

Abstract
The nuclear factor κB (NF-κB) pathway plays a central role in inflammation and immunity. In response to proinflammatory cytokines and pathogen-associated molecular patterns, NF-κB activation is controlled by IκB kinase (IKK)β. Using Cre/lox-mediated gene targeting of IKKβ, we have uncovered a tissue-specific role for IKKβ during infection with group B streptococcus. Although deletion of IKKβ in airway epithelial cells had the predicted effect of inhibiting inflammation and reducing innate immunity, deletion of IKKβ in the myeloid lineage unexpectedly conferred resistance to infection that was associated with increased expression of interleukin (IL)-12, inducible nitric oxide synthase (NOS2), and major histocompatibility complex (MHC) class II by macrophages. We also describe a previously unknown role for IKKβ in the inhibition of signal transducer and activator of transcription (Stat)1 signaling in macrophages, which is critical for IL-12, NOS2, and MHC class II expression. These studies suggest that IKKβ inhibits the “classically” activated or M1 macrophage phenotype during infection through negative cross talk with the Stat1 pathway. This may represent a mechanism to prevent the over-exuberant activation of macrophages during infection and contribute to the resolution of inflammation. This establishes a new role for IKKβ in the regulation of macrophage activation with important implications in chronic inflammatory disease, infection, and cancer.
NF-κB is a ubiquitous transcription factor that regulates expression of proinflammatory and antiapoptotic genes and is thought to play an important role in driving the inflammatory response (1). NF-κB activation in response to proinflammatory stimuli is regulated by IκB kinase (IKK)β, which has recently become a major target for the development of new antiinflammatory drugs (2). However, we have previously established both pro- and antiinflammatory roles for NF-κB in inflammation (3, 4). In this study, we have tested the hypothesis that NF-κB has tissue-specific roles in inflammation; although NF-κB activation in epithelial cells is required to drive the inflammatory response, NF-κB activity in macrophages has an opposing role in the resolution of inflammation.
Macrophages are an extremely heterogeneous lineage displaying a range of both pro- and antiinflammatory functions (5, 6). Two extremes in the spectrum of macrophage function are represented by the “classically” activated (or M1) and the alternative (or M2) phenotypes (7). The M1 phenotype is typified by IFN-γ–primed macrophages and is characterized by increased MHC class II, IL-12, and inducible nitric oxide synthase (NOS2) expression. M2 macrophages are characterized by decreased expression of NOS2, MHC class II, and IL-12, but elevated expression of antiinflammatory cytokines such as IL-10. The M2 phenotype is promoted by Th2 cytokines such as IL-4 and IL-13. Generally, M2 macrophages are considered antiinflammatory cells, whereas M1 macrophages are proinflammatory. The signaling pathways that regulate these phenotypes in vivo are not well understood.
We used Cre/lox-mediated gene targeting of IKKβ in lung epithelial cells and macrophages (9, 10) and show that IKKβ deletion in airway epithelial cells inhibits inflammation and prevents clearance of bacteria in the lung. However, IKKβ deletion in macrophages has the opposite role, increasing the inflammatory response and enhancing the clearance of bacteria. In addition, we show that IKKβ activation suppresses the classically activated or M1 macrophage phenotype, and we present evidence for negative regulation of Stat1 activation by IKKβ. These findings represent important new information regarding the role of NF-κB in macrophage biology during inflammation and infection. Furthermore, cross talk between IKKβ and Stat1 signaling pathways could have implications for the development and clinical use of IKKβ inhibitors.
RESULTS AND DISCUSSION
To investigate the tissue-specific role of IKKβ in innate immunity, we specifically targeted the Ikkβ gene using Cre/lox-mediated recombination in lung epithelial cells and myeloid cells using the clara cell–specific protein promoter (Cc10) and lysozyme promoter to express Cre in bronchial airway epithelial cells and the myeloid cell lineage, respectively. Cre transgenic mice were crossed to Ikkβf/f mice to generate tissue-specific deletion of Ikkβ. Tissue-specific targeting of the Ikkβ gene in these mice has been established (9, 10). To study innate immunity, we infected IKKβΔMye (myeloid deletion) and IKKβΔEpi (epithelial deletion) mice intranasally with group B streptococcus (GBS) and measured bacterial clearance from the lung (11). Fig. 1 A shows that IKKβΔEpi mice had impaired ability to clear the GBS infection, demonstrating significantly higher bacterial titres in the lung than littermate controls (Fig. 1 A; IKKβf/f-Otet-cre vs. IKKβΔEpi; *, P = 0.0242). Histological analysis of lung tissue from IKKβΔEpi mice infected with GBS revealed reduced neutrophil (PMN) recruitment and persistence of bacteria in the lung (Fig. 1 B). In contrast, IKKβΔMye mice showed enhanced clearance of bacteria (Fig. 1 C; IKKβf/f vs. IKKβΔMye; *, P = 0.0304), and analysis of lung tissue from IKKβΔMye mice revealed increased PMN infiltration after 4 h (Fig. 1, D and E). Furthermore, inflammation in GBS-infected control mice completely resolved after 24 h; however, in IKKβΔMye mice inflammation failed to resolve despite increased clearance of bacteria (Fig. 1 D). These experiments suggested that IKKβ played a tissue-specific role in response to infection and that IKKβ activity in airway epithelium was required for leukocyte recruitment and clearance of bacteria; however, IKKβ activation in resident macrophages or myeloid cells recruited to the lung inhibited the inflammatory response and therefore impaired clearance of bacteria.
