposed to toxic metals in the work-place and environment, it is imperative that we understand how these metals affect essential cellular processes, and ultimately, human health. Molecules containing toxic metals have found ways to mimic endogenous molecules in order to gain access to target cells via essential transporters. Furthermore, some metals have been shown to mimic endogenous intracellular molecules, thereby interfering with essential cellular processes. Understanding how toxic metals interact with extracellular and intracellular proteins will prove to be important to the development of treatment regimes of diseases induced by exposure to these metals.
Acknowledgments
This work was supported, in part, by the National Institutes of Health (NIEHS) grants, ES05980, ES05157, and ES11288, awarded to Dr. Zalups. Dr. Bridges is supported by a Ruth L. Kirschstein National Research Service Award, ES012556, funded by the National Institutes of Health (NIEHS).
References
- Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- Afonso J, deAlvarez R. Effects of mercury on human gestation. Am. J. Obstet. Gynecol. 1960;80:145–154. doi: 10.1016/s0002-9378(16)36432-8. [DOI] [PubMed] [Google Scholar]
- Agency for Toxic Substance and Disease Registry . Toxicological Profile for Mercury. U.S. Department of Health and Humans Services, Public Health Service, Centers for Disease Control; Atlanta, GA: 2003a. [Google Scholar]
- Agency for Toxic Substance and Disease Registry . Toxicological Profile for Cadmium. U.S. Department of Health and Humans Services, Public Health Service, Centers for Disease Control; Atlanta, GA: 2003b. [Google Scholar]
- Agency for Toxic Substance and Disease Registry . Toxicological Profile for Lead. U.S. Department of Health and Humans Services, Public Health Service, Centers for Disease Control; Atlanta, GA: 2003c. [Google Scholar]
- Agency for Toxic Substance and Disease Registry . Toxicological Profile for Selenium. U.S. Department of Health and Humans Services, Public Health Service, Centers for Disease Control; Atlanta, GA: 2003d. [Google Scholar]
- Agency for Toxic Substance and Disease Registry . Toxicological Profile for Arsenic. U.S. Department of Health and Humans Services, Public Health Service, Centers for Disease Control; Atlanta, GA: 2003e. [Google Scholar]
- Akerboom TP, Inoue M, Sies H, Kinne R, Arias IM. Biliary transport of glutathione disulfide studied with isolated rat-liver canalicular-membrane vesicles. Eur. J. Biochem. 1984;141:211–215. doi: 10.1111/j.1432-1033.1984.tb08177.x. [DOI] [PubMed] [Google Scholar]
- Akerboom TP, Narayanaswami V, Kunst M, Sies H. ATP-dependent S-(2,4-dinitrophenyl)glutathione transport in canalicular plasma membrane vesicles from rat liver. J. Biol. Chem. 1991;266:13147–13152. [PubMed] [Google Scholar]
- Alexander J, Aaseth J. Organ distribution and cellular uptake of methylmercury in the rat as influenced by the intra- and extracellular glutathione concentration. Biochem. Pharmacol. 1982;31:685–690. doi: 10.1016/0006-2952(82)90450-6. [DOI] [PubMed] [Google Scholar]
- Alexander J, Aaseth J, Mikalsen A. Excretion of lead in rat bile— The role of glutathione. Acta Pharmacol. Toxicol. 1986;59:486–489. doi: 10.1111/j.1600-0773.1986.tb02809.x. [DOI] [PubMed] [Google Scholar]
- Amin-Zaki L, Elhassani LS, Majeed MA, Clarkson TW, Doherty RA, Greenwood M. Intrauterine methylmercury poisoning in Iraq. Pediatrics. 1974;54:587–595. [PubMed] [Google Scholar]
- Andersen O, Nielsen JB, Sorensen JA, Scherrebeck L. Experimental localization of intestinal uptake sites for metals (Cd, Hg, Zn, Se) in vivo in mice. Environ. Health Perspect. 1994;102(Suppl 3):199–206. doi: 10.1289/ehp.94102s3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GJ, Frazer DM, McKie AT, Wilkins SJ, Vulpe CD. The expression and regulation of the iron transport molecules hephaestin and IREG1: implications for the control of iron export from the small intestine. Cell Biochem. Biophys. 2002;36:137–146. doi: 10.1385/CBB:36:2-3:137. [DOI] [PubMed] [Google Scholar]
- Andres S, Laporte JM, Mason RP. Mercury accumulation and flux across the gills and the intestine of the blue crab (Callinectes sapidus). Aquat. Toxicol. 2002;56:303–320. doi: 10.1016/s0166-445x(01)00228-4. [DOI] [PubMed] [Google Scholar]
- Anundi I, Hogberg J, Stahl A. Absorption of selenite in the rat small intestine: interactions with glutathione. Acta Pharmacol. Toxicol. (Copenh) 1984;54:273–277. doi: 10.1111/j.1600-0773.1984.tb01930.x. [DOI] [PubMed] [Google Scholar]
- Arduser F, Wolffram S, Scharrer E. Active absorption of selenate by rat ileum. J. Nutr. 1985;115:1203–1208. doi: 10.1093/jn/115.9.1203. [DOI] [PubMed] [Google Scholar]
- Armstrong CW, Stroube RB, Rubio T, Siudyla EA, Miller GB., Jr. Outbreak of fatal arsenic poisoning caused by contaminated drinking water. Arch. Environ. Health. 1984;39:276–279. doi: 10.1080/00039896.1984.10545849. [DOI] [PubMed] [Google Scholar]
- Aslamkhan AG, Han YH, Yang XP, Zalups RK, Pritchard JB. Human renal organic anion transporter 1-dependent uptake and toxicity of mercuric thiol conjugates in Madin-Darby canine kidney cells. Mol. Pharmacol. 2003;63:590–596. doi: 10.1124/mol.63.3.590. [DOI] [PubMed] [Google Scholar]
- Aschner M, Clarkson TW. Uptake of methylmercury in the rat brain: effects of amino acids. Brain Res. 1988;462:31–39. doi: 10.1016/0006-8993(88)90581-1. [DOI] [PubMed] [Google Scholar]
- Aschner M, Clarkson TW. Methylmercury uptake across bovine brain capillary endothelial cells in vitro: the role of amino acids. Pharmacol. Toxicol. 1989;64:293–297. doi: 10.1111/j.1600-0773.1989.tb00650.x. [DOI] [PubMed] [Google Scholar]
- Aschner M, Eberle NB, Goderie S, Kimelberg HK. Methyl-mercury uptake in rat primary astrocyte cultures: the role of the neutral amino acid transport system. Brain Res. 1990;521:221–228. doi: 10.1016/0006-8993(90)91546-s. [DOI] [PubMed] [Google Scholar]
- Aschner M, Eberle NB, Kimelberg HK. Interactions of methylmercury with rat primary astrocyte cultures: methylmercury efflux. Brain Res. 1991;554:10–14. doi: 10.1016/0006-8993(91)90165-r. [DOI] [PubMed] [Google Scholar]
- Ask K, Akesson A, Berglund M, Vahter M. Inorganic mercury and methylmercury in placentas of Swedish women. Environ. Health Perspect. 2002;110:523–526. doi: 10.1289/ehp.02110523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzarolo AM, Ritchie G, Quamme GA. Some characteristics of sodium-independent phosphate transport across renal basolateral membranes. Biochim. Biophys. Acta. 1991;1064:229–234. doi: 10.1016/0005-2736(91)90306-s. [DOI] [PubMed] [Google Scholar]
- Babu E, Kanai Y, Chairoungdua A, Kim DK, Iribe Y, Tangtrongsup S, Jutabha P, Yuewei L, Ahmed N, Sakamoto S, Anzai N, Nagomori S, Endou H. Identification of a novel system L amino acid transporter structurally distinct from heterodimeric amino acid transporters. J. Biol. Chem. 2003;278:43838–43845. doi: 10.1074/jbc.M305221200. [DOI] [PubMed] [Google Scholar]
- Baker EL, Goyer RA, Fowler BA, Khattr U, Bernard OB, Adler S, White R, Babayan R, Feldman RG. Occupational lead exposure, nephropathy and renal cancer. Am. J. Med. 1980;1:139–148. doi: 10.1002/ajim.4700010204. [DOI] [PubMed] [Google Scholar]
- Ballatori N. Glutathione mercaptides as transport forms of metals. Adv. Pharmacol. 1994;27:271–298. doi: 10.1016/s1054-3589(08)61036-4. [DOI] [PubMed] [Google Scholar]
- Ballatori N. Transport of toxic metals by molecular mimicry. Environ. Health Perspect. 2002;110(Suppl 5):689–694. doi: 10.1289/ehp.02110s5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N, Clarkson TW. Developmental changes in the biliary excretion of methylmercury and glutathione. Science. 1982;216:61–63. doi: 10.1126/science.7063871. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Clarkson TW. Biliary transport of glutathione and methylmercury. Am. J. Physiol. 1983;244:G435–G441. doi: 10.1152/ajpgi.1983.244.4.G435. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Clarkson TW. Dependence of biliary secretion of inorganic mercury on the biliary transport of glutathione. Biochem. Pharmacol. 1984;33:1093–1098. doi: 10.1016/0006-2952(84)90519-7. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Clarkson TW. Sulfobromophthalein inhibition of glutathione and methylmercury secretion into bile. Am. J. Physiol. 1985a;248:G238–G245. doi: 10.1152/ajpgi.1985.248.2.G238. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Clarkson TW. Biliary secretion of glutathione and of glutathione–metal complexes. Fundam. Appl. Toxicol. 1985b;5:816–831. doi: 10.1016/0272-0590(85)90165-4. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Dutczak WJ. Identification and characterization of high and low affinity transport systems for reduced glutathione in liver cell canalicular membranes. J. Biol. Chem. 1994;269:19731–19737. [PubMed] [Google Scholar]
- Ballatori N, Truong T. Multiple canalicular transport mechanisms for glutathione S-conjugates. Transport on both ATP- and voltage-dependent carriers. J. Biol. Chem. 1995;270:3594–3601. doi: 10.1074/jbc.270.8.3594. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Wang W, Lieberman MW. Accelerated methylmercury elimination in γ-glutamyltranspeptidase-deficient mice. Am. J. Pathol. 1998;152:1049–1055. [PMC free article] [PubMed] [Google Scholar]
- Bannon DI, Olivi L, Bressler JP. The role of anion exchange in the uptake of Pb by human erythrocytes and Madin-Darby canine kidney cells. Toxicology. 2000;147:101–107. doi: 10.1016/s0300-483x(00)00187-6. [DOI] [PubMed] [Google Scholar]
- Bannon DI, Portnoy ME, Olivi L, Lees PS, Culotta VC, Bressler JP. Uptake of lead and iron by divalent metal transporter 1 in yeast and mammalian cells. Biochem. Biophys. Res. Commun. 2002;295:978–984. doi: 10.1016/s0006-291x(02)00756-8. [DOI] [PubMed] [Google Scholar]
- Bannon DI, Abounader R, Lees PSJ, Bressler JP. Effect of DMT1 knockdown on iron, cadmium, and lead uptake in Caco-2 cells. Am. J. Physiol.: Cell Physiol. 2003;284:C44–C50. doi: 10.1152/ajpcell.00184.2002. [DOI] [PubMed] [Google Scholar]
- Barton JC. Active transport of lead-210 by everted segments of rat duodenum. Am. J. Physiol. 1984;247:G193–G198. doi: 10.1152/ajpgi.1984.247.2.G193. [DOI] [PubMed] [Google Scholar]
- Battell ML, Delgatty HLM, McNeill JH. Sodium selenate corrects glucose tolerance and heart function in STZ diabetic rats. Mol. Cell. Biochem. 1998;179:27–34. doi: 10.1023/a:1006819227506. [DOI] [PubMed] [Google Scholar]
- Becker DJ, Reul B, Ozcelikay AT, Buchet JP, Henquin JC, Brichard SM. Oral selenate improves glucose homeostasis and partly reverses abnormal expression of liver glycogenic and gluconeogenic enzymes in diabetic rats. Diabetologia. 1996;39:3–11. doi: 10.1007/BF00400407. [DOI] [PubMed] [Google Scholar]
- Berg EA, Wu JY, Campbell L, Kagey M, Stapleton SR. Insulin-like effects of vanadate and selenate on the expression of glucose-6-phosphate dehydrogenase and fatty acid synthase in diabetic rats. Biochimie. 1995;12:919–924. doi: 10.1016/0300-9084(95)80002-6. [DOI] [PubMed] [Google Scholar]
- Berglund M, Akesson A, Nermell B, Vahter M. Intestinal absorption of dietary cadmium in women depends on body iron stores and fiber intake. Environ. Health Perspect. 1994;102:1058–1066. doi: 10.1289/ehp.941021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt WO, Baggett J.McC., Blacker A, Houser M. Renal glutathione and mercury uptake by kidney. Fundam. Appl. Toxicol. 1985;5:832–839. doi: 10.1016/0272-0590(85)90166-6. [DOI] [PubMed] [Google Scholar]
- Betz AL, Goldstein GW. Polarity of the blood–brain barrier: neutral amino acid transport into isolated brain capillaries. Science (Washington, D.C.) 1978;202:225–227. doi: 10.1126/science.211586. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya MH, Wilson AK, Tajan SS, Jonah M. Biochemical pathways in cadmium toxicity. In: Zalups RK, Koropatnick J, editors. Molecular Biology and Toxicology of Metals. Taylor and Francis; London: 2000. pp. 34–74. [Google Scholar]
- Bickley LK, Papa CM. Chronic arsenicism with vitiligo, hyperthyroidism, and cancer. N.J. Med. 1989;86:377–380. [PubMed] [Google Scholar]
- Blackman SA. The lesions of lead encephalitis in children. Bull. Johns Hopkins Hosp. 1937;61:1–61. [Google Scholar]
- Blake KC, Mann M. Effect of calcium and phosphorus on the gastrointestinal absorption of 203Pb in man. Environ. Res. 1983;30:188–194. doi: 10.1016/0013-9351(83)90179-2. [DOI] [PubMed] [Google Scholar]
- Blazka ME, Shaikh ZA. Differences in cadmium and mercury uptakes by hepatocytes: role of calcium channels. Toxicol. Appl. Pharmacol. 1991;110:355–363. doi: 10.1016/s0041-008x(05)80018-3. [DOI] [PubMed] [Google Scholar]
