

Abstract
The inwardly rectifying potassium channel Kir4.1 has been suggested to underlie the principal K+ conductance of mammalian Müller cells and to participate in the generation of field potentials and regulation of extracellular K+in the retina. To further assess the role of Kir4.1 in the retina, we generated a mouse line with targeted disruption of theKir4.1 gene (Kir4.1 −/−). Müller cells from Kir4.1 −/− mice were not labeled with an anti-Kir4.1 antibody, although they appeared morphologically normal when stained with an anti-glutamine synthetase antibody. In contrast, in Müller cells from wild-type littermate (Kir4.1 +/+) mice, Kir4.1 was present and localized to the proximal endfeet and perivascular processes. In situ whole-cell patch-clamp recordings showed a 10-fold increase in the input resistance and a large depolarization of Kir4.1 −/− Müller cells compared with Kir4.1 +/+ cells. The slow PIII response of the light-evoked electroretinogram (ERG), which is generated by K+ fluxes through Müller cells, was totally absent in retinas from Kir4.1 −/− mice. The b-wave of the ERG, in contrast, was spared in the null mice. Overall, these results indicate that Kir4.1 is the principal K+channel subunit expressed in mouse Müller glial cells. The highly regulated localization and the functional properties of Kir4.1 in Müller cells suggest the involvement of this channel in the regulation of extracellular K+ in the mouse retina.
Keywords: Müller cell, inwardly rectifying potassium channel, Kir4.1, retina, null mouse, glia, electroretinogram, slow PIII response, b-wave, astrocyte
Radially oriented Müller glial cells span the depth of the neural retina from the inner limiting membrane at the vitreal surface to the subretinal space adjacent to the photoreceptors (Newman, 1996; Newman and Reichenbach, 1996). As in other glial cells, inwardly rectifying potassium (Kir) channels constitute the main K+ conductance in the plasma membrane of Müller cells (Newman, 1984). These channels have high open probability near the resting membrane potential and conduct currents better in the inward than in the outward direction (Newman, 1993). There is extensive evidence that Kir channels in Müller cells are vital elements for regulation of the extracellular K+ concentration ([K+]o) in the retina (Reichenbach et al., 1992; Newman and Reichenbach, 1996).
Although the Müller cell in the retina has served as an important model for investigations of glial function, the molecular identity of the Kir channels expressed in these cells is not yet known. Recently, many Kir subunits have been cloned. Structural comparisons suggest that they may be subdivided into six or seven subfamilies (Kir1–Kir6), forming either homo-oligomeric or hetero-oligomeric channels (Doupnik et al., 1995; Isomoto et al., 1997; Nichols and Lopatin, 1997). Expression of various Kir subunits in heterologous systems has shown that Kir2.1–3 and Kir4.1–2 subunits form K+ channels with biophysical properties that resemble the native channels in glial cells (Isomoto et al., 1997).
Recently, Kir4.1 expression in oligodendrocytes and Bergman glia has been reported (Takumi et al., 1995). In the retina, Kir4.1 is found mainly in Müller cells (Ishii et al., 1997) with large enrichment in the endfoot and perivascular processes (Nagelhus et al., 1999). The expression of Kir4.1 in Müller cells and its restricted subcellular distribution led to the suggestion that Kir4.1 mediates [K+]o homeostasis in the retina (Ishii et al., 1997; Nagelhus et al., 1999). However, the possibility remains that other Kir subunits are expressed in these glial cells because Kir4.1 is able to hetero-oligomerize with other Kir subunits in heterologous expression systems (Fakler et al., 1996;Pessia et al., 1996).
We have performed the genetic inactivation of the Kir4.1gene in the mouse to determine the function of Kir4.1 in the retina. We investigated the effect of lack of Kir4.1 on retinal organization, the electrical properties of Müller cells, and the electroretinogram (ERG). Collectively, our results indicate that Kir4.1 is the primary K+ conductance of Müller cells, and therefore it is likely to have an important role in the regulation of [K+]o in the mammalian retina.
MATERIALS AND METHODS
Preparation and characterization of Kir4.1 antibody.Rabbit polyclonal antibodies were made against a synthetic peptide REQAEKEGSALSVRISNV corresponding to the amino acids 362–379 in the C terminus of mouse Kir4.1. A reactive cysteine was included at the N terminus of the synthetic peptide to facilitate its conjugation to keyhole limpet hemocyanin carrier. Affinity purification of the antiserum was performed using a column with immobilized Kir4.1 peptide. Bound anti-Kir4.1 antibody was eluted with 100 mm glycine, pH 2.5, and subsequently dialyzed against PBS.
To determine the specificity of the affinity-purified anti-Kir4.1 antibody, we transfected COS cells as described previously (Doupnik et al., 1997), with the following Kir subunits cloned into pcDNA3 vector (Invitrogen, San Diego, CA): mouse Kir2.1 (kindly provided by Dr. L. Jan, University of California, San Francisco, CA), rat Kir3.1 (Dascal et al., 1993), rat Kir4.1 (kindly provided by Dr. J. P. Adelman, Oregon Health Sciences University, Portland, OR), and rat Kir6.2 (kindly provided by Dr. S. Seino, Chiba University, Chiba, Japan). The immunocytochemistry was performed as described below for the retinal sections.
PCR analysis. Total RNA from mouse retinas was extracted using the RNaqueous kit (Ambion, Austin, TX) and treated with DNase I (Ambion) to prevent contamination by genomic DNA. cDNAs were synthesized by priming with oligo-dT and using Superscript Reverse Transcriptase (Life Technologies, Rockville, MD). PCRs were performed using the following primer pairs: Kir2.1 (GenBank accession number AF021136), forward 5′-TTCTCCATCGAGACCCAGAC-3′ and reverse 5′-ATCTATTTCGTGAACGATAG-3′; Kir2.2 (GenBank accession numberX80417), forward 5′-TCCACGGCTTCATGGCAGCC-3′ and reverse 5′-GTCCAGTGGGATGTACTCAC; Kir2.3 (GenBank accession number U11075), forward 5′-CATCAAGCCCTACATGACAC-3′ and reverse 5′-AACTCGTTCTCATAGCAGAA; Kir4.1, forward 5′-TACAGTCAGACGACTCAGACA-3′ and reverse 5′-GAAGCAGTTTGCCTGTCACCT-3′; and Kir5.1 (GenBank accession numberAB016197), forward 5′-GCTATTACGGAAGTAGCTACC-3′ and reverse 5′-GGTGACACAGCGGTAACCGTA-3′. Each of the 35 cycles of PCR consisted of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C. Expected sizes for the PCR products were as follows: Kir2.1, 419 bp; Kir2.2, 361 bp; Kir2.3, 461 bp; Kir4.1, 630 bp; and Kir5.1, 415 bp.
For the genotyping, DNA was isolated from mouse tails using conventional methods (Sambrook et al., 1989), and the following pairs were used for the PCR amplifications: Kir4.1, forward 5′-TGGACGACCTTCATTGACATGCAGTGG-3′ and reverse 5′-CTTTCAAGGGGCTGGTCTCATCTACCACAT-3′; and neomycin resistance gene, forward 5′-GATTCGCAGCGCATCGCCTTCTATC-3′. Each of the 35 cycles of PCR consisted of 1 min at 94°C, 1 min at 65°C, and 1 min at 72°C. PCR primers amplify a 634 bp fragment in the +/+ allele and a 383 bp fragment in the mutant allele.