Figure 1.

Tissue-specific role for IKKβ in Streptococcal pneumonia. (A) IKKβΔEpi and IKKβf/f-Otet-cre control mice were infected intranasally with 5 × 106 CFUs GBS in PBS. Bronchialveolar lavage was performed after 4 h, and CFUs were determined by serial dilution on Todd-Hewitt agar plates (n = 11–14; *, P = 0.0242). (B) Hematoxylin and eosin stain (left, H+E) of lungs from GBS-infected mice after 4 h showed reduced leukocyte infiltration in IKKβΔEpi mice (arrows). Gram stain (right) of infected lungs showed accumulation of bacteria in alveolar spaces of IKKβΔEpi lungs (arrows). Representative panels are shown from n = 8. Bar, 100 μm. (C) IKKβΔMye and IKKβf/f control mice were infected as described above, and BAL CFUs were measured at 4 h (n = 11–14; *, P = 0.0304). (D) Hematoxylin and eosin stain of lungs from GBS-infected mice after 4 and 24 h showed resolution of peribronchial inflammation in the lungs of IKKβf/f mice at 24 h (top, arrows); however, inflammation in IKKβΔMye mice fails to resolve (bottom, arrows). Bar, 200 μm. (E) PMN recruitment in IKKβΔMye and IKKβf/f mice infected with GBS was counted under high power field after hematoxylin and eosin stain. Data are represented as mean ± SEM of n = 8 (**, P = 0.005).
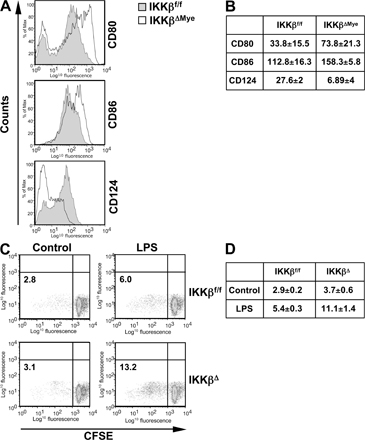
We next systemically challenged IKKβΔMye mice with a lethal dose of GBS and monitored their survival to determine if the increased inflammatory response could confer resistance to infection. IKKβΔMye mice were indeed more resistant to systemic infection with GBS. More than 60% of IKKβΔMye mice survived a lethal dose of GBS beyond 48 h, whereas >90% of littermate controls had died within 24 h, and none survived to 48 h (Fig. 2 A). Furthermore, resistance of IKKβΔMye mice to lethal GBS infection correlated with significantly lower titres of bacteria in the blood (Fig. 2 B; IKKβf/f vs. IKKβΔMye; **, P = 0.0057). To investigate the mechanism for this resistance, we measured the expression of proinflammatory cytokines in the spleen and serum of GBS-infected mice. Fig. 2 C shows a significant increase in the levels of protective Th1 cytokines IFN-γ and IL-12 in IKKβΔMye mice (Fig. 2 C; IKKβf/f vs. IKKβΔMye; IFN-γ: *, P = 0.0148; IL-12: **, P = 0.0023). Recently it has been reported that IKKβ inhibits caspase 1–dependent IL-1β release in LPS-stimulated PMNs (4); however, we found no increase in serum IL-1β in GBS-infected IKKβΔMye mice (Fig. 2 C). In fact, IL-1β levels were slightly decreased, although these data did not reach statistical significance (Fig. 2 C). To further analyze the macrophage phenotype in IKKβΔMye mice, we isolated peritoneal macrophages from GBS-infected mice and measured MHC class II expression by FACS. IKKβΔMye mice showed a threefold increase in MHC class II expression compared with control mice (Fig. 2 D). Expression of the costimulatory molecules CD80 (B7.1) and CD86 (B7.2) were also increased on macrophages from IKKβΔMye mice, whereas CD124 (IL-4Rα) expression was decreased (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20080124/DC1). We also analyzed IL-12p40 and arginase-1 mRNA expression in the spleen from GBS-infected mice; although expression of IL-12p40 was increased in IKKβΔMye mice, expression of the antiinflammatory macrophage marker arginase-1 was reciprocally decreased (Fig. 2 E). In addition, immunohistochemical analysis of lungs from GBS-infected mice revealed dramatically increased NOS2 expression in alveolar macrophages, whereas NOS2 expression in lung epithelium remained unchanged (Fig. 2 F). IL-12 is a critical cytokine for host defense (12) and, along with MHC class II and NOS2, is a marker for classically activated or M1 macrophages (7). On the other hand, increased expression of arginase-1 and IL-10 is characteristic of alternative M2 macrophage activation, which is dependent on CD124 expression (7).
