

Abstract
The mitogen-activated protein kinase (MAPK) pathway plays a critical role in Toll-like receptor (TLR) signaling. MAPK phosphatase-1 (MKP-1) inhibits the MAPK pathway and decreases TLR signaling, but the regulation of MKP-1 is not completely understood. We now show that MKP-1 is acetylated, and that acetylation regulates its ability to interact with its substrates and deactivate inflammatory signaling. We found that LPS activates acetylation of MKP-1. MKP-1 is acetylated by p300 on lysine residue K57 within its substrate-binding domain. Acetylation of MKP-1 enhances its interaction with p38, thereby increasing its phosphatase activity and interrupting MAPK signaling. Inhibition of deacetylases increases MKP-1 acetylation and blocks MAPK signaling in wild-type (WT) cells; however, deacetylase inhibitors have no effect in cells lacking MKP-1. Furthermore, histone deacetylase inhibitors reduce inflammation and mortality in WT mice treated with LPS, but fail to protect MKP-1 knockout mice. Our data suggest that acetylation of MKP-1 inhibits innate immune signaling. This pathway may be an important therapeutic target in the treatment of inflammatory diseases.
Innate immune responses play a critical role in defending the host from pathogens. Pathogen-associated molecular patterns stimulate pattern recognition receptors such as the Toll-like receptors (TLRs), which activate a set of signaling pathways, inducing expression of innate immune effectors (1–3). LPS is a pathogen-associated molecular pattern that interacts with TLR4, which in turn interacts with intracellular adaptor proteins such as MyD88 (4). The TLR4 signaling complex then activates two intracellular pathways, the NF-κB signaling pathway and the mitogen-activated protein kinase (MAPK) cascade, both of which direct an inflammatory response.
The MAPK pathway plays a critical role in innate immune signaling (5, 6). The three major families of MAPKs include extracellular signal-regulated kinases (ERKs), the p38 MAPKs, and the c-Jun NH2-terminal kinases (JNK) (7–9). These MAPKs are activated by MAPK kinases (MAPKKs) (10, 11). MAPKKs are in turn activated by a set of MAPKK kinases. The MAPK pathway that mediates innate immune signaling includes MKK3/4/6, p38, and JNK (12–14).
Negative regulators of innate immunity prevent excessive inflammation and autoimmunity (15, 16). Distinct inhibitors of TLR signaling have been identified, many of which act upon the Myd88 pathway (3, 17–24). Furthermore, endogenous inhibitors of the MAPK system may also negatively regulate TLR signaling (25–28).
MAPK phosphatases (MKPs) are dual-specificity phosphatases that inactivate MAPK members by dephosphorylating phosphotyrosine and phosphothreonine residues (29–34). The MKP family includes four types; the type II, III, and IV MKPs all include a MAPK-docking domain and a dual-specific phosphatase domain (34). The docking domain mediates interactions between MKP and its substrate MAPK. MKP binding to its MAPK target via the docking domain increases MKP catalytic activity by more than fivefold (35–38). MKP-1 can be phosphorylated to regulate its stability, but other modifications have not been reported (39). Recent studies have emphasized the importance of MKP-1 in regulating innate immune responses. Mice lacking MKP-1 are more susceptible to LPS than WT mice (28, 40–42). Furthermore, in response to TLR signals, macrophages lacking MKP-1 produce higher levels of proinflammatory cytokines.
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) can regulate gene expression by modifying histone proteins (43–45). However, HAT and HDAC can regulate specific signaling pathways and have other targets in addition to histones, including NF-κB, Stat3, and p53 (46–48). Recent reports suggest that inhibitors of HDAC can decrease inflammation (49–56). Interestingly, HDAC inhibitors repress expression of some inflammatory genes, but increase expression of others (57). This reinforces the idea that HDAC inhibitors do not regulate expression of inflammatory proteins only by a general effect on transcription, but may also have specific targets. In this study, we searched for acetylated targets in innate immune signaling, and we discovered that acetylation of MKP-1 is a negative regulator of innate immunity.
RESULTS
HDAC inhibitors decrease LPS activation of NOS2 expression
To explore the effect of global protein acetylation upon NOS2 expression, we pretreated RAW 264.7 murine macrophages with the HDAC inhibitor trichostatin A (TSA) or control, added LPS, and measured the concentration of the nitric oxide (NO) metabolite nitrite (NO2−) in the media. TSA decreases LPS-activated NO production in a dose-dependent manner (Fig. 1 A). Another HDAC inhibitor, sodium butyrate, also inhibits NO production (Fig. 1 B). To explore the mechanism by which TSA inhibits NO production, we measured the steady-state RNA and protein levels of NOS2 in LPS-stimulated macrophages. TSA decreases NOS2 mRNA levels in a dose- and time-dependent manner (Fig. 1, C and D). TSA also decreases NOS2 steady-state protein levels (Fig. 1 E). These results suggest that HDACs regulate NOS2 expression.
Figure 1.

Deacetylase inhibitors decrease LPS activation of NO synthesis and NOS2 expression. (A) TSA inhibits LPS-induced NO production in a dose-dependent manner. RAW cells were pretreated with increasing amounts of TSA for 1 h, and then treated with or without LPS 100 ng/ml for 16 h, and the amount of NO2− was measured in the supernatant by the Griess reaction. (n = 3 ± the SD). (B) Sodium butyrate inhibits LPS-induced NO production in RAW cells. RAW cells were pretreated with increasing amounts of sodium butyrate for 1 h and treated with or without LPS 100 ng/ml for 16 h, and the amount of NO2− was measured in the supernatant by the Griess reaction (n = 3 ± the SD). (C) TSA inhibits the LPS-induced increase in NOS2 RNA levels (dose–response). RAW cells were pretreated with increasing amounts of TSA for 1 h and treated with LPS 100 ng/ml for 6 h. Total RNA was analyzed by Northern blotting with a cDNA probe for NOS2 (top) or a ribosomal phosphoprotein RNA 36B4 as a control (bottom). (D) TSA inhibits LPS-induced increase in NOS2 RNA levels (time course). RAW cells were pretreated with TSA 30 ng/ml for 1 h and treated with LPS 100 ng/ml for 0–7 h. Total RNA was analyzed by Northern blotting with a cDNA probe for NOS2. (E) TSA inhibits LPS-induced increase in NOS2 protein levels (dose–response). RAW cells were pretreated with increasing amounts of TSA for 1 h, LPS was added for 6 h, and cell lysates were immunoblotted with antibody to NOS2 (top) or ERα (bottom). (F) TSA decreases LPS-induced inflammatory cytokine RNA levels. RAW cells were treated with LPS and TSA for 4 h. Total RNA was analyzed by Northern blotting with probes for TNF-α, IL-1β, IL-6, or GAPDH as a control. (G) TSA decreases LPS-, PGN-, and poly(I:C)-induced NOS2 protein levels. RAW cells were treated with the TLR agonists PGN, poly(I:C), LPS, or E. coli for 16 h. Cell lysates were immunoblotted with antibody to NOS2. (H) TSA slightly increases expression of COX-2 and CXCL2. RAW cells were treated with LPS, TSA, or both as in F, and analyzed for COX-2 and CXCL2 expression by RT-PCR.
We also explored the effect of TSA upon expression of inflammatory cytokines. TSA inhibits the expression of TNF-α, IL-6, and IL-1β in LPS-stimulated macrophages (Fig. 1 F). We examined the effect of TSA upon activation of other TLR signaling pathways. LPS or Escherichia coli, which are activators of TLR4 signaling, peptidoglycan, which is an activator of TLR2 signaling, or double-stranded RNA, which is an activator of TLR3 signaling, can activate NOS2 expression in macrophages (Fig. 1 G). TSA inhibits expression of NOS2 activated by each of these four TLR activators (Fig. 1 G). However, we found that TSA slightly increases the expression of other proinflammatory molecules, such as COX-2 and CXCL2 (Fig. 1 H and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071728/DC1). These results suggest that HDACs regulate expression of a subset of proinflammatory cytokines activated by multiple TLR activators (56, 57).