Generation of Kir4.1null (Kir4.1 −/−) mouse line. The mouse Kir4.1 gene was isolated (Sambrook et al., 1989) from a commercial mouse genomic library derived from 129/SvEvTac mice DNA (Stratagene, La Jolla, CA) using conventional methods. The gene encoding mouse Kir4.1 was cloned from a mouse 129/SvEvTac genomic library. A 6 kb fragment, which contained the entire coding sequence exon, was used for the construction of the genomic targeting vector. TheHindIII-BglII fragment was deleted in the targeting vector and replaced by the neomycin resistance gene (see Fig.1A). An additional herpes simplex virus thymidine kinase was also added for negative selection. The deleted fragment encodes the amino acids 33–266 in the Kir4.1 deduced primary sequence and thus contains the putative transmembrane domains and part of the C terminus of the Kir4.1 polypeptide. For hybridization, we used a fragment of the rat Kir4.1 cDNA kindly provided Dr. J. P. Adelman. The targeting vector was electroporated into CJ7 embryonic stem (ES) cells using a Bio-Rad (Hercules, CA) Gene Pulser set at 230 V and 500 μF capacitance. A total of 25 μg of the linearized targeting vector was used for 30 × 106 ES cells. ES cells were submitted to G418 and FIAU selection 24 hr after the electroporation. ES cell lines with targeted disruption of theKir4.1 gene were identified by Southern blot analysis ofXhoI- or BglII-digested genomic DNA; the probes used were the 5′ flanking regions. Four clones from a total of 576 G418 and FIAU double-resistant colonies contained the desired targeted allele. Chimeric mice were generated by injecting ES cells from two of these cell lines into C57BL/6J blastocysts and then implanting the blastocysts into the uteri of pseudopregnant recipients. Mice were maintained in a mixed Sv129 and C57BL/6 genetic background.
Fig. 1.

Targeted strategy for Kir4.1 gene interruption. A, Part of the wild-type Kir4.1 locus containing the coding exon (WT Allele), the targeting construct, and the targeted locus (Mutant Allele) are shown. PGKneo, Neomycin resistance gene.MC1tk, Thymidine kinase gene. The sizes of theXhoI fragments predicted to hybridize to the indicated diagnostic probe are shown. Restriction endonucleases:B, BamHI; Bl,BglII; E, EcoRI;H, HindIII; S,SacI; X, XhoI.B, Southern blot analysis ofXhoI-digested genomic DNA from wild-type (+/+), heterozygous (+/−), and homozygous mutant mice (−/−) with the diagnostic probe indicated in A. C, PCR analysis of tail genomic DNA PCR primers amplify a 634 bp fragment in the +/+ allele and a 383 bp fragment in the mutant allele. Notice the 634 bp fragment obtained from PCR analysis using tail DNA from +/+ mouse, the 383 bp fragment using tail DNA from −/− mouse, and both fragments from +/− mouse. As a control (C), the PCR analysis was performed in the absence of mouse genomic DNA
Histological and immunocytochemical analysis. Mice were deeply anesthetized with intraperitoneal injection of pentobarbital or exposure to CO2. Eyes were dissected, and the cornea and lens were removed. For retinal slices, the resulting eyecups were fixed in a 4% paraformaldehyde–PBS solution, pH 7.4, overnight at 4°C. Eyecups were sectioned at 10 μm thickness with a cryostat, placed onto slides coated with gelatin, and stored desiccated at −80°C. Sections were blocked and permeabilized with 5% donkey serum–0.2% Triton X-100 in PBS for 1 hr at room temperature and then incubated with the primary antibody for 2 hr. After being washed with PBS, retinal sections were incubated for 2 hr with FITC-conjugated donkey anti-rabbit IgG and tetramethylrhodamine isothiocyanate-conjugated donkey anti-mouse IgG (both secondary antibodies diluted 1:100 in PBS; Jackson ImmunoResearch, West Grove, PA). Sections were washed again with PBS and coverslipped with Vectashield (Vector Laboratories, Burlingame, CA). Sections were imaged with a Leica TCS4D confocal microscope, using a 50× oil immersion lens. Optical sections were collected at 0.75–1.5 μm intervals. Data are presented as projections of several optical images onto a single plane. Metamorph software (Universal Imaging Corp., West Chester, PA) was used to assign to each pixel in the final image the maximum intensity value recorded at that x–y location from among the stack of optical images. All control tissues were imaged with identical parameters to enable direct visual comparison of staining.
For the retinal whole mounts, the retinas were isolated and fixed in a 4% paraformaldehyde PBS solution, pH 7.4, overnight at 4°C. Blocking and permeabilization were performed for 2 hr in PBS solution containing 10% donkey serum–1% Triton X-100. Tissue was then incubated for 72 hr at 4°C in primary antibodies diluted in PBS, 5% powdered milk, 1% thimerosal, 0.01% anti-foam, and 1% Triton X-100. After PBS rinses, the retinas were incubated for 48–72 hr at 4°C with the secondary antibodies diluted in PBS–0.3% Triton X-100. After PBS rinses, the retinas were mounted with Vectashield and imaged as described for the retinal sections. The monoclonal antibodies anti-glutamine synthetase (GS) and anti-glial fibrillary acid protein (GFAP) were purchased from Chemicon (Temecula, CA).
Input resistance. Müller cell input resistance was measured in current-clamp experiments. Recordings were made from isolated retinal whole mounts as described previously (Zahs and Newman, 1997). Briefly, isolated retinas were digested in collagenase/dispase (2 mg/ml) and DNase (0.1 mg/ml) for 16 min at room temperature to remove the vitreous humor and the basal lamina at the retinal surface. Retinas were affixed to a polycarbonate filter membrane by suction (vitreal side up), mounted in a perfusion chamber, and superfused with oxygenated bicarbonate-buffered Ringer's solution at room temperature. Recordings were made from the endfeet of Müller cells at the vitreal surface of the retina, using patch electrodes in the whole-cell recording mode. Patch electrodes were filled with a solution containing Lucifer yellow to identify the cells. Labeled Müller cells were identified by an endfoot at the retinal surface, a process projecting distally through the inner plexiform layer (IPL), and a cell body in the inner nuclear layer (INL). Input resistance was measured by passing positive current pulses (0.02–0.5 nA) through the recording pipette and measuring the resulting voltage displacement. A bridge circuit was used to balance the voltage drop across the recording pipette.
Electroretinogram. The ERG was measured from the eyecup preparation, as described previously (Newman and Bartosch, 1999). Briefly, the back half of the mouse eye was everted over a dome and held in place by a thin plastic sheet with a hole in it. The plastic sheet prevented the retina from detaching and electrically isolated the vitreal surface of the retina from the sclera. The retinal surface of the eyecup was superfused with oxygenated Ringer's solution at ∼2.5 ml/min at 29°C.
