

Abstract
We have developed an automated procedure for aligning peaks in multiple TOF spectra that eliminates common timing errors and small variations in spectrometer output. Our method incorporates high resolution peak detection, re-binning and robust linear data fitting in the time domain. This procedure aligns label-free (uncalibrated) peaks to minimize the variation in each peak’s location from one spectrum to the next, while maintaining a high number of degrees of freedom. We apply our method to replicate pooled-serum spectra from multiple laboratories and increase peak precision (t/σt) to values limited only by small random errors (with σt less than one time count in 89 of 91 instances, 13 peaks in 7 data sets). The resulting high precision allowed for an order of magnitude improvement in peak m/z reproducibility. We show that the CV for m/z is 0.01% (100 parts per million) for 12 of the 13 peaks that were observed in all data sets between 2995 and 9297 Da.
Keywords: Algorithm, Mass Spectra, Mass Spectrometry, Reproducibility
1 Introduction
Protein expression profiling using MALDI-TOF-MS is a widely used technique for a variety of studies including microbial typing [1], semi-quantitative comparison [2], imaging MS [3], etc. While advances in signal processing and instrumentation improve the ability to resolve spectral features, the reproducibility of such spectra remains a major limitation to the precision of MALDI-TOF-MS. Precise time measurement is critical to both protein identification and pattern recognition in mass spectra. In order to achieve high precision, it is necessary to align (or synchronize) spectra so that characteristic features occur at the same time in all spectra being analyzed.
Instrument precision can be reduced by both systematic and random errors. Although it is hard to avoid random errors, systematic errors can be reduced or eliminated from individual spectra, provided they can be characterized. The most important sources of systematic instrumental error in MALDI spectra are variations in the triggering time from spectrum to spectrum and small variations in the accelerating voltage. Since these errors appear as linear effects in the TOF data, it should be straightforward to remove such errors using corrections to uncalibrated TOF data.
There are various approaches to aligning TOF data including the use of: frequent calibration [4, 5]; clustering or re-binning [6–13]; cross-correlations [14, 15]; minimizing entropy [16, 19]; and others [17–24]. Of these, only three are based in the time domain, either directly [24] or indirectly by adjusting calibration parameters [16, 23]. Corrections made in the time domain should be inherently more accurate since m/z values are derived from equations obtained by a quadratic fit to a few calibration peaks in time data. (Corrections to data after calibration in effect fit predicted values rather than measured values.)
In this study, we reanalyze the raw TOF data obtained during a multi-lab reproducibility study [25] using our high resolution peak detection and label-free alignment methods to show an improvement of more than an order of magnitude in precision. In this paper, label-free alignment refers to aligning time domain data by using the most commonly occurring peaks, without regard to the identity of those peaks. While we have used naturally occurring peaks, it would be possible to use markers that were added for calibration. However, even for added calibration markers, the actual m/z values would not be used for this alignment. This method of label-free alignment should be broadly applicable and have significant impact for researchers using TOF data for expression pattern analysis, imaging MS, and improved MS/MS protein ID.
2 Materials and Methods
2.1 Data source
A multi-institution assessment of platform reproducibility has recently characterized the performance of SELDI-TOF instruments at six different locations [25]. In this previous study, using stringent procedures for calibration/synchronization and standardization, six laboratories obtained inter-laboratory reproducibility approaching the intra-laboratory reproducibility. Quality Control (QC) samples were prepared using pooled normal human sera from 360 healthy individuals (197 women and 163 men) and provided to all the sites. Each site then collected TOF mass spectra from these QC samples and data were processed at a central site. Peaks were identified by locating local maxima and aligned by locating clusters of peaks within the ‘window of potential shift’ ( m/z = 0.2% ) for each value of m/z [6] The synchronization and standardization process included strict acceptance criteria with tolerances (for m/z resolution, intensity values and signal-to-noise ratios) prescribed for three omnipresent peaks ( m/z = 5910, 7773 and 9297 Da). Following synchronization and standardization, 96 QC replicates were run at each site to evaluate the data reproducibility. During this subsequent evaluation, the three target peaks were shown to have CVs for m/z of 0.1%. For this paper, we have reprocessed the raw post-standardization TOF data from this multi-lab reproducibility study using the steps outlined below.
2.2 Label-free alignment of a single data set
Prior to aligning data sets from different sites, we aligned the individual data sets separately. Figure 1 provides a schematic of our alignment procedure. The steps are described below.
Figure 1.