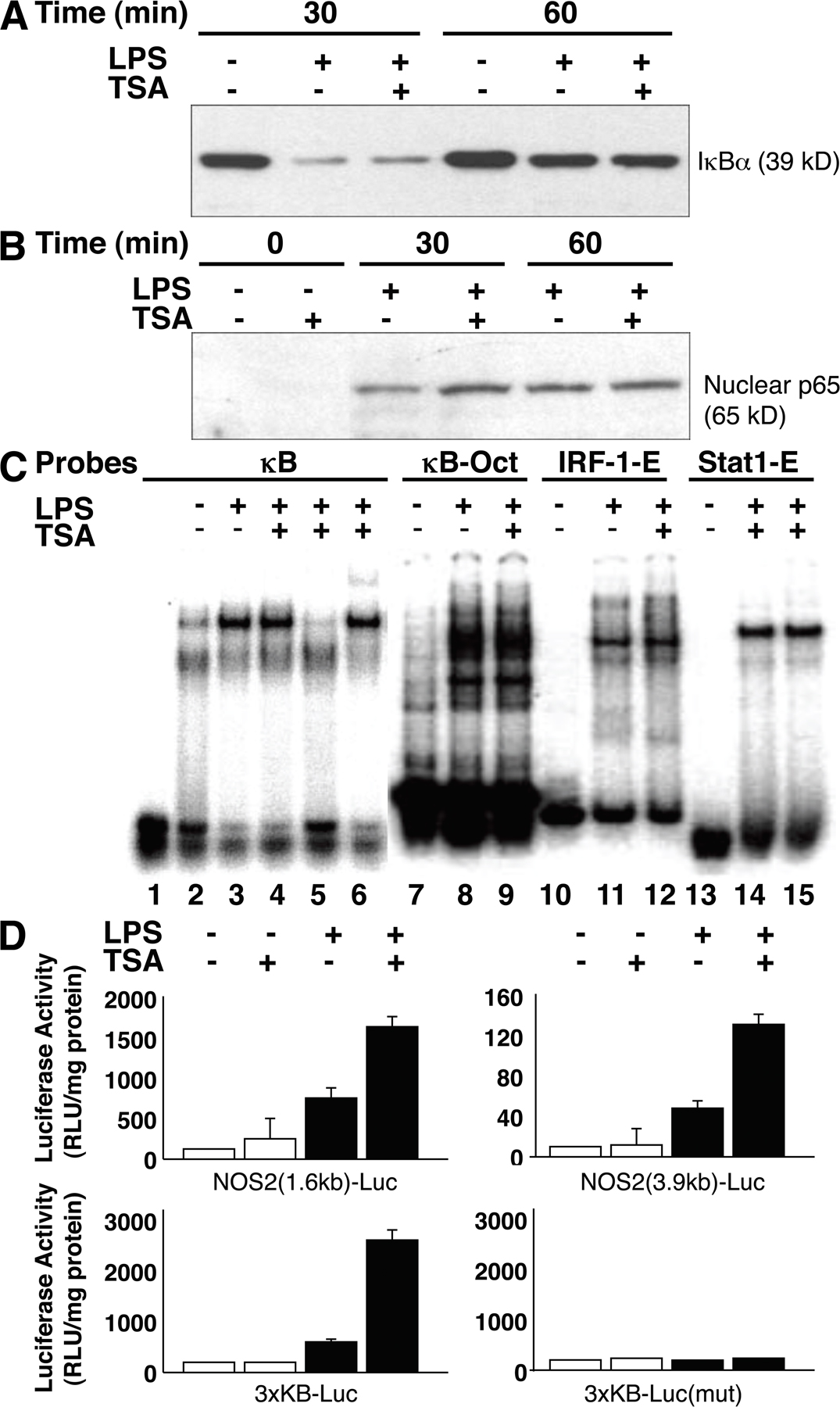
HDAC inhibitors do not regulate NOS2 expression through NF-κB
To determine the mechanism by which HDAC inhibitors decrease NOS2 expression, we examined the effect of TSA upon NF-κB, which is a transcriptional regulator of NOS2. However, TSA does not affect LPS activation of the NF-κB pathway. In fact, TSA slightly increases LPS-triggered IκBα degradation, p65 nuclear translocation, κB-binding activity, and NF-κB transactivation of the NOS2 promoter (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20071728/DC1). Although TSA slightly enhances NF-kB signaling, it also decreases NOS2 expression (Fig. 1). We therefore explored the effect of TSA upon other pathways that regulate NOS2 expression.
HDAC inhibitors inhibit MAPK pathway
Because the MAPK pathway regulates NOS2 expression (58), we explored the effect of HDAC inhibition upon the MAPK pathway. We treated RAW macrophages with SB203580 to inhibit p38, with U0126 to inhibit ERK, or with SP600125 to inhibit Jun-N-terminal kinase (JNK); we then added LPS and measured expression of NOS2 protein. MAPK inhibitors decrease LPS activation of NOS2 expression (Fig. 2 A). These MAPK inhibitors are not cytotoxic (unpublished data). To identify which MAPKs are affected by deacetylase inhibition, we added media, LPS alone, TSA alone, or LPS and TSA together to RAW cells, and then measured levels of the three major groups of phosphorylated MAPK proteins as follows: p38; ERK1 and ERK2; and JNK1 and JNK2. LPS increases phosphorylation of p38, ERK1/2, and JNK1/2 (Fig. 2 B). TSA decreases LPS-induced phosphorylation of p38 and ERK1/2, but not JNK1/2 (Fig. 2 B).
Figure 2.

Deacetylase inhibition blocks MAPK signaling. (A) The MAPK pathway regulates NOS2 expression. RAW cells were treated for 1 h with 10 μM of the p38 MAPK inhibitor SB203580, 10 μM of the MEK1/2 inhibitor UO126, or 5 μM of the JNK inhibitor SP600125, and then 100 ng/ml LPS was added for 6 h and cell lysates were immunoblotted with antibody to NOS2 (top) or MEK1/2 (bottom left) of HuR (bottom right). (B) TSA inhibits phosphorylation of the MAPKs p38 and ERK1, but not JNK. RAW cells were pretreated with 30 ng/ml TSA for 1 h, and then treated with 100 ng/ml LPS for 30 min, and cell lysates were immunoblotted with antibodies to MAPK pathway members. (C) TSA inhibits the MKK3/6–p38–Elk-1 signaling cascade between MKK3/6 and p38. RAW cells were treated with TSA and LPS as in B, and cell lysates immunoblotted with antibodies to MAPK members. (D) TSA does not inhibit MKK3 kinase activity. RAW cells were treated with LPS and TSA as in B and C, and lysates were immunoprecipitated with antibody to MKK3. The kinase activity of MKK3 was assayed by adding recombinant GST-p38 and ATP to immunoprecipitated MKK3 for 30 min at 30°C, and then immunoblotting for phospho-p38 (top) and total p38 (bottom).
The p38 pathway of MAPK signaling includes MKK3/6, which phosphorylates p38, which in turn phosphorylates ELK-1 and other targets (59). LPS increases phosphorylation of all components of this pathway, from MKK3/6 to p38 to Elk-1 (Fig. 2 C). Although TSA decreases phosphorylation of p38 and Elk-1, TSA does not affect phosphorylation of the upstream MAPKK MKK3/6 (Fig. 2 C). Furthermore, TSA does not affect MKK3 kinase activity (Fig. 2 D).
These data suggest that HDAC inhibition acts upon the MAPK signaling pathway at the level of p38, possibly upon a kinase or phosphatase that modifies p38. Furthermore, our data imply that proteins that phosphorylate p38, such as MKK3/6 or transforming growth factor β–activated protein kinase 1, are not affected by acetylation (60).
Acetylation of MKP-1
We next determined which molecules in the MAPK signal pathway are acetylated, focusing on proteins that modify p38. We reasoned that acetylated MAPK components would interact with acetylases. Accordingly, we immunoprecipitated the histone acetylase p300 from RAW cell lysates, and immunoblotted precipitants for a variety of MAPK proteins, including MKP-1. We found that MKP-1 does not associate with p300 in resting macrophages (Fig. 3 A). However, we reasoned that HDAC and HAT might compete for the same acetylation site of MKP-1, and blockade of HDAC might permit an unopposed increase in the interaction between p300 and MKP-1. For example, TSA not only increases acetylation of Sp1 but also recruits p300 into a complex that includes Sp1 and HDAC (61). Accordingly, we tested the idea that TSA might increase the interaction of p300 and MKP-1. We found that MKP-1 associates with p300 in TSA-treated macrophages (Fig. 3 A).
Figure 3.

MKP-1 is acetylated. (A) MKP-1 interacts with the histone acetyl transferase p300 in RAW cells. RAW cells were treated with TSA and LPS. Cell lysates were immunoprecipitated with antibody to p300, fractionated by SDS-PAGE, and immunoblotted with antibodies to MKP-1. Immunoblots of total MKP-1 and p300 are shown in the middle and bottom gels. (B) TSA increases MKP-1 acetylation. RAW cells were pretreated with [3H]sodium acetate for 1 h and TSA for 0.5 h and with 100 ng/ml LPS for 0–3 h, and then cell lysates were immunoprecipitated with antibody to MKP-1 and autoradiographed (top) or immunoblotted with antibody to MKP-1 (bottom). (C) p300 and PCAF acetylate MKP-1 on residue K57 in vitro. A peptide encoding the MAPK docking domain of WT (WT) MKP-1 (amino acids 47 to 76) was incubated with recombinant p300 HAT domain or recombinant PCAF in the presence of [14C]-acetyl CoA. A mutant MKP-1 peptide with lysine replaced by arginine (K57R) was used as a control. The reactions were fractionated by 16.5% Tris-Tricine gel and autoradiographed. (D) MKP-1 is acetylated on residue K57 in cells. HEK293 cells were transfected with vectors expressing a fragment of WT HA-MKP-1(WT) or mutant HA-MKP-1(K57R), and then treated with TSA and LPS as above. Cell lysates were immunoprecipitated with antibody to acetyl-lysine, fractionated by SDS-PAGE, and immunoblotted with antibody to HA (top). Total lysates were also immunoblotted for HA-MKP-1 (bottom).
We then measured the acetylation of MKP-1 by immunoprecipitating MKP-1 from RAW macrophages labeled with [3H]sodium acetate. MKP-1 is not acetylated in resting cells (Fig. 3 B). However, LPS triggers acetylation of MKP-1. Furthermore, HDAC inhibition enhances acetylation of MKP-1, peaking 2 h after LPS and then decreasing (Fig. 3 B).
Which lysine residues of MKP-1 are acetylated? MKP-1 contains a lysine K57 that is conserved among most MKP isoforms (including MKP isoforms 1, 2, 3, and X; Table I). This conserved lysine residue is located in the middle of the MKP-1 docking domain, which interacts with p38. We therefore explored whether or not this particular lysine residue of MKP-1 is acetylated, using two complementary techniques: radiolabeled cells and purified peptides (47, 48). We synthesized a peptide consisting of MKP-1 residues 47–76, designated MKP-1(47–76)(WT). We also synthesized a mutant peptide consisting of MKP-1(47–76) with an arginine substituted for lysine at residue 57, designated MKP-1(47–76)(K57R). We incubated the WT and mutant MKP-1 peptides with recombinant acetyltransferases p300 or PCAF (p300/CBP-associated factor) and [14C]acetyl-CoA. p300 acetylates MKP-1(47–76) (Fig. 3 C). However, p300 cannot acetylate MKP-1(47–76)(K57R) (Fig. 3). Similar results were obtained for PCAF and the WT and mutant MKP-1 peptides. These data suggest that these acetyltransferases acetylate MKP-1 on residue K57.