The transretinal ERG was measured between the vitreous humor (positive) and the sclera with a bandpass of direct current (DC) to 200 Hz. We found that, at 29°C, the preparation remained stable for far longer than had been reported previously (Newman and Bartosch, 1999) for eyecups maintained at 37°C. The transretinal ERG b-wave declined by only 25 ± 2% after 6 hr (n = 6). The amplitude of the transretinal b-wave was measured from the trough of the a-wave to the peak of the b-wave, ignoring oscillatory potentials.
The intraretinal ERG was measured between an intraretinal micropipette (positive) and the vitreous with a bandpass of DC to 50 Hz. The pipette (outer tip diameter of ∼2.5 μm) was filled with Ringer's solution and was advanced into the retina at a shallow angle (20°). The pipette was advanced into the distal retina in which an intraretinal a-wave was recorded. The pipette was then withdrawn until the a-wave all but disappeared, indicating that the pipette tip lay near the outer limiting membrane (Brown and Wiesel, 1961). The amplitude of the intraretinal b-wave was measured from the prestimulus baseline to the b-wave peak, ignoring oscillatory potentials. The amplitude of the slow PIII response was measured from the baseline to the maximal positive response after decay of the b-wave.
Solutions. The bicarbonate-buffered superfusion solution used in electrophysiology experiments contained (in mm): NaCl 117.0, KCl 3.0, CaCl2 2.0, MgSO4 1.0, NaH2PO4 0.5, dextrose 15.0, NaHCO3 32, and l-glutamate 0.01, and was bubbled with 5% CO2 in O2. The pipette solution for whole-cell recording contained (in mm): NaCl 5, KCl 120, CaCl2 1, MgCl2 7, EGTA 5, Na2ATP 5, HEPES 5, and 0.2% Lucifer yellow.
RESULTS
General phenotype of Kir4.1 null (Kir4.1 −/−) mice
Heterozygous animals (Kir4.1 +/−) were identified by Southern blots (Fig. 1B) and PCR analysis (Fig. 1C) and were bred with each other to obtain homozygous animals. Progeny of the heterozygous animals show no gross discernable phenotypic differences in the first postnatal week. Afterward, the homozygous animals were considerably smaller than their littermates and displayed a higher rate of mortality. In addition, they developed clear motor coordination deficits, which became obvious ∼2 weeks after birth. The animals displayed awkward and jerky movements and loss of balance and occasionally fell on their side. The general phenotype of Kir4.1 −/− and its impact in the CNS will be described in a separate report. Most of the homozygous mice survived up to 3 weeks of age, which allowed us to study their retinal physiology after eyelid opening [postnatal day 13 (P13) and P14]. All of the experiments were performed with young mice between P16 and P21 unless otherwise noted.
Analysis of Kir4.1 mRNA in retina
Wild-type (Kir4.1 +/+), heterozygous (Kir4.1 +/−), and homozygous (Kir4.1 −/−) animals were identified by PCR analysis from tail biopsies. Mouse retinas were dissected, and total RNA was extracted and subjected to reverse transcriptase-PCR amplification. PCR using mouse-specific oligonucleotide primers for Kir2.1, Kir2.2, Kir2.3, Kir4.1, and Kir5.1 showed the amplification of products of expected sizes for retinas from Kir4.1 +/+ mice. Using brain RNA from Kir4.1 +/+ mice, we could also detect the expression for all tested Kir channel subunits (Fig. 2). In contrast, Kir4.1 mRNA was not detected in retinas from Kir4.1 −/− mice using this assay, although other Kir channel subunits were detected after amplification using other Kir-specific oligonucleotide primers (Fig.2). Thus, mRNA analysis in the retina shows the expected lack of Kir4.1 mRNA expression in retinas from genotyped Kir4.1 −/− mice.
Fig. 2.

Analysis of Kir4.1 mRNA in retina. PCR analysis of total RNA extracted from P21 Kir4.1 +/+ and Kir4.1 −/− mice retinas and Kir4.1 +/+ brain. Kir-specific oligonucleotide pairs were used to determine the expression of the various Kir channels subunits in retina and brain. As a control (C), cDNA was replaced by water, and the PCR was performed using Kir5.1-specific oligonucleotide primers.
Cellular organization and distribution of Kir4.1 in retina
Cellular organization and general morphology were assessed in retinal sections from Kir4.1 −/− and Kir4.1 +/+ mice stained with hematoxylin and eosin. Figure 3 shows light micrographs of retinal sections from P11 Kir4.1 +/+ and Kir4.1−/− mice. The retinas of the Kir4.1 −/− mice appeared to be normally organized with no apparent disruption of the normal pattern of lamination. Comparable results were obtained with older mice (P18–P21).
Fig. 3.

Histological analyses of the retinas of the Kir4.1 +/+ and Kir4.1 −/− mice. Cross-sections of mouse retinas from +/+ and −/− mutant mice (P11) were stained with hematoxylin and eosin.GCL, Ganglion cell layer; OPL, outer plexiform layer. Scale bar, 25 μm.
We raised an antibody against a peptide corresponding to a sequence in the C terminus of Kir4.1 to determine the cellular and subcellular distribution of Kir4.1 in the retina. The specificity of the affinity-purified anti-Kir4.1 antibody was tested by transient transfection of COS cells with Kir2.1, Kir3.1, Kir4.1, and Kir6.2 cDNAs, followed by immunostaining using the anti-Kir4.1 antibody. As expected, only cells transfected with Kir4.1 cDNA showed immunostaining. Furthermore, this labeling was blocked upon preadsorption of the anti-Kir4.1 antibody with a large excess of the antigenic peptide (data not shown).
Double-immunofluorescence experiments were performed on retinal whole mounts from Kir4.1 +/+ mice. Labeling for Kir4.1 was revealed with secondary antibodies coupled to FITC. Müller cells were labeled with an antibody against GS and visualized with secondary antibodies coupled to Texas Red. Figure 4 shows confocal optical sections in several retinal layers. At the inner limiting membrane, staining for Kir4.1 was detected along with staining for GS (Fig. 4A). Particularly intense staining for Kir4.1 was seen along the superficial blood vessels. In addition, the cell bodies of ganglion cells clearly showed a lack of labeling for both antibodies. In the IPL (Fig. 4B), large punctate staining was apparent for Kir4.1 and GS. These puncta presumably reflects the expression of both proteins in the stalk of Müller cells. In the inner nuclear layer (INL) (Fig. 4C), overlapping expression of Kir4.1 and GS was seen surrounding neuronal cell bodies and very prominent Kir4.1 clustering along the blood vessels. Finally, in the outer nuclear layer (ONL) (Fig.4D), Kir4.1 and GS were concentrated in Müller cell processes surrounding the photoreceptors. Thus, double-immunolabeling reveals the overlapping expression of GS and Kir4.1 proteins, indicating the expression of Kir4.1 in the Müller glial cell population. Kir4.1 labeling was particularly intense in Müller cell processes contacting blood vessels. The labeling pattern for Kir4.1 for the mouse retinas agrees closely with the results reported for rat retinas (Nagelhus et al., 1999).
Fig. 4.