Figure 2.

IKKβ inhibits M1 macrophage activation during infection. (A) IKKβΔMye and IKKβf/f control mice were infected by intraperitoneal injection of 5 × 107 CFUs GBS in PBS, and survival was monitored (n = 16–17). (B) Blood was collected at 4 h by retroorbital bleed. Serial dilutions of blood samples were plated in triplicate on Todd-Hewitt agar plates, and CFUs were counted (n = 9–10; **, P = 0.0057). (C) Serum levels of IL-12p70, IL-1β, and spleen IFN-γ were measured by ELISA (n = 7–8; IL-12p70: **, P = 0.0023; IFN-γ: *, P = 0.0148). (D) Total RNA was isolated from the spleens of GBS-infected mice for real-time PCR analysis of IL-12p40 and arginase-1 expression. Data are represented as fold induction of mRNA expression compared with uninfected mice (n = 7–8). (E) MHC class II expression on peritoneal macrophages measured by FACS (percentage of positive cells indicated). (F) Immunohistochemical analysis of NOS2 expression in GBS-infected lungs shows increased expression of NOS2 in alveolar macrophages of IKKβΔMye mice (arrows), whereas expression in bronchial epithelial cells remains unchanged (asterisks). Representative panels are shown from n = 8. Bar, 200 μm.
Previous work from our group and others has demonstrated that IKKβ and NF-κB activation in bronchial epithelium is required for leukocyte recruitment and inflammation in the lung by regulating the expression of chemotactic cytokines (9, 13). The impaired ability of IKKβΔEpi mice to clear bacteria is therefore quite expected; however, the finding that IKKβ deletion in myeloid cells increases inflammation and bacterial clearance was somewhat surprising. In IKKβΔMye mice, IKKβ expression is effectively deleted in both macrophages and PMNs (14). PMNs are the major defense against extracellular bacteria such as GBS, although macrophage activation is also critical to PMN recruitment in inflammation (15). Previous work from our group has also shown that NF-κB can have a proapoptotic role in leukocytes during the resolution of inflammation (3), which was recently confirmed in IKKβΔMye mice (4); however, we found no difference in apoptosis in the lungs of GBS-infected IKKβΔMye mice (not depicted). We performed in vitro killing assays with macrophages and PMNs isolated from IKKβΔMye mice to test their direct antimicrobial activity. Although IKKβΔ macrophages showed some level of increased killing in vitro, IKKβΔ PMNs did not (Fig. 3, A and B). This suggested increased phagocytic killing by macrophages, probably through increased NOS2 expression, and macrophage-mediated PMN recruitment was responsible for enhanced clearance of bacteria rather than increased PMN activation. To test this hypothesis, we challenged IKKβΔMye mice intranasally with a noninfectious stimulus, bacterial LPS, to measure activation of alveolar macrophages and PMN recruitment. Lung tissue was collected for FACS analysis of PMN recruitment (CD11b+ Gr-1+ staining), and macrophage activation was measured by MHC class II expression (CD11b+ Gr-1− MHC class II+). Fig. 3 C shows increased MHC class II expression on alveolar macrophages from IKKβΔMye mice compared with control mice. FACS analysis of lung cells also revealed increased PMN recruitment in LPS-challenged IKKβΔMye mice (Fig. 3 D). Immunohistochemical analysis of LPS-stimulated lungs from IKKβΔMye mice also revealed increased NOS2 expression in alveolar macrophages (Fig. 3 F). In addition, cytokine analysis of bronchialveolar lavage fluid (BAL) from LPS-challenged mice showed increased IL-12 production but reduced levels of the antiinflammatory cytokine IL-10 in IKKβΔMye mice (Fig. 3 E; IKKβf/f vs. IKKβΔMye; IL-12: *, P = 0.0318), in keeping with an increase in M1 macrophage activation.
Figure 3.

IKKβ inhibits M1 macrophage activation in response to LPS in vivo. (A) Peritoneal macrophages (PM) and (B) neutrophils (PMN) were isolated from IKKβΔMye and IKKβf/f mice for in vitro killing assays with GBS (MOI of 5:1). (C) IKKβΔMye and IKKβf/f mice were challenged intranasally with 10 ng LPS in PBS. After 4 h, lungs were harvested and digested for FACS analysis of CD11b (myeloid), Gr-1 (PMN), and MHC class II expression. A representative histogram of MHC class II expression on CD11b+ Gr-1− alveolar macrophages is shown. (D) Tabulated data of mean fluorescence intensity for CD11b+ Gr-1− MHC class II+–activated macrophages in the lung and PMN recruitment as percentage of BAL cells from LPS-challenged mice (data are represented as mean ± SEM of n = 6). (E) In parallel experiments, IL-12 and IL-10 levels were measured in lung homogenates from LPS-challenged mice by ELISA. Data are represented as mean ± SEM (n = 8; *, P = 0.0381). (F) Immunohistochemical analysis of NOS2 expression in LPS-challenged mice. IKKβΔMye mice show increased expression of NOS2 in alveolar macrophages compared with IKKβf/f control mice (arrows). Representative panels are shown from n = 6 mice. Bar, 100 μm.