- Blazka ME, Shaikh ZA. Cadmium and mercury accumulation in rat hepatocytes: interactions with other metal ions. Toxicol. Appl. Pharmacol. 1992;113:118–125. doi: 10.1016/0041-008x(92)90015-k. [DOI] [PubMed] [Google Scholar]
- Blom S, Lagerkvist B, Linderholm H. Arsenic exposure to smelter workers: clinical and neurophysiological studies. Scand. J. Work. Environ. Health. 1985;11:265–269. doi: 10.5271/sjweh.2227. [DOI] [PubMed] [Google Scholar]
- Bluhm RE, Bobbitt RG, Welch LW, Wood AJ, Bonfiglio JR, Sarzen C, Heath AJ, Branch RA. Elemental mercury vapour toxicity, treatment, and prognosis after acute, intensive exposure in chloralkali plant workers: Part I, history, neuropsychological findings and chelator effects. Hum. Exp. Toxicol. 1992;11:201–210. doi: 10.1177/096032719201100308. [DOI] [PubMed] [Google Scholar]
- Bode BP. Recent molecular advances in mammalian glutamine transport. J. Nutr. 2001;131:2475S–2485S. doi: 10.1093/jn/131.9.2475S. [DOI] [PubMed] [Google Scholar]
- Bogden JD, Gertner SB, Christakos S, Kemp FW, Yang Z, Katz SR, Chu C. Dietary calcium modified concentrations of lead and other metals and renal calbindin in rats. J. Nutr. 1992;122:1351–1360. doi: 10.1093/jn/122.7.1351. [DOI] [PubMed] [Google Scholar]
- Borke JL, Penniston JT, Kumar R. Recent advances in calcium transport by the kidney. Semin. Nephrol. 1990;10:15–23. [PubMed] [Google Scholar]
- Bothwell TH, Charlton RW, Cook JD, Finch CA. Iron Metabolism in Man. Blackwell Sci.; Oxford: 1979. pp. 7–81. [Google Scholar]
- Brandman A, Eskenazi B, Sutton P, Athanasoulis M, Goldman LR. Iron deficiency associated with higher blood lead in children living in contaminated environments. Environ. Health Perspect. 2001;109:1079–1084. doi: 10.1289/ehp.011091079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges CC, Zalups RK. Homocysteine, system b0,+, and the renal epithelial transport and toxicity of inorganic mercury. Am. J. Pathol. 2004 doi: 10.1016/S0002-9440(10)63396-2. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges CC, Bauch C, Verrey F, Zalups RK. Mercuric conjugates of cysteine are transported by the amino acid transporter, system b0,+: implications of molecular mimicry. J. Am. Soc. Nephrol. 2004;15:663–673. doi: 10.1097/01.ASN.0000113553.62380.F5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeks A, Gerrard B, Allikments R, Dean M, Plasterk RH. Homologues of the human multidrug resistance genes MRP and MDR contribute to heavy metal resistance in the soil nematode Caenorhabditis elegans. EMBO J. 1996;15:6132–6143. [PMC free article] [PubMed] [Google Scholar]
- Brown JR, Shockley P. Serum albumin: structure and characterization of its ligand binding sites. In: Jost PC, Griffith OH, editors. Lipid-Protein Interactions. Vol. 1. Wiley Inc; New York: 1982. pp. 25–68. [Google Scholar]
- Buchet JP, Roels H, Bernard A, Lauwerys R. Assessment of renal function of workers exposed to inorganic lead, cadmium or mercury vapor. J. Occup. Med. 1980;22:741–750. [PubMed] [Google Scholar]
- Bunn CR, Matrone G. In vivo interactions of cadmium, copper, zinc and iron in the mouse and rat. J. Nutr. 1966;90:395–399. doi: 10.1093/jn/90.4.395. [DOI] [PubMed] [Google Scholar]
- Cannon VT, Zalups RK, Barfuss DW. Molecular homology and the luminal transport of Hg++ in the renal proximal tubule. J. Am. Soc. Nephrol. 2000;11:394–402. doi: 10.1681/ASN.V113394. [DOI] [PubMed] [Google Scholar]
- Cannon VT, Zalups RK, Barfuss DW. Amino acid transporters involved in luminal transport of mercuric conjugates of cysteine in rabbit proximal tubule. J. Pharmacol. Exp. Ther. 2001;298:780–789. [PubMed] [Google Scholar]
- Canonne-Hergaux F, Gros P. Expression of the iron transporter DMT1 in kidney from normal and anemic mk mice. Kidney Int. 2002;62:147–156. doi: 10.1046/j.1523-1755.2002.00405.x. [DOI] [PubMed] [Google Scholar]
- Canonne-Hergaux F, Gruenheid P, Ponka P, Gros P. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood. 1999;93:4406–4417. [PubMed] [Google Scholar]
- Cantley LC, Jr., Cantley LG, Josephson L. A characterization of vanadate interactions with the (Na,K)-ATPase: mechanistic and regulatory implications. J. Biol. Chem. 1978a;253:7361–7368. [PubMed] [Google Scholar]
- Cantley LC, Resh MD, Guidotti G. Vanadate inhibits the red cell (Na+, K+) ATPase from the cytoplasmic side. Nature. 1978b;272:552–554. doi: 10.1038/272552a0. [DOI] [PubMed] [Google Scholar]
- Canty AJ, Colton R, D'Agostino AP, Traeger JCA. Positive and negative ion electrospray mass spectrometric studies of some amino acids and glutathione, and their interactions with alkali metal ions and methylmercury(II). Inorg. Chim. Acta. 1994;223:103–107. [Google Scholar]
- Cardin CJ, Mason J. Sulphate transport by rat ileum. Effect of molybdate and other anions. Biochim. Biophys. Acta. 1975;394:46–54. doi: 10.1016/0005-2736(75)90203-5. [DOI] [PubMed] [Google Scholar]
- Carmignani M, Boscolo P, Artese L, Del Rosso G, Porcelli G, Felaco M, Volpe AR, Giuliano G. Renal mechanisms in the cardiovascular effects of chronic exposure to inorganic mercury in rats. Br. J. Ind. Med. 1992;49:226–232. doi: 10.1136/oem.49.4.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattori V, van Montfoort JE, Stieger B, Landmann L, Meijer DK, Winterhalter KH, Meier PJ, Hagenbuch B. Localization of organic anion transporting polypeptide 4 (Oatp4) in rat liver and comparison of its substrate specificity with Oatp1, Oatp2, and Oatp3. Pflugers Arch. 2001;443:188–195. doi: 10.1007/s004240100697. [DOI] [PubMed] [Google Scholar]
- Caverzasio J, Selz T, Bonjour JP. Characteristics of phosphate transport in osteoblast like cells. Calcif. Tissue Int. 1988;43:83–87. doi: 10.1007/BF02555151. [DOI] [PubMed] [Google Scholar]
- Cherian MG, Vostal JJ. Biliary excretion of cadmium in rat. I. Dose-dependent biliary excretion and the form of cadmium in the bile. J. Toxicol. Environ. Health. 1977;2:945–954. doi: 10.1080/15287397709529493. [DOI] [PubMed] [Google Scholar]
- Cherian MG, Goyer RA. Metallothioneins and their role in the metabolism and toxicity of metals. Life Sci. 1978;23:1–9. doi: 10.1016/0024-3205(78)90317-x. [DOI] [PubMed] [Google Scholar]
- Cherian MG, Nordberg M. Cellular adaptation in metal toxicology and metallothionein. Toxicology. 1983;28:1–15. doi: 10.1016/0300-483x(83)90101-4. [DOI] [PubMed] [Google Scholar]
- Cherian MG, Goyer RA, Delaquerriere-Richardson L. Cadmium metallothionein induced nephropathy. Toxicol. Appl. Pharmacol. 1976;38:399–408. doi: 10.1016/0041-008x(76)90146-0. [DOI] [PubMed] [Google Scholar]
- Chillaron J, Roca R, Valencia A, Zorzano A, Palacin M. Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am. J. Physiol.: Renal. Physiol. 2001;281:F995–F1018. doi: 10.1152/ajprenal.2001.281.6.F995. [DOI] [PubMed] [Google Scholar]
- Choudhury H, Harvey T, Thayer WC, Lockwood TF, Stiteler WM, Goodrum PE, Hassett JM, Diamond GL. Urinary cadmium elimination as a biomarker of exposure for evaluating a cadmium dietary exposure-biokinetics model. J. Toxicol. Environ. Health. 2001;63:321–350. doi: 10.1080/15287390152103643. [DOI] [PubMed] [Google Scholar]
- Chung J, Wessling-Resnick M. Molecular mechanisms and regulation of iron transport. Crit. Rev. Clin. Lab. Sci. 2003;40:151–182. doi: 10.1080/713609332. [DOI] [PubMed] [Google Scholar]
- Civantos DP, Rodriquez AL, Aguado-Borruey JM, Narvaez JA. Fulminant malignant arrhythmia and multiorgan failure in acute arsenic poisoning. Chest. 1995;108:1774–1775. doi: 10.1378/chest.108.6.1774-a. [DOI] [PubMed] [Google Scholar]
- Clarkson TW. Molecular and ionic mimicry of toxic metals. Annu. Rev. Pharmacol. Toxicol. 1993;33:545–571. doi: 10.1146/annurev.pa.33.040193.002553. [DOI] [PubMed] [Google Scholar]
- Clinton M., Jr. Selenium fume exposure. J. Ind. Hyg. Toxicol. 1947;29:225–226. [PubMed] [Google Scholar]
- Cole SP, Sparks KE, Fraser K, Loe DW, Grant CE, Wilson GM, Deeley RG. Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 1994;54:5902–5910. [PubMed] [Google Scholar]
- Combs GF, Jr., Gray WP. Chemopreventive agents. Selenium. Pharmacol. Ther. 1998;79:179–192. doi: 10.1016/s0163-7258(98)00014-x. [DOI] [PubMed] [Google Scholar]
- Conrad ME, Umbreit JN. Iron absorption and transport. Am. J. Hematol. 2000;64:287–296. doi: 10.1002/1096-8652(200008)64:4<287::aid-ajh9>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells, Mol. Dis. 2002;29:336–355. doi: 10.1006/bcmd.2002.0564. [DOI] [PubMed] [Google Scholar]
- Cooper WC, Wong O, Kheifets L. Mortality among employees of lead battery plants and lead-producing plants, 1947–1980. Scand. J. Work. Environ. Health. 1985;11:331–345. doi: 10.5271/sjweh.2215. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, Krasnikov BF, Zalups RK. Potent inactivation of rat liver γ-cystathionase by mercury cysteine S-conjugate. A nefarious case of molecular mimicry. 2004 Unpublished data. [Google Scholar]
- Cotzias GC, Borg DC, Selleck B. Specificity of zinc pathway in the rabbit: zinc-cadmium exchange. Am. J. Physiol. 1961;201:63–66. [Google Scholar]
- Cotzias GC, Borg DC, Selleck B. Specificity of zinc pathway through the body: turnover of Zn65 in the mouse. Am. J. Physiol. 1962;202:359–363. [Google Scholar]
- Cragg RA, Christie GR, Phillips SR, Russi RM, Kury S, Mathers JC, Taylor PM, Ford D. A novel zinc-regulated human zinc transporter, hZTL1, is localized to the enterocyte apical membrane. J. Biol. Chem. 2001;277:22789–22797. doi: 10.1074/jbc.M200577200. [DOI] [PubMed] [Google Scholar]
- Crichton RR. Inorganic Biochemistry of Iron Metabolism. Horwood; West Sussex: 1991. pp. 29–58. [Google Scholar]
- Cullen NM, Wolf LR, St. Clair D. Pediatric arsenic ingestion. Am. J. Emerg. Med. 1995;13:432–435. doi: 10.1016/0735-6757(95)90133-7. [DOI] [PubMed] [Google Scholar]
- Dave MH, Schulz N, Zecevic M, Wagner CA, Verrey F. Expression of heterodimeric amino acid transporters along the murine intestine. J. Physiol. 2004;558:597–610. doi: 10.1113/jphysiol.2004.065037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David C, Ullrich KJ. Substrate specificity of the luminal Na+-dependent sulphate transport system in the proximal renal tubule as compared to the contraluminal sulphate exchange system. Pflugers Arch. 1992;421:455–465. doi: 10.1007/BF00370256. [DOI] [PubMed] [Google Scholar]
- Dawson DC, Ballatori N. Membrane transporters as sites of action and routes of entry for toxic metals. In: Goyer RA, Cherian MG, editors. Toxicology of Metals. Springer-Verlag; Berlin: 1995. pp. 53–76. [Google Scholar]
- de Ceaurriz J, Ban M. Role of gamma-glutamyltranspeptidase and beta-lyase in the nephrotoxicity of hexachloro-1,3-butadiene and methylmercury in mice. Toxicol. Lett. 1990;50:249–256. doi: 10.1016/0378-4274(90)90017-g. [DOI] [PubMed] [Google Scholar]
- de Ceaurriz J, Payan JP, Morel G, Brondeau MT. Role of extracellular glutathione and γ-glutamyltranspeptidase in the disposition and kidney toxicity of inorganic mercury in rats. J. Appl. Toxicol. 1994;14:201–206. doi: 10.1002/jat.2550140310. [DOI] [PubMed] [Google Scholar]
- Delnomdedieu M, Basti MM, Otvos JD, Thomas DJ. Reduction and binding of arsenate and dimethylarsinate by glutathione: a magnetic resonance study. Chem. Biol. Interact. 1994;90:139–155. doi: 10.1016/0009-2797(94)90099-x. [DOI] [PubMed] [Google Scholar]
- De Master EG, Mitchell RA. A comparison of arsenate and vanadate as inhibitors or uncouplers of mitochondrial and glycolytic energy metabolism. Biochemistry. 1973;12:3616–3621. doi: 10.1021/bi00743a007. [DOI] [PubMed] [Google Scholar]
- Diamond GL, Zalups RK. Understanding renal toxicity of heavy metals. Toxicol. Pathol. 1998;26:92–103. doi: 10.1177/019262339802600111. [DOI] [PubMed] [Google Scholar]
- DiSimplicio P, Gorelli M, Ciuffreda P, Leonzio C. The relationship between γ-glutamyltranspeptidase and Hg levels in Se/Hg antagonism in mouse liver and kidney. Pharmacol. Res. 1990;22:515–526. doi: 10.1016/1043-6618(90)90757-5. [DOI] [PubMed] [Google Scholar]
- Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Positional cloning of zebrafish ferroportin 1 identifies a conserved vertebrate iron exporter. Nature (London) 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- Dorian C, Gattone VH, II, Klaassen CD. Renal cadmium deposition and injury as a result of accumulation of cadmium-metallothionein (CdMT) by the proximal convoluted tubules: a light microscopy autoradiography study with 109CdMT. Toxicol. Appl. Pharmacol. 1992;114:173–181. doi: 10.1016/0041-008x(92)90066-2. [DOI] [PubMed] [Google Scholar]
- Dudley HC, Miller JW. Toxicology of selenium. VI. Effects of subacute exposure to hydrogen selenide. J. Ind. Hyg. Toxicol. 1941;23:470–477. [Google Scholar]