Table I.
Docking domains of murine MKP isoforms
| MKP isoform | Docking domain |
|---|---|
| MKP-1/DUSP1(m) | RFSTIVRRRAKGAMGLE |
| MKP-1/DUSP1(h) | RFSTIVRRRAKGAKGAG |
| MKP-2/DUSP4/HVH2 | RCNTIVRRRAKGSVSLE |
| MKP-X/DUSP7 | IPGLMLRRLRKGNLPIR |
| MKP-3/DUSP6 | IPGIMLRRLQKGNLPVR |
| MKP-5/DUSP10 | ADKISRRRLQQGKITVL |
| MKP-7/DUSP16 | CSKLMKRRLQQDKVLIT |
| DUSP8/VH5 | CSKLVKRRLQQGKVTIA |
| MKP-4/DUSP9 | LPSLMLRRLRRGSMSVR |
| DUSP2/PAC1 | WNALLRRRARGTPAAAL |
| DUSP2/PAC1 | WNALLRRRARGTPAAAL |
The murine isoform of MKP-1 is designated by (m), the human isoform by (h). All other isoforms are murine. A di-arginine motif is shown in bold, and the adjacent lysine is italicized.
We next searched for acetylation of MKP-1 in cells. We transfected HEK293 cells with a vector expressing an HA tag fused to a fragment of WT MKP-1, designated HA-MKP-1(WT), or with an expression vector expressing an HA-tagged mutant MKP-1 with a K57R substitution, designated HA-MKP-1(K57R). We then treated cells with LPS and TSA. We precipitated cell lysates with antibody to acetyl-lysine and the precipitants were fractionated and immunoblotted with antibody to HA. MKP-1 is acetylated in response to LPS and TSA, but MKP-1(K57R) is not (Fig. 3 D).
These data suggest that MKP-1 is acetylated on residue K57 in vitro and in cells.
Acetylation of MKP-1 increases its interaction with p38
We next examined the effect of MKP-1 acetylation upon its interaction with p38. We treated RAW cells with LPS and TSA, and then immunoprecipitated cell lysates with antibody to p38 and immunoblotted precipitants with antibody to MKP-1. HDAC inhibition increases the association between p38 and MKP-1 in cells (Fig. 4 A). Immunoprecipitation using antibody to MKP-1 and immunoblotting for p38 confirm that p38 and MKP-1 form a complex within cells. It appears as if TSA alone increases the interaction between MKP-1 and p38, even in the absence of LPS (Fig. 4 A). Furthermore, TSA increases the expression of MKP-1 levels (Fig. 4 A).
Figure 4.

MKP-1 acetylation increases the interaction between MKP-1 and p38. (A) TSA increases MKP-1 interaction with p38 in cells. RAW cells were treated with LPS and TSA for 1 h, cell lysates were immunoprecipitated with antibodies to p38 or MKP-1, and immunoprecipitates were immunoblotted with antibodies as shown. (B) Acetylation increases MKP-1 peptide interaction with p38 in vitro. Biotinylated peptides from the MKP-1 docking domain (containing aa residues 46–74) were synthesized with lysine (K57) or with acetyl-lysine (K57Ac). MKP-1 peptides were incubated with recombinant p38, precipitated with streptavidin–agarose beads, and precipitants were immunoblotted with antibody to p38. (top) p38 precipitant with MKP-1 peptide. (bottom) Total p38 input. (C) Acetylation increases MKP-1 peptide interaction with p38 in vitro. Biotin-MKP-1(47–76) (blue) and biotin-MKP-1(47-K57Ac-76) (red) were immobilized onto a streptavidin sensor chip. Recombinant p38 was injected into the flow cell, and changes in SPR were measured over 15 min. This experiment was repeated twice with similar results. (D) Acetylation increases WT MKP-1(WT), but not mutant MKP-1(K57R), interaction with p38 in cells. HeLa cells were transfected with plasmids expressing MKP-1(WT)-ERα or MKP-1(_K57R)-ERα. The cells were treated with 4-HT to induce MKP-1-ERα expression, and then treated with LPS and TSA for 1 h. Cell lysates were immunoprecipitated and immunoblotted with antibodies to ERα and to p38 as indicated. (E) Acetylation increases WT MKP-1(WT) interaction with ERK, but not mutant MKP-1(K57R) interaction with ERK. HeLa cells were transfected with plasmids expressing MKP-1(WT)-ERα or MKP-1(K57R)-ERα and His6-ERK1. The cells were treated with 4-HT to induce MKP-1-ERα expression, and then treated with LPS and TSA for 1 h. Cell lysates were immunoprecipitated, and then immunoblotted with antibody to the ERα tag of MKP-1 and antibody to the (His)6 tag of (His)6-ERK1.
To explore the effects of acetylation upon the direct interaction of MKP-1 and p38, we used recombinant polypeptides. We synthesized a biotinylated MKP-1 peptide, designated biotin-MKP-1(K57), and a similar peptide with an acetylated K57 residue, designated biotin-MKP-1(K57Ac). We incubated the WT and acetylated MKP-1 peptide with recombinant p38, precipitated the mixture with streptavidin–agarose beads, and immunoblotted precipitants with antibody to p38. The MKP-1 peptide with an acetylated lysine in the docking site domain has a higher affinity for p38 than a nonacetylated MKP-1 peptide (Fig. 4 B).
We further defined the interaction between MKP-1 and p38 using surface plasmon resonance (SPR). Biotinylated MKP-1 peptide was immobilized to a streptavidin sensor chip, using either biotin-MKP-1(47–76) or biotin-MKP-1(47-K57Ac-76). We injected recombinant (His)6-p38 into the flow cell and measured changes in SPR. Recombinant p38 associates with both peptides (Fig. 4 C). However, the association rate constant for p38 with the acetylated MKP-1 peptide is greater than for p38 with the unmodified MKP-1 peptide (7.9 × 103 vs. 4.0 × 103 1/Ms); and the dissociation rate constant for p38 from the acetylated MKP-1 peptide is less than for p38 from the unmodified MKP-1 peptide (1.5 × 10−4 vs. 3.4 × 10−4 1/s; Fig. 4 C). Thus, the affinity constant between p38 and acetylated MKP-1 peptide is ∼4.5-fold greater than the affinity constant between p38 and unmodified MKP-1 peptide.
To explore the importance of the K57 residue in mediating the interaction between MKP-1 and p38 in cells, we transfected cells with an inducible vector expressing a fusion polypeptide consisting of the estrogen receptor α (ERα) fused to WT MKP-1, designated MKP-1(WT)-ERα. Other cells were transfected with a vector expressing the mutant MKP-1(K57R)-ERα. Cells were cotransfected with a vector expressing FLAG-tagged p38 (FLAG-p38). Tamoxifen was added to cells to induce expression of MKP-1-ERα. Cells expressing both FLAG-p38 and MKP-1-ERα were treated with LPS and TSA, cell lysates were immunoprecipitated with antibody to ERα, and precipitants immunoblotted with antibody to FLAG. HDAC inhibition increases the interaction between MKP-1(WT) and p38 (Fig. 4 D). However, HDAC inhibition is not able to affect the interaction between mutant MKP-1(K57R) and p38 (Fig. 4 D). TSA also increases the interaction of ERK1 and WT, but not mutant, MKP-1 in cells (Fig. 4 E).
These data suggest that acetylation of the MKP-1 lysine residue 57 regulates the interaction between MKP-1 and p38.
Acetylation of MKP-1 decreases phosphorylation of p38
We predicted that by enhancing the interaction of MKP-1 and p38, acetylation of MKP-1 would increase the dephosphorylation of p38. To test this hypothesis, we synthesized recombinant MKP-1(WT) and MKP-1(K57R) in vitro, added p300 to acetylate MKP-1, and then incubated the mixture with phospho-p38. The mixture was fractionated and then immunoblotted for phospho-p38. Recombinant MKP-1 reduces phosphorylation of p38 (Fig. 5 A, left). Adding the acetyltransferase p300 which acetylates MKP-1 leads to a further decrease in phosphorylation of p38 (Fig. 5 A, left). Although MKP-1(K57R) can decrease the phosphorylation of p38, addition of p300 has no further effect on MKP-1 dephosphorylation of p38 (Fig. 5 A, right).
Figure 5.

MKP-1 acetylation decreases phosphorylation of p38. (A) Recombinant MKP-1. Recombinant MKP-1 was treated with p300 or control, incubated with recombinant phospho-p38, and levels of phospho-p38 were measured by immunoblotting. (B) MKP-1 derived from cells. RAW cells were treated with LPS and TSA or control for 4 h, and cell lysates were immunoprecipitated with antibody to MKP-1. Immunoprecipitates were incubated with recombinant phospho-p38 or phospho-ERK1, and then immunoblotted with antibodies to phospho-p38 or phospho-ERK. The total amount of phospho-p38 or phospho-ERK1 added to the reaction is shown as input above. (C) MKP-1 catalytic assay. Recombinant MKP-1 was pretreated or not with p300 and acetyl-CoA for 30 min at 30°C, and then recombinant p38 or control was added for 1 h at 30°C. The artificial substrate OMFP was added and the A 477 nm was measured at 30°C for 0–40 min (n = 2 ± the SD; error bars too small to see).