We further analyzed the phenotype of IKKβΔ macrophages after stimulation in vitro. Both resident and elicited peritoneal macrophages from IKKβΔMye mice express normal levels of typical macrophage lineage markers (including CD11b, F4/80, and CD68) and display no distinctive morphology (not depicted). Peritoneal macrophages were elicited and stimulated in vitro with heat-killed bacteria over a 24-h time course, and protein extracts were prepared for biochemical analysis. As expected, there was a profound impairment in IKK activity and NF-κB activation in IKKβΔ macrophages, whereas activation of JNK MAPK remained unchanged (Fig. 4 A). However, although the absence of IKK activity was associated with decreased expression of pro–IL-1β and TNF-α, expression of NOS2 was in fact elevated, which correlated with increased tyrosine phosphorylation of Stat1 (pY-Stat1) (Fig. 4, B–D). Expression of the endogenous inhibitor of Stat1, suppressor of cytokine signaling (SOCS)1 (16), was unaltered in IKKβΔ macrophages (Fig. 4 B). Stat1 is a critical transcription factor for NOS2, IL-12, and MHC class II expression (17). These data suggest that IKKβ activity in macrophages suppresses NOS2 as well as MHC class II and IL-12 expression during infection by antagonizing Stat1. M1 macrophage activation is typified by IFN-γ priming (7), and IFN signaling in macrophages absolutely requires Stat1 (17). In macrophages stimulated in vitro with Toll-like receptor ligands, Stat1 is activated by the autocrine production of IFN-β; however, GBS-induced IFN-β expression was inhibited in IKKβΔ macrophages (Fig. 4 D). To investigate a role of IKKβ in the cross regulation of Stat1 signaling, we stimulated IKKβΔ macrophages with bacterial LPS and recombinant IFN-γ in vitro. As shown in Fig. 5 A, IKKβΔ macrophages had increased activation of Stat1 and NOS2 expression when stimulated with LPS/IFN-γ, whereas activation of Stat3 appeared unchanged. IFN-γ and Stat1 strongly activate MHC class II expression in macrophages. To confirm the in vivo data described above, we measured MHC class II expression on macrophages stimulated in vitro with IFN-γ. MHC class II expression was dramatically enhanced with IKKβΔ macrophages (Fig. 5 B). In addition, IKKβΔ macrophages showed an increased ability to drive T cell proliferation in an allogenic mixed lymphocyte assay (Fig. S1 B). In further experiments, we used a recombinant adenovirus to express a dominant-negative inhibitor of IKKβ (IKKβdn) in macrophages in vitro (18). Fig. 5 C shows that expression of IKKβdn increased both Stat1 activation and NOS2 expression upon stimulation with LPS. Similar results were obtained with heat-killed bacteria (not depicted). Expression of IKKβdn also dramatically increased LPS/IFN-γ–induced IL-12 production (Fig. 5 D). To confirm the role of IKKβ in the suppression of Stat1 transcriptional activity, we performed reporter assays in RAW 264.7 macrophages using a GAS luciferase reporter gene. Transient overexpression of IKKβ in RAW cells inhibited GAS reporter activity, whereas transfection of the endogenous NF-κB inhibitor IκBα increased GAS reporter activation (Fig. 5 E). As a positive control, overexpression of SOCS1 inhibited GAS activity. Interestingly, previous studies have also described sustained IL-12 expression in macrophages deficient in p50 and heterozygous for RelA expression (19), suggesting that NF-κB activation downstream of IKKβ suppresses IL-12 expression. However, these studies did not include analysis of Stat1 activation. Our data would suggest that IL-12 expression is inhibited in macrophages by the NF-κB–dependent expression of a Stat1 inhibitor other than SOCS1.
Figure 4.

IKKβ inhibits Stat-1 activation in GBS-infected macrophages. (A) Peritoneal macrophages from IKKβΔMye and IKKβf/f mice were stimulated in vitro with heat-killed GBS (MOI of 20:1). Protein extracts were prepared at the indicated time points for biochemical analysis. IKK and JNK activity was measured by IP kinase assay (KA) using recombinant substrates GST-IκBα1-54 or GST-cJun1-79, respectively. NF-κB activation was measured by electrophoretic mobility shift assay (EMSA) using 32P-labeled κB consensus oligonucleotide. (B) Expression of IKK, pro–IL-1β, NOS2, tyrosine 701 phosphorylation of Stat1 (pY-Stat1), serine 726 phosphorylation of Stat1 (pS-Stat1), total Stat1, and SOCS1 was measured by immunoblot analysis of cell lysates using actin as a loading control. (C) In parallel experiments, TNF-α production in cell culture supernatants was measured by ELISA, and (D) total RNA was isolated for real-time PCR analysis of TNF-α (Tnfa) and IFN-β (Ifnb) mRNA expression. Data are represented as mean ± SEM of n = 4. Representative data are shown from at least three independent experiments.
