

Abstract
A gene encoding Trypanosoma brucei UDP-N-acetylglucosamine pyrophosphorylase was identified, and the recombinant protein was shown to have enzymatic activity. The parasite enzyme is unusual in having a strict substrate specificity for N-acetylglucosamine 1-phosphate and in being located inside a peroxisome-like microbody, the glycosome. A bloodstream form T. brucei conditional null mutant was constructed and shown to be unable to sustain growth in vitro or in vivo under nonpermissive conditions, demonstrating that there are no alternative metabolic or nutritional routes to UDP-N-acetylglucosamine and providing a genetic validation for the enzyme as a potential drug target. The conditional null mutant was also used to investigate the effects of N-acetylglucosamine starvation in the parasite. After 48 h under nonpermissive conditions, about 24 h before cell lysis, the status of parasite glycoprotein glycosylation was assessed. Under these conditions, UDP-N-acetylglucosamine levels were less than 5% of wild type. Lectin blotting and fluorescence microscopy with tomato lectin revealed that poly-N-acetyllactosamine structures were greatly reduced in the parasite. The principal parasite surface coat component, the variant surface glycoprotein, was also analyzed. Endoglycosidase digestions and mass spectrometry showed that, under UDP-N-acetylglucosamine starvation, the variant surface glycoprotein was specifically underglycosylated at its C-terminal Asn-428 N-glycosylation site. The significance of this finding, with respect to the hierarchy of site-specific N-glycosylation in T. brucei, is discussed.
Trypanosoma brucei is the causative agent of African sleeping sickness in humans and nagana in cattle and is transmitted between mammalian hosts by the bite of the tsetse fly (Glossina spp.). T. brucei transmission occurs when the bloodstream form of the parasite is ingested by a tsetse fly during feeding, the parasite then differentiates into the procyclic form in order to colonize the tsetse fly midgut. The parasite undergoes further differentiation and migration to the salivary gland of the fly in order to infect a new mammalian host upon a subsequent bloodmeal. T. brucei and the related trypanosomatid parasites Trypanosoma cruzi and Leishmania, express an interesting array of glycoconjugates, some of which are essential to parasite survival and infectivity (reviewed in Refs. 1-4). This has led to the investigation of potential therapeutic targets in parasite glycoconjugate biosynthesis, such as enzymes of glycosylphosphatidylinositol (GPI)3 biosynthesis (5-8) and enzymes of sugar nucleotide biosynthesis. With respect to the latter, GDP-Man biosynthesis has been shown to be essential for the infectivity of Leishmania mexicana (9-13), and UDP-glucose 4′-epimerase, the only source of UDP-Gal in T. brucei, has been shown to be essential for both bloodstream form and procyclic form T. brucei (14-16) and is likely to be essential for epimastigote form T. cruzi (17). Recently, the synthesis of GDP-fucose (GDP-Fuc) has been show to be essential for flagellar adhesion and cell growth in T. brucei (18), and measurement of sugar nucleotide levels in trypanosomatids has indicated that the intracellular pools of these metabolites are highly dynamic (19).
Sugar nucleotides are activated forms of sugars that are used as the ultimate source of sugar for the majority of glycosylation reactions. Sugar nucleotides are formed in two main ways: by a salvage pathway, involving activation of the sugar using a kinase and a pyrophosphorylase, or by a de novo pathway involving the bioconversion of an existing sugar/sugar nucleotide. In most cases, sugar nucleotides are synthesized in the cytoplasm and used there and/or transported through specific transporters into the lumen of the Golgi apparatus and/or endoplasmic reticulum (ER), where they are used by glycosyltransferases as donor substrates in glycosylation reactions (20, 21).
The sugar nucleotide UDP-GlcNAc is predicted to be an important metabolite in the trypanosomatid parasites, since GlcNAc is present in glycoprotein N-linked glycans in all species and in O-linked glycans in T. cruzi (22, 23), and GlcN, derived from GlcNAc by de-N-acetylation (24), is present in all species in protein-linked and free GPI structures. In T. brucei, GlcNAc is also found in N-acetyllactosamine (LacNAc) repeats of Galβ1-4GlcNAc. These LacNAc structures are found in conventional complex N-linked glycans (25) and as part of giant poly-LacNAc-containing N-linked glycans throughout the flagellar pocket and endosomal/lysosomal system of the bloodstream form (26, 27) and as side chains of the procyclin GPI anchor and free GPIs in the procyclic form of the organism (15, 28, 29). The ability to biosynthetically radiolabel T. brucei glycoproteins with [3H]GlcN (30, 31) shows that a salvage pathway exists, presumably via the action of hexokinase (GlcN → GlcN 6-phosphate). However, most likely, the de novo pathway from glucose is the most important in vivo, since free GlcN is not an abundant sugar in either mammals or insects, and its N-acetyl derivative (GlcNAc) is not taken up by T. brucei (32).
In this work, we demonstrate that the putative T. brucei uridine-acetylglucosamine pyrophosphorylase gene (TbUAP) encodes a functional enzyme (EC 2.7.7.23) and, by making a bloodstream form T. brucei TbUAP conditional null mutant, demonstrate that TbUAP is essential in vitro and in vivo. We also characterize the effects of UDP-GlcNAc starvation on parasite protein glycosylation and uncover a hierarchy in protein N-glycosylation in T. brucei.
EXPERIMENTAL PROCEDURES
Parasite Culture—T. brucei bloodstream form parasites strain 427, variant MITat1.2 (also known as variant 221), that express T7 polymerase and tetracycline repressor protein under G418 selection were cultured in HMI-9 medium (33) up to a density of ∼2 × 106 cells/ml at 37 °C with 5% CO2.
Cloning and Sequencing of TbUAP—The TbUAP open reading frame identified in the T. brucei genome data base was amplified by PCR from genomic DNA with Pfu polymerase using forward and reverse primers containing BamHI sites (underlined) of 5′-cgcggatccaatgagtgacagggacgtgtg-3′ and 5′-cgcggatccttacatgttcgatgattcgg-3′, respectively. The products of six separate PCRs were cloned into pCR-BluntII-Topo®, and a representative clone from each PCR was sequenced. The primer 5′-cgcagcggttcttcgaggagaattcctac-3′ was also used to obtain complete sequence coverage of the ORF.
Reverse Transcription-PCR—RNA was extracted using the RNeasy extraction kits with on-column DNase digestion (RNase-free DNase; Qiagen). RNA samples (50 ng) were treated with Ominiscript reverse transcriptase (Qiagen) to generate cDNA. The cDNAs were then amplified by PCR using Taq polymerase and TbUAP ORF primers (forward, 5′-aatgagtgacagggacgtgtg-3′; reverse, 5′-ttacatgttcgatgattcgg-3′) and DPMS (Dol-P-Man synthetase) primers (forward, 5′-aatggatgcggaccttcagcacccac-3′; reverse, 5′-tagaaccgtgagcgcggtgccatac-3′) to show equal RNA addition.
Southern Blotting—Genomic DNA (5 μg) was digested with appropriate restriction endonucleases. A DNA probe was made using the TbUAP ORF and the random primer labeling kit (GE Healthcare). The probe was then detected using the CDP-Star™ detection kit (GE Healthcare).
TbUAP Protein Expression and Purification—The TbUAP ORF was cloned into the BamHI site of the expression vector pET15b (Novagen) to create pET15b-TbUAP, which incorporated a His6 tag when expressed. Expression was performed using BL21 (DE3) Escherichia coli. The cells were grown overnight at room temperature with 0.05 mm isopropyl β-d-1-thiogalactopyranoside. Cells were harvested and washed in 50 mm Tris-HCl, pH 8.0, 0.3 m NaCl, 1 mg/ml lysozyme, and Roche complete protease inhibitor mixture tablets (Roche Applied Science) and then lysed in a French press. The lysate was cleared by centrifugation (40,000 × g, 60 min, 4 °C), passed through a 0.2-μm filter, and loaded onto a precharged Ni2+ HiTrap™ chelating HP column (GE Healthcare). TbUAP-His6 was eluted with 50 mm Tris-HCl (pH 8.0), 0.3 m NaCl with 0.1-0.2 m imidazole. The protein was then dialyzed overnight using a Slide-A-Lyser® dialysis cassette (Pierce) with 10 kDa molecular mass cut-off at 4 °C in 25 mm Na2HPO4-NaH2PO4 buffer, pH 8.0. The sample was then filtered as above before being loaded onto a HiTrap™ Q HP-Sepharose column (Amersham Biosciences), preequilibrated with 25 mm Na2HPO4-NaH2PO4, pH 8.0. The column was washed with 25 mm Na2HPO4-NaH2PO4, pH 8.0, followed by a gradient to 25 mm Na2HPO4-NaH2PO4, pH 8.0, 0.5 m NaCl over 30 min. Fractions (3 ml) were collected and checked by SDS-PAGE. TbUAP-His6-containing fractions were pooled and concentrated, and the buffer was exchanged to 50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, and 20% glycerol using a Vivaspin concentrator (Vivascience) 10 kDa molecular mass cut off at 4 °C. The protein was then stored at -80%.