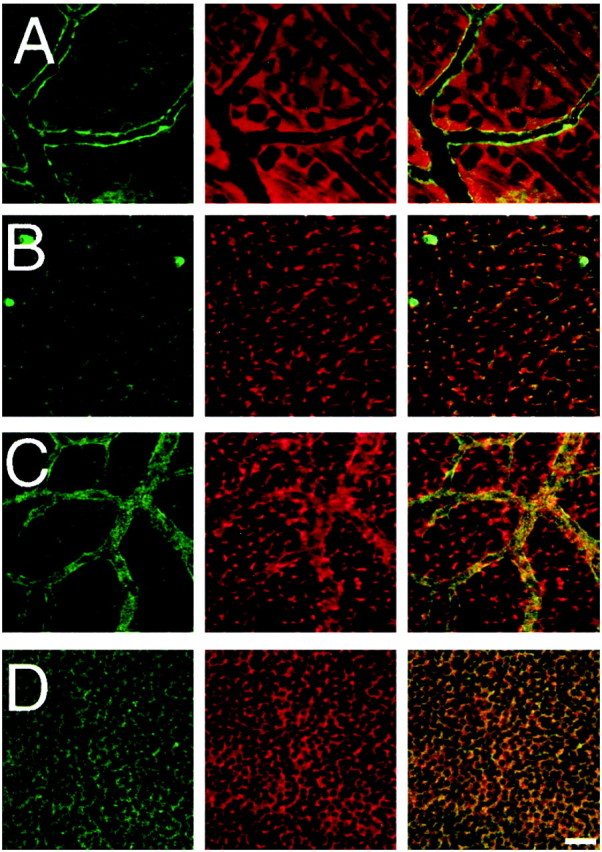
Immunohistochemistry of Kir4.1 in retinal whole mounts from Kir4.1 +/+ mouse (P18). Retinal whole mount stained with anti-Kir4.1 antibody (green) and anti-GS antibody (red). Confocal images were obtained in the ganglion cell layer (A), inner plexiform layer (B), inner nuclear layer (C), and outer nuclear layer (D). Note that the expression of Kir4.1 was clustered around neurons in the outer nuclear layer and along the blood vessels in the superficial and inner nuclear layer. Immunofluorescence in the blood vessels revealed by Texas Red donkey anti-mouse antibody (C) represents the binding of the secondary antibody to mouse IgG, because this immunoreactivity is not seen in retinas from mice transcardially perfused with PBS to wash out the blood before fixation. Scale bar, 20 μm.
Because amphibian astrocytes isolated from the optic nerve have K+ channels preferentially localized to their endfeet (Newman, 1986), we also asked whether Kir4.1 is expressed in retinal astrocytes. In retinal whole mounts, immunofluorescence labeling for GFAP revealed astrocytes in the superficial layers of retina. However, we failed to detect the labeling of Kir4.1 in astrocytes somata and proximal processes (data not shown). We could not determine whether Kir4.1 is found on the astrocytic terminal processes adjacent to blood vessels given the strong Kir4.1 immunoreactivity of adjacent Müller cell processes.
Double-immunolabeling with the anti-Kir4.1 and anti-GS antibodies was also performed on cross-sections of Kir4.1 +/+ and Kir4.1 −/− retinas. In retinal sections from Kir4.1 +/+ mice (Fig.5A,C,E), both antisera labeled all parts of Müller cells, staining the outer limiting membrane, the vitreal endfeet, and the main processes spanning the retina from the outer to the inner limiting membrane. Ganglion cells were clearly immunonegative for both anti-GS and anti-Kir4.1 antibodies (Fig. 5E). Strikingly intense signal for Kir4.1 was found at the inner limiting membrane and near blood vessels in the INL (Fig. 5A), confirming the immunolabeling pattern obtained in retinal whole mounts. In contrast to reports for albino rats (Kusaka et al., 1999a), we did not detect Kir4.1 in the retinal pigment epithelium (RPE) (data not shown).
Fig. 5.
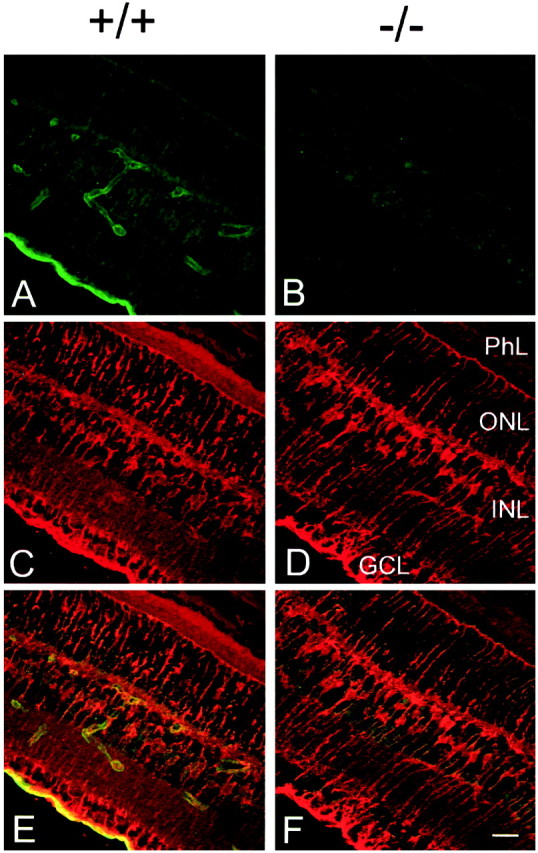
Immunohistochemical analysis of Kir4.1 in retinal sections of Kir4.1 +/+ (+/+) (A,C, E) and Kir4.1 −/− (−/−) (B, D, F) P18 mice. Sections were double-stained with affinity-purified rabbit anti-rat Kir4.1 antibody, followed by FITC-conjugated anti-rabbit IgG (A, B, green), and monoclonal anti-GS antibody, followed by Texas Red-conjugated anti-mouse IgG (C, D,red). E, F, Superposition of images A, C and B,D, respectively. Scale bar, 20 μm. GCL, Ganglion cell layer; PhL, photoreceptor layer.
In age-matched Kir4.1 −/− retinas, there was no detectable labeling with the anti-Kir4.1 antibody (Fig. 5B), although the Müller cells appeared morphologically normal as revealed by immunolabeling with the anti-GS antibody (Fig.5D,F). Because Müller cells express GFAP under some conditions, most notably in pathological states (Bignami and Dahl, 1979), we also stained retinal sections with anti-GFAP antibody. For both the Kir4.1 +/+ and Kir4.1 −/− mouse retinas, the GFAP immunolabeling was confined to cells located in the superficial layers in which the retinal astrocytes are located (data not shown). Thus, the lack of Kir4.1 did not induce the upregulation of GFAP in the Kir4.1 −/− Müller cells.
Müller cell membrane potential and input resistance
The electrophysiological properties of Müller cells in Kir4.1 +/+, Kir4.1 +/−, and Kir−/− mice were measured using whole-cell recordings in current-clamp mode. The resting membrane potential and input resistance of cells are given in Table1. In Kir4.1 +/+ and Kir4.1 +/− mice, the resting membrane potential (−85 and −88 mV, respectively) was in the normal range for Müller cells (Newman, 1987). The resting membrane potential in Kir4.1 −/− mice was significantly depolarized, to −13 mV, indicating that Müller cell K+ conductance was substantially reduced in these animals. Input resistance measurements confirmed that the K+ conductance of Müller cells in mutant mice was reduced (Fig. 6, Table1). The input resistance in Kir4.1 +/− mice (47 MΩ) was nearly double that of Kir4.1 +/+ mice (25 MΩ), whereas the input resistance in Kir4.1 −/− mice (231 MΩ) was more than nine times that of Kir4.1 +/+ animals.