We next examined the effect of MKP-1 acetylation upon dephosphorylation of p38 inside cells. RAW cells were treated with LPS and TSA, and endogenous MKP-1 was immunoprecipitated, and incubated with recombinant phospho-p38. HDAC inhibition enhances the ability of MKP-1 to remove phosphate groups from recombinant phospho-p38 (Fig. 5 B). MKP-1 derived from TSA-treated cells also removes more phosphate from recombinant phospho-ERK1 than MKP-1 from control cells (Fig. 5 B).
To explore the effect of acetylation upon MKP-1 phosphatase activity, we used an in vitro phosphatase assay. Recombinant MKP-1 was acetylated or not with p300 and acetyl-CoA, and then incubated with 3-O-methylfluorescein phosphate (OMFP) at 30°C, and the OD 477 nm was measured over 0–40 min. Acetylation of MKP-1 has a negligible effect on phosphatase activity (Fig. 5 C, closed circles vs. open circles). Recombinant p38 increases the phosphatase activity of MKP-1 (Fig. 5 C, open squares vs. open circles). This is expected because substrate binding to MKP-1 increases MKP-1 catalytic activity (35–38). However, acetylation of MKP-1 increases the phosphatase activity of MKP-1 in the presence of p38 by ∼125% (Fig. 5 C, closed vs. open squares).
These data suggest that acetylation of MKP-1 does not directly change its phosphatase activity. However, acetylation of MKP-1 indirectly increases its activity, by increasing its affinity for its substrate.
MKP-1 mediates the effects of acetylation upon MAPK signaling
To demonstrate the importance of MKP-1 in deacetylase regulation of MAPK signaling, we followed two complementary strategies. We first used RNA silencing (small interfering [si]RNA) to decrease expression of MKP-1 in macrophages. RAW cells were stably transfected with three different vectors encoding siRNA hairpin sequences directed against MKP-1 nucleotides 67–85 (Fig. 6 A, clone #1) or MKP-1 nucleotides 743–761 (clones #2 and #3) or a control siRNA sequence. Four separate stably transfected clones were isolated, and cell lysates were immunoblotted with antibody to MKP-1. The vector encoding siRNA against MKP-1 nucleotides 743–761 decreases MKP-1 expression (Fig. 6 A, clone #2 and #3). We then tested the effect of knocking down MKP-1 expression upon the ability of HDAC inhibitors to regulate phosphorylation of p38 and expression of NOS2. In control cells expressing a control hairpin RNA, LPS increases phosphorylation of p38, and TSA limits this increase as before (Fig. 6 B). However, siRNA of MKP-1 blocks the effect of TSA (Fig. 6 B). Additionally, TSA decreases LPS-induced NOS2 expression in control cells, but not in cells with decreased MKP-1 (Fig. 6 B).
Figure 6.

MKP-1 mediates the effects of acetylation upon phosphorylation of p38 and NOS2 expression in cells. (A) MKP-1 siRNA decreases MKP-1 expression in RAW cell lines. RAW cells were stably transfected with a vector encoding an siRNA hairpin sequences directed against MKP-1 nucleotides 67–85 (clone #1) or MKP-1 nucleotides 743–761 (clones #2-3) or a control siRNA sequence. Three separate stably transfected clones were isolated, and cell lysates were immunoblotted with antibody to MKP-1. (B) Knockdown of MKP-1 restores phospho-p38 levels and NOS2 expression in RAW cells treated with TSA. RAW cells stably transfected with siRNA directed against MKP-1 were treated with LPS and TSA, and cell lysates were immunoblotted with antibodies to NOS2 or MAPK family members. (C) Cells from MKP-1−/− mice maintain levels of phospho-p38 after treatment with TSA. MEFs from MKP-1−/+ or MKP-1−/− mice were treated with LPS and TSA for 30 min, and cell lysates were immunoblotted with antibodies to p38 as above. (D) Effects of TSA on p38 are restored by rescue of MKP-1−/− cells with MKP-1(WT), but not with MKP-1(K57R). Cells from MKP-1−/− were immortalized with SV40 T-antigen, and then transfected with plasmids expressing MKP-1(WT)-ERα or MKP-1(K57R)-ERα. The cells were treated with 4-HT to induce MKP-1-ERα expression, and then treated with LPS and TSA for 1 h. Cell lysates were immunoblotted with antibody to phospho-p38 (top), total p38 (middle), and the ER tag of MKP-1 (bottom). (E) Antiinflammatory effects of TSA are restored by rescue of MKP-1−/− cells with MKP-1(WT), but not with MKP-1(K57R). Cells from MKP-1−/− were transfected with plasmids expressing MKP-1(WT)-ERα or MKP-1(K57R)-ERα, treated with 4-HT, and then treated with LPS and TSA for 1 h. Total cell RNA was analyzed by RT-PCR for IL-6, TNF-α, and GAPDH. (F) MKP-1 mediates TSA inhibition of p38 in primary macrophages. Peritoneal macrophages were isolated from WT and MKP-1−/− mice, stimulated with LPS, TSA, or both, as above, and cell lysates were immunoblotted with antibody to total or phosphorylated p38.
We next used cells derived from MKP-1−/− mice to show that acetylation regulates MKP-1 dephosphorylation of p38. Murine embryonic fibroblasts (MEFs) from MKP-1−/− mice and from MKP-1+/− mice were treated with LPS and TSA, and phosphorylation of p38 was measured by immunoblotting. Inhibition of deacetylation decreases phospho-p38 levels in heterozygous cells, but not in MKP-1 KO cells (Fig. 6 C).
Can MKP-1 rescue the MKP-1 KO cells and restore sensitivity to TSA? We transfected MEF cells from MKP-1−/− mice with a vector expressing MKP-1(WT) or MKP-1(K57R). Cells were then treated with LPS and TSA, and levels of phospho-p38 were measured. Expression of ectopic MKP-1(WT) restores the ability of TSA to decrease phospho-p38 levels (Fig. 6 D, lane 3 and 4). However, expression of ectopic MKP-1(K57R) cannot restore the effects of TSA upon phospho-p38 (Fig. 6 D, lanes 7 and 8). Furthermore, ectopic expression of MKP-1(WT) in MEFs from MKP-1−/− mice also restores the ability of TSA to decrease LPS-induced TNF-α (Fig. 6 E).
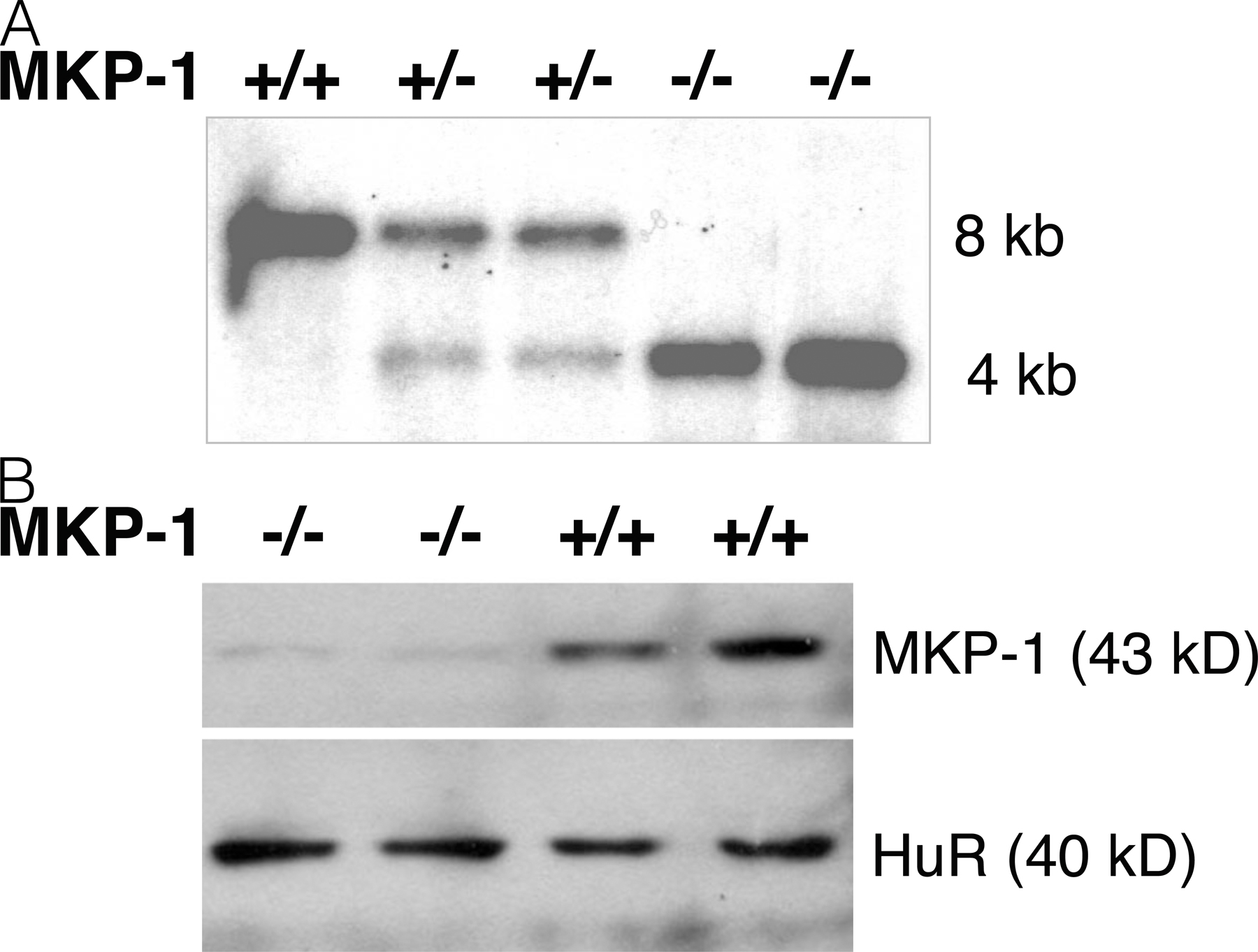
Finally, we explored the effect of TSA upon primary macrophages. We isolated primary macrophages by peritoneal lavage of resting WT or MKP-1 KO mice (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20071728/DC1). We then simulated the primary macrophages with TSA, LPS, or both, and measured phosphorylated p38. LPS induces phospho-p38 in WT macrophages and higher levels of phospho-p38 in MKP-1−/− macrophages (Fig. 6 F). However, TSA only suppresses phospho-p38 in macrophages from WT mice, not in macrophages from MKP-1−/− mice.