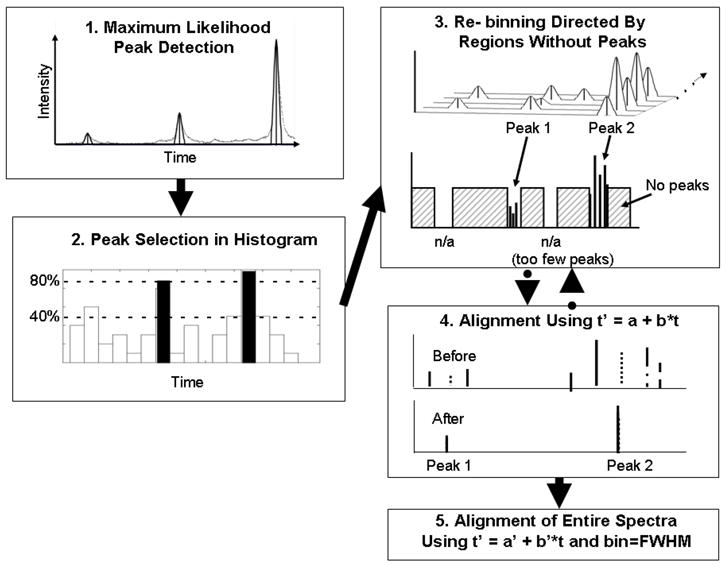
Schematic of alignment procedure incorporating high resolution peak detection (step 1), selection of isolated common peaks (step 2), re-binning reduced peak list (step 3), robust linear data fitting in the time domain (step 4), and alignment of original peak lists using corrections from step 4 and re-binning to achieve a bin size of FWHM (step 5).
Step 1: Detection of peak locations using a maximum likelihood method
We removed the slowly varying background using a charge accumulation model [15] and detected peaks using a maximum likelihood method [26]. This required fitting the data to a theoretical line shape (a symmetric Gaussian of a specified width) centered in a sliding window. Since the peak width in time units is nearly constant over a wide range of masses near the delayed extraction mass focused optimum (2–10 kDa, which covered the range of this study), we used a constant window width equal to the peak full width at half maximum (FWHM). (For higher values of m/z, the peak width increases rapidly and the method must be adapted.[27].) We used a hypothesis test to locate windows that could contain peaks, and then maximized the likelihood function to find the optimum window location (and thus peak location). To minimize the effect of line shape errors, we used only data within ±½ FWHM of the center of the line. While this method will fail if there are too many overlapping peaks, it easily detects equal intensity peaks separated by at least one FWHM. All of the peaks reported in this paper were sufficiently separated from adjacent peaks to be resolved. We had the option of using either a Gaussian noise model or a pseudo-Poisson noise model in our likelihood function. For the results presented in this paper, the latter noise model was used. A valuable byproduct of this maximum likelihood approach was the routine estimation of the uncertainty in the peak position, σt. As will be discussed, this is a meaningful estimate of the random error in the peak position and was typically a fraction of a clock count. The ratio provided an estimate of the mass precision of the series of time measurements. While we used our peak detection method, the following alignment procedure (steps 2 – 5) can be applied to peak lists obtained by other methods.
Step 2: Restricting the detected peak lists
Our peak detection method often found large numbers of peaks, including many that could be quite small. We chose not to use intensity information directly, because we expected that to vary from one patient to another within a clinical data set. However, when the peak density was too high, alignment could become unstable with peaks at almost every time value (across the data set). So, we reduced the peak list by restricting it to commonly occurring isolated peaks. We did this by creating a histogram of peaks for a data set and, using a bin width equal to our largest expected time shift in a data set (±20 time steps in this work), we identified bins that contained at least 80% of the maximum number of counts (total number of spectra) with adjacent bins containing less than 40% of the maximum number. We then created shortened peak lists for each sample by only including those peaks that were counted in the selected bins in the histogram. A sufficient number of peaks were retained to insure correct matching of peaks between spectra. For this study, eight peaks were included in the reduced peak list used to determine the correction factors. (Using eight peaks to determine two correction parameters was sufficient to prevent over-correction of the data.)
Step 3: Re - binning peak locations to construct a master list of common peaks
With the shortened peak lists, we constructed a master list of common peaks using an iterative process that delimits spectral regions without peaks. We began by choosing a large window size (typically the maximum expected shift which for this study was 8 times the FWHM) and excluding regions larger than that window in which no spectrum had peaks. This usually left some regions with peaks that were larger than the desired window size. We divided any such large regions into two parts at the largest gap between peaks within ±1 window of the average position of the peaks in the bin. We repeated this until we reached the desired widow size (for this study, 3 times FWHM), and then eliminated any bins that had contributions from too few spectra. For this study of nominally identical QC samples, we required bins to have peaks from 80% of the available spectra. Finally, we generated a master peak list of the average position of all the measured values in each remaining bin.
Step 4: Determination of corrections to minimize the differences between reduced peak lists and the master list
The reduced peak list for each spectrum was assigned a constant shift in time to minimize the average difference between its measured locations and the expected locations of the master list. The alignment procedure of Step 3 was then repeated to construct an improved master list. We then applied a robust linear fit to obtain correction factors for both offset and scale that would minimize the average difference between the reduced peak lists and the expected locations of the new master list.
Step 5: Alignment of the original peak lists
We then applied the corrections obtained in step 4 to the entire original peak lists, and then used the process of Step 3 with a desired window size of the FWHM to construct a final master list of those bins with contributions from at least 80% of the spectra. Since the alignment step introduced a scale and offset change to each spectrum, re-binning could re-assign peaks to different bins when appropriate. At the completion of this process the bin size had been reduced to the size of a peak width and the residual errors appeared to be random.