To obtain a PreScission protease cleavable His6-tagged TbUAP protein, the TbUAP open reading frame was amplified by PCR from the aforementioned pET15b-TbUAP plasmid using the forward primer, 5′-ctccatgggcagcagccatcatcatcatcatcacagcagcggcctggaagttctgttccaggggcccggatccATGAGTGACAGGGACGTGTGCATTCAG-3′, containing an NcoI restriction site (underlined), the coding sequences for MGSSHHHHHHSSG (italic type), and a PreScission protease cleavage site of LEVLFQGP (boldface type), followed by a BamHI restriction site (underlined) and a TbUAP gene-specific sequence (uppercase) and the reverse primer, 5′-gctcgagatctggatccTTACATGTTCGATGATTCGGAGACCACC-3′, containing restriction sites for BglII and BamHI (italic type) and a TbUAP gene-specific sequence (uppercase). The PCR product was cloned into the pGEM-T Easy PCR cloning vector (Promega) and subsequently digested with NcoI and BglII and inserted between the NcoI and BamHI sites of the pET15b protein expression vector (Novagen). The resulting construct, pET15b-His6-PP-TbUAP, that encodes the full TbUAP coding sequence preceded by the sequence MGSSHHHHHHSSGLEVLFQGPGS (where PreScission protease cleaves between the Q and the G) was expressed in E. coli and purified on an Ni2+ HiTrap™ chelating HP column, as described above. The sample was then digested with ∼2 mg of GST-PreScission protease (a kind gift of Bill Hunter; University of Dundee) in 50 mm Tris, pH 8.0, 100 mm NaCl, 10 mm EDTA, and 1 mm dithiothreitol at room temperature for 4-16 h at 4 °C. The sample was dialyzed for 2 h using a Slide-A-Lyser® dialysis cassette (10 kDa molecular mass cut-off) at 4 °C in 2 liters of 50 mm Tris-HCl, pH 8.0, and 50 mm NaCl to remove the EDTA, and then the sample was passed though a 0.2-μm syringe filter. The sample was passed through a GSTrap™ HP column (GE Healthcare) connected to an Ni2+ HiTrap™ chelating HP column. The flow-through was then dialyzed overnight using a Slide-A-Lyser® dialysis cassette at 4 °C in 1 liter of 25 mm Na2HPO4-NaH2PO4, pH 8.0, with two changes of buffer. The sample was then passed though a 0.2-μm syringe filter and further purified using anion exchange chromatography on a HiTrap™ Q HP-Sepharose column (GE Healthcare).
TbUAP Assays—Two methods were used to assay TbUAP. The HPLC assay used 0.05 μg of TbUAP-His6 incubated in 100 μl of the HPLC assay buffer (50 mm Tris-HCl, pH 7.5, 250 μm UTP, 10 mm MgCl2, 1 mm dithiothreitol, 20% glycerol, 250 μm GlcNAc-1-P) for 10 min, terminated by boiling for 5 min. The samples were analyzed using conditions based on Ref. 34. The HPLC assay buffer was altered to study substrate specificity, metal ion dependence, and pH dependence. For substrate specificity, GlcNAc-1-P was changed to glucose 1-phosphate, galactose 1-phosphate, or GalNAc-1-P, all at 250 μm. For metal ion dependence, MgCl2 was replaced with CaCl2, CuCl2, ZnCl2, or MnCl2. For pH dependence, the Tris-HCl buffer was replaced with a dual buffer of 50 mm Tris, 50 mm sodium acetate with the pH adjusted with HCl.
The TbUAP colorimetric assay was performed with 0.05 μg of TbUAP-His6 in a 96-well plate format (Nunc™) in 90 μl of 50 mm Tris-HCl, pH 7.5, 250 μm UTP, 250 μm GlcNAc-1-P, 10 mm MgCl2, 1 mm dithiothreitol, 20% glycerol, 0.04 units/ml pyrophosphatase (Sigma). The reaction was left for 10 min and terminated by the addition of 100 μl of the color reagent (0.2% ammonium molybdate, 0.5% Triton X-100, 0.7 n HCl, 0.03% malachite green). Absorbance at 655 nm was measured after 5 min using a SpectraMax 340 PC (Molecular Devices).
Construction of a TbUAP Conditional Null Mutant—The gene replacement cassettes were generated by PCR amplification of 500 bp of UTR immediately flanking the 5′- and 3′-ends of the TbUAP ORF with Taq polymerase using the forward and reverse primers 5′-aaggaaaaaaGCGGCCGCagatgcgtgcacaacaaaaa-3′ and 5′-gtttaaacttacggaccgtcaagctttatctataacacacggagcc-3′ and 5′-gacggtccgtaagtttaaacggatccgtggacgttgcagcgcccgg-3′ and 5′-aaggaaaaaaGCGGCCGCcaccacagttcaccatccag-3, respectively. The two PCR products were then used in a separate PCR to produce a construct containing the 5′-UTR linked to the 3′-UTR by a short HindIII, PmeI, and BamHI cloning site (italic type). The resulting PCR product was then ligated into pGEM-5Zf(+) vector (Promega) using the NotI site (uppercase). Antibiotic resistance markers were cloned into the HindIII/BamHI restriction sites between the two UTRs to produce two constructs, one containing the PAC (puromycin acetyltransferase) drug resistance gene and one containing the HYG (hygromycin phosphotransferase) drug resistance gene. To generate the tetracycline-inducible ectopic copy of the TbUAP ORF, the Nde1 site in the ORF was silenced using the primers 5′-aagcttgggatagcatacgtgcagattggaa-3′ and 5′-attccaatctgcacgtatgctatcccaagct-3′. The primers 5′-catatgatgagtgacagggacgtgtg-3′ and 5′-ttaattaattacatgttcgatgattcgg-3′ were then used to PCR-amplify the ORF, which was cloned into the vector pLew100 using the NdeI and PacI sites (italic type) (33).
These constructs were purified using the Qiagen Maxiprep kit, digested with NotI to linearize, precipitated, washed twice with 70% ethanol, and redissolved in sterile water. The linearized DNA was electroporated into T. brucei bloodstream cells (strain 427, variant 221) that were stably transformed to express T7 RNA polymerase and the tetracycline repressor protein under G418 selection. Cell culture, transformation, and selection were carried out as previously described (33).
Mouse Infection Studies—The TbUAP conditional null mutant cells were subcultured and grown without selection drugs (hygromycin, puromycin, phleomycin, and G418) for 24 h with and without 1 μg/ml tetracycline. The parasites were then introduced into groups of five mice (dosed with and without doxycycline, respectively) by intraperitoneal injection of 3 × 105 parasites in 0.2 ml of HMI-9 medium. The plus doxycycline group of animals were dosed with doxycycline in the drinking water (0.2 mg/ml in a 5% sucrose solution) for 1 week prior to infection and until the experiment was terminated. Infections were assessed by tail bleeding, diluting the blood 1:200 in HMI-9 medium and counting on a Neubauer hemocytometer.
TbUAP Localization—Two BALB/c adult mice were used to raise polyclonal antibodies against His6-tagged TbUAP protein with Freund's complete adjuvant. Each mouse received two further immunizations with Freund's incomplete adjuvant over 2 months. Antibodies were then affinity-purified on CNBr-Sepharose-immobilized TbUAP that had had its His6 tag removed with PreScission protease.
Wild type and TbUAP conditional null mutant bloodstream form T. brucei cells were grown in HMI-9 medium (with or without 1 μg/ml tetracycline for the conditional null mutant) to a density of 1 × 106 cells/ml over 48 h, harvested by centrifugation, and resuspended in trypanosome dilution buffer (0.1 m Na2HPO4, 0.01 m NaH2PO4, 0.025 m KCl, 0.4 m NaCl, 5 mm MgSO4, 0.1 m glucose adjusted to pH 7.45 with HCl) to a density of 4 × 107 cells/ml. Aliquots (15 μl) were added to 13-mm coverslips (VWR), left at room temperature for 15 min, fixed in 1 ml of 4% paraformaldehyde in phosphate-buffered saline (PBS) for 1 h followed by three 5 min washes in 2 ml of PBS. Cells were permeabilized with 0.05% Triton X-100 in PBS containing 0.5 mg/ml bovine serum albumin for 10 min at room temperature. Samples were then blocked in 2 ml of PBS, 0.5% bovine serum albumin, for 1 h at room temperature. The coverslips were incubated with mouse anti-TbUAP (1:5,000 dilution) and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antiserum (1:10,000; a kind gift of Paul Michels, Catholic University of Louvain) in PBS, 0.5% bovine serum albumin. Samples were then washed, as above, in PBS, 0.5% bovine serum albumin and incubated with 50 μl of Alexa 594-conjugated anti-mouse IgG and Alexa 488-conjugated anti-rabbit IgG (containing 4′,6-diamidino-2-phenylindole in the case of the wild type cells) for 1 h. Coverslips were mounted on glass slides (VWR), sealed with Hydromount containing 2.5% 1,4-diazabicyclo[2.2.2]octane and left to dry in the dark for 30 min. Microscopy was performed on a Zeiss Axiovert 200 M fluorescence microscope for wild type cells and on a Zeiss LSM 510 META confocal microscope for the TbUAP conditional null mutant cells.
Sugar Nucleotide Analysis—Sugar nucleotide analysis was performed as described elsewhere (19). Briefly, cells were pelleted by centrifugation, washed in ice-cold PBS, and lysed in 70% ethanol in the presence of 10 pmol of GDP-glucose internal standard. Sugar nucleotides were extracted using EnviCarb columns (35) and analyzed using multiple reaction-monitoring liquid chromatography-tandem mass spectrometry (19).
Lectin and Antibody Blotting—T. brucei cells washed with trypanosome dilution buffer and hypotonically lysed in 300 μl of water containing 0.1 mm 1-chloro-3-tosylamido-7-amino-2-heptone (TLCK) and 1 μg/ml leupeptin. Cell ghosts were harvested by centrifugation (13,000 × g for 10 min), and the pellet was resuspended in SDS-sample buffer containing 8 m urea. The lysed extracts were then subjected to electrophoresis under reducing conditions, with 1.5 × 107 or 5 × 107 cell equivalents/lane, on a NuPAGE® 4-12% BisTris gradient (Invitrogen) using MOPS SDS running buffer. Proteins were then transferred to a nitrocellulose membrane under normal Western blotting conditions. Membranes were stained with Ponceau S solution to demonstrate equal loading, blocked with 0.25% bovine serum albumin, 0.05% Igepal detergent (Sigma), 0.15 m NaCl in 50 mm Tris-HCl, pH 7.4, and then incubated with 0.33 μg/ml biotinylated tomato lectin (Vector Laboratories), with or without 3 mg/ml chitin hydrolysate (Vector Laboratories), and then with 1:10,000 diluted ExtraAvidin-horseradish peroxidase conjugate (Sigma). All membranes were then developed by chemiluminescent detection (ECL-plus; GE Healthcare).
