

Abstract
Stability parameters for individual residues in Thermus thermophilus cysteine-free RNase H were determined by native state hydrogen exchange, thus providing a unique comparison of regional thermodynamics between thermophilic and mesophilic homologues. The general distribution of stability in the thermophilic protein is similar to that of its mesophilic homologue, with a proportional increase in stability for almost all residues. As a consequence, the residue-specific stabilities of the two proteins are remarkably similar under conditions where their global stabilities are the same. These results indicate that T. thermophilus RNase H is stabilized in a delocalized fashion, preserving a finely tuned balance of stabilizing interactions throughout the structure. Therefore, although protein stability can be altered by single amino acid substitution, evolution for optimal function may require more subtle and delocalized mechanisms.
Proteins from thermophilic organisms must retain their native structures under extreme conditions, where their homologues from mesophilic organisms denature. Because these thermophilic proteins often have sequences and structures that are very similar to those of their mesophilic counterparts, it remains unclear what causes their dramatic stabilization (1). A variety of individual stabilizing interactions have been examined by mutagenesis (2–4), but little is known about the relative roles of these specific interactions and more global properties, such as how the stabilization is distributed throughout the structure and how coupling between regions of the molecule contributes to the overall stabilization. Understanding how these properties compare between thermophilic and mesophilic proteins is essential in determining how thermophilic proteins are stabilized.
The recently developed technique of native state hydrogen exchange allows these issues to be addressed, by determining the free energies of unfolding, or stabilities, of individual sites in a protein (5, 6). Although most small, single-domain proteins appear by traditional probes to unfold cooperatively, populating only the folded and unfolded conformations, native state hydrogen exchange makes it possible to detect rare, partial unfolding events under native conditions. The rates at which individual amide protons in the protein backbone exchange for solvent deuterons are used to calculate residue-specific free energies of exchange, and unfolding is distinguished from other mechanisms of exchange based on its denaturant dependence. Results from this type of experiment often show residues with similar stabilities clustered in specific secondary structural elements (5, 6) and therefore can offer a detailed description of how regions in the molecule are thermodynamically related. This technique has been used to examine the distribution of stability in a thermophilic rubredoxin (7), but to our knowledge there has been no direct comparison of such distributions between thermophilic and mesophilic homologues.
We have used native state hydrogen exchange to examine the distribution of stability in Thermus thermophilus cysteine-free RNase H (RNase H*) (8), for comparison with that of its mesophilic homologue Escherichia coli RNase H*. Although the sequence and structure of these proteins are quite similar (9), the thermophilic protein is more stable at all temperatures, because of an increased maximal stability and a decreased heat capacity of unfolding (ΔCp) (10). Previous hydrogen exchange work has demonstrated that for E. coli RNase H*, the two core helices are the most stable regions of the protein, followed by the β-sheet that packs against this core (6). In this study, we address whether this pattern of stability is intrinsic to the RNase H fold or is altered in the thermophilic enzyme, and whether the increased stability of T. thermophilus RNase H* can be localized to a specific region of the structure.
Methods
T. thermophilus RNase H*, a cysteine-free variant of the wild-type protein, was expressed in E. coli DE3 pLysS cells grown on M9 media, labeled with either 15N or 15N and 13C. Protein was purified as described (10).
Assignments for T. thermophilus RNase H* backbone amide hydrogens were determined by using 1H-15N heteronuclear single quantum coherence, CBCANH (11), and CBCA(CO)NH (12) NMR spectra. Doubly (15N and 13C) labeled T. thermophilus RNase H* was dissolved to a concentration of 1 mM in 90% H2O/10% D2O, 100 mM d3-NaAc, pH 5.9. Data were collected at 25°C on a Bruker DMX 600-MHz spectrometer and processed in azara (Wayne Boucher, University of Cambridge), using Fourier transformation for the directly detected dimension and a maximum entropy algorithm for the indirectly detected dimensions. Backbone NH peaks were assigned based on intra- and inter-residue alpha and beta carbon chemical shifts, by using the program ansig (13). These assignments are available by request.