Collectively, these complementary data from the siRNA experiments and from the MKP-1 KO cells and from the MKP-1−/− primary macrophages all suggest that MKP-1 mediates the effects of acetylation upon p38.
HDAC inhibition decreases inflammation and mortality in mice exposed to LPS
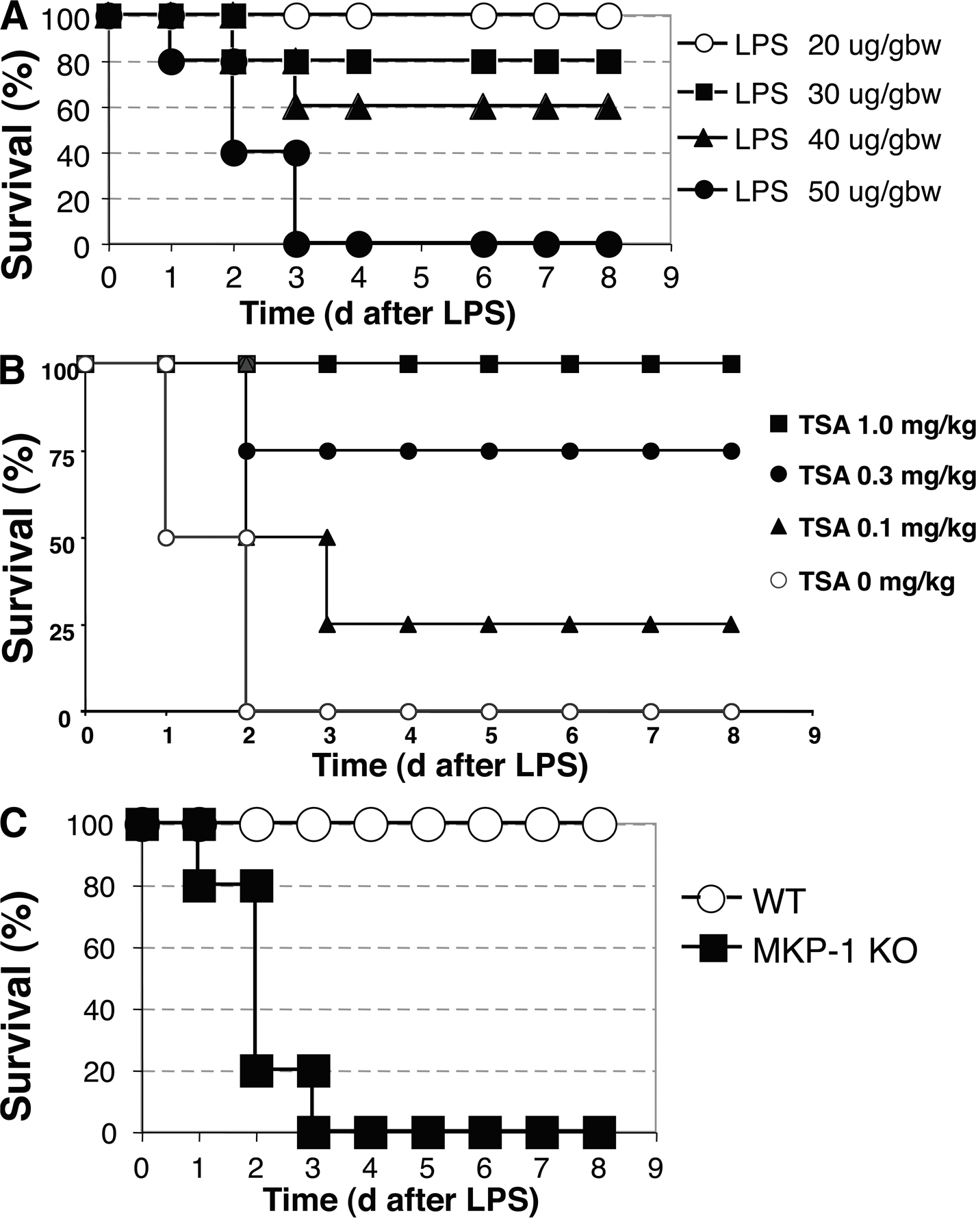
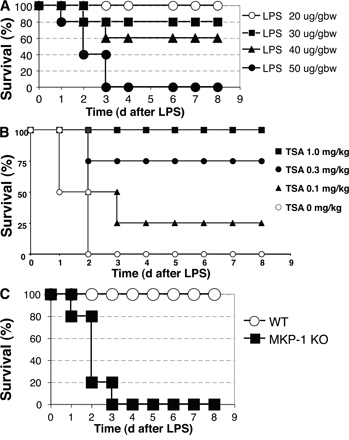
To explore the in vivo relevance of MKP-1 acetylation, we used a murine model of septic shock (Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20071728/DC1). We pretreated WT mice with an i.p. injection of 1 mg/kg TSA each day for 2 d, and then injected the mice with a single i.p. dose of 50 mg/kg LPS. TSA was given for another 3 d. LPS causes 100% mortality within 3 d (Fig. 7 A). However, TSA decreases the mortality of mice injected with LPS (Fig. 7 A). Furthermore, TSA decreases mortality after LPS in a dose-responsive manner (Fig. S4 B).
Figure 7.

HDAC inhibition decreases LPS induced mortality and inflammation in mice. (A) Mortality. Mice were injected with 1 mg/kg TSA daily for 5 d starting on day −2, injected with 50 mg/kg LPS on day 0, and their mortality was recorded each day after LPS treatment (n = 5; P < 0.00001 for LPS vs. LPS with TSA). (B) TSA decreases LPS-induced liver inflammation. Mice were injected with saline (control) or TSA each day starting on day −2, injected with LPS on day 0, and livers were harvested 2 h (top) or 2 d (bottom) after LPS, sectioned, and stained with hematoxylin and eosin. (C) TSA decreases cytokine levels in serum. WT and MKP-1−/− mice were injected with TSA each day starting on day −2 and injected with LPS on day 0, and serum was collected 2 h after LPS treatment and analyzed by ELISA for TNF-α and IL-1β (n = 2 ± the SD; *, P < 0.05 ± the SD). (D) Cytokine from primary macrophages. Macrophages were prepared from WT and MKP-1−/− mice, stimulated with TSA, LPS, or both, and after 4 h TNF-α and IL-1β levels measured in the media by ELISA (n = 2 ± SD). (E) TSA suppresses cytokine RNA in vivo. Mice were injected with TSA each day starting on day −2, injected with LPS on day 0, and liver RNA was harvested 4 h after LPS treatment and analyzed by RT-PCR for TNF-α and IL-1β. Data are shown for two representative mice. (F) TSA slightly increases COX-2 and CXCL2 in vivo. Mice were injected with TSA and LPS as above, and liver RNA was harvested 4 h after LPS treatment and analyzed by RT-PCR for COX-2 and CXCL2 (n = 2). (G) Mouse NOS2 protein expression in liver. Mice were pretreated with increasing amounts of TSA for 4 h, and then treated with LPS for 16 h. Liver was harvested and immunoblotted with antibodies to NOS2 and MEK1. (H) MKP-1 is acetylated in mice. Mice were pretreated with TSA for 4 h, and then treated with LPS for 16 h as above. Liver lysates were immunoprecipitated with antibody to MKP-1 and immunoblotted with antibody to Ac-Lys or MKP-1. (I) MKP-1 mediates the protective effects of TSA after LPS. MKP-1 KO mice and their WT littermate controls were pretreated with TSA as above, and then injected with LPS 20 mg/kg, and their mortality was recorded (n = 5; P = 0.17 for LPS vs. LPS with TSA). (J) Proposed schematic of MKP-1 acetylation and regulation of innate immune signaling.
We then analyzed the effect of TSA upon inflammation. LPS treatment activates leukocyte infiltration into the liver within 2 h, but TSA treatment decreases LPS-induced inflammation (Fig. 7 B, top). TSA also limits LPS-induced hepatocyte edema and necrosis (Fig. 7 B, bottom). TSA also decreases levels of some cytokines in vivo. For example, TSA decreases serum levels of TNF-α and IL-1β in mice; and TSA suppresses these cytokines more in WT mice than in MKP-1−/− mice (Fig. 7 C and Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20071728/DC1). TSA decreases TNF-α by 62% in WT mice, but only by 22% in MKP-1−/− mice (Fig. 7 C left). TSA decreases IL-1β by 79% in WT mice, but only by 21% in MKP-1−/− mice (Fig. 7 C right). TSA also suppresses cytokine production more in primary macrophages from WT mice than from MKP-1−/− mice; in particular, TSA suppresses TNF-α by 87% in WT macrophages, but only by 27% in MKP-1−/− macrophages; and TSA decreases IL-1β by 85% in WT macrophages, but only by 34% in MKP-1 KO macrophages (Fig. 7 D). Supporting these data, TSA decreases LPS-induced TNF-α and IL-1β mRNA levels in the liver (Fig. 7 E), and TSA decreases NOS2 steady-state protein levels in liver (Fig. 7 G). However, TSA slightly increases COX-2 and CXCL2 expression in vivo (Fig. 7 F).