To probe for p67, T. brucei was lysed with SDS-sample buffer and loaded onto a 10% SDS-polyacrylamide gel before being transferred to nitrocellulose membrane. The membrane was then probed with MAb139 (a kind gift from Jay Bangs, Madison) at a dilution of 1:2,000 as the primary antibody and then with 1:10,000 diluted anti-mouse IgG conjugated with horseradish peroxidase, followed by ECL reagent as described above.
Purification and Endoglycosidase Digestion of Soluble Form Variant Surface Glycoprotein (sVSG)—The VSG coat of trypanosomes can be conveniently released in a soluble form through osmotic cell lysis at 37 °C. This causes cleavage of the dimyristoylglycerol component of the GPI membrane anchors by the action of an endogenous GPI-specific phospholipase C (30). T. brucei cultures (100 ml) were washed in trypanosome dilution buffer and resuspended in 300 μl of lysis buffer (10 mm NaH2PO4-Na2HPO4, pH 8.0, 0.1 mm TLCK, 1 μg/ml leupeptin, and 1 μg/ml aprotinin) and incubated at 37 °C for 10 min. This was then cooled on ice for 2 min and centrifuged for 5 min at 16,000 × g, and the supernatant was applied to 200 μl of DE52 (Whatman) preequilibrated in 10 mm NaH2PO4-Na2HPO4, pH 8.0, buffer and eluted with 4 × 200 μl of fresh lysis buffer. The eluates were pooled and concentrated to ∼100 μl using a YM-10 spin concentrator (Microcon). The majority of the buffer salts were removed by diafiltration with three additions of 0.5 ml of water.
For each enzyme digestion, sVSG was dissolved at 0.2 μg/μl in 0.5% SDS, 0.1 m dithiothreitol and boiled for 10 min. For endoglycosidase H (Endo H) digests, 5 μl of the sVSG was added to 20 μl of 50 mm sodium citrate, pH 5.5, 10 mm phenylmethylsulfonyl fluoride, and 0.025 units of Endo H. For N-glycosidase F (PNGase F) digests, 5 μl of sVSG was added to 50 mm sodium phosphate, pH 7.5, 0.1% Triton X-100, 10 mm phenylmethylsulfonyl fluoride, and 0.025 units of PNGase F. The digests were then left overnight at 37 °C.
Electrospray Mass Spectrometry of sVSG—Intact sVSG was diluted to 0.05 μg/μl in 50% methanol, 1% formic acid and loaded into Micromass type-F nanotips. The sVSG was analyzed by positive ion electrospray tandem mass spectrometry using an Applied Biosystems Q-StarXL instrument, and the masses were calculated using the Bayesian protein reconstruction algorithm (ABI Analyst Software).
RESULTS
Identification, Cloning, and Expression of TbUAP—A BLASTp search of the T. brucei predicted protein data base with Homo sapiens and Saccharomyces cerevisiae UAP amino acid sequences, accession numbers NP_003106 and NP_ 010180, revealed a putative TbUAP gene (gene number Tb11.02.0120). The TbUAP open reading frame was amplified by PCR using Pfu polymerase from genomic DNA prepared from T. brucei strain 427. Two different consensus sequences emerged (accession numbers AM909685 and AM909686), suggesting that there are two slightly different TbUAP alleles, with eight base pair differences and one amino acid difference between them (Ser or Gly at position 507). Southern blot analysis using a TbUAP ORF probe confirmed that TbUAP is present as a single copy per haploid genome (supplemental Fig. 1). The gene encoding the Ser-507 variant was used throughout this study. The TbUAP amino acid sequence contains the expected pyrophosphorylase LX2GXGTX6PK motif, and 13 of 15 residues shown to be involved in substrate binding in the human UAP crystal structures (36) are conserved in the TbUAP sequence (supplemental Fig. 2).
The TbUAP ORF was cloned into a pET15b expression vector, with a His6 tag at the N terminus and a modified pET15b vector with a PreScission protease site between the protein and the His6 tag. Both were expressed in E. coli and purified as described under “Experimental Procedures” (supplemental Figs. 3 and 4). Analysis of the recombinant TbUAP-His6 protein by analytical ultracentrifugation (at 1 mg/ml) indicated that the majority (>80%) of TbUAP is monomeric at this concentration (data not shown).
Enzymatic Activity of TbUAP—The activity and substrate specificity of TbUAP was assessed by incubating recombinant TbUAP-His6 with UTP and GlcNAc-1-P, GalNAc-1-P, Glc 1-phosphate, or Gal 1-phosphate and analyzing the products by HPLC. Using GlcNAc-1-P as the substrate, a single UV-absorbing peak that co-eluted with authentic UDP-GlcNAc was observed (Fig. 1A). No sugar nucleotide product was observed in the absence of TbUAP-His6 (Fig. 1B) or when GalNAc-1-P, Glc 1-phosphate, or Gal 1-phosphate was used as a substrate (Fig. 1, C-E). These data show that GlcNAc-1-P is the preferred substrate of TbUAP under these conditions.
FIGURE 1.
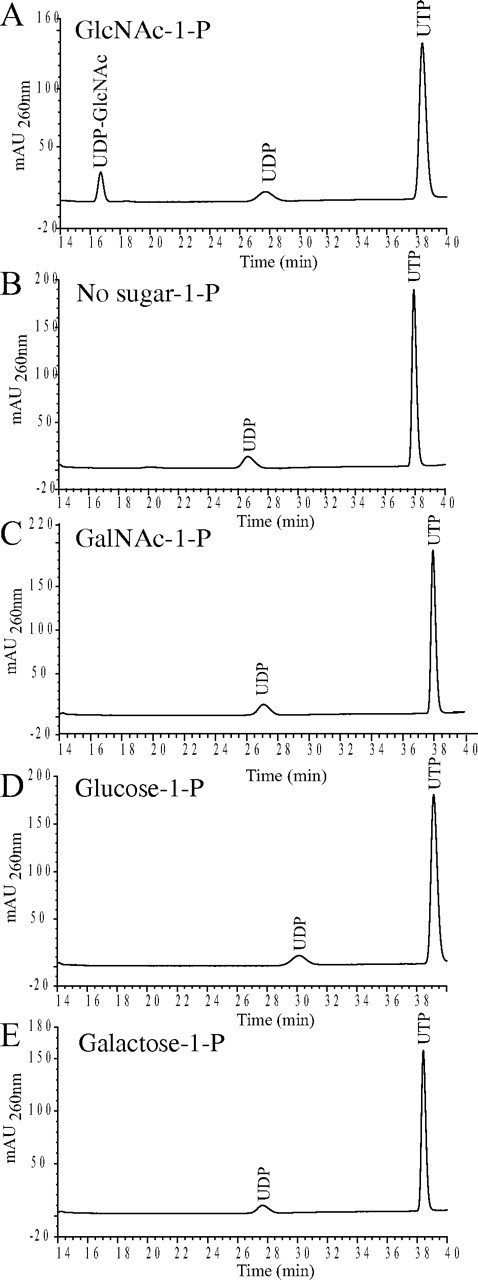
Substrate specificity of TbUAP. Recombinant TbUAP-His6 was incubated with UTP and different sugar-1-phosphate substrates, as indicated, and the products were analyzed by HPLC. A sugar nucleotide product (UDP-GlcNAc) was observed using GlcNAc-1-P (A) but not without GlcNAc-1-P (B) or with GalNAc-1-P, Glc 1-phosphate, or Gal 1-phosphate (C-E, respectively).
To test the metal ion dependence of the TbUAP-His6 activity, the enzyme was preincubated with 5 mm EDTA and then incubated with UTP, GlcNAc-1-P, and 10 mm MgCl2, MnCl2, CaCl2, CuCl2, ZnCl2, or no divalent cation. The products of triplicate experiments were analyzed by HPLC, and relative yields of UDP-GlcNAc were determined. In the presence of EDTA alone, no detectable UDP-GlcNAc was formed, suggesting that TbUAP is divalent metal ion-dependent. Similar levels of activity were restored with Mg2+ and Mn2+, whereas Ca2+, Cu2+, and Zn2+ failed to restore detectable activity (supplemental Fig. 5).
The pH dependence of the activity was studied over the pH range 5.0-9.5, using the same HPLC-based assay. The enzyme displayed a broad pH optimum between pH 6.0 and 9.0 (supplemental Fig. 6). Based on the aforementioned experiments, the enzyme was assayed at pH 7.5 in the presence of 10 mm MgCl2.
In order to measure the apparent Km values for the two substrates of TbUAP, a discontinuous, colorimetric, coupled assay was employed. The assay relies on pyrophosphatase to convert the PPi component of the UTP + GlcNAc-1-P → UDP-GlcNAc + PPi reaction to inorganic phosphate, which is subsequently measured using malachite green. The enzyme was assayed with a fixed concentration of UTP and varying concentrations of GlcNAc-1-P and with a fixed concentration of GlcNAc-1-P and varying concentrations of UTP (supplemental Figs. 7 and 8). The apparent Km values for UTP and GlcNAc-1-P are 26 ± 6 and 39 ± 13 μm, respectively, with a Vmax of 0.4 ± 0.1 nmol/min and a specific activity of 24 ± 6 μmol/min/mg. These values are compared with those of other recombinant eukaryotic UAPs (36-38) in Table 1.
TABLE 1.