Hydrogen/deuterium exchange rates for backbone amide protons were determined at 25°C in several different GdmCl concentrations, all where the folded conformation predominates. Nine samples were prepared, each containing 1 mM 15N T. thermophilus RNase H*, 100 mM NaAc, and concentrations of GdmCl ranging from 0 to 2.16 M (pH 5.9). G25 spin columns were used to exchange samples into deuterated buffer [100 mM d3-NaAc, 0.05% NaAzide, and appropriate concentrations of deuterated GdmCl, pD 5.9 (pDread 5.5)], and a series of heteronuclear single quantum coherence spectra were begun immediately. Spectra were collected rapidly (approximately 20 min each) on a Bruker 500-MHz spectrometer. Samples were monitored continuously for 3 hr after initiation of exchange, and then periodically for up to 8 months.
To test the assumption that exchange occurs in the EX2 limit, two additional samples were prepared and monitored as above, each containing 1 mM 15N T. thermophilus RNase H*, 100 mM d3-NaAc, 2.16 M GdmCl, 0.05% NaAzide, at pD 5.4 or 6.2. One further sample was prepared to probe the effects of salt on exchange rates; this sample contained 100 mM d3-NaAc, 0.9 M NaCl, 0.05% NaAzide, pD 5.9.
Heteronuclear single quantum coherence spectra were processed by using felix 97.0 (Molecular Simulations), and the intensity decrease over time for each peak was fit to a single exponential. The rate constants for exchange (kobs) then were related to free energies of exchange (ΔGhx) by using the equation: ΔGhx = −RTln(kobs/kint), where kint is the intrinsic rate of exchange for specific amide protons in unstructured peptides (14). Free energies of exchange for each residue were plotted against [GdmCl] to distinguish between local, denaturant-independent events and denaturant-dependent unfolding. These data then were fit to obtain free energies of unfolding (ΔGunf) and associated m-values, as well as free energies of local fluctuation (ΔGfl), according to the equation: ΔGhx = −RT ln (Kunf + Kfl), where Kunf = exp ((−ΔGunf + m[GdmCl])/RT) and Kfl = exp (−ΔGfl/RT) (5, 6).
Results and Discussion
Amide hydrogen/deuterium exchange rates in T. thermophilus RNase H* were determined at 25°C, in subdenaturing concentrations of GdmCl (100 mM d3-NaAc, 0–2.16 M deuterated GdmCl, pD 5.9). Under all of these conditions, the protein appears completely folded by global probes such as CD (ΔGunf = 13.1 kcal/mol, m = 3.15 kcal·mol−1·M−1 in D2O). Exchange was monitored by NMR, as the disappearance of proton peaks in 1H-15N heteronuclear single quantum coherence spectra taken periodically for up to 8 months. Of the 166 residues in the protein, 67 had amide protons that exchanged slowly enough to be monitored in these experiments, and 52 of these tractable peaks were unambiguously assigned to specific residues in the structure. Rate constants for exchange (kobs) were determined by fitting the intensity of each peak as a function of time to a single exponential, and these rate constants were related to free energies of exchange (ΔGhx) by using the equation: ΔGhx = −RTln(kobs/kint), where kint is the intrinsic rate of exchange (14) (see Methods). This analysis is based on a model involving an initial opening reaction, in which the protein adopts an “open” conformation that allows exchange at a particular amide site, followed by an exchange reaction whose rate is governed by the intrinsic chemical exchange rate of that amide proton:
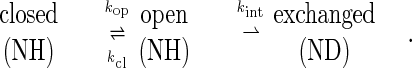 |
Under conditions where the intrinsic rate of exchange is much slower than the closing rate (EX2 conditions), the observed rate constant can be described as the product of the equilibrium constant for the opening reaction and the intrinsic chemical exchange rate constant (kobs = Kopkint), leading to the relationship between kobs and ΔGhx described above (15).