We hypothesize that MKP-1 mediates part of the antiinflammatory effects of HDAC inhibitors. Immunoprecipitation confirmed that MKP-1 is acetylated in livers of mice treated with LPS, and that TSA increases acetylation of MKP-1 in vivo (Fig. 7 H). We pretreated MKP-1 KO mice with TSA or control, and then injected them with 20 mg/kg LPS. LPS causes 100% mortality of MKP-1 KO mice within 3 d (Fig. 7 I). However, TSA has a minimal and insignificant effect on mortality (Fig. 7 I). Thus, TSA protects WT mice from LPS (Fig. 7 A), but TSA does not protect MKP-1 KO mice from LPS (Fig. 7 I).
These data confirm the findings of others that LPS induces higher levels of inflammation in MKP-1 KO mice compared with WT mice. Our data also confirm prior studies showing that HDAC inhibitors decrease inflammation caused by LPS in vivo. Furthermore, our data extend these studies by showing that TSA inhibits inflammation less in mice lacking MKP-1 than in WT mice. Therefore our data suggest that MKP-1 mediates part of the antiinflammatory effects of HDAC inhibitors in vivo.
DISCUSSION
Summary
The major finding of this study is that acetylation of MKP-1 regulates TLR signaling. Acetylation of MKP-1 increases its interaction with p38. A greater interaction of acetylated MKP-1 with p38 increases MKP-1 phosphatase activity by >100%, decreases cellular phospho-p38 levels, and inhibits the MAPK signaling cascade. Acetylation may be a negative regulator of innate immune pathways (Fig. 7 J).
Acetylation of MKP-1
Several lines of evidence suggest that MKP-1 is acetylated. The acetyltransferase p300 associates with MKP-1 in cells, and PCAF and p300 acetylate MKP-1 in vitro (Fig. 3). Treatment with the deacetylase inhibitor TSA increases levels of acetylated MKP-1 in cells and in mice (Figs. 3 and 7). Previous studies have demonstrated that regulators of transcription are acetylated, including histones, transcription factors, nuclear import factors, and tubulin (46–48, 62–66). Our data identify a member of a signal transduction cascades as a novel target of acetylation.
Acetylation of MKP-1 regulates protein–protein interactions and phosphatase activity
Our data suggest that acetylation of MKP-1 at a specific site regulates its ability to interact with its substrates. The interaction between MKP and their MAPK substrates is mediated by a docking domain in the N terminus of MKP and a docking domain in the MAPK substrate. The docking domain of murine MKP-1 extends from amino acid residues R47 to E63, and contains a central region with 4 arginine and 1 lysine residues: RFSTIVRRRAKGAMGLE (Table I) (35, 67). We identified K57 as an amino acid residue acetylated in MKP-1 (Figs. 3 and 4). This lysine residue lies within the docking domain of MKP-1. Acetylation of K57 may neutralize the positive charge within the docking domain, which may explain our observation that acetylation of MAPK K57 increases the interactions between MKP-1 and p38. Our data thus confirm other studies that found that acetylation of lysine residues can increase protein–protein interactions (47, 68). The docking domains of all MKP family members are rich in arginine and lysine residues (Table I). Some isoforms contain a lysine residue in the same relative position as MKP-1 K57, others contain a lysine residue two amino acyl residues adjacent to K57 (Table I). Other MKP isoforms lack a lysine in the docking domain. It is possible that selective MKP isoforms are acetylated in their respective docking domains, and others are not.
We also found that acetylation of MKP-1 indirectly affects its phosphatase activity. Substrate binding to MKP-1 increases the phosphatase activity (35). Other MKP isoforms are also activated by interacting with their respective substrates (69, 70). Our studies of recombinant MKP-1 show that acetylation does not affect phosphatase activity of the enzyme alone (Fig. 5 C.) However, acetylation of MKP-1 significantly increases phosphatase activity in the presence of substrate by ∼125% (Fig. 5 C). Our SPR assay of the interaction between p38 and MKP-1 peptides also suggests that acetylation of MKP-1 increases its affinity for its substrate (Fig. 4 C). Collectively, our findings suggest that acetylation indirectly increases phosphatase activity by increasing the avidity of MKP-1 for its substrate.
Acetylation of MKP-1 negatively regulates innate immune signaling
Our data provide a mechanistic explanation for the antiinflammatory effects of HDAC inhibitors administered to animals. Numerous studies have shown that HDAC inhibitors decrease inflammation in vivo (50–55). Although it is thought that HDAC inhibitors decrease inflammation by regulating histone acetylation and inflammatory gene transcription, our data suggest that HDAC inhibitors decrease inflammation in part by increasing acetylation of MKP-1. However, HDAC inhibitors probably have other antiinflammatory targets in addition to MKP-1, especially because TSA partially decreases cytokines in macrophages (Fig. 7 D) and mice (Fig. 7 C), and because TSA has a small protective effect on MKP-1−/− mouse mortality (Fig. 7 I). Other studies suggest that MKP isoforms can be regulated by other posttranslational modifications, such as oxidation of cysteine residues (33, 71). Our data demonstrate another mechanism of regulating inflammation through MKP. Acetylation of target proteins in inflammatory signaling pathways may be novel therapeutic targets in patients with sepsis or autoimmune diseases.
MATERIALS AND METHODS
Materials.
LPS, TSA, sodium butyrate, actinomycin D, and 4-hydroxytamoxifen were purchased from Sigma-Aldrich. Peptidoglycan was purchased from Fluka. Poly(I:C) was purchased from GE Healthcare. MAPK pathway inhibitor SB203580 (p38) and U0126 (ERK) were purchased from Calbiochem. E. coli DH5 was purchased from Invitrogen. Antibodies to NF-κB p65, IκBα, NOS2 (M-19), MKP-1 (V-15), MEK1/2, pan acetyl-K (C2), p300 (C20), Elk-1, and ERα were purchased from Santa Cruz Biotechnology, Inc. Antibodies to ERK1/2, phospho-ERK1/2, phospho-MEK1/2, phospho-Raf, phospho-Elk-1, phospho-p90, p38, phospho-p38, JNK, phospho-JNK, phospho-Raf and acetyl-K, phospho-MKK3/6, and MKK3 were purchased from Cell Signaling Technology. Antibody to HA tag (5C12) was purchased from Roche. Antibodies to (His)6 and FLAG tag were purchased from Sigma-Aldrich. Antibody to acetyl-histone was purchased from Millipore. The MTS assay was purchased from Promega.
The mouse MKP-1 expression plasmids pUSEHA-MKP-1 (WT) and pUSEHA-MKP-1(K57R) were generated from a mouse MKP-1 cDNA (a gift from N.K. Tonks, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). The human MKP-1 expression vector was a gift from P. Stork (Oregon Health Sciences University, Portland, OR). The bacterial expression vector for MKP-1 was a gift from S. Keyse (University of Dundee, Dundee, Scotland). The MKP-1(K57R) mutant was generated by PCR. The plasmid pCMV5 His6-ERK1 was a gift from P. Shaw (Queen's Medical Center, Nottingham, UK). The plasmid pcDNA Flag-p38 was a gift from J. Han (The Scripps Institute, San Diego, CA). We produced a plasmid vector to express MKP-1 under the control of estrogen. We cloned the MKP-1 cDNA and cloned it into the expression vector pBABE-PURO such that it encodes a fusion polypeptide consisting of ERα ligand-binding domain fused to the MKP-1 (72, 73). The addition of hydroxytamoxifen increases expression of the fusion protein.
Cell culture and transfection.
The mouse monocyte cell line RAW 264.7 (American Type Culture Collection: TIB-71), HeLa cells, HEK 293 cells, or MEFs from MKP KO mice (provided by R.P. Ryseck, Bristol-Myers Squibb, Princeton, NJ) were grown in DME supplemented with 10% FBS and penicillin/streptomycin. The cells were transfected with Lipofectamine 2000 following the manufacturer's instructions (Invitrogen). Expression of the MKP-1-ER fusion protein was induced with 200 nM of 4-hydroxyl-tamoxifen (Sigma-Aldrich). We prepared primary macrophages from resting mice (not treated with thioglycolate) by peritoneal lavage with 5 ml PBS.
In vitro kinase assay.
RAW cell lysates were immunoprecipitated with antibody to MKK3 (Cell Signaling Technology). The immunoprecipitates were washed and immunoblotted with antibody to phospho-p38.
Protein acetylation assay.
HeLa cells were transfected with plasmids pUSEHA-MKP-1(WT) or pUSEHA-MKP-1(K57R) for 16 h, incubated with 1 mCi/ml [3H]sodium acetate (ICN) for 1 h before addition of LPS and/or TSA. Cells were lysed, immunoprecipitated with antibody to MKP-1 or antibody to HA-tag, fractionated by SDS PAGE, and autoradiographed.
MKP-1 peptides (docking domain) representing MKP-1 aa 47–76 RFSTIVRRRAKGAMGLEHIVPNAELRGRLLA and a mutated peptide (K57R) RFSTIVRRRARGAMGLEHIVPNAELRGRLLA (Johns Hopkins DNA Core Facility) were incubated with recombinant p300 (HAT domain) or recombinant PCAF (Millipore) and 0.2 mM [3H]acetyl-CoA (ICN) at 30°C for 10 min, fractionated, and autoradiographed.
MKP-1 phosphatase assay.
For the in vitro MKP-1 phosphatase assay using phospho-p38 as a substrate, WT and mutant K57R MKP-1 proteins were generated from the plasmids pRSET-His-MKP-1 using an in vitro transcription and translation kit following the manufacturer's recommendation (TNT System; Promega). In vitro acetylation of MKP-1 was performed with recombinant p300 (Millipore) and cold acetyl-CoA (Sigma-Aldrich), as described in the previous section.