Figure 5.

IKKβ inhibits IFN-γ signaling in macrophages. (A) Peritoneal macrophages from IKKβΔMye and IKKβf/f mice were stimulated in vitro with 100 ng/ml LPS in combination with 100 U/ml recombinant mouse IFN-γ, and protein extracts were prepared at the indicated time points. NOS2 expression, pY-Stat1, and pY-Stat3 were measured by immunoblot analysis. (B) Peritoneal macrophages from IKKβΔMye and IKKβf/f mice were stimulated in vitro, with IFN-γ and MHC class II expression analyzed by FACS. Representative data are shown from at least three independent experiments. (C) BMDMs were infected with recombinant adenovirus expressing a dominant-negative inhibitor of IKKβ (Ad-IKKβdn) or EGFP (Ad-GFP) 48 h before stimulation with LPS. NOS2 and pY-Stat1 expression was measured by immunoblot analysis at the indicated time points. (D) BMDMs were infected with Ad-IKKβdn or Ad-GFP before stimulation with LPS/IFN-γ, and IL-12p40 production was measured in cell culture supernatants by ELISA. Data are represented as mean ± SEM of n = 4. (E) RAW 264.7 macrophages were transfected with GAS luciferase reporter and either empty vector (pCDNA) or cDNA expressing IKKβ, IκBα, or SOCS1. After LPS/IFN-γ stimulation for 6 h, GAS reporter activity was measured by dual luciferase activity assay. Data are represented as relative light units (RLU) normalized for transfection efficiency with pRLTK.
We previously described a role for IKKα, the sister kinase of IKKβ, in limiting inflammation during infection and resistance to septic shock, by regulating the magnitude and duration of NF-κB activation (13). In the current study, we describe a specific role for IKKβ in limiting the expression of Stat-1–regulated genes. There is clearly still much to learn about the relative and specific roles for IKKα and IKKβ in inflammation and immunity. Our working hypotheses, based on previously published work and data presented here, is that IKKα acts to limit inflammation by inhibiting the persistent expression of NF-κB–dependent proinflammatory genes. However, IKKβ/NF-κB also acts to limit Stat-1 activity and the overexpression of Stat-1–dependent genes as a result of autocrine and paracrine IFN signaling. These antiinflammatory roles for the IKK complex have clear implications for the clinical use of IKK inhibitors.
Here, we have described a new role for IKKβ in the suppression of M1 macrophage activation during infection through the inhibition of Stat1 activity. To further test this hypothesis, we infected IKKβΔMye mice with the fungal pathogen Cryptococcus neoformans (Cn). Cn infects macrophages and promotes an M2 phenotype to evade a protective Th1 response (20). IKKβΔMye mice infected with Cn showed increased clearance of fungi after 10 d compared with control mice (Fig. S2 B, available at http://www.jem.org/cgi/content/full/jem.20080124/DC1; IKKβf/f vs. IKKβΔMye; *, P = 0.003). FACS analysis of BAL and lung tissue from Cn-infected mice showed MHC class II expression on alveolar macrophages was significantly increased (Fig. S2 C; IKKβf/f vs. IKKβΔMye; *, P = 0.0172). In addition, the proportion of T cells in the lungs of IKKβΔMye mice was elevated, although these data did not reach statistical significance (Fig. S2 C). These changes correlated with increased IFN-γ in BAL and reduced levels of Th2 cytokines IL-4 and IL-5 (Fig. S2 D), suggesting a switch to a protective Th1 response. These data, and that described above, suggest that IKKβ inhibits M1 macrophage activation during infection with both extracellular Streptococci and the intracellular fungal pathogen Cn. The precise pathophysiological significance of M1 and M2 macrophage populations is not yet well established (6, 7). However, it is clear that M1 macrophage activation is critical to control certain infections, but a switch to an M2 phenotype may limit the innate immune response and be required for the resolution of inflammation.
In summary, IKKβ plays a tissue-specific role in inflammation. In resident tissue cells, such as lung epithelium, IKKβ-mediated NF-κB activation drives cytokine and chemokine production required to initiate the inflammatory response (9, 13). However, NF-κB activation in resident macrophages or leukocytes recruited during inflammation has an antiinflammatory role (3, 21). This may represent a mechanism to prevent the over-exuberant activation of macrophages during inflammation. In a parallel study in this issue on page 1261, Hagemann et al. (22) show that inhibition of IKKβ in tumor-associated macrophages, which have an M2 phenotype (8, 23), also leads to increased Stat1 activation and a polarized M1 phenotype (increased; NOS2, MHC class II, and IL-12). However, in this context IKKβ inhibition confers enhanced tumoricidal activity, analogous to the increased antimicrobial activity observed in IKKβΔMye mice. It will be interesting to investigate if pathogens can highjack the IKKβ–NF-κB pathway to evade M1 macrophage activation and innate immunity in a similar fashion to tumor cells.