- Dudley RE, Gammal LM, Klaassen CD. Cadmium-induced hepatic and renal injury in chronically exposed rats: likely role of hepatic cadmium-metallothionein in nephrotoxicity. Toxicol. Appl. Pharmacol. 1985;77:414–426. doi: 10.1016/0041-008x(85)90181-4. [DOI] [PubMed] [Google Scholar]
- Dunlap LG. Perforations of the nasal septum due to inhalation of arsenous oxide. J. Am. Med. Assoc. 1921;76:568–569. [Google Scholar]
- Dunn JD, Clarkson TW. Does mercury exhalation signal demethylation of methylmercury? Health Physiol. 1980;38:411–414. [PubMed] [Google Scholar]
- Dutczak WJ, Ballatori N. γ-glutamyltransferase-dependent biliaryhepatic recycling of methyl mercury in the guinea pig. J. Pharmacol. Exp. Ther. 1992;262:619–623. [PubMed] [Google Scholar]
- Dutczak WJ, Clarkson TW, Ballatori N. Biliary-hepatic recycling of a xenobiotic. Gallbladder absorption of methylmercury. Am. J. Physiol. 1991;260:G873–G880. doi: 10.1152/ajpgi.1991.260.6.G873. [DOI] [PubMed] [Google Scholar]
- Dutczak WJ, Truong AT, Ballatori N. Transport of glutathione–methylmercury complex in rat liver canalicular membrane vesicles. Toxicologist. 1993;13:191. [PubMed] [Google Scholar]
- DuVal G, Fowler BA. Toxicologist. 1990;10:160. [Google Scholar]
- Elinder CG, Kjellstrom T, Lind B, Linnman L, Friberg L. Cadmium in kidney cortex, liver and pancreas from Swedish autopsies. Estimation of biological halftime in kidney cortex, considering calorie intake and smoking habits. Arch. Environ. Health. 1976;31:292–302. doi: 10.1080/00039896.1976.10667239. [DOI] [PubMed] [Google Scholar]
- Elisma F, Jumarie C. Evidence for cadmium uptake through Nramp2: metal speciation studies with Caco-2 cells. Biochem. Biophys. Res. Commun. 2001;20:662–668. doi: 10.1006/bbrc.2001.5245. [DOI] [PubMed] [Google Scholar]
- Endo T. Transport of cadmium across the apical membrane of epithelial cell lines. Comp. Biochem. Physiol., C Toxicol. Pharmacol. 2002;131:223–239. doi: 10.1016/s1532-0456(02)00009-1. [DOI] [PubMed] [Google Scholar]
- Endo T, Nakaya S, Kimura R, Murata T. Gastrointestinal absorption of inorganic mercuric compounds in vivo and in situ. Toxicol. Appl. Pharmacol. 1984;74:223–229. doi: 10.1016/0041-008x(84)90146-7. [DOI] [PubMed] [Google Scholar]
- Endo T, Nakaya S, Kimura R, Murata T. Gastrointestinal absorption of inorganic mercuric compounds in vitro. Toxicol. Appl. Pharmacol. 1986;83:187–196. doi: 10.1016/0041-008x(86)90295-4. [DOI] [PubMed] [Google Scholar]
- Endo T, Kimura O, Sakata M. Bidirectional transport of cadmium across apical membrane of renal epithelial cell lines via H+-antiporter and inorganic anion exchanger. Toxicology. 1998a;131:183–192. doi: 10.1016/s0300-483x(98)00133-4. [DOI] [PubMed] [Google Scholar]
- Endo T, Kimura O, Sakata M. pH-dependent transport of cadmium in rat renal brush border membrane vesicles: cadmium efflux via H+-antiport. Toxicol. Lett. 1998b;99:99–107. doi: 10.1016/s0378-4274(98)00139-8. [DOI] [PubMed] [Google Scholar]
- Endo T, Kimura O, Sakata M. Cadmium uptake from apical membrane of LLC-PK1 cells via inorganic anion exchanger. Pharmacol. Toxicol. 1998c;82:230–235. doi: 10.1111/j.1600-0773.1998.tb01430.x. [DOI] [PubMed] [Google Scholar]
- Endo T, Kimura O, Hatakeyama M, Takada M, Sakata M. Effects of zinc and copper on cadmium uptake by brush border membrane vesicles. Toxicol. Lett. 1998d;91:111–120. doi: 10.1016/s0378-4274(97)03878-2. [DOI] [PubMed] [Google Scholar]
- Endo T, Kimura O, Sakata M. Further analysis of cadmium uptake from apical membrane of LLC-PK1 cells via inorganic anion exchanger. Pharmacol. Toxicol. 1999;84:187–192. doi: 10.1111/j.1600-0773.1999.tb00898.x. [DOI] [PubMed] [Google Scholar]
- Environmental Protection Agency . Supplement to the 2086 EPA air quality criteria for lead. Vol. 1, Addendum. EPA/600/8−89/049A. U.S. Office of Health and Environmental Assessment; Washington, D.C: 1989. [Google Scholar]
- Environmental Protection Agency . Integrated Risk Information System (IRIS) on Arsenic. National Center for Environmental Assessment, Office of Research and Development; Washington, DC: 1999. [Google Scholar]
- Erfurt C, Roussa E, Thevenod F. Apoptosis by Cd2+ or CdMT in proximal tubule cells: different uptake routes and permissive role of endo/lysosomal CdMT uptake. Am. J. Physiol.: Cell Physiol. 2003;285:C1367–C1376. doi: 10.1152/ajpcell.00217.2003. [DOI] [PubMed] [Google Scholar]
- Evers R, Zaman GJ, van Deemter L, Jansen H, Calafat J, Oomen LC, Oude Elferink RP, Borst P, Schinkel AH. Basolateral localization and export activity of the human multidrug resistance-associated protein in polarized pig kidney cells. J. Clin. Invest. 1996;97:1211–1218. doi: 10.1172/JCI118535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezaki O. The insulin-like effects of selenate in rat adipocytes. J. Biol. Chem. 1990;265:1124–1128. [PubMed] [Google Scholar]
- Feldman RG, Niles CA, Kelly-Hayes M, Sax DS, Dixon WJ, Thompson DJ, Landau E. Peripheral neuropathy in arsenic smelter workers. Neurology. 1979;29:939–944. doi: 10.1212/wnl.29.7.939. [DOI] [PubMed] [Google Scholar]
- Felley-Bosco E, Diezi J. Fate of cadmium in rat renal tubules: a microinjection study. Toxicol. Appl. Pharmacol. 1987;91:204–211. doi: 10.1016/0041-008x(87)90101-3. [DOI] [PubMed] [Google Scholar]
- Felley-Bosco E, Diezi J. Fate of cadmium in rat renal tubules: a micropuncture study. Toxicol. Appl. Pharmacol. 1989;98:243–251. doi: 10.1016/0041-008x(89)90229-9. [DOI] [PubMed] [Google Scholar]
- Felley-Bosco E, Diezi J. Dietary calcium restriction enhances cadmium-induced metallothionein synthesis in rats. Toxicol Lett. 1992;60:139–144. doi: 10.1016/0378-4274(92)90268-o. [DOI] [PubMed] [Google Scholar]
- Ferguson CJ, Wareing M, Ward DT, Green R, Smith CP, Riccardi D. Cellular localization of divalent metal transporter DMT-1 in rat kidney. Am. J. Physiol. 2001;280:F803–F814. doi: 10.1152/ajprenal.2001.280.5.F803. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Takikawa H, Horie T, Ookhtens M, Kaplowitz N. Canalicular transport of reduced glutathione in normal and mutant Eisai hyperbilirubinemic rats. J. Biol. Chem. 1992;267:1667–1673. [PubMed] [Google Scholar]
- Fernandez-Checa JC, Ookhtens M, Kaplowitz N. Selective induction by phenobarbital of the electrogenic transport of glutathione and organic anions in rat liver canalicular membrane vesicles. J. Biol. Chem. 1993;268:10836–10841. [PubMed] [Google Scholar]
- Ferrier B, Martin M, Roch-Ramel F. Effects of p-aminohippurate and pyrazinoate on the renal excretion of salicylate in the rat: a micropuncture study. J. Pharmacol. Exp. Ther. 1983;224:451–458. [PubMed] [Google Scholar]
- Flanagan PR, McLellan JS, Haist J, Cherian G, Chamberlain MJ, Valberg LS. Increased dietary cadmium absorption in mice and human subjects with iron deficiency. Gastroenterology. 1978;74:841–846. [PubMed] [Google Scholar]
- Flanagan PR, Hamilton DL, Haist J, Valberg LS. Interrelationships between iron and lead absorption in iron-deficient mice. Gastroenterology. 1979;77:1074–1081. [PubMed] [Google Scholar]
- Foulkes EC. Renal tubular transport of cadmium-metallothionein. Toxicol. Appl. Pharmacol. 1978;45:505–512. doi: 10.1016/0041-008x(78)90112-6. [DOI] [PubMed] [Google Scholar]
- Foulkes EC. Interactions between metals in rat jejunum: implications on the nature of cadmium uptake. Toxicology. 1985;37:117–125. doi: 10.1016/0300-483x(85)90118-0. [DOI] [PubMed] [Google Scholar]
- Foulkes EC. On the mechanism of transfer of heavy metals across cell membranes. Toxicology. 1988;52:263–272. doi: 10.1016/0300-483x(88)90131-x. [DOI] [PubMed] [Google Scholar]
- Foulkes EC. Metallothionein and glutathione as determinants of cellular retention and extrusion of cadmium and mercury. Life Sci. 1993;52:1617–1620. doi: 10.1016/0024-3205(93)90042-2. [DOI] [PubMed] [Google Scholar]
- Foulkes EC. Transport of toxic heavy metals across cell membranes. Proc. Soc. Exp. Biol. Med. 2000;223:234–240. doi: 10.1046/j.1525-1373.2000.22334.x. [DOI] [PubMed] [Google Scholar]
- Foulkes EC, Bergman D. Inorganic mercury absorption in mature and immature rat jejunum: transcellular and intercellular pathways in vivo and in everted sacs. Toxicol. Appl. Pharmacol. 1993;120:89–95. doi: 10.1006/taap.1993.1090. [DOI] [PubMed] [Google Scholar]
- Foulkes EC, McMullen DM. Kinetics of transepithelial movement of heavy metals in rat jejunum. Am. J. Physiol. 1987;253:G134–G138. doi: 10.1152/ajpgi.1987.253.2.G134. [DOI] [PubMed] [Google Scholar]
- Fowler BA. Mechanisms of kidney cell injury from metals. Environ. Health Perspect. 1993;100:57–63. doi: 10.1289/ehp.9310057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler BA, DuVal G. Effects of lead on the kidney: roles of high-affinity binding proteins. Environ. Health Perspect. 1991;91:77–80. doi: 10.1289/ehp.919177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MR, Jacobs RM, Jones AO, Fry BE, Jr., Stone CL. Effects of vitamin C and iron on cadmium metabolism. Ann. N. Y. Acad. Sci. 1980;355:249–291. doi: 10.1111/j.1749-6632.1980.tb21343.x. [DOI] [PubMed] [Google Scholar]
- Frazer DM, Vulpe CD, McKie AT, Wilkins SJ, Trinder D, Cleghorn GJ, Anderson GJ. Cloning and gastrointestinal expression or rat hephaestin: relationship to other iron transport proteins. Am. J. Physiol.: Gastrointest. Liver Physiol. 2001;281:G931–G939. doi: 10.1152/ajpgi.2001.281.4.G931. [DOI] [PubMed] [Google Scholar]
- Friedman PA, Gesek FA. Cadmium uptake by kidney distal convoluted tubule cells. Toxicol. Appl. Pharmacol. 1994;128:257–263. doi: 10.1006/taap.1994.1205. [DOI] [PubMed] [Google Scholar]
- Friberg L. Health hazards in the manufacture of alkaline accumulators with special reference to chronic cadmium poisoning. Acta Med. Stand. 1950;138(Suppl 240):1–124. [PubMed] [Google Scholar]
- Friberg L. Studies on the metabolism of mercuric chloride and methyl mercury dicyandiamide: experiments on rats given subcutaneous injections with radioactive mercury (203Hg). AMA Arch. Ind. Health. 1959;20:42–49. [PubMed] [Google Scholar]
- Friberg L, Elinder CG, Kjellstron T, et al. General summary and conclusions and some aspects of diagnosis and treatment of chronic cadmium poisoning. In: Friberg L, Elinder CG, Kjellstrom T, et al., editors. Cadmium and Health: A Toxicological and Epidemiological Appraisal. Vol. 2. CRC Press; Boca Raton, FL: 1986. pp. 247–263. [Google Scholar]
- Friedman PA. Renal calcium transport: sites and insights. News Physiol. Sci. 1988;3:17–21. [Google Scholar]
- Fuhr B, Rabenstein DL. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. IX. The binding of cadmium, zinc, lead, and mercury by glutathione. J. Am. Chem. Soc. 1973;95:6944–6950. doi: 10.1021/ja00802a013. [DOI] [PubMed] [Google Scholar]
- Furnsinn C, Englisch R, Ebner K, Nowotny P, Vogle C, Waldhausl W. Insulin-like vs. non-insulin stimulation of glucose metabolism by vanadium, tungsten, and selenium compounds in rat muscle. Life Sci. 1996;59:1989–2000. doi: 10.1016/s0024-3205(96)00550-4. [DOI] [PubMed] [Google Scholar]
- Gage JC. Distribution and excretion of methyl and phenylmercury salts. Br. J. Ind. Med. 1964;21:197–202. doi: 10.1136/oem.21.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy V, Ganapathy ME, Leibach FH. Intestinal transport of peptides and amino acids. In: Barrett KE, Donowitz M, editors. Current Topics in Membranes. Academic Press; New York: 2001. pp. 379–412. [Google Scholar]
- Garcia-Morales P, Saceda M, Kenney N, Kim N, Salomon DS, Gottardis MM, Soloman HB, Sholler PF, Jordan VC, Martin MB. Effect of cadmium on estrogen receptor levels and estrogen-induced responses in human breast cancer cells. J. Biol. Chem. 1994;269:16896–16901. [PubMed] [Google Scholar]
- Garcia-Ruiz C, Fernandez-Checa J, Kaplowitz N. Bidirectional mechanism of plasma membrane transport of reduced glutathione in intact rat hepatocytes and membrane vesicles. J. Biol. Chem. 1992;267:22256–22264. [PubMed] [Google Scholar]
- Ghosh R, Mukherjee B, Chatterjee M. A novel effect of selenium on streptozotocin-induced diabetic mice. Diabetes Res. 1994;25:165–171. [PubMed] [Google Scholar]
- Glover JR. Selenium in human urine: a tentative maximum allowable concentration for industrial and rural populations. Ann. Occup. Hyg. 1967;10:3–14. doi: 10.1093/annhyg/10.1.3. [DOI] [PubMed] [Google Scholar]
- Goldstein GW. Lead encephalopathy: the significance of lead inhibition of calcium uptake by brain mitochondria. Brain Res. 1977;136:185–188. doi: 10.1016/0006-8993(77)90145-7. [DOI] [PubMed] [Google Scholar]
- Goldstein GW. Brain capillaries: a target for inorganic lead poisoning. Neurotoxicology. 1984;5:167–175. [PubMed] [Google Scholar]
- Goldstein GW, Ar D. Lead activates calmodulin sensitive processes. Life Sci. 1983;33:1001–1006. doi: 10.1016/0024-3205(83)90757-9. [DOI] [PubMed] [Google Scholar]