Comparison of recombinant eukaryotic UAP properties
| Species | Molecular mass | GlcNAc-1-P Km | UTP Km | Specific activity | Alternative substrates | Reference/Source |
|---|---|---|---|---|---|---|
| kDa | μm | μm | μmol·min−1·mg− 1 | |||
| T. brucei (TbUAP) | 60.1 | 39 ± 13 | 26 ± 6 | 26 ± 6 | None | This study |
| G. intestinalis (GiUAP) | 49.6 | 300 ± 120 | 38 ± 17 | 9.2 ± 1.9, 55.4 ± 11.1 | Glc 1-phosphate, GalNAc-1-P | Ref. 38 |
| S. cerevisiae (ScUAP) | 53.5 | 14 | 21 | 17 | Glc 1-phosphate | Ref. 37 |
| H. sapiens (AgX1) | 57.0 | 5.3 | 53 | 69 | GalNAc-1-P | Ref. 36 |
| H. sapiens (AgX2) | 58.8 | 6.0 | 49 | 68 | GalNAc-1-P | Ref. 36 |
Construction of a TbUAP Conditional Null Mutant—In order to test the hypothesis that UDP-GlcNAc biosynthesis is essential for bloodstream form T. brucei and that there is no alternate route to this metabolite, a conditional null mutant of the final step of UDP-GlcNAc biosynthesis was created by the replacement of both endogenous copies of the TbUAP gene and the introduction of an ectopic inducible copy of TbUAP under tetracycline control. A genetically modified strain 427 T. brucei cell line was used to generate the conditional null mutant. This cell line is engineered to constitutively express T7 polymerase and tetracycline repressor protein under the control of a T7 promoter. The two transgenes are maintained under G418 selection (33). This cell line will be referred to as “wild type” from hereon.
The first TbUAP allele was replaced by homologous recombination following electroporation of the parasites in the presence of linear DNA containing a PAC gene flanked by about 500 bp of TbUAP 5′- and 3′-UTR. Following selection with puromycin, a ΔTbUAP:PAC clone was selected and transformed with an ectopic, tetracycline-inducible, copy of TbUAP, introduced into the ribosomal DNA locus under phleomycin selection (33). After tetracycline induction, the second endogenous allele was replaced by a HYG gene to yield the desired ΔTbUAP:PAC/TbUAPTi/ΔTbUAP: HYG clone (Fig. 2A). After each round of transformation, genomic DNA was extracted for Southern blot analysis using a TbUAP ORF probe and genomic DNA digested with BglII and NheI. Under these conditions, the endogenous TbUAP gene produces a fragment of ∼5.6 kb, whereas the ectopic copy produces a fragment of ∼2.5 kb. The blot (Fig. 2B) shows the successful introduction of the ectopic copy and replacement of both endogenous alleles in the ΔTbUAP:PAC/TbUA-PTi/ΔTbUAP:HYG clone used for further studies. This cell line will be referred to from hereon as the TbUAP conditional null mutant.
FIGURE 2.

Creation of the bloodstream form T. brucei TbUAP conditional null mutant. A, a schematic representation of the genetic transformations. The first endogenous allele of TbUAP was replaced with the PAC gene, creating ΔTbUAP:PAC. The ectopic copy (pLew100-TbUAP) was then inserted into the rDNA region, creating ΔTbUAP:PAC/TbUAPTi. The second allele of TbUAP was then replaced with the HYG gene, creating ΔTbUAP:PAC/TbUAPTi/ΔTbUAP:HYG. B, Southern blot of the TbUAP conditional null mutant and its intermediates. Genomic DNA from the wild type and three mutant cell lines produced during the creation of the TbUAP conditional null mutant was digested with BglII and NheI and probed with TbUAP open reading frame. Genomic DNA was from wild type (lane 1), ΔTbUAP:PAC (lane 2), ΔTbUAP:PAC/TbUAPTi (lane 3), and the final ΔTbUAP:PAC/TbUAPTi/ΔTbUAP:HYG conditional null mutant (lane 4).
The TbUAP Gene Is Essential to Bloodstream Form T. brucei in Vitro and in Vivo—Triplicate cultures of wild type and TbUAP conditional null mutant cells, under permissive and nonpermissive conditions (i.e. with and without tetracycline, respectively), were inoculated at 1 × 105 cells/ml and subcultured every 2 days. The TbUAP conditional null mutant cultures under permissive conditions had a reduced growth rate and grew to approximately half the cell density of the wild type but were otherwise healthy (Fig. 3, A and B). Under nonpermissive conditions the cells grew for 2 days but, upon subculturing, failed to grow for more than 5 days (Fig. 3C). The eventual resumption of growth is typical of bloodstream form T. brucei conditional null mutants for essential genes (6, 14, 18, 39-42), whereby the essential gene becomes constitutively active through loss of tetracycline control due to deletion of the tetracycline repressor protein gene (14). Analysis by reverse transcription-PCR confirmed that TbUAP mRNA was undetectable by 12 h of tetracycline removal but that the cells that resumed growth, 10 days after tetracycline removal, were reexpressing TbUAP mRNA (Fig. 3D).
FIGURE 3.

TbUAP expression is essential for the growth of bloodstream form T. brucei in culture. A, growth curves for wild type cells, subcultured every 2 days. B, growth curves for TbUAP conditional null mutant cells, subcultured every 2 days, grown under permissive (plus tetracycline) conditions. C, growth curves for TbUAP conditional null mutant cells grown under nonpermissive (minus tetracycline) conditions. D, ethidium bromide-stained agarose gel of reverse transcription-PCR products from RNA extracted from TbUAP conditional null mutant cells after 0, 2, 6, 12, and 48 h and 10 days without tetracycline, as indicated. The upper panel shows reverse transcription-PCR products using TbUAP primers, the middle panel is a control without reverse transcriptase, and the lower panel is a control using Dol-P-Man synthetase (DPMS) primers to show equal RNA input.
The TbUAP conditional null mutant cells were subcultured and grown for 24 h with and without tetracycline and then introduced into groups of five mice that were dosed with or without doxycycline in the drinking water. The animals dosed with the drug showed high blood parasitemias (>3 × 107/ml) within 4 days and were sacrificed, whereas the doxycycline-free animals did not show any signs of infection for up to 14 days, when the experiment was terminated. These data show that the TbUAP gene is essential to bloodstream form T. brucei both in vitro and in vivo.
Subcellular Localization of TbUAP—To obtain antibodies, TbUAP-His6 was used to inoculate mice and the resulting anti-serum was affinity-purified on a column of recombinant PreScission protease-treated (tag-free) TbUAP coupled to CNBr-Sepharose beads. The mouse anti-TbUAP affinity-purified antibody was used together with rabbit anti-GAPDH antibody as a glycosomal marker. The secondary antibodies were anti-mouse Alexa 594 (red) and anti-rabbit Alexa 488 (green). The fluorescence micrographs of wild type cells show that the anti-GAPDH co-localized with the anti-TbUAP, indicating that TbUAP is located in glycosome microbodies in bloodstream form T. brucei (Fig. 4, A-C). A similar experiment was performed with the TbUAP conditional null mutant grown with and without tetracycline for 48 h. The results show that the TbUAP signal co-localizes with the glyosomal GAPDH marker (as before) (Fig. 4, D-F) but that the intensity of the TbUAP signal is greatly reduced after 48 h without tretracycline (Fig. 4, G-I), demonstrating the strict specificity of the antibody for TbUAP in immunofluorescence microscopy.
FIGURE 4.

Subcellular localization of TbUAP. Wild type (A-C) and TbUAP conditional null mutant cells grown with tetracycline (D-F) and without tetracycline for 48 h (G-I) were stained with rabbit anti-GAPDH (a glycosomal marker) and Alexa 488 anti-rabbit (green channel; A, D, and G) and affinity-purified mouse anti-TbUAP and Alexa 594 anti-mouse (red channel; B, E, and H). Merged images are shown in C, F, and I. The merged image in I also includes a 4′,6-diamidino-2-phenylindole stain (blue) for DNA. No significant signals were obtained when primary antibodies were omitted (data not shown).
Sugar Nucleotide Levels in the TbUAP Conditional Null Mutant—To analyze the effect of the selective removal of TbUAP gene expression on parasite UDP-GlcNAc levels, sugar nucleotides were extracted from the TbUAP conditional null mutant under permissive and nonpermissive conditions, chromatographed as described in (35), and quantitated by the method described in Ref. 19. Briefly, sugar nucleotides were extracted from T. brucei, separated by reverse phase HPLC and quantitated by multiple reaction monitoring tandem mass spectrometry using an internal standard (GDP-glucose), a sugar nucleotide that is not found in trypanosomes. The multiple reaction monitoring approach exploits characteristic transitions between precursor and product ions to identify specific metabolites in complex mixtures. For example UDP-GlcNAc gives rise to an [M-H]- precursor ion at m/z 606 that fragments to produce a major product ion of [UDP-H2O]- at m/z 385. Thus, a chromatogram of the mass transition 606 → 385 is highly selective for UDP-GlcNAc. Similarly, GDP-Man and GDP-Glc can be monitored using the [GDP-Hex]- to [GDP-H2 O]- transition of m/z 604 → 424. Representative chromatograms (Fig. 5) illustrate the dramatic reduction in UDP-GlcNAc levels, relative to the GDP-Glc internal standard, in the TbUAP conditional null mutant after 48 h in the absence of tetracycline. These data and the effects on other sugar nucleotides are summarized in (Table 2). The levels of UDP-Glc, UDP-Gal, and GDP-Man in the TbUAP conditional null cell line under permissive conditions agree reasonably well with the wild type levels determined previously (19). However, even under permissive conditions, the level of UDP-GlcNAc is significantly lower in the conditional null mutant (16 pmol/1 × 107 cells) than in the wild type (80 pmol/1 × 107 cells). The reduced level may be because TbUAP expression is no longer under the control of its endogenous promoter but rather under the control of the procyclin promoter in the pLew100-TbUAP ectopic copy. Thus, the lower growth rate of the mutant under permissive conditions may be a result of the reduced level of UDP-GlcNAc. Under nonpermissive conditions, the cells stopped dividing between 48 and 60 h and died, by cell lysis, by around 72 h. At 48 h, the level of UDP-GlcNAc was 2.9 pmol/1 × 107, <5% of wild type levels.
FIGURE 5.