For most proteins under our experimental conditions, the EX2 assumption appears to be valid; we verified this for T. thermophilus RNase H* by monitoring exchange rates at two additional pDs, 5.4 and 6.2. These experiments were carried out in the highest GdmCl concentration used in the native state exchange experiments (2.16 M), where the closing rates are presumed to be the slowest. In EX2 conditions, the natural logarithm of kobs varies linearly with pD, with a slope of 1, because of the pD dependence of kint. For T. thermophilus RNase H*, this was the case only for the lower two pDs (5.4 and 5.9). Although exchange was slower than expected at pD 6.2, the EX2 condition does appear to apply in the conditions of our native state hydrogen exchange experiments (pD 5.9).
The free energies of exchange for each residue were plotted as a function of GdmCl concentration, to distinguish between denaturant-dependent unfolding events and denaturant-independent local fluctuations (Fig. 1). For some residues, ΔGhx decreased linearly with [GdmCl], as expected for an exchange process governed by an unfolding reaction that exposes hydrophobic surface area. These data were fit to obtain the free energy of unfolding in the absence of denaturant (ΔGunf) and the slope of the denaturant dependence (m-value) for each of these residues (Table 1). For other residues, ΔGhx was independent of [GdmCl] at low denaturant concentrations, implying that exchange under these conditions was being governed by a more local event not involving significant hydrophobic surface area exposure. These local fluctuations were seen for some residues in all structural elements except helix E, where unfolding is low enough in energy to dominate the exchange mechanism under all conditions tested. In almost all cases where local fluctuations were seen at low [GdmCl], unfolding occurred at higher [GdmCl], where it required less energy than the local fluctuation. Thus for these residues, values for ΔGunf and m-value as well as free energies for the local fluctuation event (ΔGfl) could be determined (Table 1). Lastly, seven residues could not be described by these categories of unfolding and local fluctuations. All of these residues were in regions of local positive charge and displayed a [NaCl] dependence to their exchange rates (data not shown). This result is consistent with other findings that intrinsic rates of exchange for positively charged residues can be influenced by salt concentration (16). Because determining free energies of unfolding for these residues would require extrapolation from concentrations of GdmCl where the kint is likely to be overestimated, these residues were excluded from our analysis.
Figure 1.

Free energy of exchange vs. GdmCl concentration for residues in T. thermophilus RNase H*. Five residues are shown: W26 (□) and L30 (▵) in strand II, A55 in helix A (■), M116 in helix D (○), and R143 in helix E (▴).
Table 1.
Stability parameters for residues in T. thermophilus RNase H*, measured by native state hydrogen exchange
| Residue | Secondary structure | ΔGunf, kcal·mol−1 | m-value, kcal·mol−1⋅M−1 | ΔGfl, kcal·mol−1 |
|---|---|---|---|---|
| A10 | Strand I | 12.2 ± 1.5 | 3.8 ± 0.8 | 6.2 ± 0.1 |
| L11 | Strand I | 13.3 ± 0.8 | 4.8 ± 0.5 | 8.3 ± 0.3 |
| T13 | Strand I | 10.4 ± 0.4 | 3.1 ± 0.2 | 7.2 ± 0.1 |
| G22 | Loop | 8.8 ± 0.8 | 1.3 ± 0.4 | 7.3 ± 0.2 |
| G24 | Strand II | 9.1 ± 0.09 | 2.33 ± 0.07 | |
| G25 | Strand II | 10.9 ± 0.4 | 3.0 ± 0.2 | 9.0 ± 0.2 |
| W26 | Strand II | 11.1 ± 0.4 | 3.5 ± 0.2 | 8.3 ± 0.1 |