- Goyer RA. The nephrotoxic effects of lead. In: Bach PH, Bonner FW, Bridges JW, Lock EA, editors. Nephrotoxicity. Wiley, Chichester; England: 1982. pp. 338–348. [Google Scholar]
- Goyer RA. Lead toxicity: current concerns. Environ. Health Perspect. 1993;100:177–187. doi: 10.1289/ehp.93100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyer RA. Nutrition and metal toxicity. Am. J. Clin. Nutr. 1995;61(3 Suppl):646S–650S. doi: 10.1093/ajcn/61.3.646S. [DOI] [PubMed] [Google Scholar]
- Goyer RA. Toxic and essential metal interactions. Annu. Rev. Nutr. 1997;17:37–50. doi: 10.1146/annurev.nutr.17.1.37. [DOI] [PubMed] [Google Scholar]
- Goyer RA, Cherian MG, Delaquerriere-Richardon L. Correlation of parameters of cadmium exposure with onset of cadmium-induced nephropathy in rats. J. Environ. Pathol., Toxicol. Oncol. 1984;5:89–100. [PubMed] [Google Scholar]
- Gregus Z, Stein AF, Klaassen CD. Effect of inhibition of gamma-glutamyl-transpeptidase on biliary and urinary excretion of glutathione-derived thiols and methyl-mercury. J. Pharmacol. Exp. Ther. 1987;242:27–32. [PubMed] [Google Scholar]
- Griffiths WJH, Kelly AL, Smith SJ, Cox TM. Localization of iron transport and regulatory proteins in human cells. Q. J. Med. 2000;93:575–587. doi: 10.1093/qjmed/93.9.575. [DOI] [PubMed] [Google Scholar]
- Groten JP, Luten JB, van Bladeren PJ. Dietary iron lowers the intestinal uptake of cadmium-metallothionein in rats. Eur. J. Pharmacol. 1992;228:23–28. doi: 10.1016/0926-6917(92)90007-y. [DOI] [PubMed] [Google Scholar]
- Gruenheid S, Cellier M, Vidal S, Gros P. Identification and characterization of a second mouse Nramp gene. Genomics. 1995;25:514–525. doi: 10.1016/0888-7543(95)80053-o. [DOI] [PubMed] [Google Scholar]
- Grundemann D, Gorboulev V, Gambaryan S, Veyhl M, Koepsell H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature. 1994;372:549–552. doi: 10.1038/372549a0. [DOI] [PubMed] [Google Scholar]
- Gunn SA, Gould TC, Anderson WA. The selective injurious response of testicular and epididymal blood vessels to cadmium and its prevention by zinc. Am. J. Pathol. 1963a;42:685–702. [PMC free article] [PubMed] [Google Scholar]
- Gunn SA, Gould TC, Anderson WA. Cadmium-induced interstitial cell tumors in rats and mice and their prevention by zinc. J. Natl. Cancer Inst. 1963b;31:745–759. [PubMed] [Google Scholar]
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- Gyurasics A, Varga F, Gregus Z. Glutathione-dependent biliary excretion of arsenic. Biochem. Pharmacol. 1991;42:465–468. doi: 10.1016/0006-2952(91)90306-p. [DOI] [PubMed] [Google Scholar]
- Habeebu SS, Liu J, Klaassen CD. Cadmium-induced apoptosis in mouse liver. Toxicol. Appl. Pharmacol. 1998;149:203–209. doi: 10.1006/taap.1997.8334. [DOI] [PubMed] [Google Scholar]
- Habermann E, Crowell K, Janicki P. Lead and other metals can substitute for Ca2+ in calmodulin. Arch. Toxicol. 1983;54:61–70. doi: 10.1007/BF00277816. [DOI] [PubMed] [Google Scholar]
- Hall RH, Laskin S, Frank P, Maynard EA, Hodge HC. Preliminary observations on toxicity of elemental selenium. A.M.A. Arch. Ind. Hyg. Assoc. 1951;4:458–464. [PubMed] [Google Scholar]
- Hamilton DL, Valberg LS. Relationship between cadmium and iron absorption. Am. J. Physiol. 1974;227:1033–1037. doi: 10.1152/ajplegacy.1974.227.5.1033. [DOI] [PubMed] [Google Scholar]
- Harada M. Congenital Minamata disease: intrauterine methylmercury poisoning. Teratology. 1978;18:285–288. doi: 10.1002/tera.1420180216. [DOI] [PubMed] [Google Scholar]
- Harada M. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit. Rev. Toxicol. 1995;25:1–24. doi: 10.3109/10408449509089885. [DOI] [PubMed] [Google Scholar]
- Heijn M, Hooijberg JH, Scheffer GL, Szabo G, Westerhoff HV, Lankelma J. Anthracyclines modulate multidrug resistance protein (MRP) mediated organic anion transport. Biochim. Biophys. Acta. 1997;1326:12–22. doi: 10.1016/s0005-2736(97)00003-5. [DOI] [PubMed] [Google Scholar]
- Hill CH, Matrone G, Payne WL, Barber CW. In vivo interactions of cadmium with copper, zinc, and iron. J. Nutr. 1963;80:227–235. doi: 10.1093/jn/80.3.227. [DOI] [PubMed] [Google Scholar]
- Hinkle PM, Kinsella PA, Osterhoudt KC. Cadmium uptake and toxicity via voltage-sensitive calcium channels. J. Biol. Chem. 1987;262:16333–16337. [PubMed] [Google Scholar]
- Hirayama K. Effect of amino acids on brain uptake of methylmercury. Toxicol. Appl. Pharmacol. 1980;55:318–323. doi: 10.1016/0041-008x(80)90093-9. [DOI] [PubMed] [Google Scholar]
- Hirayama K. Effects of combined administration of thiol compounds and methylmercury chloride on mercury distribution in rats. Biochem. Pharmacol. 1985;34:2030–2032. doi: 10.1016/0006-2952(85)90328-4. [DOI] [PubMed] [Google Scholar]
- Holmqvist I. Occupational arsenical dermatitis: a study among employees at a copper ore smelting work including investigations of skin reactions to contact with arsenic compounds. Acta Derm.- Venerol. Suppl. 1951;26:1–214. [PubMed] [Google Scholar]
- Holness DL, Taraschuk IG, Nethercott JR. Health status of copper refinery workers with specific reference to selenium exposure. Arch. Environ. Health. 1989;44:291–297. doi: 10.1080/00039896.1989.9935896. [DOI] [PubMed] [Google Scholar]
- Huebers HA, Huebers E, Csiba E, Rummel W, Finch CA. The cadmium effect on iron absorption. Am. J. Clin. Nutr. 1987;45:1007–1012. doi: 10.1093/ajcn/45.5.1007. [DOI] [PubMed] [Google Scholar]
- Hughes WL. A physiochemical rationale for the biological activity of mercury and its compounds. Ann. N. Y. Acad. Sci. 1957;65:454–460. doi: 10.1111/j.1749-6632.1956.tb36650.x. [DOI] [PubMed] [Google Scholar]
- Huisingh J, Matrone G. Interaction of molybdenum in animal nutrition. In: Chappell WR, Peterson KK, editors. Molybdenum in the Environment. The Biology of Molybdenum. Marcel Dekker; New York: 1976. pp. 125–148. [Google Scholar]
- Iioka H, Moriyama I, Oku M, Hino K, Itani Y, Okamura Y, Ichijo M. The effect of inorganic mercury on placental amino acid transport using microvillous membrane vesicles. Nippon Sanka Fujinka Gakkai Zasshi. 1987;39:202–206. [PubMed] [Google Scholar]
- Inouye M, Kajiwara Y. Developmental disturbances of the fetal brain in guinea pigs caused by methylmercury. Arch. Toxicol. 1988;62:15–21. doi: 10.1007/BF00316251. [DOI] [PubMed] [Google Scholar]
- Inouye M, Kajiwara Y. Placental transfer of methylmercury and mercuric mercury in mice. Environ. Med. 1990;34:169–172. [Google Scholar]
- Inouye M, Murao K, Kajiwara Y. Behavioral and neuropathological effects of prenatal methylmercury exposure in mice. Neurobehav. Toxicol. Teratol. 1985;7:227–232. [PubMed] [Google Scholar]
- Ishizawa T, Tsuji A, Tamai I, Teraski T, Hosoi K, Fukatsu S. Sodium and pH dependent carrier-mediated transport of antibiotic, fosfomycin, in the rat intestinal brush-border membrane. J. Pharmacobio-dyn. 1990;13:292–300. doi: 10.1248/bpb1978.13.292. [DOI] [PubMed] [Google Scholar]
- Jacobs RM, Jones AO, Fox MR, Lener J. Effects of dietary zinc, manganese, and copper on tissue accumulation of cadmium by Japanese quail. Proc. Soc. Exp. Biol. Med. 1983;172:34–38. doi: 10.3181/00379727-172-41522. [DOI] [PubMed] [Google Scholar]
- Jacobson KB, Turner JE. The interaction of cadmium and certain other metal ions with proteins and nucleic acids. Toxicology. 1980;16:1–37. doi: 10.1016/0300-483x(80)90107-9. [DOI] [PubMed] [Google Scholar]
- Jacquemin E, Hagenbuch B, Steiger C, Wolkoff AW, Meier PJ. Expression cloning of a rat liver (Na+)-independent organic anion transporter. Proc. Natl. Acad. Sci., U. S. A. 1994;91:133–137. doi: 10.1073/pnas.91.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe KM, Shurtleff DB, Robertson WO. Survival after acute mercury vapor poisoning—Role of intensive supportive care. Am. J. Dis. Child. 1983;137:749–751. doi: 10.1001/archpedi.1983.02140340033008. [DOI] [PubMed] [Google Scholar]
- Jarup L, Berglund M, Elinder CG, Nordberg G, Vahter M. Health effects of cadmium exposure: a review of the literature and a risk estimate. Scand. J. Work Environ. Health. 1998;1:1–51. [PubMed] [Google Scholar]
- Jansson T. Amino acid transporters in the human placenta. Pediatr. Res. 2001;49(Suppl):141–147. doi: 10.1203/00006450-200102000-00003. [DOI] [PubMed] [Google Scholar]
- Jensen GE, Hansen ML. Occupational arsenic exposure and glycosylated haemoglobin. Analyst. 1998;123:77–80. doi: 10.1039/a705699k. [DOI] [PubMed] [Google Scholar]
- Jernelov A. A new biochemical pathway for the methylation of mercury and some ecological implications. In: Miller MW, Clarkson TW, editors. Mercury, Mercurials, and Mercaptans. Thomas; Springfield, IL: 1973. pp. 315–324. [Google Scholar]
- Johnson MD, Kenney N, Stoica A, Hilakivi-Clarke L, Singh B, Chepko G, Clarke R, Sholler PF, Lirio AA, Foss C, Reiter R, Trock B, Paik S, Martin MB. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat. Med. 2003;9:1081–1084. doi: 10.1038/nm902. [DOI] [PubMed] [Google Scholar]
- Kägi JHR, Vallee BL. Metallothionein: a cadmium- and zinc-containing protein from equine renal cortex. J. Biol. Chem. 1960;235:3460–3465. [PubMed] [Google Scholar]
- Kajiwara Y, Inouye M. Effects of methylmercury and mercuric chloride on preimplantation mouse embryos in vivo. Teratology. 1986;33:231–237. doi: 10.1002/tera.1420330210. [DOI] [PubMed] [Google Scholar]
- Kajiwara Y, Inouye M. Inhibition of implantation caused by methylmercury and mercuric chloride in mouse embryos in vivo. Bull. Environ. Contam. Toxicol. 1992;49:541–546. doi: 10.1007/BF00196296. [DOI] [PubMed] [Google Scholar]
- Kajiwara Y, Yasutake A, Adachi T, Hirayama K. Methylmercury transport across the placenta via neutral amino acid carrier. Arch. Toxicol. 1996;70:310–314. doi: 10.1007/s002040050279. [DOI] [PubMed] [Google Scholar]
- Kala SV, Neely MW, Kala G, Prater CI, Atwood DW, Rice JS, Lieberman MW. The MRP2/cMOAT transporter and arsenic–glutathione complex formation are required for biliary excretion of arsenic. J. Biol. Chem. 2000;275:33404–33408. doi: 10.1074/jbc.M007030200. [DOI] [PubMed] [Google Scholar]
- Kala SV, Kala G, Prater CI, Sartorelli AC, Lieberman MW. Formation and urinary excretion of arsenic triglutathione and methylarsenic diglutathione. Chem. Res. Toxicol. 2004;17:243–249. doi: 10.1021/tx0342060. [DOI] [PubMed] [Google Scholar]
- Kamiyama T, Miyakawa H, Li JP, Akiba T, Liu JH, Liu J, Marumo F, Sato C. Effects of one-year cadmium exposure on livers and kidneys and their relation to glutathione levels. Res. Commun. Mol. Pathol. Pharmacol. 1995;88:177–186. [PubMed] [Google Scholar]
- Kanai Y, Segawa H, Miyamoto K, Uchino H, Takeda E, Endou H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 1998;273:23629–23632. doi: 10.1074/jbc.273.37.23629. [DOI] [PubMed] [Google Scholar]
- Kapoor SC, van Rossum GD. Effects of Pb2+ added in vitro on Ca2+ movements in isolated mitochondria and slices of rat kidney cortex. Biochem. Pharmacol. 1984;33:1771–1778. doi: 10.1016/0006-2952(84)90348-4. [DOI] [PubMed] [Google Scholar]
- Kar AB, Das RP. Testicular changes in rats after treatment with cadmium chloride. Acta Biol. Med. Ger. 1960;5:153–173. [PubMed] [Google Scholar]
- Karlish SJD, Beauge LA, Glynn IM. Vanadate inhibits (Na+, K+) ATPase by blocking conformational change of the unphosphorylated form. Nature. 1979;282:333–335. doi: 10.1038/282333a0. [DOI] [PubMed] [Google Scholar]
- Kazantzis G. Some long-term effects of cadmium on the human kidney.. Cadmium 77. Proceedings of the 1st International Cadmium Conference in San Francisco; Metal Bulletin, Ltd., London. 1977.1978. pp. 194–198.1. [Google Scholar]; its apical isoform MRP2. Chem. Biol. Interact. 24:153–161. doi: 10.1016/s0009-2797(97)00158-0. [DOI] [PubMed] [Google Scholar]
- Keppler D, Leier I, Jedlitschky G, Konig J. ATP-dependent transport of glutathione S-conjugates by the multidrug resistance Protein MRP. 1998. [DOI] [PubMed]
- Kerper LE, Ballatori N, Clarkson TW. Methylmercury transport across the blood–brain barrier by an amino acid carrier. Am. J. Physiol. 1992;262:R761–R765. doi: 10.1152/ajpregu.1992.262.5.R761. (Regulatory Integrative Comp. Physiol.) [DOI] [PubMed] [Google Scholar]
- Kerper LE, Hinkle PM. Cellular uptake of lead is activated by depletion of intracellular calcium stores. J. Biol. Chem. 1997a;272:8346–8352. doi: 10.1074/jbc.272.13.8346. [DOI] [PubMed] [Google Scholar]
- Kerper LE, Hinkle PM. Lead uptake in brain capillary endothelial cells: activation by calcium store depletion. Toxicol. Appl. Pharmacol. 1997b;146:127–133. doi: 10.1006/taap.1997.8234. [DOI] [PubMed] [Google Scholar]
- King LM, Banks WA, George WJ. Differences in cadmium transport to the testis, epididymis, and brain in cadmium-sensitive and -resistant murine strains 129/J and A/J. J. Pharmacol. Exp. Ther. 1999;289:825–830. [PubMed] [Google Scholar]