Table 1.
Resting membrane potential and input resistance of Müller cells
| Cell type | Membrane potential (mV) | Input resistance (MΩ) | n |
|---|---|---|---|
| Kir4.1 +/+ | −85.2 ± 0.9 | 24.7 ± 2.6 | 32 |
| Kir4.1 +/− | −87.6 ± 0.7 | 47.0 ± 2.4* | 44 |
| Kir4.1 −/− | −13.2 ± 2.9* | 231.3 ± 28.0* | 9 |
| Kir4.1 −/− with hyperpolarizing current injection | −84.2 ± 1.6 | 310.5 ± 97.1* | 4 |
Data are given as mean ± SEM.
indicates statistically different from Kir4.1 +/+ (Student's t test, unpaired samples, p ≤ 0.001).
Fig. 6.

Input resistance of Müller cells in Kir4.1 +/+, Kir4.1 +/−, and Kir4.1 −/− mice. Traces show the displacement of the membrane potential produced by an injected current pulse. The traces are scaled so that the amplitudes of the responses reflect the relative input resistances of the three cells (voltage divided by injected current is equal for alltraces). Note that the membrane time constant of the −/− cell is substantially longer because of its increased membrane resistance. Amplitude of current pulses: 0.5 nA for +/+ and +/−; 0.1 nA for −/−.
The membrane potential of Kir4.1 −/− cells was substantially depolarized, and it is possible that the large input resistance measured in these cells is attributable to this depolarization. To avoid this complication, we measured the input resistance of Kir4.1 −/− cells that were artificially hyperpolarized by injection of a constant negative current. Kir4.1 −/− cells hyperpolarized to an average of −84 mV had an input resistance of 310 MΩ, >12 times that of Kir4.1 +/+ cells, demonstrating that the increased input resistance of Kir4.1 −/− cells was not attributable to cell depolarization.
Electroretinogram
Several components of the ERG, including the slow PIII response, are believed to be generated by K+ current flow through Müller cells (Witkovsky et al., 1975; Bolnick et al., 1979; Newman, 2000). These ERG components should be absent or greatly reduced in mutants lacking the predominant Müller cell K+ channel. We tested this prediction by measuring the slow PIII response in Kir4.1 +/+ and Kir4.1 −/− mice. The slow PIII response was monitored with an intraretinal electrode positioned in the distal retina of the mouse eyecup. In Kir4.1 +/+ mice, a brief light flash evoked a transient negative intraretinal b-wave followed by a slower positive response, the slow PIII (Fig.7, +/+). Addition of the K+ channel blocker Ba2+ (0.4 mm) eliminated the slow PIII, as expected, but spared the b-wave (Fig. 7, +/+ plus Ba2+). When the intraretinal ERG recorded in Ba2+ was subtracted from the control ERG (Fig. 7, +/+ difference), the Ba2+-sensitive component, the slow PIII, was revealed in isolation.
Fig. 7.

The slow PIII response of the ERG in Kir4.1 +/+ and Kir4.1 −/− mice. Traces show the intraretinal ERG recorded between an electrode in the distal retina and one in the vitreous humor. In a Kir4.1 +/+ mouse, a transient negative b-wave and a slower positive slow PIII response are evident (+/+). When Ba2+ (0.4 mm) is added to the superfusate (+/+ plus Ba2+), the slow PIII response is abolished, whereas the b-wave remains primarily unchanged. Subtracting the trace in Ba2+from the other (+/+ difference) reveals the Ba2+-sensitive component of the ERG, the slow PIII response in isolation. In a Kir4.1 −/− mouse (−/−), the b-wave is present but the slow PIII response is absent. Addition of Ba2+ (−/− plus Ba2+) produces a small increase in the b-wave but no change in the response after decay of the b-wave, as illustrated by the differencetrace (−/− difference). The time course of the light stimulus is shown at the bottom.Dashed lines indicate prestimulus baseline levels.
The intraretinal ERG recorded from Kir4.1 −/− mice differed qualitatively from that of Kir4.1 +/+ mice (Fig. 7, −/−). Although the b-wave was present, the slow PIII response was absent. After decay of the b-wave, the ERG did not rise above the level of the prestimulus baseline. Addition of Ba2+ (Fig. 7, −/−plus Ba2+) produced little change in the ERG, as shown in the difference trace (Fig. 7, −/−difference), which is flat after an initial transient reflecting a small Ba2+-induced change in the amplitude of the b-wave.
Amplitudes of the intraretinal slow PIII and b-wave, as well as the transretinal b-wave, are given in Table2. The intraretinal slow PIII amplitude, measured from the prestimulus baseline to the peak of the response, was 283 μV in Kir4.1 +/+ mice and −34 μV in Kir4.1 −/− mice, demonstrating that the slow PIII response was completely absent in the mutant. A negative slow PIII amplitude indicates that the response is not present.
Table 2.
Amplitude of the slow PIII response and b-wave of the ERG
| Cell type | Intraretinal slow PIII amplitude (μV) | Intraretinal b-wave amplitude (μV) | Transretinal b-wave amplitude (μV) | n |
|---|---|---|---|---|
| Kir4.1 +/+ | 283 ± 51 | 418 ± 153 | 142 ± 32 | 4 |
| Kir4.1 +/+ plus Ba2+ | −59 ± 38* | 698 ± 163 | 291 ± 53* | 4 |
| Kir4.1 −/− | −34 ± 1* | 629 ± 65 | 117 ± 16 | 3 |
| Kir4.1 −/− plus Ba2+ | −15 ± 28* | 621 ± 82 | 131 ± 5 | 3 |
Data are given as mean ± SEM.
indicates statistically different from Kir4.1 +/+ (Student's t test, unpaired samples, p < 0.05).
The b-wave response, in contrast, was not eliminated in the mutant (Table 2). Measured intraretinally, the b-wave was 418 μV in Kir4.1 +/+ mice and 629 μV in Kir4.1 −/− mice. The larger intraretinal b-wave measured in Kir4.1 −/− mice probably arose because of the absence in these animals of the positive slow PIII, which normally would offset the negative b-wave. This would explain why b-wave amplitude in −/− animals was similar to the amplitude in +/+ animals treated with Ba2+. Measured transretinally, the b-wave was 142 μV in Kir4.1 +/+ and 117 μV in Kir4.1 −/− mice. The smaller transretinal b-wave in Kir4.1 −/− mice most likely reflects the smaller size of the eyes in mutant animals.