2.3 Label-free alignment of multiple data sets
For data sets collected on different dates or in different laboratories, preliminary steps were required to “roughly align” data sets prior to merging and aligning as a single data set. First, a master peak list was obtained for each data set (steps 1 and 2 above) independently. (For this preliminary master list construction, the inclusion criterion for peaks was relaxed from occurrence in 80% to 50% of the spectra.) Next, approximate time offsets and scale factors were obtained (using least squares fit) to “roughly align” the master peak lists. After applying these corrections, we merged the data sets and the entire group was simultaneously re-binned and aligned using the procedure above. This method was coded in C and R routines and implemented as a custom R module within a commercially available visual bioinformatics workflow environment (VIBE, http://www.incogen.com). This module as well as the independent routines can be downloaded from http://wecook.people.wm.edu.
3 Results and Discussion
3.1 Alignment errors
Two types of systematic instrumental error are observed in TOF data: variations in the triggering time from spectrum to spectrum and small variations in the accelerating voltage. Triggering time errors, or jitter between spectra, are differences in the measured TOF start times due to variations in the output from the digitizing clock and supporting analog electronics. These timing errors appear as constant time offsets in TOF spectra and are expected to be at least ±1 time count. Since a triggering time error effects all time measurements in a spectrum equally, it can easily be eliminated by subtracting a constant from each time value. We observe offsets as large at 12 clock counts within a single lab and as large as 62 clock counts between laboratories. It is important to note that we are dealing with averaged spectra which are obtained from multiple laser shots. Without access to individual spectra, it is impossible to completely remove error due to jitter. The presence of time jitter between individual shots can produce split peaks in the averaged spectra and consequentially produce errors in peak identification. The Appendix to this paper briefly describes this phenomenon. In addition to the start time jitter, any low frequency variation in the spectrometer acceleration voltage or any thermal expansion (or contraction) of the time-of-flight tube can produce an apparent linear dilation or contraction of the time measurement scale. As with the correction for jitter, a systematic error of this type can be eliminated by simultaneously correcting all the points in a spectrum. This type of error can be corrected with a simple linear scale factor. We observe scale corrections within +/− 0.05%. For data taken during the same day at a given location we typically observe correction factors of no more than 10−3. These correction factors are consistent with the expected differences in power supply outputs in different instruments (1 part in a thousand). Figure 2 illustrates the offset and scale errors in two spectra obtained at one site on two different dates. For this case, there is a 30 count offset and approximately a dilation of 0.007 (25 counts over 3600 counts). While frequent calibration can rectify these errors, it cannot remove the shot to shot voltage variations that occur due to pick-up of 60 Hz (line voltage) noise.
Figure 2.
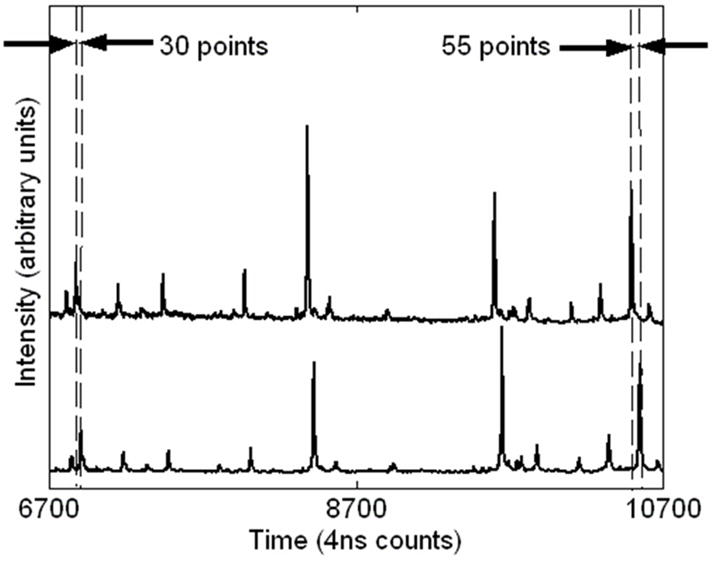
Evidence of trigger error (time offset) and voltage variation (time scale dilation) in spectra taken 3 months apart. The offset is 30 counts and the dilation is approximately 0.007 (25 counts over 3600 counts).
3.2 Typical peak displacements before and after label-free alignment
We have analyzed 7 data sets, five containing 96 spectra each from 5 laboratories, the sixth containing 42 spectra from a single laboratory (Lab 2) and a seventh containing 522 spectra from all six laboratories combined. (A number of replicates in the data set from Laboratory 2 data could not be used due to peak splitting revealed by our high resolution detection method. The appendix describes this problem.) For this study, 8 peaks in the range 2–10 kDa occur in at least 80% of the spectra prior to alignment and were used for the alignment procedure. Of the three target peaks used for the previous multi-lab study, only the m/z = 7773 Da peak was included in the restricted peak list used to obtain correction factors for alignment in this analysis. The other two target peaks were not sufficiently isolated (as described in Section 2.2 Step 2) to be included in the reduced peak list.
To illustrate the effectiveness of our alignment procedure, Figure 3 shows histograms of the peak displacements (from the aligned time value) for the m/z = 7773 Da peak before (a) and after (b) alignment. Prior to alignment, jitter between spectra appears as integer jumps in the peak locations. Data from each lab clusters in the vicinity of its corresponding mean displacement (indicated by vertical arrows) revealing systematic differences between the data sets. The mean for Lab 6 does not coincide with the center of a cluster because data from this lab is split between two clusters (at approximately −20 and +32) which were acquired on two occasions, three months apart. (Data from Lab 6 required a preliminary step in which the data obtained on the two different dates were “roughly aligned” as described in Section 2.3.) Table 1 provides a numerical summary of the mean displacements. After alignment, the residual variation in peak location is significantly smaller and appears random. This successful alignment of data from 6 different sources without the application of calibrations demonstrates the power of our time domain alignment method.