Measurement of sugar nucleotides in the TbUAP conditional null mutant under permissive and nonpermissive conditions. Representative liquid chromatography-tandem mass spectrometry chromatograms of sugar nucleotides extracted from the TbUAP conditional null mutant before (A) and after (B) withdrawal of tetracycline for 48 h. The upper chromatogram in each panel shows the peaks corresponding to GDP-Man and the GDP-Glc internal standard, and the lower chromatogram shows the peak corresponding to UDP-GlcNAc.
TABLE 2.
Sugar nucleotide levels in the TbUAP conditional null mutant under permissive and nonpermissive conditions
| Sugar nucleotide | Wild typea | TbUAP cKO (0 h)b | TbUAP cKO (48 h)b |
|---|---|---|---|
| pmol/107 cells | pmol/107 cells | pmol/107 cells | |
| UDP-GlcNAc | 80 ± 20 | 16 | 2.9 |
| GDP-Man | 5.6 ± 3.9 | 9.5 | 19 |
| UDP-Gal | 55 ± 3 | 44 | 35 |
| UDP-Glc | 123 ± 7 | 141 | 70 |
Sugar nucleotide levels for wild type T. brucei taken from Ref. 19.
Values are the means of two independent measurements.
Changes in the levels of other sugar nucleotides in the TbUAP conditional null mutant under nonpermissive conditions were less profound (Table 2). However, when the level of UDP-GlcNAc falls, there is an accumulation of GDP-Man. Since the consumption of GDP-Man for protein N-glycosylation and GPI anchor synthesis depends on the availability of UDP-GlcNAc, an accumulation of GDP-mannose is not unexpected. The reason for the reduction in UDP-Glc and UDP-Gal levels, to about 60% of wild type, is less clear. However, a comparable phenomenon was seen in the TbGalE (UDP-Glc 4′-epimerase) conditional null mutant, where rapid loss of UDP-Gal under nonpermissive conditions was followed by a reduction in UDP-Glc and UDP-GlcNAc levels (16).
Effects of UDP-GlcNAc Starvation on Protein Glycosylation—Ricin, which binds nonreducing terminal galactose residues, has been shown to bind to the flagellar pocket of T. brucei (43) and to the endosomal/lysosomal system of the parasite (27). The poly-N-acetyllactosamine-specific (Galβ1-4GlcNAc), lectin from Lycopersicon esculentum (tomato) has also been shown to bind exclusively to glycoproteins in the flagellar pocket and endosomal/lysosomal system in T. brucei (26). The ricin and tomato lectin binding oligosaccharide structures were characterized and were shown to include a family of unusually large N-linked poly-N-acetyllactosamine-containing glycans with an average of 54 N-acetyllactosamine repeats/glycan (27).
The effect of UDP-GlcNAc starvation on these structures was assessed using Western blots of whole cell lysates from wild type and the TbUAP conditional null mutant cell lines probed with tomato lectin. A large smear was detected in the wild type cell lysate, which decreased progressively from 0 to 48 h in the absence of tetracycline in the TbUAP conditional null mutant (Fig. 6A). The decrease in intensity, and a downward shift in apparent molecular weight, of the tomato lectin binding high molecular weight glycoproteins indicated a reduction in total poly-N-acetyllactosamine synthesis as the cellular levels of UDP-GlcNAc fall. The specificity of the tomato lectin blot for carbohydrate was confirmed by including chitin hydrolysate, a tomato lectin inhibitor (Fig. 6B). To distinguish whether this reduction in tomato lectin binding was specifically caused by UDP-GlcNAc starvation and not just a general phenomenon in dying cells, the same experiment was performed with a TbGPI12 conditional null mutant (6). This is another (lethal) glycosylation conditional null mutant but, this time, in the GPI biosynthetic pathway. Under nonpermissive (without tetracycline) conditions, this cell line also ceases cell division and dies, but, in this case, the cell lysate blots showed relatively little change in tomato lectin binding (supplemental Fig. 9).
FIGURE 6.

Tomato lectin blotting of wild type the TbUAP conditional null mutant extracts. A, extracts of wild type and TbUAP conditional null mutant (TbUAP cKO) cells 0, 24, and 48 h after tetracycline removal (as indicated) were subjected to SDS-PAGE, transferred to nitrocellulose, and probed with biotinylated tomato lectin followed by horseradish peroxidase-streptavidin. B, as in A, except that the tomato lectin inhibitor, chitin hydrolysate, was included with the lectin.
To observe the effects of UDP-GlcNAc starvation on a specific glycoprotein, wild type and TbUAP conditional null mutant cells, grown in the presence and absence of tetracycline for 24 and 48 h, were harvested, lysed, and analyzed by SDS-PAGE and Western blotting with the p67-specific monoclonal antibody MAb139 (supplemental Fig. 10). The intensity of the staining was slightly decreased, compared with wild type, in the TbUAP conditional null mutant under permissive conditions (0 h), which may reflect the overall difference in UDP-GlcNAc levels between the mutant and wild type cell line described earlier. After 48 h in the absence of tetracycline, both the intensity and the apparent molecular weight of the p67 smear had decreased. This result suggests that p67 glycosylation is impaired by UDP-GlcNAc starvation.
To further demonstrate that the poly-N-acetyllactosamine structures were being affected by UDP-GlcNAc starvation, wild type, and the TbUAP conditional null mutant cells, after 0, 24, and 48 h without tetracycline, were analyzed using fluorescence microscopy (Fig. 7, A-E). Cells were fixed onto coverslips and incubated with biotinylated tomato lectin and fluorescein-conjugated streptavidin. Using this approach, poly-N-acetyllactosamine structures specific to the flagellar pocket and endosomal/lysosomal region were visualized in the wild type cells (Fig. 7A). The specificity of this reaction was confirmed by including chitin hydrolysate as a tomato lectin inhibitor (Fig. 7E). Little difference in tomato lectin staining was seen in the TbUAP conditional null mutant cells after 24 h in the absence of tetracycline (Fig. 7, B and C), but after 48 h, almost no signal was seen within the flagellar pocket or endosomal/lysosomal region (Fig. 7D). These results correlate well with the tomato lectin blot data in (Fig. 6A). To assist in orienting the tomato lectin staining, results using wild type cells and including 4′,6-diamidino-2-phenylindole staining to highlight the positions of the nuclear and kinetoplast DNA, are included (Fig. 7, F-J).
FIGURE 7.

Fluorescence microscopy of tomato lectin binding to wild type and TbUAP conditional null mutant cells. Wild type (WT) and conditional null mutant (cKO) cells after 0, 24, and 48 h without tetracycline were visualized by phase-contrast and tomato lectin fluorescence microscopy, as indicated (A-E). Also shown are wild type cells stained with 4′,6-diamidino-2-phenylindole (DAPI) and tomato lectin (TL) (F-J).
To assess the effects of UDP-GlcNAc starvation on gross cellular morphology and flagellar pocket morphology, including the appearance of the flagellar pocket lumenal contents, wild type cells and TbUAP conditional null mutant cells, grown in the absence of tetracycline for 48 h, were subjected to scanning and transmission electron microscopy (supplemental Figs. 11 and 12, respectively). However, no significant differences in the images were recorded.
To observe the effects UDP-GlcNAc starvation had on VSG glycosylation, sVSG was purified from wild type and TbUAP conditional null mutant cells, grown in the presence and absence of tetracycline for 48 h. Analysis by SDS-PAGE and Coomassie Blue staining revealed that sVSG from wild type and TbUAP conditional null cells grown under permissive conditions appear as single bands, whereas sVSG from the TbUAP conditional null grown for 48 h in the absence of tetracycline appeared as a doublet (Fig. 8A). The formal possibility that the altered sVSG profile may have been caused by the cells switching VSG expression to a new VSG variant was eliminated by tryptic mass fingerprinting, by which both bands were positively identified as VSG221 (data not shown).
FIGURE 8.

Endoglycosidase digestions of sVSG221 from wild type and TbUAP conditional null mutant cells. A, sVSG221 purified from wild type and TbUAP conditional null mutant (TbUAP cKO) cells before (0 h) and after 48 h without tetracycline (48 h) were subjected to SDS-PAGE and Coomassie Blue staining. B, aliquots of wild type sVSG221 were digested with Endo H (that removes only oligomannose N-linked glycans) or PNGase F (that removes all N-linked glycans), and the products were subjected to SDS-PAGE and Coomassie Blue staining. Species containing two, one, and no N-linked glycans were resolved, as indicated. C, aliquots of sVSG221 from TbUAP conditional null cells grown for 48 h without tetracycline were digested with Endo H or PNGase F, and the products were subjected to SDS-PAGE and Coomassie Blue staining. Species containing two, one, and no N-linked glycans were resolved, as indicated.
Aliquots of sVSG from wild type cells and the TbUAP conditional null mutant grown for 48 h in the absence of tetracycline were digested with the enzymes Endo H and PNGase F and analyzed by SDS-PAGE and Coomassie Blue staining. As previously described (44), wild type sVSG showed a small shift in molecular weight when digested with Endo H, due to the removal of the C-terminal oligomannose N-linked glycan, and a greater shift when digested with PNGase F, due to the removal of both N-linked glycans (Fig. 8B). When the sVSG from the TbUAP conditional null mutant was digested with Endo H, the upper band collapsed into the lower band, and when digested with PNGase F, both bands collapsed into a fully deglycosylated form (Fig. 8C). These data suggest that the VSG doublet present in the mutant represents VSG molecules that are similar to those found in wild type (i.e. the upper band) and VSG molecules that have selectively lost an Endo H-sensitive N-linked glycan (i.e. the lower band).