DISCUSSION
Distribution of Kir4.1 in the retina
Phylogenetic analysis shows that Kir channels encompass at least 13 members subdivided into six or seven structurally related subfamilies (Isomoto et al., 1997; Nichols and Lopatin, 1997). The Kir4.1 cDNA was first isolated from a brain cDNA library and named BIR10 (Bond et al., 1994). Subsequently, other groups isolated the same cDNA and named it BIRK (Bredt et al., 1995) and KAB-2 (Takumi et al., 1995). The widespread distribution of Kir4.1 mRNA in brain has been noted (Bredt et al., 1995), but some uncertainty persists as to whether Kir4.1 is expressed predominantly in neuronal (Ma et al., 1998) or glial (Takumi et al., 1995) populations. In the periphery, Kir4.1 is localized to several cell types involved in transport of K+ions. Thus, Kir4.1 has been shown to be expressed in the marginal (Hibino et al., 1997) or intermediate (Ando and Takeuchi, 1999) cells in the cochlea stria vascularis. In this tissue, Kir4.1 may be critically involved in the generation of the endocochlear potential. Kir4.1 is also found in distal tubules in kidney (Ito et al., 1996) in which these channels probably secrete K+ions at the basolateral membrane.
Electrophysiological experiments demonstrated that the Kir channels are not uniformly distributed on the plasma membrane of salamander Müller cells (Newman, 1984). Instead, these channels are concentrated in the proximal endfeet adjacent to the vitreous (Newman, 1984; Brew et al., 1986). Because of such non-uniform distribution of K+ conductance, it has been hypothesized that K+ ions released from depolarized retinal neurons enter Müller cells and exit preferentially via their endfeet into the vitreous fluid (“K+ siphoning ”) (Newman et al., 1984). In mouse and other species with vascularized retinas, Kir conductances also cluster in the apical and perivascular processes of Müller cells, suggesting additional K+ siphoning to the blood vessels and subretinal space (Newman and Reichenbach, 1996).
Ishii et al. (1997) were the first to describe the presence of Kir4.1 in rat retina and its restricted localization to Müller cells. In their study, Kir4.1 was found to be expressed in Müller cells byin situ hybridization and immunocytochemical techniques. Immunogold techniques were used to demonstrate that Kir4.1 is selectively expressed in the endfoot of the rat Müller cell and in those processes enveloping the blood vessels (Nagelhus et al., 1999). The present study confirms the cellular and subcellular distribution of Kir4.1 in mouse retinas using confocal immunocytochemical techniques.
Thus, in both rat and mouse, the spatial distribution of Kir4.1 within Müller cells is in remarkable agreement with the distribution of K+ conductances determined by electrophysiological techniques (Newman, 1987) and argues for the involvement of this Kir subunit in K+siphoning in the mammalian retina.
The highly restricted distribution of Kir4.1 within Müller cells raises new questions concerning the molecular mechanisms responsible for the localized distribution of these channels. It is interesting to note that Kir4.1 and aquaporin-4 channels primarily colocalize in Müller cells and that both proteins contain a PDZ [Postsynaptic density-95 (PSD-95)/Discs large/zona occludens-1] domain recognition sequence that mediates binding to members of the PSD-95/SAP90 (synapse-associated protein-90) protein family (Nagelhus et al., 1998). Thus, PSD-95/SAP90-like proteins may play roles in regulating the abundance and distribution of channels within glial cells as has been demonstrated for synapses and other neuronal membrane specializations (Sheng and Wyszynsky, 1997). It is also interesting to speculate that such large macromolecular structures underlie the orthogonal arrays of intramembranaceous particles (OAPs). These OAPs are localized to Müller cell and astrocyte endfeet (Wolburg, 1995) and are thought to arise from aquaporin-4 (Yang et al., 1996). It is not known, however, whether Kir4.1 also participates in the formation of these particles. Freeze fracture studies in Kir4.1 −/− Müller cells may answer some of these questions.
In retina, Kir4.1 has also been demonstrated in the RPE (Kusaka et al., 1999a), which has been implicated in the transport of K+ in the subretinal space (Immel and Steinberg, 1986; Newman, 2000). The expression of Kir4.1 in the RPE was demonstrated in albino but not in pigmented rats, possibly because pigmentation in the RPE prevents the immunodetection of Kir4.1 in these cells (Kusaka et al., 1999a). Because our studies were performed in pigmented mice, we could not verify the expression of Kir4.1 to the RPE.
Biophysical and functional properties of Kir4.1
Several biophysical features of Kir4.1, as revealed by heterologous expression studies, are similar to those reported for Kir channels in Müller cells. For example, the single-channel conductance for Kir4.1 expressed in HEK 293 cells is 25 pS (Tada et al., 1998). This value is similar to the conductance of 28 pS in salamander Müller cells (Newman, 1993) and of 20 pS in monkey Müller cells (Kusaka and Puro, 1997). In addition, Kir channels in human Müller cells are activated by intracellular ATP (Kusaka and Puro, 1997), as are Kir4.1 channels expressed in Xenopusoocytes (Takumi et al., 1995). These features are, however, shared by several members of the Kir channel superfamily (Isomoto et al., 1997;Nichols and Lopatin, 1997), and thus a more detailed comparison of the single-channel properties and modulation of Kir currents between Kir4.1 and the native Kir channels in Müller cells is warranted. In particular, it will be interesting to determine whether Kir4.1 currents in heterologous expression systems are modulated by protein kinases and serum factors as reported for the native Kir currents in Müller cells (Schwartz, 1993; Kusaka et al., 1998, 1999b). In rabbit Müller cells, Kir channels of 60 and 105 pS (Nilius and Reichenbach, 1988) have also been found, and the molecular identity of these channels is presently unclear.
In our study, in situ measurements of the input resistance of Müller cells shows that Kir4.1 −/− cells have an ∼10-fold higher resistance than Kir4.1 +/+ cells. These results indicate that Kir4.1 is the predominant channel in mouse Müller cells, comprising ∼90% of the conductance of the cell at the resting membrane potential. In addition, the input resistance of Kir4.1 +/− cells is almost precisely double that of Kir4.1 +/+ cells, indicating that there is a gene dosage effect. Only one-half as many channels are expressed in cells in which one of the two channel genes is knocked out.
Although these measurements indicate that Kir4.1 underlies the main conductance of Müller cells, we do not yet know whether other Kir subunits are expressed in these cells. In particular, it is interesting that PCR amplification of several Kir channels revealed the presence of Kir5.1 in the retina. The Kir5.1 subunits are not functional by themselves, but in heterologous expression systems, these subunits are able to coassemble with Kir4.1 and modify their biophysical properties (Pessia et al., 1996). Heteromultimeric Kir4.1/Kir5.1 channels display a steeper degree of inward rectification and altered gating properties (Pessia et al., 1996). It is conceivable that channel composition varies among the different regions of Müller cells to allow regional specialization of K+ conductance properties.
Electroretinogram
Several components of the ERG are believed to be generated by Müller cells in response to light-evoked changes in [K+]o (Newman, 2000). A case in point, the slow PIII response is thought to be generated by a light-evoked decrease in [K+]o in the distal retina arising from photoreceptor hyperpolarization (Witkovsky et al., 1975; Bolnick et al., 1979; Fujimoto and Tomita, 1979; Dick et al., 1985). This [K+]o decrease establishes a K+ efflux from the distal end of Müller cells and a K+ influx into their proximal end (Newman and Odette, 1984). These K+ currents, flowing through Müller cell K+ channels, establish a dipole recorded as a positive field potential in the distal retina.