Figure 3.
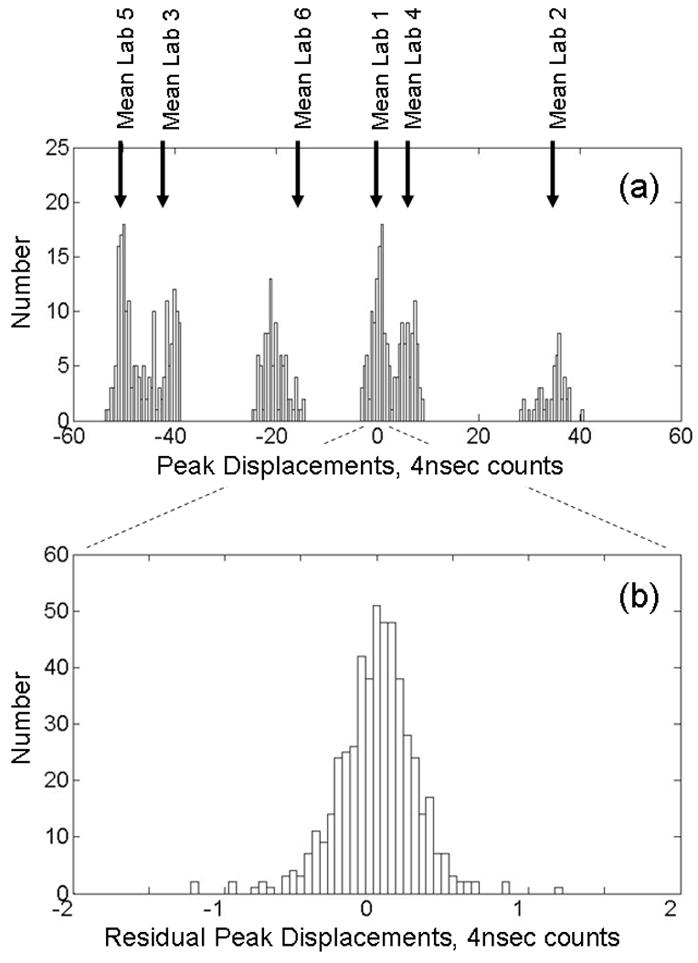
Histograms for m/z = 7773 Da peak displacements from aligned peak location for combined data set. (a) Before alignment, integer jumps show clock jitter. Mean displacements for individual labs are indicated by vertical arrows. (See Table 1 for numerical summary of means and standard deviations.) (b) After alignment, residual displacements are small and random. Note the change of scale. (This peak was used for alignment in both reference [25] and this study).
Table 1.
Mean peak displacements prior to alignment for m/z = 7773 Da.
| Lab 1 | Lab 2 | Lab 3 | Lab 4 | Lab 5 | Lab 6 | |
|---|---|---|---|---|---|---|
| Mean, 4 nsec Time counts | −0.01 | 34.46 | −42.20 | 5.85 | 50.15 | −15.47 |
This large multi-lab data set allows us to obtain statistics on the performance of our alignment procedure. While comparable data sets from other instruments are not available to us, we have also observed, and corrected, systematic time shift errors in standard spectra obtained using an Ultraflex III MALDI-TOF (Bruker Daltonik) data [27].
3.3 Standard deviations for peak time locations following label-free alignment
Figure 4 presents standard deviations for peak time locations after alignment for 13 peaks that are detected in all the data sets. The m/z values are obtained using a quadratic fit to the three target peaks identified in reference [25]. Peak location variability is reduced to below 1 time count in 89 of 91 instances. The inter-laboratory variability (solid symbols) is comparable with the intra-lab variability indicating that the alignment procedure has reached the limit of precision given the uncertainty predicted by maximum likelihood peak detection. The higher variability seen in the Lab 6 data set may be the result of the combination of two smaller data sets acquired on different days. The lower variability seen in the Lab 4 data results from a higher sample rate used for data acquisition (twice the rate used by the other labs).
Figure 4.
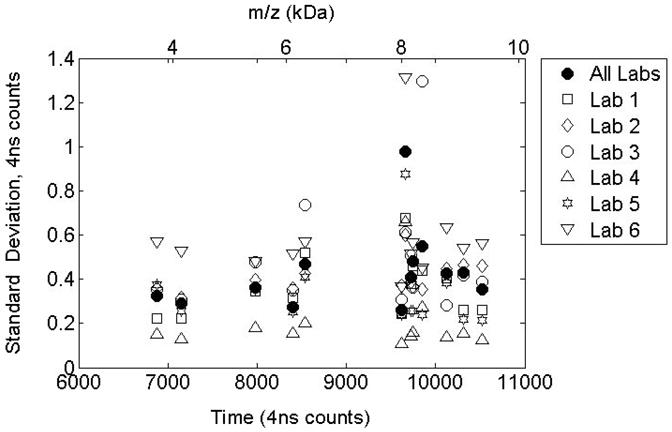
Standard Deviation in peak time locations following alignment. Variability is reduced to below 1 time count for most cases. Inter-laboratory variability is comparable to intra-laboratory variability.