Considering the dominant role of Stat1 in regulation of NOS2, MHC class II, and IL-12 expression, it is likely that activation of this pathway is a major determinant of the M1 macrophage phenotype. Collectively, these data suggest that IKKβ activation plays an important role in suppression of this phenotype, contributing to both the resolution of inflammation during infection and the suppression of macrophage tumoricidal activity in cancer. The potential to manipulate the inflammatory phenotype of macrophages by targeting IKKβ may offer therapeutic opportunities for the treatment of inflammatory diseases, infection, and cancer.
MATERIALS AND METHODS
Mice.
The generation and characterization of IKKβf/f, IKKβΔMye, IKKβf/f-Otet-cre, and IKKβΔEpi mice has been described (9, 10). IKKβf/f littermate control mice on a C57Bl6/J genetic background were used for experiments with IKKβf/f-LysM-cre (IKKβΔMye) mice, and IKKβf/f-Otet-cre littermate controls were used in experiments with IKKβf/f-Otet-cre-CC10-rtta (IKKβΔEpi) mice. Animal husbandry and procedures were performed in accordance with Queen Mary University of London UK Home Office guidelines.
Infections.
The clinical GBS isolate, NCTC10/84 (serotype V), was grown in Todd-Hewitt Broth (Difco) without agitation at 37°C to an OD600 of 0.4, equivalent to 108 CFUs/ml. Bacteria collected by centrifugation were washed with sterile PBS. Mice were inoculated intranasally with 3 × 107 CFUs NCTC in 30 μl PBS (11). Cn strain 52 was obtained from the American Type Culture Collection and grown to stationary phase (48–72 h) at room temperature in Sabouraud dextrose broth (Difco). The cultures were washed in saline, counted on a hemocytometer, and diluted in sterile PBS. Stock concentrations and viability were confirmed by plating on Sabouraud agar plates and incubation for 48 h at room temperature before colony counts. Mice were inoculated intranasally with 105 CFUs Cn in 30 μl PBS. Mice were killed when indicated, and their tracheas were cannulated for BAL with 3 aliquots of 0.8 ml ice-cold PBS. Serial dilutions of BAL fluid were plated in triplicate, and CFUs were determined. Lung tissue was removed and digested at 37°C in collagenase solution, and single cell suspensions were prepared for FACS analysis. In parallel experiments, lung tissue was collected for histological and biochemical analysis. For survival experiments, mice were inoculated by intraperitoneal injection with 5 × 107 CFUs NCTC in 0.3 ml PBS, as described above. Lung, liver, and spleen were collected postmortem for histological and biochemical analysis.
LPS lung inflammation.
Mice were challenged intranasally with 10 μg LPS (Escherichia coli serotype B5:055; Sigma-Aldrich) in PBS. BAL fluid was collected as described above. Lung tissue was removed and digested at 37°C in collagenase solution, and single cell suspensions were prepared for FACS analysis. In parallel experiments, lung tissue was collected for histological and biochemical analysis.
Macrophage and PMN isolation and stimulation.
Bone marrow–derived macrophages (BMDMs) were generated as described previously (11). Peritoneal macrophages were elicited by intraperitoneal injection of 1.5 ml of 3% thioglycollate (Difco) or 1% Biogel (Bio-Rad Laboratories). After 3 d, cells were harvested and plated in RPMI 1640 supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin/streptomycin, and 2 mM glutamine. Peritoneal PMNs were isolated 4 h after thioglycollate injection and maintained in RPMI 1640 supplemented with 10% heat-inactivated autologous serum, 100 U/ml penicillin/streptomycin, and 2 mM glutamine. For infection experiments, bacteria were prepared as described above, macrophages were washed with antibiotic-free media, and bacteria were added at a multiplicity of infection (MOI) of 5:1. Killing assays were performed as described previously (11, 24). In some experiments, macrophages were treated with heat-killed bacteria, and bacteria were prepared as described above and heat killed for 30 min at 80°C. For other experiments, LPS was used at a concentration of 100 ng/ml and recombinant mouse IFN-γ was used at 100 U/ml (PeproTech).
Adenovirus preparation.
Recombinant, replication-deficient adenoviral vectors were developed to express cDNA in primary macrophages. Virus-encoding GFP was used to assess infection efficiency. Virus expressing a kinase-defective dominant-negative form of IKKβ (IKKβdn) was used to inhibit IKKβ activity and NF-κB activation. These viruses are E1/E3 deleted, belong to the Ad5 serotype, and have been used in other studies (18). Approximately 80–90% of BMDMs were found to express GFP when infected with virus at an MOI of 200:1. IKKβf/f BMDMs were infected with recombinant adenovirus expressing Cre recombinase, and IKKβf/f BMDMs infected with Adv-GFP were used as a control. Infections were performed at day 4 of macrophage differentiation in the presence of 10 ng/ml recombinant murine M-CSF (PeproTech).