If the slow PIII response is actually generated by K+ current flow through Müller cells, one expects its absence in mutant lacking the predominant Müller cell K+ channel. This proved to be the case. The slow PIII, present in Kir4.1 +/+ mice, was completely absent in Kir4.1 −/− animals. The response was also missing in Kir4.1 +/+ preparations treated with Ba2+ to block Müller cell K+ channels. The results provide strong support for a Müller cell origin of the slow PIII response.
The origin of another ERG component, the b-wave, has provoked controversy over the past 50 years (Newman, 2000). The b-wave was initially believed to be generated by retinal neurons (Tomita and Funaishi, 1952), then attributed to a K+current generated by Müller cells (Miller and Dowling, 1970;Newman, 1980; Newman and Odette, 1984), and, most recently, to retinal neurons once again (Xu and Karwoski, 1994). Our results strongly support a neuronal origin of the response. The amplitude of the b-wave recorded in Kir4.1 −/− mice did not differ from the response in Kir4.1 +/+ animals. This finding demonstrates conclusively that the b-wave is not generated by K+ currents flowing through Müller cells.
Conclusions
Together, our results indicate that Kir4.1 is the principal Kir channel subtype in Müller cells. The Kir4.1−/− mouse will facilitate future studies on the glial regulation of [K+]o in the mammalian retina.
Footnotes
This work was supported by National Institute of Health Grants GM-29836, MH-49176, EY04077, EY10383, and EY12949. We thank S. Pease, M. Larabee, and T. Wu for expert technical help.
Correspondence should be addressed to Paulo Kofuji, Department of Neuroscience, University of Minnesota, 6–145 Jackson Hall, 321 Church Street SE, Minneapolis, MN 55455. E-mail:kofuj001@tc.umn.edu.
REFERENCES
- 1.Ando M, Takeuchi S. Immunological identification of an inward rectifier K+ channel (Kir4.1) in the intermediate cell (melanocyte) of the cochlear stria vascularis of gerbils and rats. Cell Tissue Res. 1999;298:179–183. doi: 10.1007/s004419900066. [DOI] [PubMed] [Google Scholar]
- 2.Bignami A, Dahl D. The radial glia of Müller in the rat retina and their response to injury. An immunofluorescence study with antibodies to the glial fibrillary acidic (GFA) protein. Exp Eye Res. 1979;28:63–69. doi: 10.1016/0014-4835(79)90106-4. [DOI] [PubMed] [Google Scholar]
- 3.Bolnick DA, Walter AE, Sillman AJ. Barium suppresses slow PIII in perfused bullfrog retina. Vision Res. 1979;19:1117–1119. doi: 10.1016/0042-6989(79)90006-3. [DOI] [PubMed] [Google Scholar]
- 4.Bond C, Pessia M, Xia X, Lagrutta A, Kavanaugh M, Adelman J. Cloning and expression of a family of inward rectifier potassium channels. Receptors Channels. 1994;2:183–191. [PubMed] [Google Scholar]
- 5.Bredt D, Wang T, Cohen N, Guggino W, Snyder S. Cloning and expression of two brain-specific inwardly rectifying potassium channels. Proc Natl Acad Sci USA. 1995;92:6753–6757. doi: 10.1073/pnas.92.15.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brew H, Gray P, Mobbs P, Attwell D. Endfeet of retinal glial cells have higher densities of ion channels that mediate K+ buffering. Nature. 1986;324:466–468. doi: 10.1038/324466a0. [DOI] [PubMed] [Google Scholar]
- 7.Brown KT, Wiesel TN. Localization of origins of electroretinogram components by intraretinal recording in the intact cat eye. J Physiol (Lond) 1961;158:911–931. doi: 10.1113/jphysiol.1961.sp006768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dascal N, Schreibmayer W, Lim NF, Wang W, Chavkin C, DiMagno L, Labarca C, Kieffer BL, Gaveriaux-Ruff C, Trollinger D, Lester HA, Davidson N. Atrial G protein-activated K+ channel: expression cloning and molecular properties. Proc Natl Acad Sci USA. 1993;90:10235–10239. doi: 10.1073/pnas.90.21.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dick E, Miller RF, Bloomfield S. Extracellular K+ activity changes related to electroretinogram components. II. Rabbit (E-type) retinas. J Gen Physiol. 1985;85:911–931. doi: 10.1085/jgp.85.6.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doupnik C, Davidson N, Lester H. The inward rectifier potassium channel family. Curr Opin Neurobiol. 1995;5:268–277. doi: 10.1016/0959-4388(95)80038-7. [DOI] [PubMed] [Google Scholar]
- 11.Doupnik C, Davidson N, Lester H, Kofuji P. RGS proteins reconstitute the rapid gating kinetics of Gβγ-activated inwardly rectifying K+ channels. Proc Natl Acad Sci USA. 1997;16:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fakler B, Bond C, Adelman J, Ruppersberg J. Hetero-oligomeric assembly of inward-rectifier K+ channels from subunits of different subfamilies: Kir2.1 (IRK1) and Kir4.1 (BIR10). Eur J Physiol. 1996;433:77–83. doi: 10.1007/s004240050251. [DOI] [PubMed] [Google Scholar]
- 13.Fujimoto M, Tomita T. Reconstruction of the slow PIII from the rod potential. Invest Ophthalmol Vis Sci. 1979;18:1091–1093. [PubMed] [Google Scholar]
- 14.Hibino H, Horio Y, Inanobe A, Doi K, Ito M, Yamada M, Gotow T, Uchiyama Y, Kawamura M, Kubo T, Kurachi Y. An ATP-dependent inwardly rectifying potassium channel, KAB-2 (Kir4.1), in cochlear stria vascularis of inner ear: its specific subcellular localization and correlation with the formation of endocochlear potential. J Neurosci. 1997;17:4711–4721. doi: 10.1523/JNEUROSCI.17-12-04711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Immel J, Steinberg RH. Spatial buffering of K+ by the retinal pigment epithelium in frog. J Neurosci. 1986;6:3197–3204. doi: 10.1523/JNEUROSCI.06-11-03197.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishii M, Horio Y, Tada Y, Hibino H, Inanobe A, Ito M, Yamada M, Gotow T, Uchiyama Y, Kurachi Y. Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Müller cell membrane: their regulation by insulin and laminin signals. J Neurosci. 1997;20:7725–7735. doi: 10.1523/JNEUROSCI.17-20-07725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isomoto S, Kondo C, Kurachi Y. Inwardly rectifying potassium channels: Their molecular heterogeneity and function. Jpn J Physiol. 1997;47:11–39. doi: 10.2170/jjphysiol.47.11. [DOI] [PubMed] [Google Scholar]
- 18.Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonosaki A, Tomoike H, Kurachi Y. Immunolocalization of an inwardly rectifying K+ channel, KAB-2 (Kir4.1), in the basolateral membrane of renal distal tubular epithelia. FEBS Lett. 1996;388:11–15. doi: 10.1016/0014-5793(96)00502-9. [DOI] [PubMed] [Google Scholar]