For the in vitro MKP-1 phosphatase assay using an artificial substrate, acetylation of MKP-1 was performed by mixing recombinant MKP-1 0.66 μg, recombinant p300 2.5 μg, and 5 μl of 1 mM acetyl-CoA for 30 min at 30°C. 2 μg of recombinant p38 was added, and the mixture was incubated for 1 h at 30°C. The artificial substrate OMFP (Sigma-Aldrich) was added, and the A 477 nm was monitored at 30°C for 0–60 min.
MKP-1 peptide/p38 binding assay.
MKP-1 peptides encoding the MKP-1 docking site aa residues 47–76 and conjugated to an N-terminal biotin (biotin-RFSTIVRRRAKGAMGLE, designated MKP-1[47–76] and biotin-RFSTIVRRRAKacGAMGLE, designated MKP-1[47-K57Ac-76]), were incubated with 10 μM recombinant p38 for 2 h at 22°C, and then streptavidin agarose beads (Stratagene) were added for 16 h at 4°C. The beads were washed, eluted, and precipitants were analyzed by immunoblotting.
SPR was performed using a BIACORE 2000 (Biacore AB) with a sensor chip preimmobilized with streptavidin (Sensor Chip SA; Biacore AB). The streptavidin sensor chip was loaded either with biotin-MKP-1(47–76) or with biotin-MKP-1(47-K57Ac-76; 150 μg in 100 μl PBS). Recombinant (His)6-p38 at 3.0 μM were injected over 6 min at a rate of 10 μl/min into flow cells, and the SPR angle was measured and reported in resonance units. Association and disassociation rates were calculated.
Northern and Southern blotting.
Total cellular RNA was prepared from RAW cells treated with or without LPS and TSA using TRIZOL (Invitrogen). 20 μg of RNA was separated and transferred to a NYTRAN membrane (Nytran; Schleicher & Schuell). Northern and Southern blot analysis was then performed, as previously described (73).
Animal studies.
C57BL6/J male 4–6-wk-old mice were purchased from The Jackson Laboratory and housed at The Johns Hopkins University School of Medicine animal care facilities. All animal studies were performed under protocols approved by The Johns Hopkins University School of Medicine Animal Care and Use Committee. TSA was injected i.p. twice a day for a total of 5 d. A single dose of LPS (50 μg/gram body weight) was injected i.p. on day 3 for the WT mice on the C57BL/6J background; these doses of LPS were determined to kill 100% of the mice by day 2 (unpublished data). Mouse sera were tested by ELISA for levels of TNF-α and IL-1β (R&D Systems). Because the background of the original MKP-1 KO mice is not well defined, we bred the MKP-1 KO mice (The Jackson Laboratory) onto a C57BL/6J background, and then interbred the heterozygotes for use as WT controls and MKP-1 KO mice.
Online supplemental materials.
Fig. S1 shows the effect of TSA on COX-2. Fig. S2 shows that TSA does not affect the NF-κB pathway. Fig. S3 characterizes the MKP-1 KO mice. Fig. S4 shows that TSA only alters the mortality after LPS of WT mice, not of MKP-1 KO mice. Fig. S5 shows that TSA decreases inflammation only in WT mice, not MKP-1 KO mice. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071728/DC1.
Supplementary Material
Acknowledgments
We gratefully acknowledge Aeryon Kim and Scheherazade Sadegh-Nasseri of The Johns Hopkins University School of Medicine for assistance with SPR measurements.
Supported by grants from the National Institutes of Health (R01 HL63706-04, R01 HL074061, P01 HL65608, P01 HL56091), the American Heart Association (EIG 0140210N), the Ciccarone Center, the John and Cora H. Davis Foundation, and the Clarence P. Doodeman Professorship to CJL.
The authors have no conflicting financial interests.
Abbreviations used: ER, estrogen receptor; ERK, extracellular signal–regulated kinase; HAT, histone acetyltransferase; HDAC, histone deacetylase; JNK, Jun-N-terminal kinase; MAPK, mitogen-activated protein kinase; MAPKK, MAPK kinase; MEF, murine embryonic fibroblast; MKP, MAPK phosphatase; NO, nitric oxide; si, small interfering; OMFP, 3-O-methylfluorescein phosphate; SPR, surface plasmon resonance; TLR, Toll-like receptor; TSA, trichostatin A.
References
- 1.Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–145. [DOI] [PubMed] [Google Scholar]
- 2.Gordon, S. 2002. Pattern recognition receptors: doubling up for the innate immune response. Cell. 111:927–930. [DOI] [PubMed] [Google Scholar]
- 3.Barton, G.M., and R. Medzhitov. 2003. Toll-like receptor signaling pathways. Science. 300:1524–1525. [DOI] [PubMed] [Google Scholar]
- 4.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. [DOI] [PubMed] [Google Scholar]
- 5.Dong, C., R.J. Davis, and R.A. Flavell. 2001. Signaling by the JNK group of MAP kinases. c-jun N-terminal kinase. J. Clin. Immunol. 21:253–257. [DOI] [PubMed] [Google Scholar]
- 6.Dong, C., R.J. Davis, and R.A. Flavell. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 20:55–72. [DOI] [PubMed] [Google Scholar]
- 7.Han, J., and R.J. Ulevitch. 1999. Emerging targets for anti-inflammatory therapy. Nat. Cell Biol. 1:E39–E40. [DOI] [PubMed] [Google Scholar]
- 8.Davis, R.J. 2000. Signal transduction by the JNK group of MAP kinases. Cell. 103:239–252. [DOI] [PubMed] [Google Scholar]
- 9.Chang, L., and M. Karin. 2001. Mammalian MAP kinase signalling cascades. Nature. 410:37–40. [DOI] [PubMed] [Google Scholar]
- 10.Morrison, D.K., and R.J. Davis. 2003. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 19:91–118. [DOI] [PubMed] [Google Scholar]
- 11.Whitmarsh, A.J., and R.J. Davis. 1999. Signal transduction by MAP kinases: regulation by phosphorylation-dependent switches. Sci. STKE. 1999:PE1. [DOI] [PubMed] [Google Scholar]
- 12.Lu, H.T., D.D. Yang, M. Wysk, E. Gatti, I. Mellman, R.J. Davis, and R.A. Flavell. 1999. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J. 18:1845–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blander, J.M., and R. Medzhitov. 2004. Regulation of phagosome maturation by signals from toll-like receptors. Science. 304:1014–1018. [DOI] [PubMed] [Google Scholar]
- 14.Doyle, S.E., R.M. O'Connell, G.A. Miranda, S.A. Vaidya, E.K. Chow, P.T. Liu, S. Suzuki, N. Suzuki, R.L. Modlin, W.C. Yeh, et al. 2004. Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 199:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liew, F.Y., D. Xu, E.K. Brint, and L.A. O'Neill. 2005. Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5:446–458. [DOI] [PubMed] [Google Scholar]
- 16.Wells, C.A., T. Ravasi, and D.A. Hume. 2005. Inflammation suppressor genes: please switch out all the lights. J. Leukoc. Biol. 78:9–13. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi, K.S., and R.A. Flavell. 2004. Shielding the double-edged sword: negative regulation of the innate immune system. J. Leukoc. Biol. 75:428–433. [DOI] [PubMed] [Google Scholar]
- 18.Burns, K., J. Clatworthy, L. Martin, F. Martinon, C. Plumpton, B. Maschera, A. Lewis, K. Ray, J. Tschopp, and F. Volpe. 2000. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat. Cell Biol. 2:346–351. [DOI] [PubMed] [Google Scholar]
- 19.Kinjyo, I., T. Hanada, K. Inagaki-Ohara, H. Mori, D. Aki, M. Ohishi, H. Yoshida, M. Kubo, and A. Yoshimura. 2002. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 17:583–591. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa, R., T. Naka, H. Tsutsui, M. Fujimoto, A. Kimura, T. Abe, E. Seki, S. Sato, O. Takeuchi, K. Takeda, et al. 2002. SOCS-1 participates in negative regulation of LPS responses. Immunity. 17:677–687. [DOI] [PubMed] [Google Scholar]
- 21.Sly, L.M., M.J. Rauh, J. Kalesnikoff, C.H. Song, and G. Krystal. 2004. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity. 21:227–239. [DOI] [PubMed] [Google Scholar]
- 22.Wald, D., J. Qin, Z. Zhao, Y. Qian, M. Naramura, L. Tian, J. Towne, J.E. Sims, G.R. Stark, and X. Li. 2003. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 4:920–927. [DOI] [PubMed] [Google Scholar]
- 23.Garlanda, C., F. Riva, N. Polentarutti, C. Buracchi, M. Sironi, M. De Bortoli, M. Muzio, R. Bergottini, E. Scanziani, A. Vecchi, et al. 2004. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. USA. 101:3522–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diehl, G.E., H.H. Yue, K. Hsieh, A.A. Kuang, M. Ho, L.A. Morici, L.L. Lenz, D. Cado, L.W. Riley, and A. Winoto. 2004. TRAIL-R as a negative regulator of innate immune cell responses. Immunity. 21:877–889. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, Y., J.N. Blattman, N.J. Kennedy, J. Duong, T. Nguyen, Y. Wang, R.J. Davis, P.D. Greenberg, R.A. Flavell, and C. Dong. 2004. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature. 430:793–797. [DOI] [PubMed] [Google Scholar]
- 26.Shepherd, E.G., Q. Zhao, S.E. Welty, T.N. Hansen, C.V. Smith, and Y. Liu. 2004. The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. J. Biol. Chem. 279:54023–54031. [DOI] [PubMed] [Google Scholar]
- 27.Zhao, Q., E.G. Shepherd, M.E. Manson, L.D. Nelin, A. Sorokin, and Y. Liu. 2005. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J. Biol. Chem. 280:8101–8109. [DOI] [PubMed] [Google Scholar]