- Koh AS, Simmons-Willis TA, Pritchard JB, Grassl SM, Ballatori N. Identification of a mechanism by which the methylmercury antidotes N-acetylcysteine and dimercaptopropanesulfonate enhance urinary metal excretion: transport by the renal organic transporter-1. Mol. Pharmacol. 2002;62:921–926. doi: 10.1124/mol.62.4.921. [DOI] [PubMed] [Google Scholar]
- Kojima R, Sekine T, Kawachi M, Cha SH, Suzuki Y, Endou H. Immunolocalization of multispecific organic anion transporters, OAT1, OAT2, OAT3 in rat kidney. J. Am. Soc. Nephrol. 2002;13:848–857. doi: 10.1681/ASN.V134848. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Boyd CAR. Human placental amino acid transporter genes: expression and function. Reproduction. 2002;124:593–600. doi: 10.1530/rep.0.1240593. [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick GA, Hagenbuch B, Steiger B, Wolkoff AW, Meier PJ. Functional characterization of the basolateral rat liver organic anion transporting polypeptide. Hepatology. 1994;20:411–416. [PubMed] [Google Scholar]
- Kutsuna M, editor. Study Group of Minamata Disease. Kumamoto University; Japan: Minamata disease. pp. 1–4. [Google Scholar]
- Landner L. Biochemical model for the biological methylation of mercury suggested from methylation studies in vivo with Neurospora crassa. Nature (London) 1971;230:452–454. doi: 10.1038/230452a0. [DOI] [PubMed] [Google Scholar]
- Lagerkvist B, Linderholm H, Nordberg GF. Vasospastic tendency and Raynaud's phenomenon in smelter workers exposed to arsenic. Environ. Res. 1986;39:465–474. doi: 10.1016/s0013-9351(86)80070-6. [DOI] [PubMed] [Google Scholar]
- Lagerkvist BEA, Linderholm H, Nordberg GF. Arsenic and Raynaud's phenomenon: vasospastic tendency and excretion of arsenic in smelter workers before and after the summer vacation. Int. Arch. Occup. Environ. Health. 1988;60:361–364. doi: 10.1007/BF00405671. [DOI] [PubMed] [Google Scholar]
- Lagerkvist BJ, Zetterlund B. Assessment of exposure to arsenic among smelter workers: a five-year follow up. Am. J. Ind. Med. 1994;25:477–488. doi: 10.1002/ajim.4700250403. [DOI] [PubMed] [Google Scholar]
- Laporte J-M, Andres S, Mason RP. Effect of ligands and other metals on the uptake of mercury and methylmercury across the gills and the intestines of the blue crab (Callinectes sapidus). Comp. Biochem. Physiol., C. 2002;131:185–196. doi: 10.1016/s1532-0456(01)00289-7. [DOI] [PubMed] [Google Scholar]
- Leaner JJ, Mason RP. Methylmercury accumulation and fluxes across the intestine of channel catfish, Ictalurus punctatus. Comp. Biochem. Physiol., C Toxicol. Pharmacol. 2002;132:247–259. doi: 10.1016/s1532-0456(02)00072-8. [DOI] [PubMed] [Google Scholar]
- Leazer TM, Klaassen CD. The presence of xenobiotic transporters in rat placenta. Drug Metab. Dispos. 2003;31:153–167. doi: 10.1124/dmd.31.2.153. [DOI] [PubMed] [Google Scholar]
- Leazer TM, Liu Y, Klaassen CD. Cadmium absorption and its relationship to divalent metal transporter-1 in the pregnant rat. Toxicol. Appl. Pharmacol. 2002;185:18–24. doi: 10.1006/taap.2002.9505. [DOI] [PubMed] [Google Scholar]
- Lecoeur S, Huynh-Delerme C, Blais A, Duche A, Tome D, Kolf-Clauw M. Implication of distinct proteins in cadmium uptake and transport by intestinal cells HT-29. Cell Biol. Toxicol. 2002;18:409–423. doi: 10.1023/a:1020867707079. [DOI] [PubMed] [Google Scholar]
- Leier I, Jedlitschky G, Buchholz U, Center M, Cole SPC, Deeley RG, Keppler D. ATP-dependent glutathione disulfide transport mediated by the MRP gene-encoded conjugate export pump. Biochem. J. 1996;314:433–437. doi: 10.1042/bj3140433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon L, Johnson DR. Role of iron in jejunal uptake of cadmium in the newborn rat. J. Toxicol. Environ. Health. 1985;15:687–696. doi: 10.1080/15287398509530696. [DOI] [PubMed] [Google Scholar]
- Leslie EM, Deeley RG, Cole SP. Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters. Toxicology. 2001;167:3–23. doi: 10.1016/s0300-483x(01)00454-1. [DOI] [PubMed] [Google Scholar]
- Leslie EM, Haimeur A, Waalkes MP. Arsenic transport by the human multidrug resistance protein (MRP1/ABCC1): evidence that a tri-glutathione conjugate is required. J. Biol. Chem. 2004;279:32700–32708. doi: 10.1074/jbc.M404912200. [DOI] [PubMed] [Google Scholar]
- Lewis GF, Coughlin L, Jusko W, Hartz S. Contribution of cigarette smoking of cadmium accumulation in man. Lancet. 1972;1:291–292. doi: 10.1016/s0140-6736(72)90294-2. [DOI] [PubMed] [Google Scholar]
- Lilis R. Long-term occupational lead exposure: chronic nephropathy and renal cancer: a case report. Am. J. Ind. Med. 1981;2:293–297. doi: 10.1002/ajim.4700020309. [DOI] [PubMed] [Google Scholar]
- Lin JL, Lim PS. Massive oral ingestion of elemental mercury. J. Toxicol. Clin. Toxicol. 1993;31:487–492. doi: 10.3109/15563659309000417. [DOI] [PubMed] [Google Scholar]
- Liu J, Habeebu SS, Liu Y, Klaassen CD. Acute CdMT injection is not a good model to study chronic Cd nephropathy: comparison of chronic CdCl2 and CdMT exposure with acute CdMT injection in rats. Toxicol. Appl. Pharmacol. 1998a;153:48–58. doi: 10.1006/taap.1998.8506. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu Y, Habeebu SS, Klaassen CD. Susceptibility of MT-null mice to chronic CdCl2-induced nephrotoxicity indicated that renal injury is not mediated by the CdMT complex. Toxicol. Sci. 1998b;46:197–203. doi: 10.1006/toxs.1998.2541. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu Y, Habeebu SM, Waalkes MP, Klaassen CD. Chronic combined exposure to cadmium and arsenic exacerbates nephrotoxicity, particularly in metallothionein-I/II null mice. Toxicology. 2000;147:157–166. doi: 10.1016/s0300-483x(00)00194-3. [DOI] [PubMed] [Google Scholar]
- Liu J, Chen H, Miller DS, Saavedra JE, Keefer LK, Johnson DR, Klaassen CD, Waalkes MP. Overexpression of glutathione S-transferase II and multidrug resistance transport proteins is associated with acquired tolerance to inorganic arsenic. Mol. Pharmacol. 2001;60:302–309. doi: 10.1124/mol.60.2.302. [DOI] [PubMed] [Google Scholar]
- Lundgren KD. Damage to respiratory organs in workers in a smelting plant. Nord. Hyg. Tidskr. 1954;3:66–82. [PubMed] [Google Scholar]
- Lundgren KD, Swensson A. Occupational poisoning by alkyl mercury compounds. J. Ind. Hyg. Toxicol. 1949;31:190–200. [PubMed] [Google Scholar]
- Mack U, Powell LW, Halliday JW. Detection and isolation of a hepatic membrane receptor for ferritin. J. Biol. Chem. 1983;258:4672–4675. [PubMed] [Google Scholar]
- Madsen KM. Mercury accumulation in kidney lysosomes of proteinuric rats. Kidney Int. 1980;18:445–453. doi: 10.1038/ki.1980.157. [DOI] [PubMed] [Google Scholar]
- Magnuson MA, Andreone TL, Printz RL, Koch S, Granner DK. Rat glucokinase gene: structure and regulation by insulin. Proc. Natl. Acad. Sci. U.S.A. 1989;86:4838–4842. doi: 10.1073/pnas.86.13.4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magos L, Butler WH. The kinetics of methylmercury administered repeatedly to rats. Arch. Toxicol. 1976;35:25–39. doi: 10.1007/BF00333983. [DOI] [PubMed] [Google Scholar]
- Magos L, Clarkson TW, Allen J. The interrelationship between non-protein bound thiols and the biliary excretion of methylmercury. Biochem. Pharmacol. 1978;27:2203–2208. doi: 10.1016/0006-2952(78)90078-3. [DOI] [PubMed] [Google Scholar]
- Magos L, Peristianis GG, Clarkson TW, Brown A, Preston S, Snowden RT. Comparative study of the sensitivity of male and female rats to methylmercury. Arch. Toxicol. 1981;48:11–20. doi: 10.1007/BF00297071. [DOI] [PubMed] [Google Scholar]
- Magos L, Brown AW, Sparrow S, Bailey E, Snowden RT, Skipp WR. The comparative toxicology of ethyl- and methyl-mercury. Arch. Toxicol. 1985;57:260–267. doi: 10.1007/BF00324789. [DOI] [PubMed] [Google Scholar]
- Mahaffey KR, Gartside PS, Glueck CJ. Blood lead levels and dietary calcium intake in 1–11 year-old children: the Second National Health and Nutrition Examination Survey, 1976–1980. Pedatrics. 1986;78:257–262. [PubMed] [Google Scholar]
- Martin MB, Reiter R, Pham T, Avellanet YR, Camara J, Lahm M, Pentecost E, Pratap K, Gilmore BA, Divekar S, Dagata RS, Bull JL, Stoica A. Estrogen-like activity of metals in Mcf-7 breast cancer cells. Endocrinology. 2003;144:2425–2436. doi: 10.1210/en.2002-221054. [DOI] [PubMed] [Google Scholar]
- Martini LA, Tchack L, Wood RJ. Iron treatment downregulates DMT1 and IREG1 mRNA expression in Caco-2 cells. J. Nutr. 2002;132:693–696. doi: 10.1093/jn/132.4.693. [DOI] [PubMed] [Google Scholar]
- Mason J, Cardin CJ. The competition of molybdate and sulphate ions for a transport system in the ovine small intestine. Res. Vet. Sci. 1977;22:313–315. [PubMed] [Google Scholar]
- Matsumoto H, Koya G, Tekeuchi T. Fetal Minamata disease. A neuropathological study of two cases of intrauterine intoxication by a methylmercury compound. J. Neuropathol. Exp. Neurol. 1965;24:563–574. [PubMed] [Google Scholar]
- McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell. 2002;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- McMahon RJ, Cousins RJ. Regulation of the zinc transporter ZnT-1 by dietary zinc. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4841–4846. doi: 10.1073/pnas.95.9.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil SI, Bhatnagar MK, Turner CJ. Combined toxicity of ethanol and methylmercury in rat. Toxicology. 1988;53:345–363. doi: 10.1016/0300-483x(88)90226-0. [DOI] [PubMed] [Google Scholar]
- McNeill JH, Delgatty HLM, Battell ML. Insulin-like effects of sodium selenate in streptozotocin-induced diabetic rats. Diabetes. 1991;40:1675–1678. doi: 10.2337/diab.40.12.1675. [DOI] [PubMed] [Google Scholar]
- Min KS, Kobayashi K, Onosaka S, Ohta N, Okada Y, Tanaka K. Tissue distribution of cadmium and nephropathy after administration of cadmium in several chemical forms. Toxicol. Appl. Pharmacol. 1986;86:262–270. doi: 10.1016/0041-008x(86)90057-8. [DOI] [PubMed] [Google Scholar]
- Min KS, Onosaka S, Tanaka K. Renal accumulation of cadmium and nephropathy following long-term administration of cadmiummetallothionein. Toxicol. Appl. Pharmacol. 1996;141:102–109. doi: 10.1006/taap.1996.0265. [DOI] [PubMed] [Google Scholar]
- Mohamed KB. Occupational contact dermatitis from arsenic in a tin-smelting factory. Contact Dermatitis. 1998;38:224–225. doi: 10.1111/j.1600-0536.1998.tb05721.x. [DOI] [PubMed] [Google Scholar]
- Mokrzan EM, Kerper LE, Ballatori N, Clarkson TW. Methylmercury-thiol uptake into cultured brain capillary endothelial cells on amino acid system L. J. Pharmacol. Exp. Ther. 1995;272:1277–1284. [PubMed] [Google Scholar]
- Montessuit C, Caverzasio J, Bonjour JP. Characterization of a Pi transport system in cartilage matrix vesicles. Potential role in the calcification process. J. Biol. Chem. 1991;266:17791–17797. [PubMed] [Google Scholar]
- Morgan EH, Baker E. Iron uptake and metabolism by hepatocytes. Fed. Proc. 1986;45:2810–2816. [PubMed] [Google Scholar]
- Morton WE, Caron GA. Encephalopathy: an uncommon manifestation of workplace arsenic poisoning? Am. J. Ind. Med. 1989;15:1–5. doi: 10.1002/ajim.4700150102. [DOI] [PubMed] [Google Scholar]
- Motohashi H, Sakurai Y, Saito H, Masuda S, Urakami Y, Goto M, Fukatsu A, Ogawa O, Inui K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002;13:866–874. doi: 10.1681/ASN.V134866. [DOI] [PubMed] [Google Scholar]
- Moustaid N, Beyer RS, Sul HS. Identification of an insulin response element in the fatty acid synthase promoter. J. Biol. Chem. 1994;269:5629–5634. [PubMed] [Google Scholar]
- Mulder KM, Kostyniak PJ. Effect of L-(αS, 5S)-α-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid on urinary excretion of methylmercury in the mouse. J. Pharmacol. Exp. Ther. 1985;234:156–160. [PubMed] [Google Scholar]
- Murakami M, Webb M. A morphological and biochemical study of the effects of l-cysteine on the renal uptake and nephrotoxicity of cadmium. Br. J. Exp. Pathol. 1981;62:115–130. [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Cain K, Webb M. Cadmium-metallothionein-induced nephropathy: a morphological and autoradiographic study of cadmium distribution, the development of tubular damage and subsequent cell regeneration. J. Appl. Toxicol. 1983;3:237–244. doi: 10.1002/jat.2550030504. [DOI] [PubMed] [Google Scholar]
- Murakami M, Sano K, Webb M. The effect of l-cysteine on the portion-selective uptake of cadmium in the renal proximal tubule. Arch. Toxicol. 1987;60:365–369. doi: 10.1007/BF00295756. [DOI] [PubMed] [Google Scholar]
- Murphy MJ, Culliford EJ, Parsons V. A case of poisoning with mercuric chloride. Resuscitation. 1979;7:35–44. doi: 10.1016/0300-9572(79)90013-3. [DOI] [PubMed] [Google Scholar]