- 19.Kusaka S, Puro D. Intracellular ATP activates inwardly rectifying K+ channels in human and monkey retinal Müller (glial) cells. J Physiol (Lond) 1997;500:593–604. doi: 10.1113/jphysiol.1997.sp022045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kusaka S, Kapousta-Bruneau N, Green D, Puro D. Serum-induced changes in the physiology of mammalian retinal glial cells: role of lysophosphatidic acid. J Physiol (Lond) 1998;506:445–458. doi: 10.1111/j.1469-7793.1998.445bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kusaka S, Horio Y, Fujita A, Matsushita K, Inanobe A, Gotow T, Uchiyama Y, Tano Y, Kurachi Y. Expression and polarized distribution of an inwardly rectifying K+ channel, Kir4.1, in rat retinal pigment epithelium. J Physiol (Lond) 1999a;520:373–381. doi: 10.1111/j.1469-7793.1999.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kusaka S, Kapousta-Bruneau N, Puro D. Plasma-induced changes in the physiology of mammalian retinal glial cells: role of glutamate. Glia. 1999b;25:205–215. doi: 10.1002/(sici)1098-1136(19990201)25:3<205::aid-glia1>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 23.Ma W, Zhang L, Xing G, Hu Z, Iwasa K, Clay J. Prenatal expression of inwardly rectifying potassium channel mRNA (Kir4.1) in rat brain. NeuroReport. 1998;9:223–227. doi: 10.1097/00001756-199801260-00009. [DOI] [PubMed] [Google Scholar]
- 24.Miller RF, Dowling JE. Intracellular responses of the Müller (glial) cells of mudpuppy retina: their relation to b-wave of the electroretinogram. J Neurophysiol. 1970;33:323–341. doi: 10.1152/jn.1970.33.3.323. [DOI] [PubMed] [Google Scholar]
- 25.Nagelhus E, Veruki M, Torp R, Haug F, Laake J, Nielsen S, Agre P, Ottersen O. Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Müller cells and fibrous astrocytes. J Neurosci. 1998;18:2506–2519. doi: 10.1523/JNEUROSCI.18-07-02506.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagelhus E, Horio Y, Inanobe A, Fujita A, Haug F, Nielsen S, Kurachi Y, Ottersen O. Immnunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 27.Newman E. Regional specialization of retinal glial cell membrane. Nature. 1984;309:157–159. doi: 10.1038/309155a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newman E. Distribution of potassium conductance in mammalian Müller (glial) cells: a comparative study. J Neurosci. 1987;7:2423–2432. [PMC free article] [PubMed] [Google Scholar]
- 29.Newman E. Inward-rectifying potassium channels in retinal glial (Müller) cells. J Neurosci. 1993;13:3333–3345. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman E. Regulation of extracellular K+ and pH by polarized ion fluxes in glial cells: the retinal Müller cell. The Neuroscientist. 1996;2:109–117. [Google Scholar]
- 31.Newman E, Frambach D, Odette L. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newman E, Reichenbach A. The Müller cell: a functional element of the retina. Trends Neurosci. 1996;19:307–312. doi: 10.1016/0166-2236(96)10040-0. [DOI] [PubMed] [Google Scholar]
- 33.Newman EA. Current source-density analysis of the b-wave of frog retina. J Neurophysiol. 1980;43:1355–1366. doi: 10.1152/jn.1980.43.5.1355. [DOI] [PubMed] [Google Scholar]
- 34.Newman EA. High potassium conductance in astrocyte endfeet. Science. 1986;233:453–454. doi: 10.1126/science.3726539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newman EA. Müller cells and the retinal pigment epithelium. In: Albert DM, Jakobiec FA, editors. Principles and practice of ophthalmology. Saunders; Philadelphia: 2000. pp. 1763–1785. [Google Scholar]
- 36.Newman EA, Bartosch R. An eyecup preparation for the rat and mouse. J Neurosci Methods. 1999;93:169–175. doi: 10.1016/s0165-0270(99)00138-7. [DOI] [PubMed] [Google Scholar]
- 37.Newman EA, Odette LL. Model of electroretinogram b-wave generation: a test of the K+ hypothesis. J Neurophysiol. 1984;51:164–182. doi: 10.1152/jn.1984.51.1.164. [DOI] [PubMed] [Google Scholar]
- 38.Nichols C, Lopatin A. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 39.Nilius B, Reichenbach A. Efficient K+ buffering by mammalian retinal glial cells is due to cooperation of specialized ion channels. Pflügers Arch. 1988;411:654–660. doi: 10.1007/BF00580862. [DOI] [PubMed] [Google Scholar]
- 40.Pessia M, Tucker S, Lee K, Bond C, Adelman J. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 1996;15:2980–2987. [PMC free article] [PubMed] [Google Scholar]
- 41.Reichenbach A, Henke A, Eberhardt W, Reichelt W, Dettmer D. K+ ion regulation in retina. Can J Physiol Pharmacol Suppl. 1992;70:S239–S247. doi: 10.1139/y92-267. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Plainview, NY: 1989. [Google Scholar]
- 43.Schwartz E. l-glutamate conditionally modulates the K+ current of Müller glial cells. Neuron. 1993;10:1141–1149. doi: 10.1016/0896-6273(93)90062-v. [DOI] [PubMed] [Google Scholar]
- 44.Sheng M, Wyszynsky M. Ion channel targeting in neurons. BioEssays. 1997;19:847–853. doi: 10.1002/bies.950191004. [DOI] [PubMed] [Google Scholar]
- 45.Tada Y, Horio Y, Kurachi Y. Inwardly rectifying K+ channel in retinal Müller cells: comparison with the KAB-2/Kir4.1 channel expressed in HEK293T cells. Jpn J Physiol. 1998;48:71–80. doi: 10.2170/jjphysiol.48.71. [DOI] [PubMed] [Google Scholar]
- 46.Takumi T, Ishii T, Horio Y, Morishige K, Takahashi N, Yamada M, Yamashita T, Kiyami H, Sohmiya K, Nakanishi S, Kurachi Y. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem. 1995;270:16339–16346. doi: 10.1074/jbc.270.27.16339. [DOI] [PubMed] [Google Scholar]
- 47.Tomita T, Funaishi A. Studies on intraretinal action potential with low-resistance microelectrode. J Neurophysiol. 1952;15:75–84. doi: 10.1152/jn.1952.15.1.75. [DOI] [PubMed] [Google Scholar]
- 48.Witkovsky P, Dudek FE, Ripps H. Slow PIII component of the carp electroretinogram. J Gen Physiol. 1975;65:119–134. doi: 10.1085/jgp.65.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolburg H. Orthogonal arrays of intramembranous particles: a review with special reference to astrocytes. J Hirnforsch. 1995;36:239–258. [PubMed] [Google Scholar]
- 50.Xu X, Karwoski CJ. Current source density analysis of retinal field potentials. II. Pharmacological analysis of the b-wave and M-wave. J Neurophysiol. 1994;72:96–105. doi: 10.1152/jn.1994.72.1.96. [DOI] [PubMed] [Google Scholar]
- 51.Yang B, Brown D, Verkman AS. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J Biol Chem. 1996;271:4577–4580. [PubMed] [Google Scholar]
- 52.Zahs KR, Newman EA. Asymmetric gap junctional coupling between glial cells in the rat retina. Glia. 1997;20:10–22. [PubMed] [Google Scholar]