| A27 | Strand II | 11.4 ± 0.6 | 3.6 ± 0.4 | 8.4 ± 0.2 |
| A28 | Strand II | 10.3 ± 0.2 | 2.7 ± 0.1 | |
| L29 | Strand II | 9.3 ± 0.3 | 2.7 ± 0.2 | |
| L30 | Strand II | 11.4 ± 0.2 | 3.6 ± 0.1 | 7.54 ± 0.06 |
| L39 | Strand III | 10.2 ± 0.6 | 3.3 ± 0.4 | 7.3 ± 0.2 |
| G41 | Strand III | 10.3 ± 0.3 | 2.7 ± 0.2 | 9.3 ± 0.3 |
| E43 | Strand III | 9.8 ± 0.3 | 3.1 ± 0.2 | 6.88 ± 0.06 |
| E52 | Helix A | 13.2 ± 0.9 | 4.4 ± 0.5 | |
| L53 | Helix A | 12.9 ± 1.1 | 4.3 ± 0.7 | |
| A55 | Helix A | 15.2 ± 0.6 | 5.1 ± 0.4 | |
| A56 | Helix A | 14.5 ± 0.7 | 4.9 ± 0.4 | |
| I57 | Helix A | 15.9 ± 1.9 | 5.7 ± 1.0 | |
| E58 | Helix A | 13.7 ± 1.2 | 5.0 ± 0.7 | |
| G59 | Helix A | 13.5 ± 1.4 | 4.3 ± 0.8 | |
| L60 | Helix A | 15.7 ± 1.3 | 5.4 ± 0.7 | |
| A62 | Helix A | 12.6 ± 1.5 | 3.8 ± 0.8 | 6.08 ± 0.07 |
| L63 | Helix A | 10.7 ± 0.4 | 3.2 ± 0.2 | |
| E68 | Strand IV | 3.46 ± 0.09 | ||
| V69 | Strand IV | 13.6 ± 0.7 | 4.8 ± 0.4 | 6.97 ± 0.08 |
| D70 | Strand IV | 13.3 ± 0.6 | 4.6 ± 0.4 | |
| L71 | Strand IV | 13.1 ± 0.9 | 4.6 ± 0.5 | |
| T73 | Strand IV | 4.66 ± 0.05 | ||
| L108 | Helix D | 4.04 ± 0.07 | ||
| W109 | Helix D | 16.5 ± 0.4 | 5.8 ± 0.2 | 7.67 ± 0.05 |
| A111 | Helix D | 14.6 ± 2.0 | 4.7 ± 1.1 | 6.31 ± 0.09 |
| L113 | Helix D | 13.5 ± 1.9 | 4.3 ± 1.0 | 7.0 ± 0.2 |
| L114 | Helix D | 13.4 ± 1.3 | 4.4 ± 0.6 | 6.52 ± 0.09 |
| A115 | Helix D | 8.6 ± 0.5 | ||
| M116 | Helix D | 16.1 ± 1.0 | 5.6 ± 0.6 | 8.6 ± 0.5 |
| A117 | Helix D | 11.2 ± 1.0 | 3.1 ± 0.5 | 6.51 ± 0.08 |
| R122 | Strand V | 14.5 ± 1.0 | 4.9 ± 0.5 | 8.4 ± 0.1 |
| E141 | Helix E | 5.5 ± 0.1 | 1.03 ± 0.07 | |
| R143 | Helix E | 7.11 ± 0.07 | 1.57 ± 0.06 | |
| Q145 | Helix E | 6.6 ± 0.2 | 1.6 ± 0.2 | |
| A146 | Helix E | 6.46 ± 0.05 | 1.48 ± 0.05 | |
| Q147 | Helix E | 5.69 ± 0.02 | 2.0 ± 0.04 |
Free energies of exchange vs. [GdmCl] for each residue were fit as described in Methods.
The residue-specific free energies of unfolding (ΔGunf) for T. thermophilus RNase H*, which range from 5.5 to 16.5 kcal/mol, are on average higher than those measured for E. coli RNase H*. These values, however, are distributed throughout the structure in a pattern that is remarkably similar to that of the mesophilic protein (Fig. 2). In both proteins, residues in the central helices (A and D) and strand IV have the highest ΔGunfs, followed by residues in the remainder of the β-sheet. This similarity between the proteins also can be described in terms of their conformational ensembles (Fig. 3). Although most (>99.99%) of the molecules are folded under the conditions of these experiments, the unfolded conformation is represented in a very small population, according to the global ΔGunf. Free energies of unfolding less than the global ΔGunf represent exchange from partially unfolded forms present in slightly larger but still extremely small populations. Residues with similar ΔGunfs cluster in regions that define such conformations and have allowed us to assign each secondary structural element to the lowest energy conformation in which it unfolds (Fig. 3). In addition to the folded conformation, there are three forms populated by T. thermophilus RNase H*. With one exception (see below), each has a structurally homologous counterpart in E. coli RNase H*, again demonstrating the conserved pattern of stability. Overall, these results suggest that the stability distribution in a protein is an intrinsic characteristic of its fold, and that proteins with drastically different global thermodynamic properties can display very similar structural patterns of stability.