- Mykk7nen HM, Wasserman RH. Gastro-intestinal absorption of lead (203Pb) in chicks: influence of lead, calcium and age. J. Nutr. 1981;111:1757–1765. doi: 10.1093/jn/111.10.1757. [DOI] [PubMed] [Google Scholar]
- Naganuma A, Oda-Urano N, Tanaka T, Imura N. Possible role of hepatic glutathione in transport of methylmercury into mouse kidney. Biochem. Pharmacol. 1988;21:291–296. doi: 10.1016/0006-2952(88)90731-9. [DOI] [PubMed] [Google Scholar]
- National Research Council . Measuring Lead Exposure in Infants, Children and other Sensitive Populations. Natl. Acad.; Washington, DC: 1993. p. 337. [PubMed] [Google Scholar]
- Needleman HL, Schell A, Bellinger D, Leviton A, Allred EN. The long-term effects of exposure to low doses of lead in childhood. N. Engl. J. Med. 1990;322:83–88. doi: 10.1056/NEJM199001113220203. [DOI] [PubMed] [Google Scholar]
- Nordberg M, Nordberg GF. Toxicological aspects of metallothionein. Cell. Mol. Biol. 2000;46:451–463. [PubMed] [Google Scholar]
- Nordberg GF, Goyer R, Nordberg M. Comparative toxicity of cadmium-metallothionein and cadmium chloride on mouse kidney. Arch. Pathol. 1975;99:192–197. [PubMed] [Google Scholar]
- Nordberg GF, Kjellstrom T, Nordberg M. Kinetics and metabolism. In: Friberg L, Elinder CG, Kjellstrom T, editors. Cadmium and Health: A Toxicological and Epidemiological Appraisal, Exposure, Dose, and Metabolism. Vol. 1. CRC Press; Boca Raton, FL: 1985. pp. 103–178. [Google Scholar]
- Norseth T, Clarkson TW. Studies on the biotransformation of 203Hg-labeled methylmercury chloride in rats. Arch. Environ. Health. 1970a;21:717–727. doi: 10.1080/00039896.1970.10667325. [DOI] [PubMed] [Google Scholar]
- Norseth T, Clarkson TW. Biotransformation of methylmercury salts in the rat studied by specific determination of inorganic mercury. Biochem. Pharmacol. 1970b;19:2775–2783. doi: 10.1016/0006-2952(70)90104-8. [DOI] [PubMed] [Google Scholar]
- Norseth T, Clarkson TW. Intestinal transport of 203Hg-labeled methylmercury chloride. Arch. Environ. Health. 1971;22:568–577. doi: 10.1080/00039896.1971.10665903. [DOI] [PubMed] [Google Scholar]
- O'Brien RM, Granner DK. Regulation of gene expression by insulin. Physiol. Rev. 1996;76:1109–1161. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- O'Brien RM, Lucas PC, Forest C, Magnuson MA, Granner DK. Identification of a sequence in the PEPCK gene that mediates a negative effect of insulin on transcription. Science. 1990;249:533–537. doi: 10.1126/science.2166335. [DOI] [PubMed] [Google Scholar]
- Oberdorster G. Pulmonary deposition, clearance and effects of inhaled soluble and insoluble cadmium compounds. IARC Sci. Publ. 1992;118:189–204. [PubMed] [Google Scholar]
- Oka JA, Christensen MD, Weigel PH. Hyperosmolarity inhibits galactosyl receptor-mediated but not fluid phase endocytosis in isolated rat hepatocytes. J. Biol. Chem. 1989;264:12016–12024. [PubMed] [Google Scholar]
- Okubo M, Yamada K, Hosoyamada M, Shibasaki T, Endou H. Cadmium transport by human Nramp 2 expressed in Xenopus laevis oocytes. Toxicol. Appl. Pharmacol. 2003;187:162–167. doi: 10.1016/s0041-008x(02)00078-9. [DOI] [PubMed] [Google Scholar]
- Oldendorf WH. Sterospecificity of blood–brain barrier permeability to amino acids. Am. J. Physiol. 1973;224:967–969. doi: 10.1152/ajplegacy.1973.224.4.967. [DOI] [PubMed] [Google Scholar]
- Olivi L, Sisk J, Bressler J. Involvement of DMT1 in uptake of Cd in MDCK cells: role of protein kinase C. Am. J. Physiol. 2001;281:C793–C800. doi: 10.1152/ajpcell.2001.281.3.C793. [DOI] [PubMed] [Google Scholar]
- Omata S, Sakimura K, Ishii T, Sugano H. Chemical nature of a methylmercury complex with a low molecular weight in the liver cytosol of rats exposed to methylmercury chloride. Biochem. Pharmacol. 1978;27:1700–1702. doi: 10.1016/0006-2952(78)90184-3. [DOI] [PubMed] [Google Scholar]
- Omata S, Sato M, Sakimura K, Sugano H. Time-dependent accumulation of inorganic mercury in subcellular fractions of kidney, liver, and brain or rats exposed to methylmercury. Arch. Toxicol. 1980;44:231–241. doi: 10.1007/BF00278031. [DOI] [PubMed] [Google Scholar]
- Ormos G, Manyai S. Anion transport of the red blood cell under nonequilibrium conditions. J. Physiol. 1978;276:501–513. doi: 10.1113/jphysiol.1978.sp012249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson A, Squibb KS, Fowler BA. Intracellular binding of lead in the kidney: the partial isolation and characterization of postmitochondrial lead binding components. Biochem. Biophys. Res. Commun. 1982;104:290–298. doi: 10.1016/0006-291x(82)91973-8. [DOI] [PubMed] [Google Scholar]
- Osterloh K, Aisen P. Pathways in the binding and uptake of ferritin by hepatocytes. Biochim. Biophys. Acta. 1989;1011:40–45. doi: 10.1016/0167-4889(89)90075-x. [DOI] [PubMed] [Google Scholar]
- Oteiza PI, Adonaylo VN, Keen CL. Cadmium-induced testes oxidative damage in rats can be influenced by dietary zinc intake. Toxicology. 1999;137:13–22. doi: 10.1016/s0300-483x(99)00067-0. [DOI] [PubMed] [Google Scholar]
- Palacin M, Estevez R, Bertran J, Zorzano A. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol. Rev. 1998;78:969–1954. doi: 10.1152/physrev.1998.78.4.969. [DOI] [PubMed] [Google Scholar]
- Palacin M, Fernandez E, Chillaron J, Zorzano A. The amino acid transport system bo,+ and cystinuria. Mol. Membr. Biol. 2001;18:21–26. [PubMed] [Google Scholar]
- Palmiter RD, Findley SD. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995;14:639–649. doi: 10.1002/j.1460-2075.1995.tb07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parizek J. The destructive effect of cadmium ion on testicular tissue and its prevention by zinc. J. Endocrinol. 1957;15:56–63. doi: 10.1677/joe.0.0150056. [DOI] [PubMed] [Google Scholar]
- Parizek J, Zahor Z. Effect of cadmium salts on testicular tissue. Nature. 1956;177:1036. doi: 10.1038/1771036b0. [DOI] [PubMed] [Google Scholar]
- Park JD, Cherrington NJ, Klaassen CD. Intestinal absorption of cadmium is associated with divalent metal transporter 1 in rats. Toxicol. Sci. 2002;68:288–294. doi: 10.1093/toxsci/68.2.288. [DOI] [PubMed] [Google Scholar]
- Perry K, Bowler RG, Buckell HM, et al. Studies in the incidence of cancer in factory handling inorganic compounds of arsenic—II: clinical and environmental investigations. Br. J. Ind. Med. 1948;5:6–15. [PMC free article] [PubMed] [Google Scholar]
- Pfleger H, Wolf HU. Activation of membrane-bound high-affinity calcium ion-sensitive adenosine triphosphatase of human erythrocytes by bivalent metal ions. Biochem. J. 1975;147:359–361. doi: 10.1042/bj1470359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard V, Govoni G, Jabado N, Gros P. Nramp 2 (DCT1/DMT1) expressed at the plasma membrane transports iron and other divalent cations into a calcein-accessible cytoplasmic pool. J. Biol. Chem. 2000;275:35738–35745. doi: 10.1074/jbc.M005387200. [DOI] [PubMed] [Google Scholar]
- Pineda M, Fernandez E, Torrents D, Estevez R, Lopez C, Camps M, Lloberas J, Zorzano A, Palacin M. Identification of a membrane protein, LAT-2, that co-expresses with 4F2 heavy chain, an L-type amino acid transport activity with broad specificity for small and large zwitterionic amino acids. J. Biol. Chem. 1999;274:19738–19744. doi: 10.1074/jbc.274.28.19738. [DOI] [PubMed] [Google Scholar]
- Pinto SS, McGill CM. Arsenic trioxide exposure in industry. Ind. Med. Surg. 1953;22:281–287. [PubMed] [Google Scholar]
- Pounds JG, Long GJ, Rosen JF. Cellular and molecular toxicity of lead in bone. Environ. Health Perspect. 1991;91:17–32. doi: 10.1289/ehp.919117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad PD, Wang H, Huang W, Kekuda R, Rajan DP, Leibach FH, Ganapathy V. Human LAT1, a subunit of system L amino acid transporter: molecular cloning and transport function. Biochem. Biophys. Res. Commun. 1999;255:283–288. doi: 10.1006/bbrc.1999.0206. [DOI] [PubMed] [Google Scholar]
- Price DJ, Joshi JG. Ferritin. Binding of beryllium and other divalent metal ions. J. Biol. Chem. 1983;258:10873–10880. [PubMed] [Google Scholar]
- Prickett CS, Laug EP, Kunze FM. Distribution of mercury in rats following oral and intravenous administration of mercuric acetate and phenylmercuric acetate. Proc. Soc. Exp. Biol. Med. 1950;73:585–588. doi: 10.3181/00379727-73-17752. [DOI] [PubMed] [Google Scholar]
- Pritchard JB. Coupled transport of p-aminohippurate by rat kidney basolateral membrane vesicles. Am. J. Physiol. 1988;255:F596–F604. doi: 10.1152/ajprenal.1988.255.4.F597. [DOI] [PubMed] [Google Scholar]
- Quamme GA, Walker JJ, Yan TS. pH gradient stimulated phosphate transport in outer medullary brush-border membranes. Am. J. Physiol. 1989;257:F639–F648. doi: 10.1152/ajprenal.1989.257.4.F639. [DOI] [PubMed] [Google Scholar]
- Rabenstein DL. Metal complexes of glutathione and their biological significance. In: Dolphin D, Auramovibc O, Poulson R, editors. Glutathione: Chemical, Biochemical, and Medical Aspects of Coenzymes and Cofactors. Vol. 3. Wiley; New York: 1989. pp. 147–186. [Google Scholar]
- Rabenstein DL, Isab AA, Kadima W, Mohanakrishnan P. A proton nuclear magnetic resonance study of the interaction of cadmium with human erythrocytes. Biochim. Biophys. Acta. 1983;762:531–541. doi: 10.1016/0167-4889(83)90057-5. [DOI] [PubMed] [Google Scholar]
- Refsvik T. Excretion of methylmercury in rat bile: the effect of diethylmaleate, cyclohexene oxide and acrylamide. Acta Pharmacol. Toxicol. 1978;42:135–141. doi: 10.1111/j.1600-0773.1978.tb02181.x. [DOI] [PubMed] [Google Scholar]
- Refsvik T. Excretion of methyl mercury in rat bile: the effect of thioctic acid, thionalide, hexadecyl- and octadecylmercaptoacetate. Acta Pharmacol. Toxicol. (Copenh) 1982;50:196–205. doi: 10.1111/j.1600-0773.1982.tb00962.x. [DOI] [PubMed] [Google Scholar]
- Refsvik T, Norseth T. Methylmercuric compounds in rat bile. Acta Pharmacol. Toxicol. 1975;36:67–78. doi: 10.1111/j.1600-0773.1975.tb00772.x. [DOI] [PubMed] [Google Scholar]
- Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569–574. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Murphy SD. Effect of glutathione depletion on tissue deposition of methylmercury in rats. Toxicol. Appl. Pharmacol. 1975;31:505–519. doi: 10.1016/0041-008x(75)90274-4. [DOI] [PubMed] [Google Scholar]
- Robinson MK, Barfuss DW, Zalups RK. Cadmium transport and toxicity in isolated perfused segments of the renal proximal tubule. Toxicol. Appl. Pharmacol. 1993;121:103–111. doi: 10.1006/taap.1993.1134. [DOI] [PubMed] [Google Scholar]
- Roch-Ramel F, Besseghir K, Murer H. Renal excretion and tubular transport of organic anions and cations. In: Windhager EE, editor. Handbook of Physiology, Section 8, Renal Physiology. Oxford Univ. Press; New York: 1992. pp. 2189–2262. [Google Scholar]
- Rolfs A, Bonkovsky HL, Kohlroser JG, McNeal K, Sharma A, Berger UV, Hediger MA. Intestinal expression of genes involved in iron absorption in humans. Am. J. Physiol.: Gastrointest. Liver Physiol. 2002;282:G598–G607. doi: 10.1152/ajpgi.00371.2001. [DOI] [PubMed] [Google Scholar]
- Rowens B, Geurrero-Betancourt D, Gottlieb CA, Boyes RJ, Eichenhorn MS. Respiratory failure and death following acute inhalation of mercury vapor. A clinical and histologic perspective. Chest. 1991;99:185–190. doi: 10.1378/chest.99.1.185. [DOI] [PubMed] [Google Scholar]
- Rubino FM, Verduci C, Giampiccolo R, Pulvirenti S, Brambilla G, Colombi A. Molecular characterization of homo- and heterodimeric mercury(II)-bis-thiolates of some biologically relevant thiols by electrospray ionization and triple quadrupole tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2004;15:288–300. doi: 10.1016/j.jasms.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Ryan J, McKillen M, Mason J. Sulphate/molybdate interactions: in vivo and in vitro studies on the group VI oxyanion transport system in ovine renal tubule epithelial cells. Ann. Rech. Vet. 1987;18:47–55. [PubMed] [Google Scholar]
- Samuels ER, Heick HMC, McLain PN, Farant JP. A case of accidental inorganic mercury poisoning. J. Anal. Toxicol. 1982;6:120–122. doi: 10.1093/jat/6.3.120. [DOI] [PubMed] [Google Scholar]
- Sandstrom AIM, Wall SGI, Taube A. Cancer incidence and mortality among Swedish smelter workers. Br. J. Ind. Med. 1989;46:82–89. doi: 10.1136/oem.46.2.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Yadav P, Bhatnagar D. Lipid peroxidative damage on cadmium exposure and alterations in antioxidant system in rat erythrocytes: a study with relation to time. BioMetals. 1998;11:153–157. doi: 10.1023/a:1009286130324. [DOI] [PubMed] [Google Scholar]
- Schafer SG, Forth W. Effect of acute and subchronic exposure to cadmium on the retention of iron in rats. J. Nutr. 1984;114:1989–1996. doi: 10.1093/jn/114.11.1989. [DOI] [PubMed] [Google Scholar]