To analyze this further, the sVSG samples were analyzed by electrospray mass spectrometry (Fig. 9). Wild type and TbUAP conditional null mutant cells grown under permissive conditions gave the expected glycoform mass ranges for sVSG221 (16, 44) (Table 3). However, sVSG from the TbUAP conditional null mutant grown under nonpermissive conditions for 48 h displayed two discrete sets of sVSG glycoforms, corresponding to the two bands seen by SDS-PAGE. One set was similar to that of wild type sVSG, except that it lacks the higher molecular weight glycoforms that contain five GlcNAc residues (Table 3). The other set of glycoforms have masses consistent with the absence of oligomannose structures at the C-terminal (Asn-428) N-glycosylation site (consistent with the Endo H resistance of the lower VSG band on SDS-PAGE). Analysis of the glycopeptide fraction of a Pronase digest of the mutant VSG sample, prepared and analyzed by electrospray tandem mass spectrometry according to Ref. 54, revealed a range of Asn-263 and Asn-428 N-linked glycopeptide and Ser-433 GPI glycopeptide species4 similar to those of wild type VSG (supplemental Fig. 13). These results rule out the possibility that lower molecular weight VSG glycoforms arise from changes at the N-linked and GPI glycosylation sites that happen to be equivalent in mass to the loss of the oligomannose structures from Asn-428.
FIGURE 9.

Electrospray mass spectrometry of sVSG221 from wild type and TbUAP conditional null mutant cells. Aliquots of sVSG221 from wild type cells (A) and TbUAP conditional null mutant cells grown in the presence of tetracycline (B) and the absence of tetracycline for 48 h (C) were analyzed by positive ion electrospray mass spectrometry, and the data were processed by Baysian protein reconstruction to produce mass graphs of isobaric glycoforms. Models of some of the principal glycoforms are indicated, and the compositions of the detected glycoforms are shown in Table 3.
TABLE 3.
Compositions of isobaric forms of sVSG
The measured masses (from Fig. 9) for sVSG221 samples from wild type (WT) and TbUAP conditional null cells grown with (cKO + Tet) or without tetracycline (cKO - Tet) for 48 h are tabulated in that order, followed by (in parentheses) the theoretical mass of the assigned VSG composition (Theo.). NA, not available. The abundance of each isobaric group of VSG molecules is indicated as follows: +++, mass peaks >70% of the biggest species; ++, mass peaks >40% of the biggest species; +, mass peaks <40% of the biggest species.
| Measured (and theoretical) molecular mass of WT/+Tet/−Tet (Theo.) | Proteina | GlcN-Ino-cPb | EtNPb | HexNAc | Hexose | WT | CKO + Tet | CKO − Tet |
|---|---|---|---|---|---|---|---|---|
| Da | ||||||||
| 51,542/51,546/NA (51,531) | 1 | 1 | 1 | 5 | 23 | + | + | − |
| 51,500/51,505/NA (51,490) | 1 | 1 | 1 | 4 | 24 | + | + | − |
| 51,379/51,384/NA (51,369) | 1 | 1 | 1 | 5 | 22 | + | + | − |
| 51,338/51,343/NA (51,328) | 1 | 1 | 1 | 4 | 23 | + | + | − |
| 51,215/51,219/NA (51,207) | 1 | 1 | 1 | 5 | 21 | + | + | − |
| 51,174/51,179/NA (51,166) | 1 | 1 | 1 | 4 | 22 | + | + | − |
| 51,053/51,058/NA (51,045) | 1 | 1 | 1 | 5 | 20 | + | + | − |
| 51,012/51,017/51,016 (51,004) | 1 | 1 | 1 | 4 | 21 | + | + | + |
| 50,891/50,897/50,895 (50,883) | 1 | 1 | 1 | 5 | 19 | + | + | − |
| 50,850/50,856/50,854 (50,842) | 1 | 1 | 1 | 4 | 20 | +++ | +++ | +++ |
| 50,688/50,694/50,691 (50,680) | 1 | 1 | 1 | 4 | 19 | ++ | ++ | ++ |
| 50,526/50,531/50,530 (50,518) | 1 | 1 | 1 | 4 | 18 | + | + | + |
| 50,364/50,369/50,368 (50,356) | 1 | 1 | 1 | 4 | 17 | + | + | + |
| NA/NA/49,311 (49,302) | 1 | 1 | 1 | 2 | 13 | − | − | + |
| NA/NA/49,149 (49,140) | 1 | 1 | 1 | 2 | 12 | − | − | +++ |
| NA/NA/48,987 (48,978) | 1 | 1 | 1 | 2 | 11 | − | − | +++ |
| NA/NA/48,826 (48,816) | 1 | 1 | 1 | 2 | 10 | − | − | + |
| NA/NA/48,663 (48,654) | 1 | 1 | 1 | 2 | 9 | − | − | + |
| NA/NA/48,501 (48,492) | 1 | 1 | 1 | 2 | 8 | − | − | + |
The average molecular weight of sVSG221 polypeptide (46,284 Da) minus amino acids 1-27 (signal peptide) and residues 460-476 (GPI attachment signal sequence) with four disulphide bonds (44).
Components of GPI common to all glycoforms of sVSG221: GlcN-Ino-cP, glucosamine-α1-6-myo-inositol-1,2-cyclic phosphate; EtNP, ethanolamine phosphate.
Taken together, these data show that under UDP-GlcNAc starvation, the elaboration of the Man3GlcNAc2 and Man4GlcNAc2 glycans at the fully occupied Asn-263 N-glycosylation site glycans with GlcNAc and Galβ1-4GlcNAc is significantly reduced and that the occupancy of the C-terminal Asn-428 site is selectively and dramatically reduced.
DISCUSSION
Whereas some enzymes of carbohydrate metabolism in T. brucei are closer to their prokaryote counterparts (45, 46), this is not the case for TbUAP, which has 31% sequence identity and 50% sequence similarity to its human counterpart and, unlike its bacterial counterpart, glmU, is not fused to a glucosamine-1-phosphate N-acetyltransferase domain (47).
Analysis of the recombinant UAP from T. brucei demonstrated that TbUAP is a conventional divalent cation-dependent pyrophosphorylase with kinetic parameters similar to those reported for other eukaryotic UAPs. However, TbUAP differs from most other UAPs in that it is highly selective for its sugar phosphate substrate, accepting only GlcNAc 1-phosphate, and that it is compartmentalized in a microbody, the glycosome.
With respect to the former, the inability of TbUAP to accept GalNAc-1-P reflects the fact that T. brucei, like all of the trypanosomatids, does not incorporate GalNAc into its glycoconjugates (reviewed in Ref. 19). Thus, the organism has no need to make UDP-GalNAc either from GalN or GalNAc or by epimerization of UDP-GlcNAc. Accordingly, both TbUAP (this study) and TbGalE (15) are exclusively GlcNAc-1-phosphate- and UDP-Glc/Gal-specific, respectively. This is unlike their mammalian counterparts that can also utilize GalNAc-1-P (36) and UDP-GlcNAc/GalNAc (48), respectively. Comparison of the TbUAP sequence with those of the human AgX1 and AgX2 UAP splice variants, for which there are crystal structures (36), indicates that TbUAP has a conserved active site, containing only a 2-amino acid difference among the 15 identified substrate-interacting residues (i.e. Arg-313 in place of Pro-288 and Ala-489 in place of Lys-455). Peneff et al. (36) reported that the equatorial GlcNAc C4 hydroxyl of UDP-GlcNAc forms one hydrogen bond with Gly-290 and one with Asn-327 of AgX1 and that the axial GalNAc C4 hydroxyl of UDP-GalNAc forms two hydrogen bonds with Asn-327. The equivalent residues in TbUAP are Gly-315 and Asn-352. However, the context of the key Asn residue (underlined) in TbUAP is KFNCANISSNLC whereas in the AgX enzymes it is LFNAGNIANHFF. Nonequivalent adjacent residues (in italic type), particularly the bulkier CA versus AG sequence immediately before the Asn residue, may affect the latter hydrogen bond network and account for the observed selectivity of TbUAP for GlcNAc-1-P over GalNAc-1-P. We are attempting to crystallize TbUAP to resolve this issue.
With respect to the glycosomal, rather than cytosolic, location of the native enzyme, a recent computational search of the T. brucei, T. cruzi, and Leishmania major genomes did not identify their UAPs as glycosomal enzymes (45). However, that study used the PTS1 Prosite pattern PS00342 of (STA-GCN)-(RKH)-(LIVMAFY)$ (where $ denotes the end of the protein) supplemented with the pattern S-S-(LIF)$. Thus, the TbUAP C-terminal tripeptide sequence, SNM$, did not pass this filter. On the other hand, the same authors noted that the most abundant residues, in order of frequency, in T. brucei PTS1 sequences are (SAGPYN)-(KHRNQ)-(LM-AVY)$, indicating that although the SNM$ permutation has not been identified previously as a T. brucei PTS1 sequence, it appears to be one. Significantly, the C-terminal tripeptide sequences of the T. cruzi and L. major UAPs are GNM$ and ANM$, respectively, which also fit the aforementioned potential PTS1 permutations. It remains to be determined experimentally whether a C-terminal (SGA)-NM$ sequence is sufficient for glycosomal import. The alternative is that TbUAP is targeted to the glycosome through association with another PTS1 or PTS2 targeted protein, as has been described previously for peroxisomal import (49).