3.4 Relation between uncertainty in peak time location and S/N
One peak in Figure 4, t = 9663 counts, has a notably higher standard deviation than the others. This is due to a low S/N (peak signal divided by the root-mean-square noise in the window.) Figure 5 presents a portion of a typical spectrum that contains this peak and a neighboring peak with a high S/N (t = 9621 counts, one of the peaks used for alignment process). Histograms for the residual peak displacements for both peaks provide insight into the alignment results. For these peaks, lower S/N is associated with higher uncertainty in the peak detection and greater residual variation after alignment. Studies with simulated data show an inverse relationship between σt and Here n is the number of time points in FWHM and C is a constant of order 1. This agrees with the theory of the peak detection method.
Figure 5.
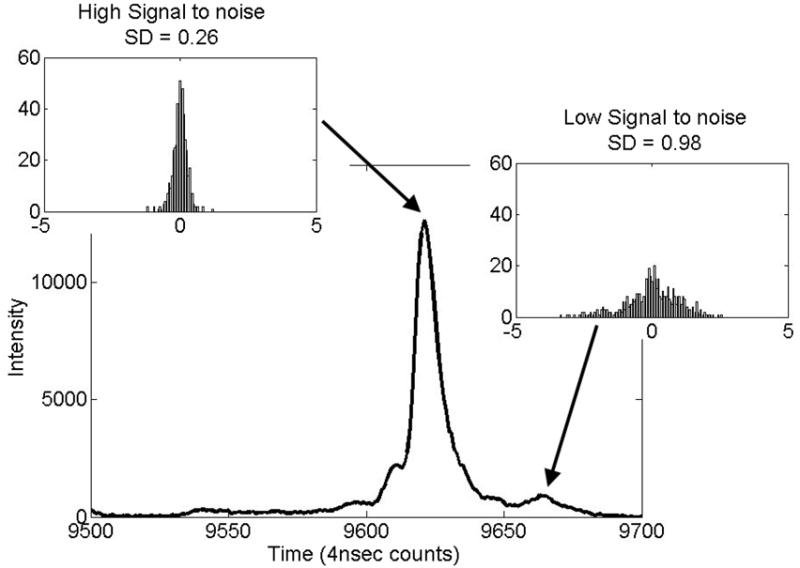
Histograms illustrating successful alignment of two peaks with different S/Ns. The peak at t = 9621 was used for alignment in reference [25] and in this study. The peak at t = 9663 has a significantly lower S/N and associated higher location uncertainty.
3.5 Random residual errors after label-free alignment
In order to determine if the residual peak displacements are random or might still contain systematic errors, displacement histograms are fit to probability density functions. Figure 6 presents histograms for four peaks with a t probability density function overlaid. The two plots on the left, a) and c), correspond to peaks used for label-free alignment (included in the reduced peak list) and the two plots on the right, b) and d), correspond to peaks to which corrections were applied (included in the original peak lists but not in the reduced peak list used for determination of correction coefficients). All show good agreement with a t -distribution which supports our claims that the residual displacements after alignment are random and systematic timing errors between spectra have been removed. The use of a t -distribution rather than a normal distribution is appropriate for modeling aligned peak values which are effectively mean quantities. [28]
Figure 6.
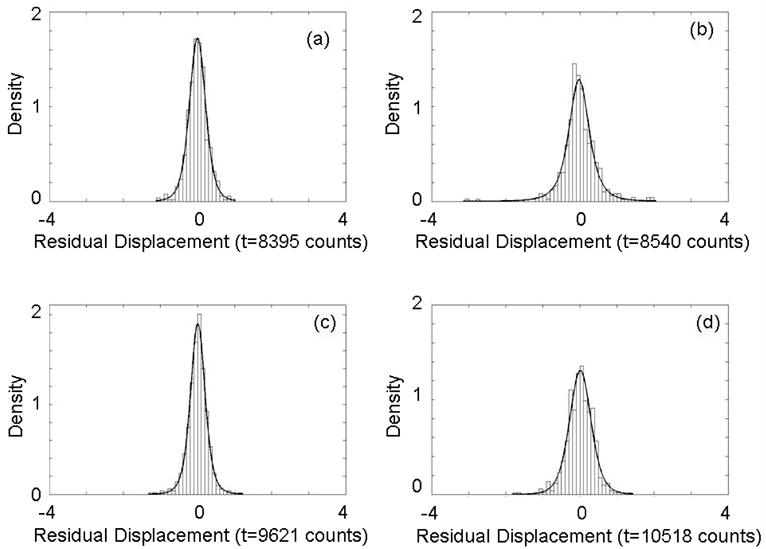
t -distribution functions fit to peak displacement histograms for four peaks after alignment show residual displacements are random. (a) m/z = 5659 Da peak. (b) m/z = 5910 Da peak. (c) m/z = 7773 Da peak (d) m/z = 9297 Da peak. Peaks at m/z = 5659 and 7773 Da were used for alignment (included in reduced peak list) and the peaks at 5910 and 9297 Da had corrections applied (included in original peak list). All peaks show good agreement between the histograms and a t -distribution consistent with removing all systematic timing errors.