- 28.Chi, H., S.P. Barry, R.J. Roth, J.J. Wu, E.A. Jones, A.M. Bennett, and R.A. Flavell. 2006. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. USA. 103:2274–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keyse, S.M. 2000. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr. Opin. Cell Biol. 12:186–192. [DOI] [PubMed] [Google Scholar]
- 30.Camps, M., A. Nichols, and S. Arkinstall. 2000. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 14:6–16. [PubMed] [Google Scholar]
- 31.Theodosiou, A., and A. Ashworth. 2002. MAP kinase phosphatases. Genome Biol. 3:S3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keyse, S.M. 1998. Protein phosphatases and the regulation of MAP kinase activity. Semin. Cell Dev. Biol. 9:143–152. [DOI] [PubMed] [Google Scholar]
- 33.Tonks, N.K. 2005. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 121:667–670. [DOI] [PubMed] [Google Scholar]
- 34.Tonks, N.K., and B.G. Neel. 2001. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 13:182–195. [DOI] [PubMed] [Google Scholar]
- 35.Slack, D.N., O.M. Seternes, M. Gabrielsen, and S.M. Keyse. 2001. Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J. Biol. Chem. 276:16491–16500. [DOI] [PubMed] [Google Scholar]
- 36.Dowd, S., A.A. Sneddon, and S.M. Keyse. 1998. Isolation of the human genes encoding the pyst1 and Pyst2 phosphatases: characterisation of Pyst2 as a cytosolic dual-specificity MAP kinase phosphatase and its catalytic activation by both MAP and SAP kinases. J. Cell Sci. 111(Pt 22):3389–3399. [DOI] [PubMed] [Google Scholar]
- 37.Tanoue, T., T. Moriguchi, and E. Nishida. 1999. Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J. Biol. Chem. 274:19949–19956. [DOI] [PubMed] [Google Scholar]
- 38.Camps, M., A. Nichols, C. Gillieron, B. Antonsson, M. Muda, C. Chabert, U. Boschert, and S. Arkinstall. 1998. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 280:1262–1265. [DOI] [PubMed] [Google Scholar]
- 39.Brondello, J.M., J. Pouyssegur, and F.R. McKenzie. 1999. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science. 286:2514–2517. [DOI] [PubMed] [Google Scholar]
- 40.Hammer, M., J. Mages, H. Dietrich, A. Servatius, N. Howells, A.C. Cato, and R. Lang. 2006. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med. 203:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao, Q., X. Wang, L.D. Nelin, Y. Yao, R. Matta, M.E. Manson, R.S. Baliga, X. Meng, C.V. Smith, J.A. Bauer, et al. 2006. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med. 203:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salojin, K.V., I.B. Owusu, K.A. Millerchip, M. Potter, K.A. Platt, and T. Oravecz. 2006. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J. Immunol. 176:1899–1907. [DOI] [PubMed] [Google Scholar]
- 43.Peterson, C.L. 2002. HDAC's at work: everyone doing their part. Mol. Cell. 9:921–922. [DOI] [PubMed] [Google Scholar]
- 44.Minucci, S., and P.G. Pelicci. 2006. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer. 6:38–51. [DOI] [PubMed] [Google Scholar]
- 45.Yang, X.J., and E. Seto. 2007. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 26:5310–5318. [DOI] [PubMed] [Google Scholar]
- 46.Chen, L.f., W. Fischle, E. Verdin, and W.C. Greene. 2001. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 293:1653–1657. [DOI] [PubMed] [Google Scholar]
- 47.Yuan, Z.L., Y.J. Guan, D. Chatterjee, and Y.E. Chin. 2005. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 307:269–273. [DOI] [PubMed] [Google Scholar]
- 48.Gu, W., and R.G. Roeder. 1997. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 90:595–606. [DOI] [PubMed] [Google Scholar]
- 49.Koyama, Y., M. Adachi, M. Sekiya, M. Takekawa, and K. Imai. 2000. Histone deacetylase inhibitors suppress IL-2-mediated gene expression prior to induction of apoptosis. Blood. 96:1490–1495. [PubMed] [Google Scholar]
- 50.Leoni, F., A. Zaliani, G. Bertolini, G. Porro, P. Pagani, P. Pozzi, G. Dona, G. Fossati, S. Sozzani, T. Azam, et al. 2002. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. USA. 99:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mishra, N., C.M. Reilly, D.R. Brown, P. Ruiz, and G.S. Gilkeson. 2003. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Invest. 111:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chung, Y.L., M.Y. Lee, A.J. Wang, and L.F. Yao. 2003. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol. Ther. 8:707–717. [DOI] [PubMed] [Google Scholar]
- 53.Reilly, C.M., N. Mishra, J.M. Miller, D. Joshi, P. Ruiz, V.M. Richon, P.A. Marks, and G.S. Gilkeson. 2004. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J. Immunol. 173:4171–4178. [DOI] [PubMed] [Google Scholar]
- 54.Reddy, P., Y. Maeda, K. Hotary, C. Liu, L.L. Reznikov, C.A. Dinarello, and J.L. Ferrara. 2004. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc. Natl. Acad. Sci. USA. 101:3921–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi, J.H., S.W. Oh, M.S. Kang, H.J. Kwon, G.T. Oh, and D.Y. Kim. 2005. Trichostatin A attenuates airway inflammation in mouse asthma model. Clin. Exp. Allergy. 35:89–96. [DOI] [PubMed] [Google Scholar]
- 56.Bode, K.A., K. Schroder, D.A. Hume, T. Ravasi, K. Heeg, M.J. Sweet, and A.H. Dalpke. 2007. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology. 122:596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aung, H.T., K. Schroder, S.R. Himes, K. Brion, W. van Zuylen, A. Trieu, H. Suzuki, Y. Hayashizaki, D.A. Hume, M.J. Sweet, and T. Ravasi. 2006. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 20:1315–1327. [DOI] [PubMed] [Google Scholar]
- 58.Singh, K., J.L. Balligand, T.A. Fischer, T.W. Smith, and R.A. Kelly. 1996. Regulation of cytokine-inducible nitric oxide synthase in cardiac myocytes and microvascular endothelial cells. Role of extracellular signal-regulated kinases 1 and 2 (ERK1/ERK2) and STAT1 alpha. J. Biol. Chem. 271:1111–1117. [DOI] [PubMed] [Google Scholar]
- 59.Zarubin, T., and J. Han. 2005. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15:11–18. [DOI] [PubMed] [Google Scholar]
- 60.Ge, B., H. Gram, F. Di Padova, B. Huang, L. New, R.J. Ulevitch, Y. Luo, and J. Han. 2002. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 295:1291–1294. [DOI] [PubMed] [Google Scholar]
- 61.Huang, W., S. Zhao, S. Ammanamanchi, M. Brattain, K. Venkatasubbarao, and J.W. Freeman. 2005. Trichostatin A induces transforming growth factor beta type II receptor promoter activity and acetylation of Sp1 by recruitment of PCAF/p300 to a Sp1.NF-Y complex. J. Biol. Chem. 280:10047–10054. [DOI] [PubMed] [Google Scholar]
- 62.Munshi, N., M. Merika, J. Yie, K. Senger, G. Chen, and D. Thanos. 1998. Acetylation of HMG I(Y) by CBP turns off IFN beta expression by disrupting the enhanceosome. Mol. Cell. 2:457–467. [DOI] [PubMed] [Google Scholar]
- 63.Bereshchenko, O.R., W. Gu, and R. Dalla-Favera. 2002. Acetylation inactivates the transcriptional repressor BCL6. Nat. Genet. 32:606–613. [DOI] [PubMed] [Google Scholar]
- 64.Boyes, J., P. Byfield, Y. Nakatani, and V. Ogryzko. 1998. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 396:594–598. [DOI] [PubMed] [Google Scholar]
- 65.Zhang, W., and J.J. Bieker. 1998. Acetylation and modulation of erythroid Kruppel-like factor (EKLF) activity by interaction with histone acetyltransferases. Proc. Natl. Acad. Sci. USA. 95:9855–9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martinez-Balbas, M.A., U.M. Bauer, S.J. Nielsen, A. Brehm, and T. Kouzarides. 2000. Regulation of E2F1 activity by acetylation. EMBO J. 19:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanoue, T., T. Yamamoto, and E. Nishida. 2002. Modular structure of a docking surface on MAPK phosphatases. J. Biol. Chem. 277:22942–22949. [DOI] [PubMed] [Google Scholar]
- 68.Mujtaba, S., Y. He, L. Zeng, A. Farooq, J.E. Carlson, M. Ott, E. Verdin, and M.M. Zhou. 2002. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol. Cell. 9:575–586. [DOI] [PubMed] [Google Scholar]
- 69.Mandl, M., D.N. Slack, and S.M. Keyse. 2005. Specific inactivation and nuclear anchoring of extracellular signal-regulated kinase 2 by the inducible dual-specificity protein phosphatase DUSP5. Mol. Cell. Biol. 25:1830–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen, P., D. Hutter, X. Yang, M. Gorospe, R.J. Davis, and Y. Liu. 2001. Discordance between the binding affinity of mitogen-activated protein kinase subfamily members for MAP kinase phosphatase-2 and their ability to activate the phosphatase catalytically. J. Biol. Chem. 276:29440–29449. [DOI] [PubMed] [Google Scholar]
- 71.Kamata, H., S. Honda, S. Maeda, L. Chang, H. Hirata, and M. Karin. 2005. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 120:649–661. [DOI] [PubMed] [Google Scholar]
- 72.Morgenstern, J.P., and H. Land. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18:3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cao, W., C. Bao, and C.J. Lowenstein. 2003. Inducible nitric oxide synthase expression inhibition by adenovirus E1A. Proc. Natl. Acad. Sci. USA. 100:7773–7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}