In all trypanosomatids, the two-step conversion of glucose to fructose 6-phosphate, via hexokinase and glucose-6-phosphate isomerase, occurs in the glycosome, and it is conceivable that the entire UDP-GlcNAc biosynthetic pathway is located in this organelle. Although glucosamine-fructose-6-phosphate transaminase, glucosamine-phosphate N-acetyltransferase, and phosphoacetylglucosamine mutase lack obvious PTS1 or PTS2 sequences, the T. cruzi and L. major glucosamine-6-phosphate deaminase sequences do contain PTS1 sequences (46). This enzyme catalyzes the reverse reaction to glucosamine-fructose-6-phosphate transaminase. Our current hypothesis, which we are currently testing, is that glucosamine-fructose-6-phosphate transaminase, glucosamine-phosphate N-acetyltransferase, and phosphoacetylglucosamine mutase are piggybacked into the glycosome via oligomerization with glucose-6-phosphate isomerase and/or glucosamine-6-phosphate deaminase and/or TbUAP to provide functional UDP-GlcNAc synthesis machinery in a single location. Indeed, it may be that all sugar nucleotide biosynthesis occurs in this location in trypanosomes, since both UDP-Glc 4′-epimerase and GDP-Man 4,6-dehydratase, required for in UDP-Gal and GDP-Fuc synthesis, respectively, have also been shown to be glycosomal in T. brucei (15, 18). A glycosomal location for the synthesis of sugar nucleotides further suggests that there may be specific transporters or antiporters in the glycosome membrane to, for example, exchange UTP for UDP-sugars and GTP for GDP-sugars or NDP-sugars for PPi. In this regard, it is worth noting that, whereas most sugar nucleotide antiporters exchange NDP-sugars for NMPs (50) (requiring nucleoside diphosphatases to convert NDPs to NMPs), NDP-sugar/NDP and NDP-sugar/NDP-sugar antiporter activities have also been recently reported (51).
The phenotype generated through UDP-GlcNAc starvation with regard to tomato lectin binding is similar to that caused by UDP-Gal starvation (16) (i.e. a reduction in lectin binding in Western blots and fluorescence microscopy), indicating a reduction in both size and quantity of the giant poly-LacNAc structures normally found throughout the flagellar pocket and endosomal/lysosomal system (26, 27). This is not a general phenomenon of dying trypanosomes. For example, tomato lectin blots remain unchanged for a TbGPI12 conditional null mutant (6) under nonpermissive conditions (supplemental Fig. 9). Transmission electron microscopy images of gold-conjugated ricin binding in the flagellar pocket had suggested that the bloodstream form-specific fibrous material in the flagellar pocket might correspond to ricin-binding glycoconjugates (43). However, although a significant reduction in tomato lectin binding was observed in this study, transmission electron microscopy images did not show a significant reduction in the fibrous material when compared with wild type cells. This suggests that the fibrous material is either unrelated to the poly-LacNAc-containing glycoproteins of the flagellar pocket or that the fibrous appearance of the glycoproteins in transmission electron microscopy is unaffected by reducing their poly-LacNAc content. Hopefully, future studies on the composition of the lumenal contents of the flagellar pocket will resolve this issue.
Another way of assessing the glycosylation phenotype of bloodstream form T. brucei mutants is to use the abundant VSG as a reporter and to assess the status of its two N-glycosylation sites and GPI anchor (16, 44). In the case of UDP-Gal starvation, the copy number and integrity of the VSG coat is not affected, although the GPI anchors, which normally sport an average of five Gal side chain residues, are free of galactose (16). The effect on VSG glycosylation was very different under UDP-GlcNAc starvation. Mass spectrometry and endoglycosidase digestion of VSG221 isolated from the TbUAP conditional null mutants under nonpermissive conditions revealed the presence of two major species of VSG221 in approximately equal amounts. One form was almost indistinguishable from wild-type VSG221, whereas the other, lower molecular mass form, specifically lacked the C-terminal (Asn-428) N-linked glycan. From a hierarchical point of view, it is not surprising that GPI anchor synthesis is maintained, even when protein N-glycosylation and N-glycan elaboration are affected, since GPI synthesis and transfer to protein are clearly essential to bloodstream form T. brucei (5-8). Furthermore, from a structural point of view, the Asn-263 N-linked glycan may be more important to the correct folding of the VSG. Blum et al. (52) analyzed the crystal structures of two VSG variants, VSG ILTat.1.24 and VSG221, and found that a short α-helix in ILTat.1.2 is absent in VSG221 but replaced by the protein-proximal sugars of the Asn-263 N-linked glycan. However, this does not provide a mechanistic explanation for the hierarchy of Asn-263 N-glycosylation in strict preference to the N-glycosylation of Asn-428 under UDP-GlcNAc starvation. Radiolabeling studies suggest that bloodstream form T. brucei contain very low steady-state levels of Man9GlcNAc2-PP-dolichol and a relatively large pool of Man5GlcNAc2-PP-dolichol (53). Despite their differences in abundance, the organism appears to use both of these oligosaccharyl-PP-dolichol species for transfer to protein, the former to sites destined to contain oligomannose glycans and the latter to sites destined to become complex type structures (44). In most eukaryotes, a single type of dolichol-PP-linked oligosaccharide precursor (Glc3Man9GlcNAc2-PP-dolichol) is transferred to the nascent peptide in the ER through the action of oligosaccharyltransferase (OST), a complex of typically eight different proteins (54). Trypanosomatids lack candidate genes for all but the catalytic subunit, STT3, of this complex. However, there are three copies of the TbSTT3 gene in the T. brucei genome. Two of the TbSTT3 genes encode almost identical proteins, and we speculate that the unique TbSTT3 utilizes either exclusively Man5GlcNAc2-PP-dolichol or Man9GlcNAc2-PP-dolichol and that one or both of the two similar TbSTT3s utilizes the other donor. Under UDP-GlcNAc starvation, GDP-Man levels rise slightly, and presumably Dol-P-Man is also present at greater than normal levels. From this point of view, one might expect the ratio of Man9GlcNAc2-PP-dolichol/Man5GlcNAc2-PP-dolichol to increase slightly. However, the absolute levels of both precursors are presumably significantly reduced as UDP-GlcNAc becomes limiting for the synthesis of the common GlcNAc2-PP-dolichol core. Thus, the simplest explanation for the selective loss of the C-terminal oligomannose N-linked glycans from VSG221 under UDP-GlcNAc starvation is that the effective concentration of Man9GlcNAc2-PP-dolichol falls so far below the effective Km for the relevant TbSTT3 that most VSG molecules appearing in the ER fail to receive a C-terminal N-glycan. Comparative studies on the substrate specificities and kinetic parameters of the two classes of TbSTT3 should resolve this issue. Meanwhile, additional support for such a dual oligosaccharyltransferase model comes from the analysis of a T. brucei ALG3 mutant that can only make Man5GlcNAc2 and that also underglycosylates the C-terminal Asn-428 site of VSG221 (55) and from pulse-chase studies that show, in at least two different VSGs, that Endo H-sensitive oligomannose sites (but not Endo H-resistant sites) can be N-glycosylated post-translationaly (30, 56).
Finally, through the creation of the conditional null mutant, TbUAP was shown to be essential for the growth and survival of bloodstream T. brucei in culture, suggesting that this enzyme might be considered as a potential therapeutic target for human African sleeping sickness. Further in vivo support for this notion was obtained when the TbUAP conditional null was shown to be infectious to mice dosed with doxycycline in their drinking water but not to doxycycline-free mice. Although the essentiality of UDP-GlcNAc biosynthesis via TbUAP was expected, experimental validation in vitro and in vivo is, nevertheless, important, since it rules out any possible metabolic and/or nutritional bypasses. Compared with other conditional null mutants generated in this laboratory, the ablation of UDP-GlcNAc synthesis led to more rapid killing of the parasite (<72 h) than ablation of either UDP-Gal synthesis (14, 16) or GPI biosynthesis (6), which both take 4-5 days. The rapidity of cell death presumably reflects the fact that UDP-GlcNAc is simultaneously required for GPI anchor biosynthesis, core protein N-glycosylation, and the subsequent decoration of complex N-glycans with lactosamine units. Of course, UDP-GlcNAc is also essential to mammalian cells (57), and selective inhibition of the parasite UAP would be a therapeutic requirement.
Supplementary Material
Acknowledgments
We thank Adel Ibrahim (Dundee College of Life Sciences Cloning Service) for making the PreScission TbUAP construct, John James and Martin Kierans (Dundee Centre for High-Resolution Imaging and Processing) for electron and light microscopy, Doug Lamont and Kenny Beattie for proteomics and mass spectrometry support, and Jay Bangs and Paul Michels for kindly providing antibodies.
Author's Choice—Final version full access.
This work was supported in part by Wellcome Trust Programme Grant 071463. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1-13.
Footnotes
The abbreviations used are: GPI, glycosylphosphatidylinositol; ER, endoplasmic reticulum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Lac-NAc, N-acetyllactosamine; PBS, phosphate-buffered saline; Fuc, fucose; ORF, open reading frame; GlcNAc-1-P, GlcNAc 1-phosphate; GalNAc-1-P, GalNAc 1-phosphate; UTR, untranslated region; TLCK, 1-chloro-3-tosylamido-7-amino-2-heptone; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; MOPS, 4-morpholinepropanesulfonic acid; UAP, uridine-acetylglucosamine pyrophosphorylase.
The range of GPI glycoforms in wild type sVSG221 contain up to nine hexose residues (i.e. up to Gal6Man3 (44, 55)). However, in the case of the TbUAP conditional null sVSG221, GPI glycoforms that contain up to 10 hexose residues were detected (see supplemental Fig. 13). It is possible that the lack of a C-terminal N-glycan in about half of the VSG molecules in this preparation allows some additional processing of VSG-linked GPIs up to Gal7Man3.