- Schali C, Vaughn DA, Fanestil DD. Reconstitution of the partially purified renal phosphate (Pi) transporter. Biochem. J. 1986;235:189–197. doi: 10.1042/bj2350189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub TP, Kartenbeck J, Konig J, Spring H, Dorsam J, Staehler G, Storkel S, Thon WF, Keppler D. Expression of the conjugate export pump encoded by the mrp2 gene in the apical membrane of kidney proximal tubules. J. Am. Soc. Nephrol. 1997;8:1213–1221. doi: 10.1681/ASN.V881213. [DOI] [PubMed] [Google Scholar]
- Schaub TP, Kartenbeck J, Konig J, Vogel O, Witzgall R, Kriz W, Keppler D. Expression of the MRP2 gene-encoded conjugate export pump in human kidney proximal tubules and in renal cell carcinoma. J. Am. Soc. Nephrol. 1999;10:1159–1169. doi: 10.1681/ASN.V1061159. [DOI] [PubMed] [Google Scholar]
- Schell LM, Denham M, Stark AD, Gomez M, Ravenscroft J, Parsons PJ, Aydermir A, Samelson R. Maternal blood lead concentration, diet during pregnancy, and anthropometry predict neonatal blood lead in a socio-economically disadvantaged population. Environ. Health Perspect. 2003;111:195–200. doi: 10.1289/ehp.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuhammer AM, Cherian MG. Quantification of metallothioneins by a silver-saturation method. Toxicol. Appl. Pharmacol. 1986;82:417–425. doi: 10.1016/0041-008x(86)90277-2. [DOI] [PubMed] [Google Scholar]
- Schoenmakers TJ, Klaren PH, Flik G, Lock RA, Pang PK, Bonga SE. Actions of cadmium on basolateral plasma membrane proteins involved in calcium uptake by fish intestine. J. Membr. Biol. 1992;127:161–172. doi: 10.1007/BF00231504. [DOI] [PubMed] [Google Scholar]
- Scott N, Hatlelid KM, MacKenzie NE, Carter DE. Reactions of arsenic(III) and arsenic(V) species with glutathione. Chem. Res. Toxicol. 1993;6:102–106. doi: 10.1021/tx00031a016. [DOI] [PubMed] [Google Scholar]
- Selevan SG, Landrigan PJ, Stern FB, Jones JH. Mortality of lead smelter workers. Am. J. Epidemiol. 1985;122:673–683. doi: 10.1093/oxfordjournals.aje.a114146. [DOI] [PubMed] [Google Scholar]
- Senn E, Scharrer E, Wolffram S. Effects of glutathione and of cysteine on intestinal absorption of selenium from selenite. Biol. Trace Elem. Res. 1992;33:103–108. doi: 10.1007/BF02783998. [DOI] [PubMed] [Google Scholar]
- Shennan DB. Selenium (selenate) transport by human placental brush border membrane vesicles. Br. J. Nutr. 1988;59:13–19. doi: 10.1079/bjn19880005. [DOI] [PubMed] [Google Scholar]
- Shepherd PR, Navé, Siddle K. Insulin stimulation of glycogen synthesis and glycogen synthase activity is blocked by wortmannin and rapamycin in 3T3-L1 adipocytes: evidence for the involvement of phosphoinositide-3 kinase and p70 ribosomal protein-S6 kinase. Biochem. J. 1995;305:25–28. doi: 10.1042/bj3050025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura A, Chonko AM, Grantham JJ. Basis for heterogeneity for para-aminohippurate secretion in rabbit proximal tubules. Am. J. Physiol. 1981;241:F430–F436. doi: 10.1152/ajprenal.1981.240.5.F430. [DOI] [PubMed] [Google Scholar]
- Shoemaker DG, Bender CA, Gunn RB. Sodium-phosphate cotransport in human red blood cells. Kinetics and role in membrane metabolism. J. Gen. Physiol. 1988;92:449–474. doi: 10.1085/jgp.92.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short JM, Wynshaw-Boris A, Short HP, Hanson RW. Characterization of the phosphoenolpyruvate carboxykinase (GTP) promoter-regulatory region. J. Biol. Chem. 1986;261:9721–9726. [PubMed] [Google Scholar]
- Simmons-Willis TA, Koh AS, Clarkson TW, Ballatori N. Transport of a neurotoxicant by molecular mimicry: the methylmercury-l-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT 2. Biochem. J. 2002;367:239–246. doi: 10.1042/BJ20020841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons TJ. Cellular interactions between lead and calcium. Br. Med. Bull. 1986a;42:431–434. doi: 10.1093/oxfordjournals.bmb.a072162. [DOI] [PubMed] [Google Scholar]
- Simons TJ. The role of anion transport in the passive movement of lead across the human red cell membrane. J. Physiol. 1986b;378:287–312. doi: 10.1113/jphysiol.1986.sp016220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons TJ. Active transport of lead by the calcium pump in human red cell ghosts. J. Physiol. 1988;405:105–113. doi: 10.1113/jphysiol.1988.sp017323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons TJ. Lead transport and binding by human erythrocytes in vitro. Pfluegers Arch. 1993;423:307–313. doi: 10.1007/BF00374410. [DOI] [PubMed] [Google Scholar]
- Simons TJB, Pocock G. Lead enters bovine adrenal medullary cells through calcium channels. J. Neurochem. 1987;48:383–389. doi: 10.1111/j.1471-4159.1987.tb04105.x. [DOI] [PubMed] [Google Scholar]
- Simovich MJ, Conrad ME, Umbreit JN, Moore EG, Hainsworth LN, Smith HK. Cellular location of proteins related to iron absorption and transport. Am. J. Hematol. 2002;70:3123–3129. doi: 10.1002/ajh.10052. [DOI] [PubMed] [Google Scholar]
- Six KM, Goyer RA. Experimental enhancement of lead toxicity by low dietary calcium. J. Lab. Clin. Med. 1970;76:933–942. [PubMed] [Google Scholar]
- Six KM, Goyer RA. The influence of iron deficiency on tissue content and toxicity of ingested lead in the rat. J. Lab. Clin. Med. 1972;79:128–136. [PubMed] [Google Scholar]
- Smith DR, Kahng MW, Quintanilla-Vega B, Fowler BA. Lead-binding polypeptides in human kidney cytosol: diazepam binding inhibitor and thymosin h4. Toxicologist. 1994;14:84. [Google Scholar]
- Smith DR, Kahng MW, Quintanilla-Vega B, Fowler BA. High affinity renal lead-binding proteins in environmentally exposed humans. Chem. Biol. Interact. 1998;115:39–52. doi: 10.1016/s0009-2797(98)00060-x. [DOI] [PubMed] [Google Scholar]
- Smith AH, Lingas EO, Rahman M. Contamination of drinking water by arsenic in Bangladesh: a public health emergency. Bull. World Health Organ. 2000;78:1093–1103. [PMC free article] [PubMed] [Google Scholar]
- Soni JP, Singhania RU, Bansal A, Rathi G. Acute mercury vapor poisoning. Indian Pediatr. 1992;29:365–368. [PubMed] [Google Scholar]
- St. Pierre MV, Serrano ME, Macias RIR, Dubs U, Hoechli M, Lauper U, Meier PJ, Marin JJG. Expression of members of the multidrug resistance protein family in human term placenta. Am. J. Physiol. 2000;279:R1495–R1503. doi: 10.1152/ajpregu.2000.279.4.R1495. [DOI] [PubMed] [Google Scholar]
- Stapleton SR. Selenium: an insulin-mimetic. Cell. Mol. Life Sci. 2000;57:1874–1879. doi: 10.1007/PL00000669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton SR, Mitchell DA, Salati LM, Goodridge AG. Triiodothyronine stimulates transcription of the fatty acid synthase gene in chick embryo hepatocytes in culture. J. Biol. Chem. 1990;265:18442–18446. [PubMed] [Google Scholar]
- Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-α by the heavy metal cadmium. Mol. Endocrinol. 2000a;14:545–553. doi: 10.1210/mend.14.4.0441. [DOI] [PubMed] [Google Scholar]
- Stoica A, Pentecost E, Martin MB. Effects of selenite on estrogen receptor-α and activity in MCF-7 breast cancer cells. J. Cell. Biochem. 2000b;79:282–292. doi: 10.1002/1097-4644(20001101)79:2<282::aid-jcb110>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Stoica A, Pentecost E, Martin MB. Effects of arsenite on estrogen receptor-α expression and activity in MCF-7 breast cancer cells. Endocrinology. 2000c;141:3595–3602. doi: 10.1210/endo.141.10.7704. [DOI] [PubMed] [Google Scholar]
- Souza V, Bucio L, Gutierrez-Ruiz MC. Cadmium uptake by a human hepatic cell line (WRL-68 cells). Toxicology. 1997;120:215–220. doi: 10.1016/s0300-483x(97)00057-7. [DOI] [PubMed] [Google Scholar]
- Stoclet JC, Derard D, Kilhoffer MC, Lugnier C, Miller R, Schaefer P. Calmodulin and its role in intracellular calcium regulation. Prog. Neurobiol. 1987;23:321–364. doi: 10.1016/0301-0082(87)90018-9. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Sugiyama Y. Excretion of GSSG and glutathione conjugates mediated by MRP1 and cMOAT/MRP2. Semin. Liver Dis. 1998;18:359–376. doi: 10.1055/s-2007-1007170. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Matsumoto N, Miyama T, Katsunuma H. Placental transfer of mercuric chloride, phenyl mercury acetate and methylmercury acetate in mice. Ind. Health. 1967;5:149–155. [Google Scholar]
- Svanberg B. Absorption of iron in pregnancy. Acta Obstet. Gynecol. Scand., Suppl. 1975;48:1–108. [PubMed] [Google Scholar]
- Tacnet F, Watkins DW, Ripoche P. Studies of zinc transport into brush-border membrane vesicles isolated from pig small intestine. Biochim. Biophys. Acta. 1990;1024:323–330. doi: 10.1016/0005-2736(90)90361-q. [DOI] [PubMed] [Google Scholar]
- Tacnet F, Watkins DW, Ripoche P. Zinc binding in intestinal brush-border membrane isolated from pig. Biochim. Biophys. Acta. 1991;1063:51–59. doi: 10.1016/0005-2736(91)90352-9. [DOI] [PubMed] [Google Scholar]
- Tallkvist J, Bowlus CL, Lonnerdal B. DMT1 gene expression and cadmium absorption in human absorptive enterocytes. Toxicol. Lett. 2001;122:171–177. doi: 10.1016/s0378-4274(01)00363-0. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Naganuma A, Imura N. Role of γ-glutamyltranspeptidase in renal uptake and toxicity of inorganic mercury in mice. Toxicology. 1990;60:187–198. doi: 10.1016/0300-483x(90)90142-4. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Naganuma A, Kobayashi K, Imura N. An explanation for strain and sex differences in renal uptake of methylmercury in mice. Toxicology. 1991;69:317–329. doi: 10.1016/0300-483x(91)90190-c. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Naganuma A, Miura N, Imura N. Role of testosterone in γ-glutamyltranspeptidase-dependent renal methylmercury uptake in mice. Toxicol. Appl. Pharmacol. 1992;112:58–63. doi: 10.1016/0041-008x(92)90279-2. [DOI] [PubMed] [Google Scholar]
- Tanaka-Kagawa T, Naganuma A, Imura N. Tubular secretion and reabsorption of mercury compounds in mouse kidney. J. Pharmacol. Exp. Ther. 1993;264:776–782. [PubMed] [Google Scholar]
- Tandy S, Williams M, Leggett A, Lopez-Jimenez M, Dedes M, Ramesh B, Srai SK, Sharp P. Nramp2 expression is associated with pH-dependent iron uptake across the apical membrane of human intestinal Caco-2 cells. J. Biol. Chem. 2000;275:1023–1029. doi: 10.1074/jbc.275.2.1023. [DOI] [PubMed] [Google Scholar]
- Thomas DJ, Smith JC. Partial characterization of a low-molecular weight methylmercury complex in rat cerebrum. Toxicol. Appl. Pharmacol. 1979;47:547–556. doi: 10.1016/0041-008x(79)90525-8. [DOI] [PubMed] [Google Scholar]
- Thomas DJ, Smith JC. Effect of coadministered low-molecular-weight thiol compounds on short-term distribution of methylmercury in the rat. Toxicol. Appl. Pharmacol. 1982;62:104–110. doi: 10.1016/0041-008x(82)90106-5. [DOI] [PubMed] [Google Scholar]
- Timmer RT, Gunn RB. Phosphate transport by the human renal cotransporter NaPi-3 expressed in HEK-293 cells. Am. J. Physiol. 1998;274:C757–C769. doi: 10.1152/ajpcell.1998.274.3.C757. [DOI] [PubMed] [Google Scholar]
- Toews AD, Kolber A, Hayward J, Krigman MR, Morell P. Experimental lead encephalopathy in the suckling rat: concentration of lead in cellular fractions enriched in brain capillaries. Brain Res. 1978;147:131–138. doi: 10.1016/0006-8993(78)90777-1. [DOI] [PubMed] [Google Scholar]
- Trinder D, Oates PS, Thomas C, Sadleir J, Morgan EH. Localization of divalent metal transporter 1 (DMT1) to the microvillus membrane of rat duodenal enterocytes in iron deficiency, but to hepatocytes in iron overload. Gut. 2000;46:270–276. doi: 10.1136/gut.46.2.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubaki T, Takahashi H. Recent Advances in Minamata Disease Studies. Kodansha, Ltd; Tokyo, Japan: 1986. [Google Scholar]
- Ullrich KJ, Rumrich G, Fasold G, Kloss S. Contraluminal para-aminohippurate (PAH) transport in the proximal tubule of the rat kidney. I. Kinetics, influence of cations, anions, and capillary preperfusion. Pflugers Arch. 1987a;409:229–235. doi: 10.1007/BF00583470. [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Rumrich G, Fasold G, Kloss S. Contraluminal para-aminohippurate (PAH) transport in the proximal tubule of the rat kidney. II. Specificity: aliphatic dicarboxylic acids. Pflugers Arch. 1987b;408:38–45. doi: 10.1007/BF00581838. [DOI] [PubMed] [Google Scholar]
- Urano T, Iwasaki A, Himeno S, Naganuma A, Imura N. Absorption of methylmercury compounds from rat intestine. Toxicol. Lett. 1990;50:159–164. doi: 10.1016/0378-4274(90)90006-8. [DOI] [PubMed] [Google Scholar]