3.6 CVs for peak m/z after label-free alignment
Low peak position uncertainty leads to remarkably high precision, as shown in Figure 7. For this figure, we translate the values for time precision into coefficients of variation for mass using . (The factor of 2 arises from the fact that m/z values are obtained by applying a quadratic calibration equation.) For reference, Figure 7 shows the previously published CV values (marked as X’s) for the three target peaks used for synchronization in the multi-lab standardization study [25]. A logarithmic scale is used to highlight the order of magnitude improvement is precision. For convenience, Table 2 reproduces the CV values for the 13 peaks detected in all data groups. Published values for the three target peaks from [25] are included and the peaks used for label-free alignment are indicated.
Figure 7.
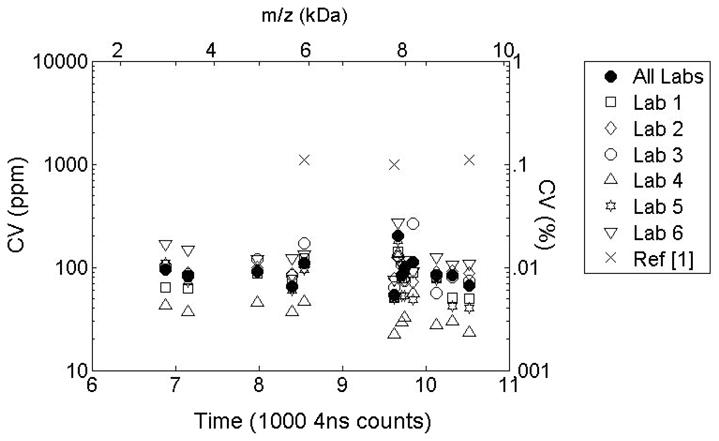
Coefficient of Variability for aligned peaks m/z values. Time domain alignment improves precision by at least an order of magnitude.
Table 2.
Coefficients of variation for peak m/z.
| Peaksa) | m/z CV, % | ||
|---|---|---|---|
| m/z, Da | Time counts | Label-free alignment | Ref [25] |
| 2995 | 6879 b) | 0.0094 | |
| 3485 | 7155 b) | 0.0081 | |
| 4941 | 7983 b) | 0.0090 | |
| 5659 | 8395 b) | 0.0065 | |
| 5910 c) | 8540 | 0.0110 | 0.11 |
| 7773 c) | 9621 b) | 0.0054 | 0.10 |
| 7845 | 9663 | 0.0202 | |
| 7939 | 9719 | 0.0084 | |
| 7989 | 9748 | 0.0099 | |
| 8164 | 9850 | 0.0111 | |
| 8630 | 10124 b) | 0.0084 | |
| 8955 | 10316 | 0.0084 | |
| 9297 c) | 10518 | 0.0067 | 0.11 |
Peaks detected in all data sets.
Peaks used for label-free alignment.
Target peaks from reference [25]
4 Concluding Remarks
We have shown significant improvements to TOF precision and, consequently, reproducibility in m/z using linear time domain methods. Peak detection with superior resolution is demonstrated and systematic instrument errors are identified and removed using linear offset and scale corrections. We expect that our post-acquisition data processing will have significant impact for researchers utilizing TOF data for expression pattern analysis, imaging MS, and improved MS/MS protein ID.
Acknowledgments
The authors gratefully acknowledge D. Manos at the College of William and Mary for stimulating discussions. This work was supported by NIH-National Cancer Institute grants CA101479 (MS), CA126118 (DIM) and CA085067 (OJS).
Abbreviations
- FWHM
peak full width at half maximum amplitude
- ppm
parts per million
- QC
quality control pooled-serum
m/z precision, the inverse of CV
- S/N
peak signal divided by root-mean-square noise in window
- t/σt
time precision
- σm/z
uncertainty (SD) in peak location in m/z
- σt
uncertainty (SD) in peak location in time
Appendix: Split spectra due to jitter
Triggering time error cannot be completely removed because each spectrum is itself the average of many laser shots, each with its associated jitter.[15] Without access to data from individual laser shots, it is therefore impossible to completely remove all triggering time error. Small timing jitter in averaged spectra usually appears as peak broadening, but sometimes the triggering jitter can jump between two widely separated vales, and this will split each peak into two (or more). Peak splitting due to timing jitter is distinguished from that resulting from the presence of chemical adducts by the fact that each peak is split by the same constant time increment over the entire TOF spectrum (from the low mass sodium peak, through the matrix region and throughout the mass focusing range). Figure A1 illustrates an egregious example of this timing jitter error in averaged spectra obtained at one site. The dotted line represents two peaks from one spectrum that appears to have little jitter, while the solid line represents the corresponding peaks from another spectrum that exhibits significant jitter. The clearly split peaks suggest that there are two main trigger start times associated with the single shot spectra and that the split spectra could be modeled as the average of two subgroups with different start times. A simulated split spectrum similar to the one in the figure is constructed by averaging 110 copies of the unsplit spectrum and 90 copies with a delayed start time of 12 clock ticks. The agreement we see between the simulated peaks (shown as X markers) and the observed split peaks is consistent for all the peaks in the split spectrum. Differences in amplitude are to be expected due to normal peak amplitude variations among replicates. To date, such peak splitting has not challenged the vendor software for peak detection and clustering alignment. However, our new high resolution peak detection method exposes this problem as an obstacle to alignment and improved precision. For this paper, spectra with split peaks are not included in the analysis and as a result, only 42 spectra for Lab 2 are analyzed.