References
- 1.Ferguson, M. A. (1999) J. Cell Sci. 112 2799-2809 [DOI] [PubMed] [Google Scholar]
- 2.Guha-Niyogi, A., Sullivan, D. R., and Turco, S. J. (2001) Glycobiology 11 45R-59R [DOI] [PubMed] [Google Scholar]
- 3.McConville, M. J., Mullin, K. A., Ilgoutz, S. C., and Teasdale, R. D. (2002) Microbiol. Mol. Biol. Rev. 66 122-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendonca-Previato, L., Todeschini, A. R., Heise, N., and Previato, J. O. (2005) Curr. Opin. Struct. Biol. 15 499-505 [DOI] [PubMed] [Google Scholar]
- 5.Nagamune, K., Nozaki, T., Maeda, Y., Ohishi, K., Fukuma, T., Hara, T., Schwarz, R. T., Sutterlin, C., Brun, R., Riezman, H., and Kinoshita, T. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 10336-10341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang, T., Milne, K. G., Guther, M. L., Smith, T. K., and Ferguson, M. A. (2002) J. Biol. Chem. 277 50176-50182 [DOI] [PubMed] [Google Scholar]
- 7.Lillico, S., Field, M. C., Blundell, P., Coombs, G. H., and Mottram, J. C. (2003) Mol. Biol. Cell 14 1182-1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith, T. K., Crossman, A., Brimacombe, J. S., and Ferguson, M. A. (2004) EMBO J. 23 4701-4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garami, A., and Ilg, T. (2001) EMBO J. 20 3657-3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garami, A., and Ilg, T. (2001) J. Biol. Chem. 276 6566-6575 [DOI] [PubMed] [Google Scholar]
- 11.Garami, A., Mehlert, A., and Ilg, T. (2001) Mol. Cell. Biol. 21 8168-8183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis, A. J., Perugini, M. A., Smith, B. J., Stewart, J. D., Ilg, T., Hodder, A. N., and Handman, E. (2004) J. Biol. Chem. 279 12462-12468 [DOI] [PubMed] [Google Scholar]
- 13.Stewart, J., Curtis, J., Spurck, T. P., Ilg, T., Garami, A., Baldwin, T., Courret, N., McFadden, G. I., Davis, A., and Handman, E. (2005) Int. J. Parasitol. 35 861-873 [DOI] [PubMed] [Google Scholar]
- 14.Roper, J. R., Guther, M. L., Milne, K. G., and Ferguson, M. A. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 5884-5889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roper, J. R., Guther, M. L., Macrae, J. I., Prescott, A. R., Hallyburton, I., Acosta-Serrano, A., and Ferguson, M. A. (2005) J. Biol. Chem. 280 19728-19736 [DOI] [PubMed] [Google Scholar]
- 16.Urbaniak, M. D., Turnock, D. C., and Ferguson, M. A. (2006) Eukaryot. Cell 5 1906-1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacRae, J. I., Obado, S. O., Turnock, D. C., Roper, J. R., Kierans, M., Kelly, J. M., and Ferguson, M. A. (2006) Mol. Biochem. Parasitol. 147 126-136 [DOI] [PubMed] [Google Scholar]
- 18.Turnock, D. C., Izquierdo, L., and Ferguson, M. A. (2007) J. Biol. Chem. 282 28853-28863 [DOI] [PubMed] [Google Scholar]
- 19.Turnock, D. C., and Ferguson, M. A. (2007) Eukaryot. Cell 6 1450-1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berninsone, P. M., and Hirschberg, C. B. (2000) Curr. Opin. Struct. Biol. 10 542-547 [DOI] [PubMed] [Google Scholar]
- 21.Gerardy-Schahn, R., Oelmann, S., and Bakker, H. (2001) Biochimie (Paris) 83 775-782 [DOI] [PubMed] [Google Scholar]
- 22.Previato, J. O., Jones, C., Xavier, M. T., Wait, R., Travassos, L. R., Parodi, A. J., and Mendonca-Previato, L. (1995) J. Biol. Chem. 270 7241-7250 [DOI] [PubMed] [Google Scholar]
- 23.Serrano, A. A., Schenkman, S., Yoshida, N., Mehlert, A., Richardson, J. M., and Ferguson, M. A. (1995) J. Biol. Chem. 270 27244-27253 [DOI] [PubMed] [Google Scholar]
- 24.Doering, T. L., Masterson, W. J., Englund, P. T., and Hart, G. W. (1989) J. Biol. Chem. 264 11168-11173 [PubMed] [Google Scholar]
- 25.Zamze, S. E., Ashford, D. A., Wooten, E. W., Rademacher, T. W., and Dwek, R. A. (1991) J. Biol. Chem. 266 20244-20261 [PubMed] [Google Scholar]
- 26.Nolan, D. P., Geuskens, M., and Pays, E. (1999) Curr. Biol. 9 1169-1172 [DOI] [PubMed] [Google Scholar]
- 27.Atrih, A., Richardson, J. M., Prescott, A. R., and Ferguson, M. A. (2005) J. Biol. Chem. 280 865-871 [DOI] [PubMed] [Google Scholar]
- 28.Treumann, A., Zitzmann, N., Hulsmeier, A., Prescott, A. R., Almond, A., Sheehan, J., and Ferguson, M. A. (1997) J. Mol. Biol. 269 529-547 [DOI] [PubMed] [Google Scholar]
- 29.Nagamune, K., Acosta-Serrano, A., Uemura, H., Brun, R., Kunz-Renggli, C., Maeda, Y., Ferguson, M. A., and Kinoshita, T. (2004) J. Exp. Med. 199 1445-1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferguson, M. A., Duszenko, M., Lamont, G. S., Overath, P., and Cross, G. A. (1986) J. Biol. Chem. 261 356-362 [PubMed] [Google Scholar]
- 31.Guther, M. L., Lee, S., Tetley, L., Acosta-Serrano, A., and Ferguson, M. A. (2006) Mol. Biol. Cell 17 5265-5274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azema, L., Claustre, S., Alric, I., Blonski, C., Willson, M., Perie, J., Baltz, T., Tetaud, E., Bringaud, F., Cottem, D., Opperdoes, F. R., and Barrett, M. P. (2004) Biochem. Pharmacol. 67 459-467 [DOI] [PubMed] [Google Scholar]
- 33.Wirtz, E., Leal, S., Ochatt, C., and Cross, G. A. (1999) Mol. Biochem. Parasitol. 99 89-101 [DOI] [PubMed] [Google Scholar]
- 34.Tomiya, N., Ailor, E., Lawrence, S. M., Betenbaugh, M. J., and Lee, Y. C. (2001) Anal. Biochem. 293 129-137 [DOI] [PubMed] [Google Scholar]
- 35.Rabina, J., Maki, M., Savilahti, E. M., Jarvinen, N., Penttila, L., and Renkonen, R. (2001) Glycoconj. J. 18 799-805 [DOI] [PubMed] [Google Scholar]
- 36.Peneff, C., Ferrari, P., Charrier, V., Taburet, Y., Monnier, C., Zamboni, V., Winter, J., Harnois, M., Fassy, F., and Bourne, Y. (2001) EMBO J. 20 6191-6202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mio, T., Yabe, T., Arisawa, M., and Yamada-Okabe, H. (1998) J. Biol. Chem. 273 14392-14397 [DOI] [PubMed] [Google Scholar]
- 38.Mok, M. T., and Edwards, M. R. (2005) J. Biol. Chem. 280 39363-39372 [DOI] [PubMed] [Google Scholar]
- 39.Krieger, S., Schwarz, W., Ariyanayagam, M. R., Fairlamb, A. H., Krauth-Siegel, R. L., and Clayton, C. (2000) Mol. Microbiol. 35 542-552 [DOI] [PubMed] [Google Scholar]
- 40.Milne, K. G., Guther, M. L., and Ferguson, M. A. (2001) Mol. Biochem. Parasitol. 112 301-304 [DOI] [PubMed] [Google Scholar]
- 41.Martin, K. L., and Smith, T. K. (2006) Biochem. J. 396 287-295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin, K. L., and Smith, T. K. (2006) Mol. Microbiol. 61 89-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brickman, M. J., and Balber, A. E. (1990) J. Protozool. 37 219-224 [DOI] [PubMed] [Google Scholar]
- 44.Jones, D. C., Mehlert, A., Guther, M. L., and Ferguson, M. A. (2005) J. Biol. Chem. 280 35929-35942 [DOI] [PubMed] [Google Scholar]
- 45.Opperdoes, F. R., and Szikora, J. P. (2006) Mol. Biochem. Parasitol. 147 193-206 [DOI] [PubMed] [Google Scholar]
- 46.Opperdoes, F. R., and Michels, P. A. (2007) Trends Parasitol. 23 470-476 [DOI] [PubMed] [Google Scholar]
- 47.Mengin-Lecreulx, D., and van Heijenoort, J. (1994) J. Bacteriol. 176 5788-5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thoden, J. B., Wohlers, T. M., Fridovich-Keil, J. L., and Holden, H. M. (2001) J. Biol. Chem. 276 15131-15136 [DOI] [PubMed] [Google Scholar]
- 49.Titorenko, V. I., Nicaud, J. M., Wang, H., Chan, H., and Rachubinski, R. A. (2002) J. Cell Biol. 156 481-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caffaro, C. E., and Hirschberg, C. B. (2006) Acc. Chem. Res. 39 805-812 [DOI] [PubMed] [Google Scholar]
- 51.Muraoka, M., Miki, T., Ishida, N., Hara, T., and Kawakita, M. (2007) J. Biol. Chem. 282 24615-24622 [DOI] [PubMed] [Google Scholar]
- 52.Blum, M. L., Down, J. A., Gurnett, A. M., Carrington, M., Turner, M. J., and Wiley, D. C. (1993) Nature 362 603-609 [DOI] [PubMed] [Google Scholar]
- 53.Low, P., Dallner, G., Mayor, S., Cohen, S., Chait, B. T., and Menon, A. K. (1991) J. Biol. Chem. 266 19250-19257 [PubMed] [Google Scholar]
- 54.Kelleher, D. J., and Gilmore, R. (2006) Glycobiology 16 47R-62R [DOI] [PubMed] [Google Scholar]
- 55.Manthri, S., Guther, M. L., Izquierdo, L., Acosta-Serrano, A., and Ferguson, M. A. (2008) Glycobiology 18 367-383 [DOI] [PubMed] [Google Scholar]
- 56.Bangs, J. D., Doering, T. L., Englund, P. T., and Hart, G. W. (1988) J. Biol. Chem. 263 17697-17705 [PubMed] [Google Scholar]
- 57.Boehmelt, G., Wakeham, A., Elia, A., Sasaki, T., Plyte, S., Potter, J., Yang, Y., Tsang, E., Ruland, J., Iscove, N. N., Dennis, J. W., and Mak, T. W. (2000) EMBO J. 19 5092-5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.