Gene expression analysis and ELISA.
Total cellular RNA was isolated using TRIzol (Invitrogen) and analyzed by RT- PCR with a SyBr Green (5700 thermocycler; PE Biosystems) or RNase protection assay (RPA). Primer sequences are available upon request. For RT-PCR analyses, all values were normalized to the level of cyclophilin mRNA. For RPA analyses, total RNA was hybridized with RNA probes using the Riboquant Multiprobe RPA System (BD Biosciences) according to the manufacturer's instructions. Cytokine levels in cell culture supernatants were determined by sandwich ELISA (R&D Systems and BD Biosciences) performed according to the manufacturer's instructions.
Kinase assay, electrophoretic mobility shift assay, and immunoblotting.
Whole cell lysates were prepared, and IKK or JNK kinase activity was measured after immunoprecipitation with anti-IKKγ or anti-JNK1 antibody (BD Biosciences) using recombinant GST-IκBα (1–54) or GST-cJun (1–29), respectively, as described previously (11). IKK and JNK recovery was determined by immunoblotting with anti-IKKα (BD Biosciences) or anti-JNK antibody (Santa Cruz Biotechnology, Inc.). Immunoblotting was performed on SDS-PAGE gel–separated whole cell lysates or nuclear and cytoplasmic extracts (11). Electrophoretic mobility shift assays were performed with 2 μg of protein extract as described previously (3, 11).
Flow cytometry.
Murine Fc receptors were blocked using anti–mouse CD16/CD32 Fc Block (BD Biosciences). For staining, cells were washed and resuspended in PBS supplemented with 1% heat-inactivated FBS and 0.01% NaN3. Antibodies were diluted in this buffer and used at a final concentration of between 2 and 20 μg/ml. The following antibodies were used: MHC class II (Ia)-PE, CD11b-APC, CD86-PE, CD80-PE, CD124 (IL-4Rα)-PE, and CD4-FITC (BD Biosciences). Incubations with antibodies were performed for 30 min on ice. Labeled cells were analyzed by flow cytometry on a FACScan flow cytometer and analyzed using CellQuest software (Becton Dickinson).
Mixed lymphocyte reaction.
103 elicited macrophages from C57Bl6/J mice were plated in 96-well plates and stimulated overnight with 100 ng/ml LPS. CD4+ T cells were negatively selected from the lymph nodes of BALB/c mice and labeled with 5 μM CSFE. 5 × 105 T cells were added to macrophages and co-cultured for 4 d. Cells were harvested, 7-AAD was added to exclude nonviable cells, and CFSE dilution in the viable cell population was measured by FACS.
Transfections and reporter assays.
3T3 or RAW264.7 cells were transiently transfected using FuGene HD (Roche). Cells were plated in 12-well plates and grown until 80–90% confluency. Cells were cotransfected with 500 ng of GAS reporter gene plasmid, 500 ng of an expression vector, and 10 (3T3) or 50 ng (RAW264.7) pRLTK (Promega). 22 h after transfection, cells were treated with LPS and IFN-γ for 6 h. Cells were harvested in 75 μl Passive Lysis Buffer (Promega). Luciferase activity was determined with the Dual-Luciferase Reporter Assay System according to the manufacturer's instructions (Promega).
Statistical analysis.
At least three independent experiments were performed, and representative data are shown. Data are represented as mean ± SEM. Student's t test or Mann-Whitney test was performed where appropriate to test the statistical significance between datasets and p-values indicated.
Online supplemental material.
Fig. S1 shows the increased immunostimulatory activity of IKKβΔ macrophages. Fig. S2 shows that macrophage IKKβ activity inhibits protective immunity to Cn infection. Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20080124/DC1.
Supplementary Material
Acknowledgments
This work was supported by funding from the Wellcome Trust and Medical Research Council to T. Lawrence.
The authors have no conflicting financial interests.
C.H.Y. Fong and M. Bebien contributed equally to this work.