- Vachon V, Delisle MC, Laprade R, Beliveau R. Reconstruction of the renal brush-border membrane sodium/phosphate co-transporter. Biochem. J. 1991;278:543–548. doi: 10.1042/bj2780543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendeland SC, Deagen JT, Whanger PD. Uptake of selenotrisulfides of glutathione and cysteine by brush border membranes from rat intestines. J. Inorg. Biochem. 1992;47:131–140. doi: 10.1016/0162-0134(92)84049-s. [DOI] [PubMed] [Google Scholar]
- Verbost PM, Senden MH, van Os CH. Nanomolar concentrations of Cd2+ inhibit Ca2+ transport systems in plasma membranes and intracellular Ca2+ stores in intestinal epithelium. Biochim. Biophys. Acta. 1987;902:247–253. doi: 10.1016/0005-2736(87)90302-6. [DOI] [PubMed] [Google Scholar]
- Vernhet L, Seite MP, Allain N, Guillouzo A, Fardel O. Arsenic induces expression of the multidrug resistance-associated protein 2 (MRP2) gene in primary rat and human hepatocytes. J. Pharmacol. Exp. Ther. 2001;298:234–239. [PubMed] [Google Scholar]
- Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999;21:195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Perantoni A. In vitro assessment of target cell specificity in cadmium carcinogenesis: interactions of cadmium and zinc with isolated interstitial cells of the rat testes. In Vitro Cell Dev. Biol. 1988;24:558–565. doi: 10.1007/BF02629091. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Poirier LA. Interactions of cadmium with interstitial tissue of the rat testes. Uptake of cadmium by isolated interstitial cells. Biochem. Pharmacol. 1985;34:2513–2518. doi: 10.1016/0006-2952(85)90535-0. [DOI] [PubMed] [Google Scholar]
- Wagle A, Jivraj S, Garlock G, Stapleton SR. Insulin regulation of glucose-6-phosphate dehydrogenase gene expression is rapamycin sensitive and requires phosphatidylinositol 3-kinase expression. J. Biol. Chem. 1998;273:14968–14974. doi: 10.1074/jbc.273.24.14968. [DOI] [PubMed] [Google Scholar]
- Wagner CA, Lang F, Brfer S. Function and structure of heterodimeric amino acid transporters. Am. J. Physiol.: Cell Physiol. 2001;281:C1077–C1093. doi: 10.1152/ajpcell.2001.281.4.C1077. [DOI] [PubMed] [Google Scholar]
- Wakita Y. Hypertension induced by methylmercury in rats. Toxicol. Appl. Pharmacol. 1987;89:144–147. doi: 10.1016/0041-008x(87)90185-2. [DOI] [PubMed] [Google Scholar]
- Wang W, Clarkson TW, Ballatori N. γ-glutamyl transpeptidase and l-cysteine regulate methylmercury uptake by HepG2 cells, a human hepatoma cell line. Toxicol. Appl. Pharmacol. 2000;168:72–78. doi: 10.1006/taap.2000.9018. [DOI] [PubMed] [Google Scholar]
- Wareing M, Ferguson CJ, Delannoy M, Cox AG, McMahon RF, Green R, Riccardi D, Smith CP. Altered dietary iron intake is a strong modulator of renal DMT1 expression. Am. J. Physiol.: Renal. Physiol. 2003;285:F1050–F1059. doi: 10.1152/ajprenal.00064.2003. [DOI] [PubMed] [Google Scholar]
- Warkany J, Hubbard DM. Acrodynia and mercury. J. Pediat. 1953;42:365–386. doi: 10.1016/s0022-3476(53)80195-2. [DOI] [PubMed] [Google Scholar]
- Webb M. Role of metallothionein in cadmium metabolism. In: Foulkes EC, editor. Cadmium. Springer-Verlag; Berlin: 1986. pp. 281–337. [Google Scholar]
- Gove PB, editor. Unabridged. Merriam-Webster, Inc.; Springfield, MS: 2002. Webster's 3rd New International Dictionary of the English Language; p. 1436. [Google Scholar]
- Wei H, Qiu L, Divine KK, Ashbaugh MD, McIntyre LC, Jr., Fernando Q, Gandolfi AJ. Toxicity and transport of three synthesized mercury–thiol-complexes in isolated rabbit renal proximal tubule suspensions. Drug Chem. Toxicol. 1999;22:323–341. doi: 10.3109/01480549909017838. [DOI] [PubMed] [Google Scholar]
- Wetterhahn-Jennette K. The role of metals in carcinogenesis: biochemistry and metabolism. Environ. Health Perspect. 1981;40:233–252. doi: 10.1289/ehp.8140233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson HM. Selenium oxide poisoning. N. C. Med. J. 1962;23:73–75. [PubMed] [Google Scholar]
- Wright SH, Dantzler WH. Molecular and cellular physiology of renal organic cation and anion transport. Physiol. Rev. 2003;84:987–1049. doi: 10.1152/physrev.00040.2003. [DOI] [PubMed] [Google Scholar]
- World Health Organization . WHO Regional Office for Europe. second ed. Copenhagen; Denmark: 2000. Mercury. Air Quality Guidelines. Chap. 6.9. [Google Scholar]
- Wu G. Screening of potential transport systems for methyl mercury uptake in rat erythrocytes at 5° by use of inhibitors and substrates. Pharmacol. Toxicol. 1995;77:169–176. doi: 10.1111/j.1600-0773.1995.tb01008.x. [DOI] [PubMed] [Google Scholar]
- Wu G. Methylmercury-cysteine uptake by rat erythrocytes: evidence for several transport systems. J. Appl. Toxicol. 1996;16:77–83. doi: 10.1002/(SICI)1099-1263(199601)16:1<77::AID-JAT319>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Wu G. Effect of probenecid on the transport of methyl mercury in erythrocytes by the organic anion transport system. Arch. Toxicol. 1997;71:218–222. doi: 10.1007/s002040050379. [DOI] [PubMed] [Google Scholar]
- Wurmli R, Wolffram S, Stingelin T, Scharrer E. Stimulation of mucosal uptake of selenium from selenite by l-cysteine in sheep small intestine. Biol. Trace Elem. Res. 1989;20:75–85. doi: 10.1007/BF02919100. [DOI] [PubMed] [Google Scholar]
- Yasutake A, Hirayama K, Inoue M. Mechanism of urinary excretion of methylmercury in mice. Arch. Toxicol. 1989;63:479–483. doi: 10.1007/BF00316452. [DOI] [PubMed] [Google Scholar]
- Yasutake A, Hirayama Y, Inouye M. Sex differences of nephrotoxicity by methylmercury in mice. In: Bach PH, et al., editors. Nephrotoxicity: Mechanisms, Early Diagnosis, and Therapeutic Management. Fourth International Symposium on Nephrotoxicity, 1989. Guilford; Marcel Dekker, Inc.; England, UK.: New York, NY: 1991. pp. 389–396. [Google Scholar]
- Zalups RK. Early aspects of the intrarenal distribution of inorganic mercury in the rabbit: effect of unilateral nephrectomy and dose of mercuric chloride. J. Toxicol. Environ. Health. 1993;33:213–228. doi: 10.1080/15287399109531519. [DOI] [PubMed] [Google Scholar]
- Zalups RK. Organic anion transport and action of γ-glutamyltranspeptidase in kidney linked mechanistically to renal tubular uptake of inorganic mercury. Toxicol. Appl. Pharmacol. 1995;132:289–298. doi: 10.1006/taap.1995.1110. [DOI] [PubMed] [Google Scholar]
- Zalups RK. Enhanced renal outer medullary uptake of mercury associated with uninephrectomy: implication of a luminal mechanism. J. Toxicol. Environ. Health. 1997;50:173–194. doi: 10.1080/009841097160564. [DOI] [PubMed] [Google Scholar]
- Zalups RK. Basolateral uptake of inorganic mercury in the kidney. Toxicol. Appl. Pharmacol. 1998a;150:1–8. doi: 10.1006/taap.1998.8416. [DOI] [PubMed] [Google Scholar]
- Zalups RK. Basolateral uptake of mercuric conjugates of N-acetylcysteine and cysteine in the kidney involves the organic anion transport system. J. Toxicol. Environ. Health. 1998b;54:101–117. doi: 10.1080/009841098158593. [DOI] [PubMed] [Google Scholar]
- Zalups RK. Intestinal handling of mercury in the rat: implications of intestinal secretion of inorganic mercury following biliary ligation or cannulation. J. Toxicol. Environ. Health A. 1998c;53:615–636. doi: 10.1080/009841098159079. [DOI] [PubMed] [Google Scholar]
- Zalups RK. Molecular interactions with mercury in the kidney. Pharmacol. Rev. 2000a;52:113–143. [PubMed] [Google Scholar]
- Zalups RK. Evidence for basolateral uptake of cadmium in the kidneys of rats. Toxicol. Appl. Pharmacol. 2000b;164:15–23. doi: 10.1006/taap.1999.8854. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Ahmad S. Molecular handling of cadmium in transporting epithelia. Toxicol. Appl. Pharmacol. 2003;186:163–188. doi: 10.1016/s0041-008x(02)00021-2. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Ahmad S. Homocysteine and the renal epithelial transport of inorganic mercury: role of basolateral transporter OAT1. J. Am. Soc. Nephrol. 2004;15:2023–2031. doi: 10.1097/01.ASN.0000135115.63412.A9. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Barfuss DW. Transport and toxicity of methylmercury along the proximal tubule of the rabbit. Toxicol. Appl. Pharmacol. 1993a;121:176–185. doi: 10.1006/taap.1993.1143. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Barfuss DW. Intrarenal distribution of inorganic mercury and albumin after coadministration. Toxicol. Appl. Pharmacol. 1993b;40:77–103. doi: 10.1080/15287399309531777. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Barfuss DW. Pretreatment with p-aminohippurate inhibits the renal uptake and accumulation of injected inorganic mercury in the rat. Toxicology. 1995;103:23–35. doi: 10.1016/0300-483x(95)03099-2. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Barfuss DW. Participation of mercuric conjugates of cysteine, homocysteine, and N-acetylcysteine in mechanisms involved in the renal tubular uptake of inorganic mercury. J. Am. Soc. Nephrol. 1998a;9:551–561. doi: 10.1681/ASN.V94551. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Barfuss DW. Small aliphatic dicarboxylic acids inhibit renal uptake of administered mercury. Toxicol. Appl. Pharmacol. 1998b;148:183–193. doi: 10.1006/taap.1997.8320. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Barfuss DW. Renal organic anion transport system: a mechanism for the basolateral uptake of mercury-thiol conjugates along the pars recta of the proximal tubule. Toxicol. Appl. Pharmacol. 2000;182:234–243. doi: 10.1006/taap.2002.9448. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Lash LH. Advances in understanding the renal transport and toxicity of mercury. J. Toxicol. Environ. Health. 1994;42:1–44. doi: 10.1080/15287399409531861. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Lash LH. Depletion of glutathione in the kidney and the renal disposition of administered inorganic mercury. Drug Metab. Dispos. 1997a;25:516–523. [PubMed] [Google Scholar]
- Zalups RK, Lash LH. Binding of mercury in renal brush-border and basolateral membrane-vesicles: implication of a cysteine conjugate of mercury involved in the luminal uptake of inorganic mercury. Biochem. Pharmacol. 1997b;53:1889–1900. doi: 10.1016/s0006-2952(97)00138-x. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Minor KH. Luminal and basolateral mechanisms involved in the renal tubular uptake of inorganic mercury. J. Toxicol. Environ. Health. 1995;46:73–100. doi: 10.1080/15287399509532019. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Robinson MK, Barfuss DW. Factors affecting inorganic mercury transport and toxicity in the isolated perfused proximal tubule. J. Am. Soc. Nephrol. 1991;2:866–878. doi: 10.1681/ASN.V24866. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Gelein RM, Cherian MG. Shifts in the dose–effect relationship for the nephropathy induced by cadmium-metallothionein in rats after a reduction of renal mass. J. Pharmacol. Exp. Ther. 1992;262:1256–1266. [PubMed] [Google Scholar]
- Zalups RK, Knutson KL, Schnellmann RG. In vitro analysis of the accumulation and toxicity of inorganic mercury in segments of the proximal tubule isolated from the rabbit kidney. Toxicol. Appl. Pharmacol. 1993;119:221–227. doi: 10.1006/taap.1993.1063. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Parks L, Cannon VT, Barfuss DW. Mechanisms of action of 2,3-dimercaptopropane-1-sulfonate and the transport, disposition, and toxicity of inorganic mercury in isolated perfused segments of rabbit proximal tubules. Mol. Pharmacol. 1998;54:353–363. doi: 10.1124/mol.54.2.353. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Aslamkhan AG, Ahmad S. Human organic anion transporter 1 mediated cellular uptake of cysteine-S-conjugates of inorganic mercury. Kidney Int. 2004;66:251–261. doi: 10.1111/j.1523-1755.2004.00726.x. [DOI] [PubMed] [Google Scholar]
- Zaman GJR, Lankelma J, van Tellingen O, Beijnen J, Dekker H, Paulusma C, Oude Elferink RPJ, Baas F, Borst P. Role of glutathione in the export of compounds from cells by the multidrug-resistance associated protein. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7690–7694. doi: 10.1073/pnas.92.17.7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler EE, Edwards BB, Jensen RL, Mahaffey KR, Formon SJ. Absorption and retention of lead by infants. Pediatr. Res. 1978;12:29–34. doi: 10.1203/00006450-197801000-00008. [DOI] [PubMed] [Google Scholar]
- Zoller H, Koch RO, Theurl I, Obrist P, Pietrangelo A, Montosi G, Haile DJ, Vogel W, Weiss G. Expression of the duodenal iron transporters divalent-metal transporter 1 and ferroportin 1 in iron deficiency and iron overload. Gastroenterology. 2001;120:1412–1419. doi: 10.1053/gast.2001.24033. [DOI] [PubMed] [Google Scholar]
- Zoller H, Theurl I, Koch R, Kaser A, Weiss G. Mechanisms of iron mediated regulation of the duodenal iron transporters divalent metal transporter 1 and ferroportin 1. Blood Cells, Mol. Dis. 2002;29:488–497. doi: 10.1006/bcmd.2002.0587. [DOI] [PubMed] [Google Scholar]