Figure A1.
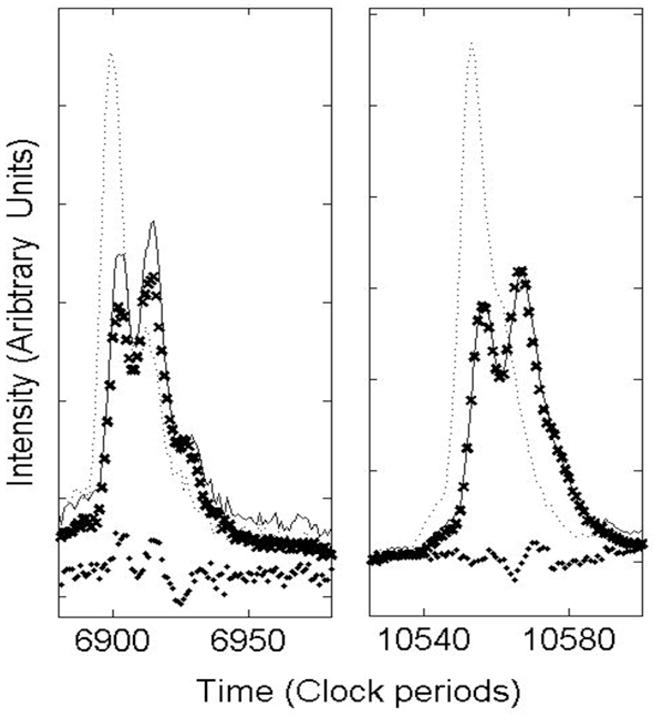
Timing jitter in averaged MALDI-TOF spectra. The dotted and solid lines represent two replicate spectra (averages of 200 shots each). To illustrate that the split spectra can arise from bimodal jitter, a simulated spectrum (represented by X’s) was generated by averaging copies of the dotted spectrum with one of two start times. The solid dots show the difference between the observed (solid line) and the simulated spectra.
References
- 1.Fenselau C, Demirev PA. Characterization of intact microorganisms by MALDI mass spectrometry. Mass Spectrom Rev 2001. 20;157–171 doi: 10.1002/mas.10004. [DOI] [PubMed] [Google Scholar]
- 2.Oleschuk RD, McComb ME, Chow A, Ens W, et al. Characterization of plasma proteins adsorbed onto biomaterials by MALDI-TOFMS. Biomaterials. 2000;21:1701–1710. doi: 10.1016/s0142-9612(00)00054-5. [DOI] [PubMed] [Google Scholar]
- 3.Caprioli RM, Farmer TB, Gile J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. Anl Chem. 1997;69(23):4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 4.Chaurand P, DaGue BB, Pearsall RS, Threadgill DW, Caprioli RM. Profiling proteins from azoxymethane-induced colon tumors at the molecular level by matrix-assisted laser desorption/ionization mass spectrometry. Proteomics. 2001;1:1320–1326. doi: 10.1002/1615-9861(200110)1:10<1320::AID-PROT1320>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 5.Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–577. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 6.Yasui Y, McLerran D, Adam BL, Winget M, et al. An automated peak identification/calibration procedure for high-dimensional protein measures from mass spectrometers. J Biomed Biotechnol. 2003;4:242–248. doi: 10.1155/S111072430320927X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adam BL, Qu Y, Davis JW, Ward MD, et al. Serum protein fingerprinting coupled with a pattern-matching algorithm distinguishes prostate cancer from benign prostate hyperplasia and healthy men. Cancer Res. 2002;62(13):3609–3614. [PubMed] [Google Scholar]
- 8.Won Y, Song H-J, Kang TW, Kim J-J, et al. Pattern analysis of serum proteome distinguishes renal cell carcinoma from other urologic diseases and healthy persons. Proteomics. 2003;3:2310–2316. doi: 10.1002/pmic.200300590. [DOI] [PubMed] [Google Scholar]
- 9.Ressom HW, Varghese RS, Abdel-Hamid M, Eissa SAL, et al. Analysis of mass spectral serum profiles for biomarker selection. Bioinformatics. 2005;21(21):4039–4045. doi: 10.1093/bioinformatics/bti670. [DOI] [PubMed] [Google Scholar]
- 10.Yanagisawa K, Shyr Y, Xu BJ, Massion PP, et al. Proteomic patterns of tumour subsets in non-small-cell lung cancer. Lancet. 2003;362:433–439. doi: 10.1016/S0140-6736(03)14068-8. [DOI] [PubMed] [Google Scholar]
- 11.Tibshirani R, Hastie T, Narasimhan B, Soltys S, et al. Sample classification from protein mass spectrometry, by ‘peak probability contrasts’. Bioinformatics. 2004;20(17):3034–3044. doi: 10.1093/bioinformatics/bth357. [DOI] [PubMed] [Google Scholar]
- 12.Kazmi SA, Ghosh S, Shin DG, Hill DW, Grant DF. Alignment of high resolution mass spectra: development of a heuristic approach for metabolomics. Metabolomics. 2006;2(2):75–83. doi: 10.1007/s11306–006–0021–7. [DOI] [Google Scholar]
- 13.Beyer S, Walter Y, Hellmann J, Kramer PJ, et al. Comparision of software tools to improve the detection of carcinogen induced changes in the rat liver proteome by analyzing SELDI-TOF-MS spectra. J Proteome Res. 2006;5:254–261. doi: 10.1021/pr050279o. [DOI] [PubMed] [Google Scholar]
- 14.Rai AJ, Stemmer PM, Zhang Z, Adam BL, et al. Analysis of human proteome organization plasma proteome project (HUPO PPP) reference specimens using surface enhanced laser desorption/ionization-time of flight (SELDI-TOF) mass spectrometry: multi-institution correlation of spectra and identification of biomarkers. Proteomics. 2005;5:3467–3474. doi: 10.1002/pmic.200401320. [DOI] [PubMed] [Google Scholar]