Figure 2.

Free energies of unfolding for residues in T. thermophilus and E. coli RNases H* mapped onto the x-ray crystal structures (9, 23). Data for E. coli RNase H* were obtained from Chamberlain et al. (6). Each sphere represents a backbone amide site for which hydrogen exchange rates could be measured; these spheres are colored according to their ΔGunf values. Stabilities in 0.88 M GdmCl are shown for T. thermophilus RNase H*, as this is where the highest regional ΔGunf is the same as that for E. coli RNase H* in the absence of denaturant; these values were calculated by using ΔGunf and m-values from Table 1.
Figure 3.

Free energies of unfolding for partially and fully unfolded forms of T. thermophilus and E. coli RNases H*. Each line represents the difference in free energy between the native conformation and a form in which the structural elements indicated are unfolded; the form with the highest ΔGunf represents the fully unfolded conformation. Values of ΔGunf were averaged for each secondary structural unit, and regions were grouped together if the average of one region overlapped with the SD for another. Strand I is not represented for T. thermophilus RNase H*; one of its three tractable residues had a ΔGunf of 10.4 kcal/mol, whereas others were closer to the core region, both in their measured ΔGunfs and in their location in the structure. Stabilities in 0.88 M GdmCl were calculated for T. thermophilus RNase H* residues by using the parameters in Table 1, and values for coupled regions were averaged together.
The C-terminal E-helix comprises the one obvious exception to this conservation of stability distribution. Although helix E is in the least stable region of both proteins, in T. thermophilus RNase H* it exchanges at a lower free energy than the β-sheet, whereas in E. coli RNase H* the helix and sheet cluster together. Whether this difference is important to the increased stability of the thermophilic protein is not clear. It appears from the crystal structures of the two proteins that helix E in E. coli RNase H* forms more contacts with the sheet. Despite this, a deletion mutant of E. coli RNase H* lacking helix E is cooperatively folded and retains weak enzymatic activity (17), demonstrating that the helix is not essential for folding. It is possible that the thermophilic protein is stabilized in part by favoring inter-strand contacts over contacts between the sheet and helix E, thus stabilizing a more central region of the protein at the expense of a helix that is not essential to the fold.
Two more subtle alterations in the patterns of stability emerge from the grouping of structural elements, although the data are not sufficient to determine whether either reflects a significant difference. First, in E. coli RNase H*, strand IV and helix B have been defined as a separate partially unfolded form. The absence of a corresponding form in T. thermophilus RNase H* is partially caused by the fact that residues in helix B were affected by salt concentration and therefore were ignored in the analysis. Residues in strand IV do appear to be slightly lower in stability than helices A and D, but were within the SD for the higher energy region. The second difference is that strand V appears to cluster with the most stable region in T. thermophilus RNase H*, whereas in E. coli RNase H* it unfolds with a free energy closer to that of strands I, II, and III. There is only one residue in strand V that could be monitored in T. thermophilus RNase H*, however, and this residue is near the N terminus of the strand, which is more stable than the C-terminal end in E. coli RNase H*. Therefore, although this strand may be more stable in the thermophilic enzyme, the small amount of data in this region makes it difficult to draw conclusions about its true stability.