References
- 1.Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 18:621–663. [DOI] [PubMed] [Google Scholar]
- 2.Karin, M., Y. Yamamoto, and Q.M. Wang. 2004. The IKK NF-kappa B system: a treasure trove for drug development. Nat. Rev. Drug Discov. 3:17–26. [DOI] [PubMed] [Google Scholar]
- 3.Lawrence, T., D.W. Gilroy, P.R. Colville-Nash, and D.A. Willoughby. 2001. Possible new role for NF-kappaB in the resolution of inflammation. Nat. Med. 7:1291–1297. [DOI] [PubMed] [Google Scholar]
- 4.Greten, F.R., M.C. Arkan, J. Bollrath, L.C. Hsu, J. Goode, C. Miething, S.I. Goktuna, M. Neuenhahn, J. Fierer, S. Paxian, et al. 2007. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 130:918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor, P.R., and S. Gordon. 2003. Monocyte heterogeneity and innate immunity. Immunity. 19:2–4. [DOI] [PubMed] [Google Scholar]
- 6.Gordon, S., and P.R. Taylor. 2005. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5:953–964. [DOI] [PubMed] [Google Scholar]
- 7.Gordon, S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3:23–35. [DOI] [PubMed] [Google Scholar]
- 8.Mantovani, A., S. Sozzani, M. Locati, P. Allavena, and A. Sica. 2002. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23:549–555. [DOI] [PubMed] [Google Scholar]
- 9.Broide, D.H., T. Lawrence, T. Doherty, J.Y. Cho, M. Miller, K. McElwain, S. McElwain, and M. Karin. 2005. Allergen-induced peribronchial fibrosis and mucus production mediated by IkappaB kinase beta-dependent genes in airway epithelium. Proc. Natl. Acad. Sci. USA. 102:17723–17728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greten, F.R., L. Eckmann, T.F. Greten, J.M. Park, Z.W. Li, L.J. Egan, M.F. Kagnoff, and M. Karin. 2004. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 118:285–296. [DOI] [PubMed] [Google Scholar]
- 11.Lawrence, T., M. Bebien, G.Y. Liu, V. Nizet, and M. Karin. 2005. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 434:1138–1143. [DOI] [PubMed] [Google Scholar]
- 12.Watford, W.T., M. Moriguchi, A. Morinobu, and J.J. O'Shea. 2003. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 14:361–368. [DOI] [PubMed] [Google Scholar]
- 13.Poynter, M.E., C.G. Irvin, and Y.M. Janssen-Heininger. 2003. A prominent role for airway epithelial NF-kappa B activation in lipopolysaccharide-induced airway inflammation. J. Immunol. 170:6257–6265. [DOI] [PubMed] [Google Scholar]
- 14.Clausen, B.E., C. Burkhardt, W. Reith, R. Renkawitz, and I. Forster. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8:265–277. [DOI] [PubMed] [Google Scholar]
- 15.Cailhier, J.F., M. Partolina, S. Vuthoori, S. Wu, K. Ko, S. Watson, J. Savill, J. Hughes, and R.A. Lang. 2005. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J. Immunol. 174:2336–2342. [DOI] [PubMed] [Google Scholar]
- 16.Alexander, W.S., R. Starr, J.E. Fenner, C.L. Scott, E. Handman, N.S. Sprigg, J.E. Corbin, A.L. Cornish, R. Darwiche, C.M. Owczarek, et al. 1999. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 98:597–608. [DOI] [PubMed] [Google Scholar]
- 17.Durbin, J.E., R. Hackenmiller, M.C. Simon, and D.E. Levy. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 84:443–450. [DOI] [PubMed] [Google Scholar]
- 18.Andreakos, E., C. Smith, C. Monaco, F.M. Brennan, B.M. Foxwell, and M. Feldmann. 2003. Ikappa B kinase 2 but not NF-kappa B-inducing kinase is essential for effective DC antigen presentation in the allogeneic mixed lymphocyte reaction. Blood. 101:983–991. [DOI] [PubMed] [Google Scholar]
- 19.Tomczak, M.F., S.E. Erdman, T. Poutahidis, A.B. Rogers, H. Holcombe, B. Plank, J.G. Fox, and B.H. Horwitz. 2003. NF-kappa B is required within the innate immune system to inhibit microflora-induced colitis and expression of IL-12 p40. J. Immunol. 171:1484–1492. [DOI] [PubMed] [Google Scholar]
- 20.Muller, U., W. Stenzel, G. Kohler, C. Werner, T. Polte, G. Hansen, N. Schutze, R.K. Straubinger, M. Blessing, A.N. McKenzie, et al. 2007. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J. Immunol. 179:5367–5377. [DOI] [PubMed] [Google Scholar]
- 21.Gadjeva, M., M.F. Tomczak, M. Zhang, Y.Y. Wang, K. Dull, A.B. Rogers, S.E. Erdman, J.G. Fox, M. Carroll, and B.H. Horwitz. 2004. A role for NF-kappa B subunits p50 and p65 in the inhibition of lipopolysaccharide-induced shock. J. Immunol. 173:5786–5793. [DOI] [PubMed] [Google Scholar]
- 22.Hagemann, T., T. Lawrence, I. McNeish, K.A. Charles, H. Kulbe, R.G. Thompson, S.C. Robinson, and F.R. Balkwill. 2008. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 205:1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagemann, T., J. Wilson, F. Burke, H. Kulbe, N.F. Li, A. Pluddemann, K. Charles, S. Gordon, and F.R. Balkwill. 2006. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J. Immunol. 176:5023–5032. [DOI] [PubMed] [Google Scholar]
- 24.Liu, G.Y., K.S. Doran, T. Lawrence, N. Turkson, M. Puliti, L. Tissi, and V. Nizet. 2004. Sword and shield: linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc. Natl. Acad. Sci. USA. 101:14491–14496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}