- 15.Malyarenko DI, Cooke WE, Adam B-L, Malik G, et al. Enhancement of sensitivity and resolution of surface-enhanced laser desorption/ionization time-of-flight mass spectrometric records for serum peptides using time-series analysis techniques. Clin Chem. 2005;51(1):65–74. doi: 10.1373/clinchem.2004.037283. previously published online at DOI:10.1373/clinchem.2004.037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villanueva J, Philip J, DeNoyer L, Tempst P. Data analysis of assorted serum peptidome profiles. Nature Protocols. 2007;2(3):588 – 602. doi: 10.1038/nprot.2007.57. previously published online at DOI:10.1038/nprot.2007.57. [DOI] [PubMed] [Google Scholar]
- 17.Baggerly KA, Morris JS, Coombes KR. Reproducibiliy of SELDI-TOF protein patterns in serum: comparing data sets from different experiments. Bioinformatics. 2004;20(5):777–785. doi: 10.1093/bioinformatics/btg484. [DOI] [PubMed] [Google Scholar]
- 18.Dekker LJ, Dalebout JC, Siccama I, Jenster G, et al. A new method to analyze matrix-assisted laser desorption/ionization time-of-flight peptide profiling mass spectra. Rapid Commun Mass Spectrom. 2005;19:865–870. doi: 10.1002/rcm.1864. 10.1002/rcm.1864. [DOI] [PubMed] [Google Scholar]
- 19.Villanueva J, Philip J, Chaparro CA, Li Y, et al. Correcting common errors in identifying cancer-specific serum peptide signatures. J Proteome Res. 2005;4:1060–1072. doi: 10.1021/pr050034b. 10.1021/pr050034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong JWH, Cagney G, Cartwright HM. SpecAlign-processing and alignment of mass spectra datasets. Bioinformatics. 2005;21(9):2088–2090. doi: 10.1093/bioinformatics/bti300. 10.1093/bioinformatics/bti300. [DOI] [PubMed] [Google Scholar]
- 21.Yu W, Wu B, Lin N, Stone K, et al. Detecting and aligning peaks in mass spectrometry data with applications to MALDI. Comput Biol Chem. 2006;30:27–38. doi: 10.1016/j.compbiolchem.2005.10.006. 10.1016/j.compbiolchem.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Yu W, Li X, Liu J, Wu B, et al. Multiple peak alignment in sequential data analysis: a scale-space-based approach. IEEE/ACM Trans Comput Biol Bioinform. 2006;3(3):208–219. doi: 10.1109/TCBB.2006.41. [DOI] [PubMed] [Google Scholar]
- 23.Jeffries N. Algorithms for alignment of mass spectrometry proteomic data. Bioinformatics. 2005;21(14):3066–3073. doi: 10.1093/bioinformatics/bti482. 10.1093/bioinformatics/bti482. [DOI] [PubMed] [Google Scholar]
- 24.Lin SM, Haney RP, Campa MJ, Fitzgerald MC, et al. Characterizing phase variations in MALDI-TOF data and correcting them by peak alignment. Cancer Informatics. 2005;1:32–40. http://la-press.com/cr_data/files/f_CI-1-1-LinSc_222.pdf. [PMC free article] [PubMed] [Google Scholar]
- 25.Semmes OJ, Feng Z, Adam BL, Banez LL, et al. Evaluation of serum protein profiling by surface-enhanced laser desorption/ionization time-of-flight mass spectrometry for the detection of prostate cancer: I. assessment of platform reproducibility. Clin Chem. 2005;51:1,102–112. doi: 10.1373/clinchem.2004.038950. previously published online at DOI:10.1373/clinchem.2004.038950. [DOI] [PubMed] [Google Scholar]
- 26.Tracy ER, Chen H, Cooke WE. Automatic peak identification method. U.S. Patent No. 7,219,038, May 15, 2007 (Assigned to the College of William and Mary) [Google Scholar]
- 27.Gatlin-Bunai CL, Cazares LH, Cooke WE, Semmes OJ, Malyarenko DI. Optimization of MALDI-TOF MS detection for enhanced sensitivity of affinity-captured proteins spanning a 100 kDa mass range. J Proteome Res. 2007 doi: 10.1021/pr0703526. ASAP Article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sivia DS. Data Analysis: A Bayesian Tutorial. Oxford University Press; New York: 1996. [Google Scholar]