In addition to the general conservation of the pattern of stability in these proteins, the magnitude and range of ΔGunfs allow us to gain insight into how the increased stability of the thermophilic protein is partitioned. As shown in Figs. 2 and 3, the ΔGunfs for most regions are higher in T. thermophilus RNase H*. This result was expected for the most stable region (helices A and D and strand IV), as exchange of these amide protons is governed by global unfolding, and the global stability for T. thermophilus RNase H* is higher at 25°C (13.1 kcal/mol in D2O, unpublished results). However, partially unfolded conformations are not governed by the global stability, and therefore changes in these regions could not be predicted. With the exception of helix E, each region in T. thermophilus RNase H* has a higher ΔGunf than its counterpart in E. coli RNase H*. Moreover, each region in the thermophilic protein is stabilized not by the same amount, but in a way that is proportional to its stability: the ΔGunf values for both the core helices and the strands are increased by 41–42%. This finding indicates that the stabilization of this thermophilic protein is not localized to a particular region of the molecule, for each region contributes the same relative amount to the overall stability in both proteins.
This delocalized stabilization suggests that the distribution as well as the number and type of stabilizing interactions in thermophilic proteins is important. It is well established that proteins can be stabilized by introducing local changes (2, 3). For example, substituting certain residues from T. thermophilus RNase H into the corresponding positions in E. coli RNase H increases the thermostability of the molecule, by filling a hydrophobic cavity (18) or stabilizing a turn with a proline residue (4). Experiments of this type have implicated a variety of local interactions, such as hydrogen bonds, salt bridges, and hydrophobic interactions, as important in stabilizing thermophilic proteins (1). Our results, however, demonstrate not only that T. thermophilus RNase H* must be stabilized by a collection of such local interactions, but also that these interactions are structurally organized so as to maintain a balance of stability throughout the protein, and that this balance of stability is conserved between proteins operating in very different environments.
The observation that each region of RNase H* contributes the same relative stability in both proteins leads to the hypothesis that under conditions where the global stability of T. thermophilus RNase H* is the same as that seen for E. coli RNase H*, the ΔGunf for each region also will be the same. Because residues with higher ΔGunfs also have higher m-values (Table 1), the partially unfolded forms are closer together in stability at higher concentrations of denaturant. Specifically, at 0.88 M GdmCl, where the average ΔGunf for the most stable region in T. thermophilus RNase H* is 9.8 kcal/mol (i.e., the same as in E. coli RNase H* in the absence of denaturant), the ΔGunf of the strands is 7.7 kcal/mol, only 0.3 kcal/mol different from the strands in E. coli RNase H* (Fig. 3). This similarity between the proteins can be seen at the residue level in Fig. 2. Furthermore, because both m-values and ΔCps are thought to reflect hydrophobic surface area burial (19), we expect that the ΔCps for the partially unfolded forms also will correlate with their ΔGunf values, leading to the same merging of regional ΔGunfs as the temperature is increased. Therefore, at a temperature (approximately 60°C) where the stability of T. thermophilus RNase H* is the same as that for E. coli RNase H* at 25°C, it is likely that the free energy pattern of T. thermophilus RNase H* is the same as that for E. coli RNase H* at 25°C.
Because E. coli and T. thermophilus RNases H* have comparable stabilities at the optimal growth temperatures of their host organisms (10), it is interesting to note that their regional stabilities also may be similar. Thermophilic proteins are often indistinguishable from their mesophilic homologues when properties such as global stability (20) and flexibility (21, 22) are evaluated at the temperatures where the proteins function. Our results suggest that this observation may be extended to a more structurally detailed level, implicating regional stability and flexibility as conserved features of enzymes at their optimal temperatures.
In conclusion, we have shown by native state hydrogen exchange that T. thermophilus RNase H* has three regions of structure with distinct free energies of unfolding. With the exception of the least stable, C-terminal helix, this pattern is similar to that seen in a mesophilic homologue of this protein, E. coli RNase H*, indicating that the structural distribution of stability is an intrinsic property of a protein’s fold. In addition, the increased stability of the thermophilic RNase H* is not localized to any particular region; almost every region of the molecule contributes the same relative amount to each protein. All of these findings suggest that although local effects, even single amino acid substitutions, can make mesophilic proteins more thermostable, natural thermophilic proteins are stabilized by more subtle and delocalized mechanisms, preserving a balance of stability, dynamics, and function.
Acknowledgments
We thank Tracy Handel and Julie Lougheed for critical assistance with three-dimensional NMR data collection and assignment of T. thermophilus RNase H* backbone amides, the Marqusee lab for discussion, and Navin Pokala, Eric Goedken, and Kael Fischer for critical reading of the manuscript. This work was supported by the National Institutes of Health (GM50945) and the Helman Faculty Fund.
Abbreviation
- RNase H*
cysteine-free RNase H
References
- 1.Jaenicke R, Bohm G. Curr Opin Struct Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 2.Jaenicke R. FEMS Microbiol Rev. 1996;18:215–224. doi: 10.1111/j.1574-6976.1996.tb00238.x. [DOI] [PubMed] [Google Scholar]
- 3.Aoshima M, Oshima T. Protein Eng. 1997;10:249–254. doi: 10.1093/protein/10.3.249. [DOI] [PubMed] [Google Scholar]
- 4.Kimura S, Nakamura H, Hashimoto T, Oobatake M, Kanaya S. J Biol Chem. 1992;267:21535–21542. [PubMed] [Google Scholar]
- 5.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamberlain A K, Handel T M, Marqusee S. Nat Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 7.Hiller R, Zhou Z H, Adams M W, Englander S W. Proc Natl Acad Sci USA. 1997;94:11329–11332. doi: 10.1073/pnas.94.21.11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanaya S, Itaya M. J Biol Chem. 1992;267:10184–10192. [PubMed] [Google Scholar]
- 9.Ishikawa K, Okumura M, Katayanagi K, Kimura S, Kanaya S, Makamura H, Morikawa K. J Mol Biol. 1993;230:529–542. doi: 10.1006/jmbi.1993.1169. [DOI] [PubMed] [Google Scholar]
- 10.Hollien J, Marqusee S. Biochemistry. 1999;38:3831–3836. doi: 10.1021/bi982684h. [DOI] [PubMed] [Google Scholar]
- 11.Grzesiek S, Bax A. J Magn Reson. 1992;99:201–207. [Google Scholar]
- 12.Grzesiek S, Bax A. J Am Chem Soc. 1992;114:6291–6293. [Google Scholar]
- 13.Kraulis P J, Domaille P J, Campbell-Burk S L, Van Aken T, Laue E D. Biochemistry. 1994;33:3515–3531. doi: 10.1021/bi00178a008. [DOI] [PubMed] [Google Scholar]
- 14.Bai Y, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hvidt A, Nielsen S. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 16.Kim P S, Baldwin R L. Biochemistry. 1982;21:1–5. doi: 10.1021/bi00530a001. [DOI] [PubMed] [Google Scholar]
- 17.Goedken E R, Raschke T M, Marqusee S. Biochemistry. 1997;36:7256–7263. doi: 10.1021/bi970060q. [DOI] [PubMed] [Google Scholar]
- 18.Ishikawa K, Nakamura H, Morikawa K, Kanaya S. Biochemistry. 1993;32:6171–6178. [PubMed] [Google Scholar]
- 19.Myers J K, Pace C N, Scholtz J M. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Grayling R A, Sandman K, Ednondson S, Shriver J W, Reeve J N. Biochemistry. 1998;37:10563–10572. doi: 10.1021/bi973006i. [DOI] [PubMed] [Google Scholar]
- 21.Zavodszky P, Kardos J, Svingor A, Petsko G A. Proc Natl Acad Sci USA. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wrba A, Schweiger A, Schultes V, Jaenicke R, Zavodsky P. Biochemistry. 1990;29:7584–7592. doi: 10.1021/bi00485a007. [DOI] [PubMed] [Google Scholar]
- 23.Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Nakamura H, Ikehara M, Matsuzaki T, Morikawa K. J Mol Biol. 1992;223:1029–1052. doi: 10.1016/0022-2836(92)90260-q. [DOI] [PubMed] [Google Scholar]